

Abstract
Neuronal interleukin-34 (IL-34) promotes the expansion of microglia in the central nervous system—microglial activation and expansion are in turn implicated in the pathogenesis of Huntington‘s disease (HD). We thus examined whether the accumulation of an amyloidogenic exon-1 fragment of mutant huntingtin (mHTTx1) modulates the expression of IL-34 in dopaminergic neurons derived from a human embryonic stem cell line. We found that mHTTx1 aggregates induce IL-34 production selectively in post-mitotic neurons. Exposure of neurons to DNA damaging agents or the excitotoxin NMDA elicited similar results suggesting that IL-34 induction may be a general response to neuronal stress including the accumulation of misfolded mHTTx1. We further determined that knockdown or blocking the activity of IκB kinase beta (IKKβ) prevented the aggregation of mHTTx1 and subsequent IL-34 production. While elevated IL-34 itself had no effect on the aggregation or the toxicity of mHTTx1 in neuronal culture, IL-34 expression in a rodent brain slice model with intact neuron-microglial networks exacerbated mHTTx1-induced degeneration of striatal medium-sized spiny neurons. Conversely, an inhibitor of the IL-34 receptor reduced microglial numbers and ameliorated mHTTx1-mediated neurodegeneration. Together, these findings uncover a novel function for IKKβ/mHTTx1 interactions in regulating IL-34 production, and implicate a role for IL-34 in non-cell-autonomous, microglial-dependent neurodegeneration in HD.
Introduction
Huntington's disease (HD) is an inherited neurodegenerative disorder caused by expansion of a CAG repeat in the exon-1 of IT-15 gene, which translates into an abnormal polyglutamine (polyQ) domain in the huntingtin protein (HTT) (1). While the exact mechanisms by which polyQ expansion in HTT promotes clinical disease remain incompletely understood, mHTT is proteolytically cleaved and ultimately generates an exon-1 (mHTTx1) fragment, which is amyloidogenic (2). Aberrant splicing of HTT mRNA also contributes to the production and accumulation of mHTTx1 in patients and in animal models (3). Mutant HTTx1 is neurotoxic in experimental settings and produces severe HD-like symptoms in a transgenic mouse model of HD (4). The toxic functions of mHTTx1 include aberrant activation of various signaling pathways, altered gene expression, and neurodegeneration (2,5).
Recent studies suggest that mHTTx1 may also induce the proliferation and activation of microglia (6). Microglia are the principal resident CNS immune cells and serve many beneficial functions including defense against pathogens, producing regulatory and neurotrophic factors, and modulating synaptogenesis (7,8). However, persistent microglial activation in HD may exacerbate neurodegeneration. Microglia are activated in the brains of pre-manifest HD patients and their presence coincides with the loss of neurons in various brain regions (9–11). Indeed, activation of microglia has been used to predict the age of disease onset in the pre-manifest HD patients (11). Pre-symptomatic HD patients also display elevated levels of proinflammatory cytokines including TNF-α and IL-6 in the circulation and in the cerebrospinal fluid (CSF), which is suggestive of systemic inflammation (12). Moreover, microglial activation in the CNS of HD patients is linked to the elevation of inflammatory cytokines in the circulation thus supporting defects in the neuro-immune signaling pathways (13).
While the molecular circuits influencing the activation and expansion of microglia and neuroinflammation in HD are poorly understood, interleukin-34 (IL-34) is known to be essential for the proliferation of microglia and other immune cells (14,15). In the CNS, IL-34 is produced and secreted by neurons, and subsequently binds to and activates colony stimulating-factor 1 receptor (CSF1R) predominantly expressed on the surface of microglia (16). Deletion of IL-34 in the CNS of rodents significantly reduces microglial numbers and ameliorates virus-induced synapse loss and cognitive impairment (14,15,17). Conversely, persistent production of IL-34 causes pathological expansion of microglia and macrophages (18,19).
Neuronal IB Kinase (IKK)/NF-κB pathway regulates neuron-microglial communication in rodents (20). Aberrant activation of the (IKK)/NF-κB has also been implicated in the neurotoxicity of mHTT (21,22). Mutant HTT activates the IKK/NF-κB pathway in animal models of HD and in immune cells of HD patients promoting the expression of pro-inflammatory cytokines (12,21,23). Modules of the IKK complex, which consists of IKKα, IKKβ, and IKKγ, also have NF-κB-independent functions (24). In the context of HD, IKKβ promotes proteolytic cleavage of mHTT thus generating amyloidogenic N-terminal fragments (25). Conversely, inhibition of IKKβ ameliorates the neurotoxicity of mHTT in rodent brain slice and animal models of HD (21,26,27).
Here, we report the novel finding that mHTTx1 aggregation activates the expression of neuronal IL-34. Inhibition of IKKβ blocks mHTTx1 aggregation and thereby prevents mHTTx1-driven production of IL-34 in dopaminergic human neurons. In addition, we provide evidence that elevated levels of IL-34 exacerbate mHTTx1-induced neurodegeneration of striatal medium spiny neurons (MSNs) in a rodent brain tissue model, whereas inhibition of the receptor for IL-34 is protective. Together, these results suggest a central role for neuronal IKKβ/mHTTx1 interactions in regulating a non-cell-autonomous, IL-34-mediated neurodegenerative pathway in HD.
Results
Mutant HTTx1 and chemical stressors induce IL-34 expression in human dopaminergic neurons derived from embryonic stem cells
To determine whether the expression of mHTTx1 may be sufficient to increase the production of IL-34, we engineered a human embryonic mesencephalic stem cell line (MESC2.10) to express HTTx1-EGFP with 25 or 103Qs (Supplementary Material, Fig. S1). The MESC2.10 line was originally isolated from the midbrain of an 8-week-old human embryo and subsequently transduced with a tet-off v-myc to prevent senescence. Shutdown of v-myc and addition of growth factors in the medium generates fully functional dopaminergic neurons within ∼6 days post-differentiation (DPD) (28). We found that the transcription of IL-34 is minimal in proliferating neuronal progenitor cells (NPCs) but slightly increases over time in developing neurons (Fig. 1A). The levels of IL-34 maximize at ∼ 8 days post differentiation (DPD). This is consistent with previous findings that the CNS IL-34 is predominantly expressed by neurons (14,15).
Figure 1.

