

Abstract
CblX (MIM309541) is an X-linked recessive disorder characterized by defects in cobalamin (vitamin B12) metabolism and other developmental defects. Mutations in HCFC1, a transcriptional co-regulator which interacts with multiple transcription factors, have been associated with cblX. HCFC1 regulates cobalamin metabolism via the regulation of MMACHC expression through its interaction with THAP11, a THAP domain-containing transcription factor. The HCFC1/THAP11 complex potentially regulates genes involved in diverse cellular functions including cell cycle, proliferation, and transcription. Thus, it is likely that mutation of THAP11 also results in biochemical and other phenotypes similar to those observed in patients with cblX. We report a patient who presented with clinical and biochemical phenotypic features that overlap cblX, but who does not have any mutations in either MMACHC or HCFC1. We sequenced THAP11 by Sanger sequencing and discovered a potentially pathogenic, homozygous variant, c.240C > G (p.Phe80Leu). Functional analysis in the developing zebrafish embryo demonstrated that both THAP11 and HCFC1 regulate the proliferation and differentiation of neural precursors, suggesting important roles in normal brain development. The loss of THAP11 in zebrafish embryos results in craniofacial abnormalities including the complete loss of Meckel’s cartilage, the ceratohyal, and all of the ceratobranchial cartilages. These data are consistent with our previous work that demonstrated a role for HCFC1 in vertebrate craniofacial development. High throughput RNA-sequencing analysis reveals several overlapping gene targets of HCFC1 and THAP11. Thus, both HCFC1 and THAP11 play important roles in the regulation of cobalamin metabolism as well as other pathways involved in early vertebrate development.
Introduction
Mutations in the transcriptional co-regulator HCFC1 have been recently associated with cblX (MIM309541), an X-linked recessive disorder, characterized by defects in cobalamin metabolism, nervous system development, neurological impairment, and failure to thrive (1). Further, functional analyses have demonstrated that mutations in HCFC1 regulate cobalamin metabolism via MMACHC, one of its many downstream target genes (1). In addition to patients with cblX, mutations in HCFC1 have also been reported in individuals with X-linked intellectual disability (XLID) (2) and other multiple congenital anomalies (3,4), both with and without the metabolic phenotype characteristic of cblX.
HCFC1 is a transcriptional co-regulator that does not have a DNA binding domain and requires interaction with other transcription factors to modulate changes in gene expression (4–9). These interactions with transcription factors and other regulatory complexes allows HCFC1 to regulate many genes involved in diverse cellular functions including cell cycle, proliferation, and transcription (4,6,8). HCFC1 has a conserved kelch domain with 5 kelch repeats/motifs, which mediate protein-protein interactions (10). CblX-associated mutations in HCFC1 occur primarily within the kelch domain potentially disrupting its interactions with other transcription factors (1). Further, HCFC1 appears to regulate the expression of MMACHC through its interaction with THAP11, a THAP domain-containing transcription factor (11). Thus, mutations in the HCFC1 kelch domain presumably disrupt the interaction with THAP11, subsequently resulting in down-regulation of MMACHC expression in cblX patients (1,12,13).
A majority of the inborn errors of cobalamin metabolism have been associated with mutations in genes that encode proteins involved in the synthesis or intracellular transport of active cobalamin-derived cofactors (14). However, the discovery that mutations in HCFC1 can lead to phenotypes associated with cblX suggests that mutations in genes outside the central cobalamin pathway can also result in biochemical outcomes characteristic of an inborn error of cobalamin metabolism. Thus, it is likely that mutations in transcription factors or other regulatory proteins that interact with HCFC1, to regulate MMACHC or other genes in the cobalamin pathway, may also result in biochemical and other phenotypes similar to those observed in patients with cblX. THAP11 is one such candidate gene because the HCFC1/THAP11 complex binds to regulatory elements controlling the expression of MMACHC as well as a diverse array of downstream target genes (11,15).