Induction of IL-34 by mHTTx1 is mimicked by neuronal chemical stressors. (A) NPCs of each line were induced to differentiate and harvested at the indicated days post differentiation (DPD). Isolated RNAs were quantified for the levels of IL-34 mRNA using standard RT-qPCR with Taqman probes and normalized to GAPDH mRNA levels. (B) WB analysis of lysates from 8-day old neuronal cultures for levels of IL-34 protein. Glycosylated and unmodified forms of IL-34 with apparent molecular weights of ∼55 kDa and ∼27 kDa, respectively, can be seen to be selectively induced by Q-expanded mHTTx1(Q103) but not by control HTTx1(Q25). (C) DNA damage activates IL-34 transcription and exacerbates the effects of mHTTx1. 8-day old cultures were treated with vehicle (V) or 0.5 μM of etoposide (ETO) for 48 hrs. Extracted RNA was tested for IL-34 mRNA levels as in (A). (D) Control and IKKβ KD cultures were treated with NMDA or staurosporine (ST) for 8 hrs. Extracted RNA was tested for IL-34 as in (A). All experiments were repeated three times. Data are presented as means ± SD. ***P < 0.001. C=Control NPCs differentiated for the indicated days.
Expression of mHTTx1-EGFP (103Q) significantly stimulated the transcription of IL-34 in developing neurons, whereas WT HTTx1-EGFP (25Q) had no effect on IL-34 mRNA levels (Fig. 1A). We confirmed these findings at the protein level where IL-34 is heavily glycosylated producing a slow-migrating product (∼55 kDa) in mammalian cells (29); in addition, we also observed low levels of non-glycosylated IL-34 species (∼27 kDa) (Fig. 1B, arrow).
Next, we examined striatal mRNA extracted from the brains of HD mice expressing mHTTx1 (R6/2) (4) or full-length mHTT (YAC-128) (30) and found in both models that IL-34 transcripts are elevated compared to age-matched controls, with greater fold-induction in the R6/2 model (Supplementary Material, Fig. S2A and B). Thus, expression of mHTT isoforms appears to induce neuronal IL-34 levels in both human and mouse models of HD.
We predicted that stress induced by the accumulation of mHTTx1 may induce the expression of IL-34. Thus, we asked whether IL-34 induction could be mimicked by treatment of MESC2.10-derived neurons with chemical stressors. We found that exposure to sub-lethal concentrations of the DNA-damaging agent etoposide (ETO) or other chemical stressors such as staurosporine (ST) and N-methyl-D-aspartic acid (NMDA; a model of excitotoxicity in HD (31) also elevated IL-34 mRNA expression levels substantially (Fig. 1C, leftmost columns and D). In addition, etoposide induction of IL-34 was additive to that induced by mHTTx1-Q103 (Fig. 1C, rightmost columns). These findings suggest that neuronal IL-34 can be induced by a range of stressful and/or proteotoxic conditions, including by accumulation of misfolded mHTTx1 itself.
mHTTx1- and stress-induced IL-34 expression requires IKKβ
Since IKKβ activity has previously been implicated in mHTT-induced neurodegeneration and has also been shown to ameliorate the toxic effects of DNA damaging agents in neurons (21–22,25,27), we next asked whether shRNA-mediated knockdown (KD) of IKKβ influences IL-34 levels in MESC2.10-derived neurons (Supplementary Material, Fig. S3) (25). We found that KD of IKKβ blocked IL-34 induction by etoposide, NMDA, and staurosporine nearly completely (Fig. 1C, middle columns, and D). These results indicate that IKKβ is required for stress-activated IL-34 expression in neurons.
To evaluate the effects of IKKβ on mHTTx1-induced IL-34 production, we next expressed an untagged mHTTx1(73Q) in the MESC2.10 NPCs with KD of IKKβ using lentiviral transduction. We found that mHTTx1-mediated induction of IL-34 mRNA levels upon differentiation into neurons was completely blocked by KD of IKKβ (Fig. 2A). Induction of IL-34 by mHTTx1(Q73) and its inhibition by IKKβ KD was confirmed at the protein level (Fig. 2B). These findings indicate that IKKβ is also required for mHTTx1-induced IL-34 production and are consistent with the results observed above for chemical neuronal stressors (Fig. 1C and D).
Figure 2.

IKKβ regulates mHTTx1-induced IL-34. (A) NPCs for each line were differentiated for the indicated days (DPD). RNA was extracted and IL-34 mRNA was quantified as in Figure 1A. All experiments were confirmed at least three times. Data are presented as means ± SD. ***P < 0.001. (B) Confirmation using WB analysis that IKKβ blocks induction of IL-34 by mHTTx1 at the protein level. C=Control NPCs differentiated for the indicated days.
KD of IKKβ prevents the aggregation of mHTTx1
We next sought to determine at what level IKKβ may be involved in the induction of IL-34 by mHTTx1. Given that chemical stressors could also induce IL-34 to similar levels and in an additive manner, one possibility is that IKKβ regulates the assembly of one or more species such as misfolded monomers, multimers, oligomers, and inclusion bodies derived from mHTTx1 protein (32–36). While proliferating NPCs predominantly express soluble monomeric mHTTx1, monomers begin to aggregate upon neuronal differentiation, coinciding with the time course of elevation of IL-34 (Figs 3A and 2A). Surprisingly, KD of IKKβ inhibited the accumulation of mHTTx1 aggregates in differentiating neurons (Fig. 3A, right 4 lanes), while having no effect on the levels monomeric mHTTx1 in proliferating NPCs (Fig. 3A, arrow, lanes 0). The absence of mHTTx1 aggregates in neurons with KD expression of IKKβ was verified by 3 additional independent methods: 1) in probing lysates from differentiated neurons with two polyclonal antibodies recognizing various conformations of mHTTx1 oligomers, we were unable to detect significant amounts of mHTTx1 aggregates in neurons in which IKKβ was KD (Supplementary Material, Fig. S4); 2) we solubilized the SDS-insoluble neuronal pellets in a buffer containing 8-molar urea and probed with two antibodies against mHTTx1, detecting no high molecular weight species of mHTTx1 in neuronal lysates in which IKKβ was knocked down (Fig. 3B); and 3) using immunocytochemistry with an aggregate-specific antibody (MW8) we found that formation of nuclear/peri-nuclear aggregates of mHTTx1 was dramatically reduced in IKKβ KD neurons (Fig. 3C and D). Together, these findings support a novel role for IKKβ regulating mHTTx1 aggregation. The transcription of mHTTx1 mRNA quantified by RT-qPCR does not alter in the IKKβ KD neurons (Supplementary Material, Fig. S5A and B). Moreover, an antibody recognizing the phospho-Ser13 epitope of mHTTx1 also identified similar levels of monomeric mHTTx1 in proliferating control and IKKβ KD NPCs (Supplementary Material, Fig. S5C). Finally, probing with an antibody, which reacts with some intermediates of mHTTx1 also revealed similar levels of mHTTx1-related products (Supplementary Material, Fig. S5D). All together, these findings further confirm that mHTTx1 is expressed equally in control and IKKβ KD NPCs. That mHTTx1 monomers do not accumulate in IKKβ KD neurons is consistent with a broad protective impact on neuronal stress response, including promoting the proteostatic clearance of misfolded monomeric mHTTx1 (22,25). However, we cannot rule out the formation of mHTTx1 assemblies, which may not be recognized by our antibody repertoire.