Here, we report a patient who presented with phenotypic features that overlap cblX, but did not have any mutation in either MMACHC or HCFC1. We sequenced THAP11 by Sanger sequencing and discovered a potentially pathogenic, homozygous variant in THAP11, c.240C > G (p.Phe80Leu). Further, we provide functional data in model organisms that suggests that both HCFC1 and THAP11 are essential for normal brain development and neural precursor differentiation. Additionally, the loss of THAP11 results in craniofacial abnormalities, consistent with our previous work demonstrating a role for HCFC1 in craniofacial development (13). Furthermore, our high throughput RNA-sequencing analysis reveals several overlapping gene targets of HCFC1 and THAP11 that likely contribute to the biochemical as well as other phenotypes associated with cblX. Taken together, our results provide evidence that both HCFC1 and THAP11 play important roles in the regulation of cobalamin metabolism as well as other genes involved in early vertebrate development.
Results
Mutations in THAP11 cause a cblX-like disorder
We have previously reported HCFC1 mutations in 14/17 male subjects with cblX, who had initially been diagnosed with cblC but had no mutation in MMACHC (1). Based on the known interaction between HCFC1 and THAP11 and their role in the regulation of gene expression (11), we hypothesized that the three subjects with no detectable mutation in HCFC1, but with phenotypic features overlapping cblX, have mutations in THAP11. Sanger sequencing of THAP11 (NM_020457.2) revealed a homozygous, missense variant in one subject, while no variations in THAP11 were detected in the other two subjects. This novel variant in THAP11, c.240C > G, resulted in a single amino acid change, p.Phe80Leu (Fig. 1A). The subject with this variant presented with a cblX-like disorder characterized by seizures, which started at 2 months of age, in addition to mild methylmalonic acidemia. Treatment with hydroxocobalamin injections, pyridoxine, folic acid and betaine, were unable to prevent the progression of the child’s disease. The child developed very severe intellectual disability and seizures and ultimately died at the age of ten. The severity of the child’s brain involvement is consistent with the clinical presentation of cblX patients.
Figure 1.

THAP11 mutation in an individual with a cblX-like disorder. (A) Chromatograph of Sanger sequencing shows a novel homozygous c.240C>G (p.Phe80Leu) missense mutation in the subject. (B) Protein domains of THAP11 identified using UniProt include THAP-type zinc-finger DNA-binding domain (THAP), Glutamine-rich domain (Gln) domain, Alanine-rich domain (Ala), HCFC1 binding motif (HBM), and a coiled-coil domain (CC). (C) Evolutionary conservation of Phe80 (highlighted in red) in THAP11 demonstrated using comparative analysis of orthologs from multiple species. Orthologs were identified by using BLASTP, and the alignments were performed by using ClustalW. Protein accession numbers are in parentheses.
The c.240C > G, p.Phe80Leu variant (chr16:67,876,697, GRCh37/hg19) is novel and was not found in public databases, including dbSNP, HapMap, 1000 Genomes, Exome Variant Server (EVS), and Exome Aggregation Consortium (ExAC). Multiple mutation prediction algorithms also predict this variant to be deleterious (CADD: 23.1; PolyPhen-2: probably damaging (0.997); SIFT/PROVEAN: deleterious (−4.37); MutationTaster: disease causing (0.99997); FATHMM: damaging (−4.08)). Furthermore, the p.Phe80Leu alteration affects an amino acid that is highly conserved across vertebrate species and localizes to the functionally conserved THAP domain, an important zinc-finger DNA binding domain involved in transcriptional regulation (16) (Fig. 1B and C). These data provide strong evidence that the detected variant in THAP11 is likely to be pathogenic and is the underlying cause of the cblX-like disorder in this subject.