Figure 3.

IKKβ-dependent aggregation of mHTTx1 in post-mitotic neurons. (A) WB analysis in developing neurons showing the accumulation of the mHTTx1 aggregates over days post differentiation (DPD) using an aggregate-specific antibody MW8. KD of IKKβ inhibits accumulation of high-molecular weight mHTTx1 aggregates to undetectable levels (right lanes). N.S.=non-specific. (B) Confirmation of inhibition of mHTTx1 aggregation by IKKβ KD using WB analysis of SDS-resistant insoluble materials dissolved in 8 M urea probed with anti-aggregate MW8 antibody (left) and a mouse polyclonal anti-HTTx1 antibody (right). (C) Inclusion body (IB) formation is inhibited neurons expressing mHTTx1 (left) with IKKβ KD (right). MW8 antibody was used to visualize IBs using immunohistochemistry. Anti-MAP2 antibody was used to stain neurons (red) and TOPRO-3 to mark the nuclei (blue). Images taken under confocal microscopy. (D) Quantification of IBs from the studies in (C). Data shown are means ± SD for 6 microscopic fields in each condition. ***P < 0.001.
To confirm these findings, we asked whether small molecule inhibitors of IKKβ could mimic the effects of IKKβ KD. We tested 3 reported compounds, luteolin, 2-[(aminocarbonyl) amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA), and sodium salicylate, which block the kinase activity of IKKβ (37–39). We found that all three IKKβ inhibitors were also able to reduce the oligomerization of mHTTx1 significantly (Fig. 4A and B). Such chemical inhibition of mHTTx1 aggregation was concomitant with reduced levels of IL-34 protein expression, as was also observed for genetic KD of IKKβ (Fig. 4A and B middle ). Inhibition of IL-34 by luteolin and sodium salicylate in neurons expressing mHTTx1 was also seen at the mRNA level, again as was observed with KD of IKKβ. (Figs 2A, 4C and D). These studies are consistent with the notion that the enzymatic activity of IKKβ is critical for mHTTx1 aggregation and IL-34 expression in human NPC-derived neurons.
Figure 4.

IKKβ inhibitors reduce mHTTx1 oligomerization and IL-34 production. (A) Inhibition of mHTTx1 protein aggregation by the small molecule IKKβ inhibitors luteolin and TPCA (top) and of IL-34 expression (middle) were determined by WB analysis. (B) Concentration-dependent inhibition of mHTTx1 aggregation by sodium salicylate (top) and of IL-34 expression (middle). MW8 was used to detect mHTTx1 aggregates and a rabbit anti-IL-34 was used to detect IL-34. (C) Luteolin was further examined for its effect on IL-34 mRNA expression by qRT-PCR. (C = control, Lut = luteolin, V= vehicle, TPCA = 2-[(aminocarbonyl) amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide). All experiments were repeated three times. Data presented are means ± SD. ***P < 0.001. (D) Sodium salicylate was also examined for its effect on IL-34 mRNA expression by qRT-PCR. Data presented are means ± SD. ***P < 0.001. The apparent IC50 for luteolin is ∼2 μg/ml and ∼100 μg/ml for sodium salicylate (Fig. 4C and D).
Aggregation of mHTTx1 drives caspase-3 expression in NPC-derived human neurons
The aggregation of mHTTx1(73Q) in MESC2.10-derived neurons coincided with expression of the apoptotic enzyme procaspase-3, which was also significantly reduced by KD of IKKβ (Fig. 5A and Supplementary Material, Fig. S6). As procaspase-3 cleavage is critical for the execution of apoptosis (40); induction of procaspase-3 also signifies a stress response to the accumulation of mHTTx1 aggregates. In these NPC-derived neurons, however, procaspase-3 is not cleaved and does not lead directly to cell death suggesting that additional signals may be required for the mHTTx1 neurotoxicity.
Figure 5.

Procaspase-3 induction by mHTTx1 aggregation is inhibited by IKKβ knockdown. (A) WB analysis of lysates from neurons expressing mHTTx1 (left lanes) and with IKKβ KD (right lanes) for the accumulation of procaspase-3. (B) IL-34 does not inhibit mHTTx1 aggregation or procaspase-3 induction. NPCs expressing mHTTx1 were co-cultured with 0, 5%, or 10% of another NPC line expressing IL-34 resulting in up to 10-fold increases in expression of secreted IL-34 protein in the co-cultures. Supernatants of 8-day-old cultures were quantified for IL-34 secretion by ELISA. All experiments were reproduced at least three times. Data presented are means ± SD. ***P <0.001. (C) WB analysis of neuronal lysates of co-cultures from (B) showing that neither mHTTx1 aggregation (top panel) nor procaspase-3 induction (middle panel) was affected by increased levels of soluble IL-34 in the co-cultures. N. S.= Non-specific.
IL-34 does not influence the oligomerization of mHTTx1
The MESC2.10 NPCs and neurons-derived from them express the CSF1R (Supplementary Material, Fig. S7). Thus, we asked whether increased IL-34 could feed back to affect the aggregation and/or the neurotoxicity of mHTTx1 in NPC-derived neurons. We added recombinant IL-34 to the culture medium of developing neurons and incubated for several days; however, this did not affect the aggregation or the toxicity of mHTTx1. We hypothesized that continuous secretion of IL-34 might have a different outcome. To test this hypothesis, we engineered MESC.2.10 NPCs to constitutively express and secrete human IL-34. In this model, IL-34 accumulates up to ∼250 ng/ml in the growth medium and had no effect on the proliferation or the differentiation of NPCs. We then co-cultured different ratios of NPCs expressing IL-34 with those expressing mHTTx1, and induced neuronal differentiation. We adjusted ratios of the co-cultured neurons to produce levels of IL-34 ∼5–10-fold above native levels (Fig. 5B). We found, again, that continuous secretion of IL-34 did not influence the aggregation of mHTTx1 nor the level or cleavage of procaspase-3 (Fig. 5C, top and middle, respectively). These data suggest that IL-34 may not act directly on neurons to alter the principal phenotypic effects of mHTTx1 expression.
IL-34 exacerbates neurodegeneration induced by mHTTx1 in a brain tissue slice model
The findings from the previous two sections suggests that increased levels of IL-34 production induced by mHTTx1 expression and aggregation may have minimal effects on neurons themselves, at least in the context of mono-cultures of NPC-derived neurons which may be dominated by cell-autonomous signaling pathways. However, as IL-34 is a potent inducer of microglia (16), we surmised that in a more complex, native brain tissue environment the functional consequences of increased IL-34 production may become more evident.
We thus turned to a rat brain tissue slice model that we have previously established for mHTTx1-induced neurodegeneration, in which biolistic gene delivery is used to introduce mHTTx1 into MSNs in the striatum (21,26). Transfection of sufficient levels of mHTTx1(Q73) can drive substantial degrees of neurodegeneration in brain slice explants over a 3–5-day period (26,35). Interestingly, we found that the effects of transfecting a sub-threshold amount of mHTTx1, which by itself did not induce measurable neurodegeneration, could be exacerbated to drive overt degeneration of MSNs by co-transfection with IL-34 (Fig. 6A and B).
Figure 6.

IL-34 exacerbates mHTTx1-induced degeneration of MSNs. Corticostriatal brain slice explants were biolistically co-transfected with YFP and/or mHTTx1 expression constructs as described in the Materials and Methods to induce degeneration of striatal medium spiny neurons (MSNs). (A) Representative fluorescent micrographs of MSNs transfected with a sub-threshold amount of mHTTx1 showing no degeneration by 3 days in culture (compare upper left and right panels) unless further co-transfected with an expression construct for IL-34 (lower right panel). Co-transfection with IL-34 only had no obvious effects on MSNs viability (lower left panel). Scale bar, 200 μm. (B) Quantification of numbers of healthy MSNs per brain slice explants as identified by the co-transfected YFP marker in each condition as described in (A). Averaged means ± SD are shown for 4 independent experimental runs, with the YFP positive control condition set to 100%. *P <0.05 by ANOVA followed by Dunnett's post hoc comparison test. (C) Treatment of brain slice explants with the CSF1R inhibitor PLX3397 shows concentration-dependent inhibition of microglial numbers/activation as visualized using anti-Iba1 immunostaining. (D) In contrast, MSNs in corticostriatal brain slices transfected with a supra-threshold amount of mHTTx1 (twice the amount of mHTTx1 DNA loaded onto the biolistic gold particles as for Parts A and B above) undergo overt and significant neurodegeneration by 4 days after transfection (compare first two bars). Treatment with PLX3397 in the same concentration range that showed microglial inhibition (c.f. panel C) provided significant neuroprotection to mHTTx1-transfected MSNs. Averaged means ± SD are shown for five independent experimental runs, with the mHTTx1 DMSO-treated condition set to 100%; third bar shows positive control condition in which mHTTx1-transfected brain slices are treated with a cocktail of 50 µM KW-6002 and 30 µM SP600125. *P <0.05 by ANOVA followed by Dunnett's post hoc comparison test.
Conversely, treatment with the CSF1R receptor inhibitor PLX3397, in a concentration range which reduces activation and expansion of resident microglia in brain slices (Fig. 6C), was able to provide partial but significant rescue against neurodegeneration of MSNs induced by a supra-threshold level of mHTTx1 transfection (Fig. 6D). As mature neurons do not express detectable levels of CSF1R in vivo, together these findings are consistent with microglial activation by mHTTx1-induced production of neuronal IL-34 contributing to the rate/extent of MSN neurodegeneration in the brain slice model. Such a non-cell autonomous mechanism is supported by our previous observations that activated microglia can be recruited to the neurites of neurons expressing mHTTx1 in primary and brain slice culture models (41).
Discussion
The molecular pathways regulating persistent activation of microglia in HD remain largely unknown. Here, we have presented evidence that the accumulation of misfolded mHTTx1 aggregates in post-mitotic neurons promotes production of neuronal IL-34, which is a major driver of microglial expansion. Elevated IL-34 levels do not appear to feed back directly onto neurons to influence the aggregation or the toxicity of mHTTx1. However, in a brain tissue model with intact neuronal-microglial interactions, elevated IL-34 promotes mHTTx1-induced neurodegeneration, whereas blocking IL-34 receptor signaling rescues the neurotoxicity of mHTTx1. Our studies also uncover a novel role for IKKβ influencing the aggregation and proteostasis of mHTTx1 monomers. Taken together, these findings suggest that IKKβ/mHTTx1 interactions regulate IL-34 production, which in turn induces a non-cell-autonomous, microglial-mediated response that exacerbates neurodegeneration triggered by mHTTx1 (Fig. 7).
Figure 7.
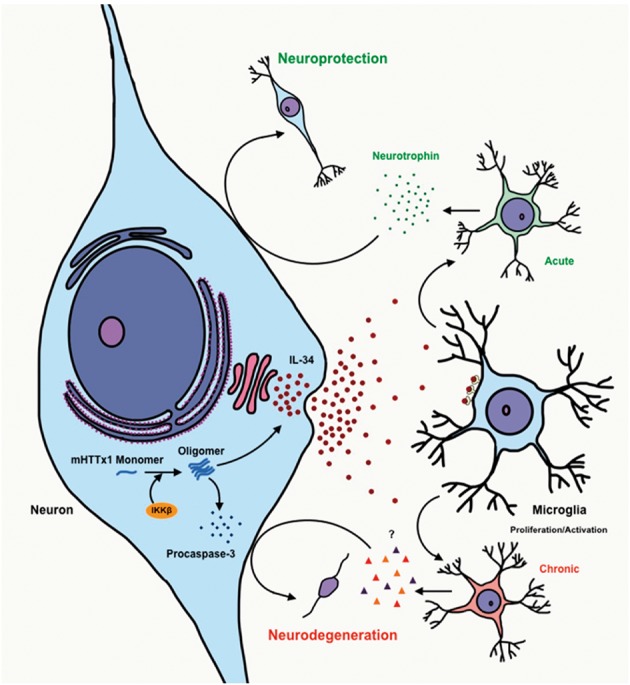
A model for mHTTx1-induced IL-34 signaling driving neurodegeneration. Accumulation of mHTTx1 creates a stressed neuronal environment activating IKKβ, which in turn promotes the aggregation of mHTTx1 and subsequent induction of procaspase-3 (ProCas-3) and IL-34. In turn, increased IL-34 secretion may initially signal to microglia to express neuroprotective factors and maintain survival. However, chronic elevation of IL-34 by neurons accumulating mHTT may lead instead to pathological activation of microglia, producing inflammatory cytokines and necrotic factors to drive neuronal degeneration and death via non-cell autonomous interactions.
Although neurons are the primary source of IL-34 in the CNS, little is known about the stimuli that regulate IL-34 production. Here, we provide evidence that IL-34 expression is induced by neuronal stress such as DNA damage, NMDA, and, importantly, by the accumulation of mHTTx1 aggregates. Interestingly, we found that mHTTx1 aggregate accumulation in pure cultures of MESC-derived neurons increased procaspase-3 levels but did not lead directly to cell death. We suggest that induction of procaspase-3 by buildup of misfolded mHTT stresses neurons and puts them at risk for a second signal to drive to overt neurodegeneration—subsequent IL-34-mediated activation of microglia, a non-cell-autonomous event, may provide this second signal leading to neurodegeneration in HD as observed in our brain slice model. In fact, microglial activation is known to overlap with neuronal loss in the striatum of HD patients several years before the onset of symptoms (9,10). Moreover, we found that levels of IL-34 mRNA are higher in an aggressive model of HD (R6/2), which may be related to the accumulation of more aggregates, compared to YAC-128 HD model expressing full-length mHTT where aggregates are less abundant (Supplementary Material, Fig. S2) (4,30).
Early induction of IL-34 by the mHTT aggregates may also signal cell autonomous as well as non-cell autonomous neuroprotective responses (Fig. 7). It has been demonstrated that IL-34 has neuroprotective properties in acute chemically-driven neurodegeneration as well as in an Alzheimer's disease (AD) mouse model (42,43). However, other studies indicate that IL-34 expands microglia by activating the myeloid-cell-specific transcription factors PU.1 and C/EBPα and promotes neuroinflammation in AD and prion disorders (19). Reduction of IL-34 level is neuroprotective in an encephalopathy viral model where blocking microglial expansion preserves synapse loss and prevents virus-mediated cognitive impairments, which also occurs in patients infected with similar viruses (17). It is relevant that the level and activity of PU.1 and its downstream gene targets are increased in the brains of HD patients and mouse models (6). Activation of microglial PU.1 coincides with the mHTTx1-mediated neurotoxicity supporting a role for non-cell-autonomous neurodegeneration in HD (6). Thus, the link between neuronal mHTT aggregation and IL-34 signaling reported here may contribute to aberrant microglial activation occurring many years before the onset of motor symptoms in HD patients (9–11). The observation that a CSF1R antagonist, which reduces microglial expansion, ameliorates mHTTx1-induced neurodegeneration (Fig. 6) raises optimism that reducing the expression of IL-34 or blocking its downstream effects on microglial activation may be a viable therapeutic strategy for HD. IL-34-mediated microglial activation may also influence neurotoxicity indirectly. A recent study indicates that pro-inflammatory microglia induce the production and maturation of a novel neurotoxic astrocytic lineage common to most amyloid-driven neurodegenerative disorders including PD, AD, and HD (44). The notion that amyloid-mediated IL-34 synthesis is an immediate stress response common to most brain disorders and controls neurotoxic pathways is intriguing and may merit further investigation.
A novel finding here was the identification of IKKβ as a key regulator of mHTTx1 aggregation and subsequent IL-34 expression. While the mechanism of IKKβ-driven mHTTx1 aggregation remains to be elucidated, our findings are consistent with the notion that low IKKβ levels and activity in neurons may promote the turnover of mHTTx1 since monomeric mHTTx1 does not accumulate. The absence of aggregation in neurons with KD of IKKβ is not due to differences in the expression of mHTTx1 (Supplementary Material, Fig. S5 A–D). Posttranslational modifications may play a role since IKKβ has been implicated in the phosphorylation of Ser13 of WT and mHTT and subsequent turnover by the proteasome-lysosome pathways (45). In our model, mHTTx1 monomers are phosphorylated at Ser13 independent of IKKβ levels in proliferating NPCs; however S13 phosphorylated mHTTx1 could not be detected in neurons (Supplementary Material, Fig. S5C). Whether IKKβ modifies other residues such as Threonine-3 to influence the turnover and/or aggregation of mHTTx1 remains to be identified (46).
KD of IKKβ expression also makes dopaminergic human neurons resilient to the damaging effects of genotoxic compounds and prevents stress-induced caspase-dependent cleavage of HTT (22,25). In rodents, selective knockout of IKKβ in neurons lowers neuroinflammation and delays systemic aging (20). Conversely, excess IKKβ activity promotes focal neurodegeneration by reducing the expression of BDNF (47). An intriguing possibility consistent with our findings is that neurons with low IKKβ levels are more efficient at removing misfolded mHTTx1 and thus less likely to display the downstream effects of mHTT aggregation such as procaspase-3 and IL-34 induction.
Previously, we demonstrated that mHTT directly activates IKKβ, which coincides with neurotoxicity and development of symptoms in HD mice (21). Studies using the immune cells of HD patients confirm these findings and further implicate mHTT-mediated IKKβ activation as an inducer of inflammation in HD (23). The data presented here reinforce the viability of IKKβ as a potential therapeutic target for HD. Indeed, inhibition of IKKβ activity reduces the neurotoxicity of mHTT in various models (21,26,27). However, translation to humans is lagging since most of the small molecule inhibitors of IKKβ have undesired side effects or are unable to cross the blood brain barrier (22). Testing of safe, natural compounds may be advantageous to systemically lower IKKβ activity and thus minimize inflammation and potentially microglial activation in HD. One compound tested here is the flavone luteolin, which reduces mHTTx1 oligomerization and IL-34 expression. Luteolin is beneficial in autistic children and inhibits microglial activity in animal models of AD and senescent aging (48–51). Mechanistically, luteolin reduces the levels of cytokines such as IL-6 and TNF-α in the sera of treated subjects (49,50). Luteolin is also an attractive therapeutic since it crosses the blood brain and thus could potentially lower microglial activation as well as delaying the oligomerization of mHTT (51). Considering that microglia and inflammatory cytokines are elevated in the pre-manifest and symptomatic HD patients (9–13), examination of luteolin or other natural inhibitors of IKKβ in HD models may merit further investigation.
Materials and Methods
Antibodies and reagents
Antibodies to detect various conformations of misfolded mHTTx1 were generated in house (Khoshnan et al., unpublished data) or described previously (52). Mouse anti-HTT polyclonal antibodies were generated by standard immunization protocols using recombinant mHTTx1 protein. Antibodies were validated for reactivity to mHTT by WBs and IHC. Antibodies to detect GAPDH, EGFP, and IL-34 were purchased from Santa Cruz Biotechnology (Dallas, TX). The anti-caspase-3 antibodies recognizing both the procaspase-3 and the cleaved P17 product were purchased from Abcam (ab32351, Cambridge, MA) and Cell Signaling (#9662, Danvers, MA). Rabbit anti-microtubule-associated protein-2 (Anti-MAP2) and mouse anti-IKKβ antibodies were purchased from Cell Signaling (Danvers, MA). DMEM/F12, bFGF-2, N-2 and B-27 neuronal supplements were obtained from ThermoFisher Scientific (Carlsbad, CA). Luteolin and TPCA were obtained from Cayman Chemicals (Ann Arbor, Michigan). Sodium salicylate, etoposide, NMDA, and staurosporine were obtained from Sigma-Aldrich (St. Louis, MO). IL-34 ELISA kit and recombinant human IL-34 were purchased from R&D Systems (Minneapolis, MN). PLX3397 was obtained from Selleckchem (Houston, TX, USA).
Lentiviral production
The construction and production of recombinant lentiviruses was as described previously (25). The cDNA for HTTx1 with 73Q was generously provided by Dr. Judith Frydman at Stanford. The HTTx1-EGFP (25Q and 103Q) constructs were described previously (21,22). Human IL-34 cDNA (NM_152456) was obtained from Origene (Rockville, MD). All cDNAs were cloned into the FUGW lentiviral backbone, provided by Dr. David Baltimore at the California Institute of Technology (Addgene, plasmid # 14883). Virus production was carried out in HEK 293 cells with plasmids encoding the structural genes and the vesicular stomatitis virus glycoprotein (VSVG) (25). Viral particles were concentrated from the supernatant using Amicon columns (Millipore, MA) with a size cutoff of 100 kDa. Viral titers were determined with an EGFP virus.
Culturing of MESC2.10 human neurons
The generation of human MESC2.10 neuronal stem line has been reported (28). Briefly, NPCs were obtained from an 8-week-old human embryo and transduced with a retrovirus encoding a tetracycline-regulated (tet-off) v-myc to prevent senescence. Propagation was done in serum-free medium containing bFGF-2 (25). In our studies, NPCs were propagated in dishes coated with poly-lysine and laminin in DMEM/F12 in the presence N2 and B-27 neuronal supplements and 40 ng/ml bFGF-2 (Invitrogen).
Transduction of MESC2.10
Proliferating MESC2.10 cells were transduced at multiplicity of infection (MOI) of ∼2 for each recombinant virus to avoid multiple copy insertion. Under these conditions, we find that ∼ 90–95% of the cells are transduced using an EGFP lentivirus as a marker. EGFP NPCs were used controls (C). NPCs with KD of IKKβ were reported previously (25). Transduced NPCs were maintained similarly to parental NPCs.
NPC differentiation
To differentiate MESC2.10 NPCs, a DMEM/F12 proliferation medium was used containing N2 supplement, 2 μg/ml of doxycycline and 20 mM cAMP. This formulation produces mature dopaminergic neurons, which are electrically active, in ∼6 days (28). Developing neurons were harvested at different time points for Western blotting or RNA extraction.
Compound treatment
8-day old developing neurons were treated with the following chemical stressors: NMDA (100 μM), staurosporine (2 μM), or etoposide (2 μM) for 8 hrs. For extended treatments with etoposide, 6-day old developing neurons were treated with 0.5 μM etoposide and incubated for 48 h. Cells were harvested and stored at −80 °C or immediately processed for RNA extraction. For examining the effects of luteolin, TPCA (2-[(aminocarbonyl) amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide), and sodium salicylate on mHTTx1 aggregation, 5-day old developing neurons were treated with the indicated concentrations of each compound and incubated for 3 additional days. Cells were harvested and processed for Western blots or examination by qRT-PCR.
RNA extraction and quantification
Total RNA was extracted by TRIzol and was further purified by RNA purification columns (Qiagen, Valencia, CA). For cDNA synthesis, 1 μg of RNA was reverse transcribed with Superscript II cDNA synthesis kit (New England Biolabs, Ipswich, MA). To quantify IL-34, Taqman probes synthesized by Thermo Fisher Scientific (Carlsbad, CA) were used for real-time PCR using a 7300 real-time PCR system. Data were analyzed comparatively by the formula 2(-ΔΔct). GAPDH was used for the normalization of different mRNAs. Each sample was compared to the mRNA levels of proliferating control cells (day 0). Results are shown as fold-changes. A Student‘s t-test was used to calculate P values.
Western blot analysis
Cells were harvested and lysed in RIPA buffer and processed for Western blotting as described previously (25). For most experiments, we used ∼100 μg of protein lysate. To denature SDS-insoluble materials, pellets were dissolved in buffers containing 8 M urea and processed as above. We used several antibodies to detect various species of mHTTx1. All HD antibodies were produced in house. For most antibodies, we used a 1: 000 dilutions. IL-34 antibody (Santa Cruz Biotechnology) was used at 1: 2000. All other antibodies used for monitoring loading were used at 1: 000. All HRP-conjugated secondary antibodies were used at 1: 10000 dilutions.
Quantification of IL-34 in supernatant
IL-34 secretion in the supernatants was quantified by an ELISA method kit following the manufacturer‘s instructions (R&D Systems).
Immunohistochemistry
NPCs were cultured on cover slips and induced to differentiate into neurons for 8 days. Cells were fixed in 4% paraformaldehyde followed by permeabilization in 70% methanol and stained with an anti-GFP antibody to visualize HTTx1-EGFP with 25 or 103 Q. Mutant HTTx1 (73Q) neurons or those with KD of IKKβ were stained with the MW8 antibody (1: 1000), which is selective for mHTTx1 aggregates (52). For a neuronal marker, an antibody to microtubule–associated protein-2 (MAP2) was used at (1: 1000). Secondary antibodies conjugated to FITC or Alexa 568 were used to detect the respective primary antibodies. All images were captured with a confocal microscope. Organotypic brain slices were fixed with 4% paraformaldehyde. Following blocking with a permeability mixture containing 10% goat serum, 20% DMSO, and 2% Triton X-100 in PBS (53), brain slices were incubated with a primary rabbit polyclonal anti-Iba1 antibody (1: 1000; Wako) followed by an Alexa Fluor 568 secondary antibody to visualize resident microglia. Images were captured under conventional epifluorescence (Axioskop, Zeiss).
Brain slice culture model
All animal experiments and care complied with federal regulations and were reviewed and approved by the Duke University Medical Center Institutional Animal Care and Use Committee. Brain slice preparation and biolistic transfection were done as previously described (26). Briefly, brain tissue was dissected from CD Sprague Dawley rats killed on postnatal day 10 (P10) (Charles River Laboratory) and placed in ice-cold culture medium containing 15% heat-inactivated horse serum, 10 mM KCl, 10 mM HEPES, 100 U/ml penicillin/streptomycin, 1 mM sodium pyruvate, and 1 mM L-glutamine in Neurobasal A (Themofisher). Brain tissue was cut into 250-μm thick coronal slices using a Vibratome and incubated for 1 h at 37 °C under 5% CO2 before biolistic transfection. Gold particles (1.6 μm gold microcarriers; Bio-Rad) were coated with the appropriate DNAs (see below) as per the manufacturer's instructions and loaded into Tefzel tubing (McMaster-Carr) for use with the Helios biolistic device (Bio-Rad), which was used at a delivery pressure of 95 psi. Gold particles were coated with expression constructs encoding yellow fluorescent protein (YFP) as a morphometric marker, mHTTx1Q-73, with or without mouse IL-34. The cDNA for mouse IL-34 was cloned from a mouse brain and subcloned into the pCDNA3.1 mammalian expression vector (ThermoFisher). The IL-34 sequence was similar to NM_001135100 in Genebank. For each condition, transfections were done on 12 brain slices, and total numbers of healthy medium spiny neurons (MSNs) expressing the YFP reporter in the entire striatal region of each brain slice were assessed using fluorescence microscopy 3 or 4 days after brain slice preparation and transfection. MSNs with normal-sized cell bodies, even and continuous expression of YFP in the cell body and dendrites, and having more than two discernable primary dendrites more than two cell bodies long were scored as healthy. Statistical significance was determined by ANOVA followed by Dunnett's post hoc comparison test at the P < 0.05 confidence level.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We are grateful to Dr. Judith Frydman and Dr. Koning Shen for providing the HTTx1 constructs.
Conflict of Interest statement. None declared.
Funding
NINDS grant R01 NS074374 awarded to AK and PHP, and R21 NS098323 to DCL.
References
- 1. Huntington‘s Disease Collaborative Research Group. (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington‘s disease chromosomes. Cell, 72, 971–983. [DOI] [PubMed] [Google Scholar]
- 2. Kim Y.J., Yi Y., Sapp E., Wang Y., Cuiffo B., Kegel K.B., Qin Z.-H., Aronin N., DiFiglia M. (2001) Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington's disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc. Natl Acad. Sci. U S A, 98, 12784–12789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sathasivam K., Neueder A., Gipson T.A., Landles C., Benjamin A.C., Bondulich M.K., Smith D.L., Faull R.L.M., Roos R.A.C., Howland D.. et al. (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U S A, 110, 2366–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Davies S.W., Turmaine M., Cozens B.A., DiFiglia M., Sharp A.H., Ross C.A., Scherzinger E., Wanker E.E., Mangiarini L., Bates G.P. (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell, 90, 537–548. [DOI] [PubMed] [Google Scholar]
- 5. Zuccato C., Valenza M., Cattaneo E. (2010) Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol. Rev., 90, 905–981. [DOI] [PubMed] [Google Scholar]
- 6. Crotti A., Benner C., Kerman B.E., Gosselin D., Lagier-Tourenne C., Zuccato C., Cattaneo E., Gage F.H., Cleveland D.W., Glass C.K. (2014) Mutant huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat. Neurosci., 17, 513–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Prinz M., Priller J. (2014) Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci., 15, 300–312. [DOI] [PubMed] [Google Scholar]
- 8. Parkhurst C.N., Yang G., Ninan I., Savas J.N., Yates J.R. 3rd, Lafaille J.J., Hempstead B.L., Littman D.R., Gan W.B. (2013) Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell, 155, 1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pavese N., Gerhard A., Tai Y.F., Ho A.K., Turkheimer F., Barker R.A., Brooks D.J., Piccini P. (2006) Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology, 66, 1638–1643. [DOI] [PubMed] [Google Scholar]
- 10. Tai Y.F., Pavese N., Gerhard A., Tabrizi S.J., Barker R.A., Brooks D.J., Piccini P. (2007) Microglial activation in presymptomatic Huntington's disease gene carriers. Brain, 130, 1759–1766. [DOI] [PubMed] [Google Scholar]
- 11. Politis M., Pavese N., Tai Y.F., Kiferle L., Mason S.L., Brooks D.J., Tabrizi S.J., Barker R.A., Piccini P. (2011) Microglial activation in regions related to cognitive function predicts disease onset in Huntington's disease: a multimodal imaging study. Hum Brain Mapp., 32, 258–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Björkqvist M., Wild E.J., Thiele J., Silvestroni A., Andre R., Lahiri N., Raibon E., Lee R.V., Benn C.L., Soulet D. (2008) A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J. Exp. Med., 205, 1869–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Politis M., Lahiri N., Niccolini F., Su P., Wu K., Giannetti P., Scahill R.I., Turkheimer F.E., Tabrizi S.J., Piccini P. (2015) Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington's disease gene carriers. Neurobiol. Dis., 8383, 115–121. [DOI] [PubMed] [Google Scholar]
- 14. Greter M., Lelios I., Pelczar P., Hoeffel G., Price J., Leboeuf M., Thomas M.K., Frei K., Ginhoux F., Merad M.. et al. (2012) Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity, 37, 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Y., Szretter K.J., Vermi W., Gilfillan S., Rossini C., Cella M., Barrow A.D., Michael S.D., Colonna M. (2012) IL-34 is a tissue-restricted ligand of CSF1R required for the development of langerhans cells and microglia. Nat. Immunol., 13, 753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chitu V., Gokhan S., Nandi S., Mehler M.F., Stanley E.R. (2016) Emerging roles for CSF-1 receptor and its ligands in the nervous system. Trends Neurosci., 39, 378–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vasek M.J., Garber C., Dorsey D., Durrant D.M., Bollman B., Soung A., Yu J., Perez-Torres C., Frouin A., Wilton D.K.. et al. (2016) A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature, 534, 538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Masteller E.L., Wong B.R. (2014) Targeting IL-34 in chronic inflammation. Drug Discov. Today, 19, 1212–1216. [DOI] [PubMed] [Google Scholar]
- 19. Gómez-Nicola D., Fransen N.L., Suzzi S., Perry V.H. (2013) Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci., 33, 2481–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang G., Li J., Purkayastha S., Tang Y., Zhang H., Yin Y., Li B., Gang Liu G., Cai D. (2013) Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature, 497, 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Khoshnan A., Ko J., Watkin E.E., Paige L.A., Reinhart P.H., Patterson P.H. (2004) Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J. Neurosci., 24, 7999–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khoshnan A., Patterson P.H. (2011) The role of IκB kinase complex in the neurobiology of Huntington's disease. Neurobiol. Dis., 43, 305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Träger U., Andre R., Lahiri N., Magnusson-Lind A., Weiss A., Grueninger S., McKinnon C., Sirinathsinghji E., Kahlon S., Pfister E.L.. et al. (2014) HTT-lowering reverses Huntington's disease immune dysfunction caused by NFκB pathway dysregulation. Brain, 137, 819–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chariot A. (2009) The NF-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol., 19, 404–413. [DOI] [PubMed] [Google Scholar]
- 25. Khoshnan A., Ko J., Tescu S., Brundin P., Patterson P.H. (2009) IKKα and IKKβ regulation of DNA damage-induced cleavage of huntingtin. PLoS One, 4, e5768.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reinhart P.H., Kaltenbach L.S., Essrich C., Dunn D.E., Joshua A., Eudailey J.A., DeMarco C.T., Turmel G.J., Whaley J.C., Wood A.. et al. (2011) Identification of anti-inflammatory targets for Huntington's disease using a brain slice-based screening assay. Neurobiol. Dis., 43, 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hsiao H.Y., Chen Y.C., Chen H.M., Tu P.H., Chern Y. (2013) A critical role of astrocyte-mediated nuclear factor-κB-dependent inflammation in Huntington's disease. Hum. Mol. Genet., 22, 1826–1842. [DOI] [PubMed] [Google Scholar]
- 28. Lotharius J., Barg S., Wiekop P., Lundberg C., Raymon H.K., Brundin P. (2002) Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J. Biol. Chem., 277, 38884–38894. [DOI] [PubMed] [Google Scholar]
- 29. Felix J., Elegheert J., Gutsche I., Shkumatov A.V., Wen Y., Bracke N., Pannecoucke E., Vandenberghe I., Devreese B., Svergun D.I.. et al. (2013) Human IL-34 and CSF-1 establish structurally similar extracellular assemblies with their common hematopoietic receptor. Structure, 21, 528–539. [DOI] [PubMed] [Google Scholar]
- 30. Graham R.K., Deng Y., Carroll J., Vaid K., Cowan C., Pouladi M.A., Metzler M., Bissada N., Wang L., Faull R.M.. et al. (2010) Cleavage at the 586 amino acid caspase-6 site in mutant huntingtin influences caspase-6 activation in vivo. J. Neurosci., 30, 15019–15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parsons M.P., Raymond L.A. (2014) Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron, 82, 279–293. [DOI] [PubMed] [Google Scholar]
- 32. Arrasate M., Finkbeiner S. (2012) Protein aggregates in Huntington's disease. Exp. Neurol., 238, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim Y.E., Hosp F., Frottin F., Ge H., Mann M., Manajit H., Hartl F.U. (2016) Soluble oligomers of polyQ-expanded Huntingtin target a multiplicity of key cellular factors. Mol Cell, 63, 951–964. [DOI] [PubMed] [Google Scholar]
- 34. Leitman J., Hartl F.U., Lederkremer G.Z. (2013) Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat. Commun., 4, 2753.. [DOI] [PubMed] [Google Scholar]
- 35. Shen K., Calamini B., Fauerbach J.A., Ma B., Shahmoradian S.H., Lachapel I.S., Chiu W., Lo D.C., Frydman J. (2016) Control of the structural landscape and neuronal proteotoxicity of mutant Huntingtin by domains flanking the polyQ tract. eLife, 5, pii: e18065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Caron N.S., Hung C.L., Atwal R.S., Truant R. (2014) Live cell imaging and biophotonic methods reveal two types of mutant huntingtin inclusion. Hum. Mol. Genet., 23, 2324–2338. [DOI] [PubMed] [Google Scholar]
- 37. Yin M.J., Yamamoto Y., Gaynor R.B. (1998) The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature, 396, 77–80. [DOI] [PubMed] [Google Scholar]
- 38. Podolin P.L., Callahan J.F., Bolognese B.J., Li Y.H., Carlson K., Davis T.G., Mellor G.W., Evans C., Roshak A.K. (2005) Attenuation of murine collagen-induced arthritis by a novel, potent, selective small molecule inhibitor of IkappaB Kinase 2, TPCA-1 (2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide), occurs via reduction of proinflammatory cytokines and antigen-induced T cell Proliferation. J. Pharmacol. Exp. Ther., 312, 373–381. [DOI] [PubMed] [Google Scholar]
- 39. Nguyen T.Y., To D.C., Tran M.H., Lee J.S., Lee J.H., Kim J.A., Woo M.H., Min B.S. (2015) Anti-inflammatory Flavonoids Isolated from Passiflora foetida. Nat. Prod. Commun., 10, 929–931. [PubMed] [Google Scholar]
- 40. Brentnall M., Weir D.B., Rongvaux A., Marcus A.I., Boise L.H. (2014) Procaspase-3 regulates fibronectin secretion and influences adhesion, migration and survival independently of catalytic function. J. Cell Sci., 127, 2217–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kraft A.D., Kaltenbach L.S., Lo D.C., Harry G.J. (2012) Activated microglia proliferate at neurites of mutant huntingtin-expressing neurons. Neurobiol. Aging, 33, 621 e17–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Luo J., Elwood F., Britschgi M., Villeda S., Zhang H., Ding Z., Zhu L., Alabsi H., Getachew R., Narasimhan R.. et al. (2013) Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J. Exp. Med., 210, 157–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mizuno T., Doi Y., Mizoguchi H., Jin S., Noda M., Sonobe Y., Takeuchi H., Suzumur A. (2011) Interleukin-34 selectively enhances the neuroprotective effects of microglia to attenuate oligomeric amyloid-β neurotoxicity. Am. J. Pathol., 179, 2016–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liddelow S.A., Guttenplan K.A., Clarke L.E., Bennett F.C., Bohlen C.J., Schirmer L., Bennett M.L., Münch A.E., Chung W.S., Peterson T.C.. et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thompson L.M., Aiken C.T., Kaltenbach L.S., Agrawal N., Illes K., Khoshnan A., Martinez-Vincente M., Arrasate M., O'Rourke J.G., Khashwji H.. et al. (2009) IKK phosphorylates huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol., 187, 1083–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bustamante M.B., Annalisa A., Pedersen J.F., Azzollini L., Cariulo C., Wang Z., Petricca L., Verani M., Puglisi F., Park H.. et al. (2015) Detection of huntingtin exon 1 phosphorylation by Phos-Tag SDS-PAGE: predominant phosphorylation on threonine 3 and regulation by IKKβ. Biochem. Biophys. Res. Commun., 463, 1317–1322. [DOI] [PubMed] [Google Scholar]
- 47. Maqbool A., Lattke M., Wirth T., Baumann B. (2013) Sustained, neuron-specific IKK/NF-κB activation generates a selective neuroinflammatory response promoting local neurodegeneration with aging. Mol. Neurodegener., 8, 40.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tsilioni I., Taliou A., Francis K., Theoharides T.C. (2015) Children with autism spectrum disorders, who improved with a luteolin-containing dietary formulation, show reduced serum levels of TNF and IL-6. Transl. Psychiatry, 5, e647.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Burton M.D., Rytych J.L., Amin R., Johnson R.W. (2016) Dietary luteolin reduces proinflammatory microglia in the brain of senescent mice. Rejuvenation Res., 19, 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang H., Wang H., Cheng H., Che Z. (2016) Ameliorating effect of luteolin on memory impairment in an Alzheimer's disease model. Mol. Med. Rep., 13, 4215–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sawmiller D., Li S., Shahaduzzaman M., Smith A.J., Obregon D., Giunta B., Borlongan C.V., Sanberg P.R., Tan J. (2014) Luteolin reduces Alzheimer's disease pathologies induced by traumatic brain injury. Int. J Mol. Sci., 15, 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ko J., Ou S., Patterson P.H. (2001) New anti-huntingtin monoclonal antibodies: implications for huntingtin conformation and its binding proteins. Brain Res. Bull., 56, 319–329. [DOI] [PubMed] [Google Scholar]
- 53. Dissing-Olesen L., MacVicar B.A. (2015) Fixation and immunolabeling of brain slices: SNAPSHOT method. Curr. Protoc. Neurosci., 71, 1.23.1–1.23.12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.