Knockdown of thap11 results in craniofacial abnormalities
We have previously demonstrated that patients with cblX have mild facial abnormalities and that knockdown of hcfc1b, a zebrafish ortholog of HCFC1, results in craniofacial defects (13). Further, these defects in facial development, resulted from deficits in mmachc expression (13). Since HCFC1 and THAP11 interact to modulate downstream gene expression, we hypothesized that a loss of thap11 in zebrafish would also result in craniofacial abnormalities. To address this hypothesis, we first used anti-sense morpholino oligonucleotides to reduce the expression of the zebrafish thap11. We phenotyped thap11 morphants for defects in craniofacial development with Alcian blue:Alizarin red to visualize the developing cartilage and bone, respectively. At 4 days post fertilization (dpf), knockdown of thap11 resulted in severe craniofacial abnormalities (Fig. 2B). Specifically, morphant embryos (n = 21) did not develop the major components of the viscerocranium, including Meckel’s cartilage, the ceratohyal, and the ceratobranchial cartilages (P < 0.0001) (Fig. 2B). Co-injection of THAP11 mRNA with thap11 morpholino (n=14) effectively restored all structures including Meckel’s cartilage, the ceratohyal, and the ceratobranchial cartilages (P = 0.0041) (Fig. 2D). Importantly, embryos injected with THAP11 mRNA alone had normal facial development (Fig. 2C). Thus, the knockdown of thap11 resulted in craniofacial abnormalities in the zebrafish.
Figure 2.
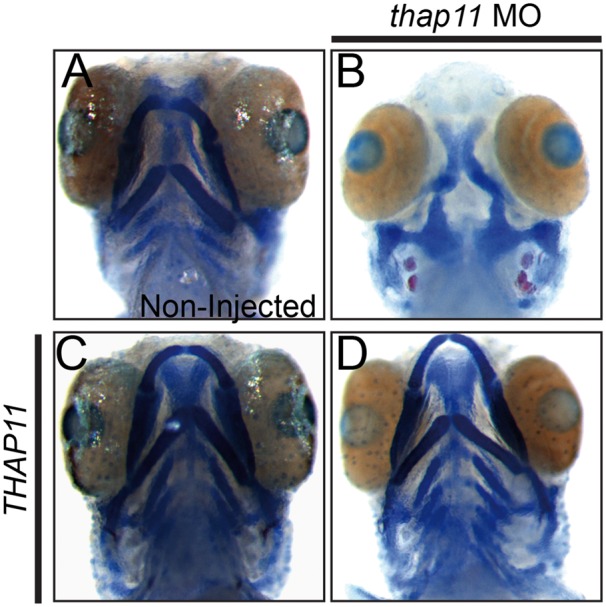
Thap11 regulates craniofacial development in zebrafish. (A–D) Alcian Blue-Alizarin Red staining of 4 day post fertilization (dpf) zebrafish embryos with the following treatment; (A) non-injected, (B) thap11 morpholino (thap11 Mo)-injected, (C) injected with wild-type THAP11 mRNA or (D) co-injected with thap11 morpholino and wild-type THAP11 mRNA.
Knockdown of thap11 results in structural brain malformations
cblX is also characterized by severe neurological impairment and brain abnormalities (1). Therefore, we hypothesized that knockdown of thap11 in the developing zebrafish would result in structural brain abnormalities. To test this hypothesis, we analysed the brains from 2-day-old zebrafish embryos injected with thap11 morpholino, using immunohistochemistry with antibodies against acetylated tubulin. Acetylated tubulin, which is found in stable axonal microtubules, can be used to visualize the structure of the brain and identify brain malformations (17). The knockdown of thap11 expression resulted in an abnormal pattern of acetylated tubulin in the morphant embryos when compared to non-injected embryos (Fig. 3A and B). The wildtype non-injected embryos (n = 9) demonstrate patterns of acetylated tubulin extending from the midbrain towards the fourth ventricle. However, in the morphants (n = 9) the extension of the axon tracks was significantly reduced in the presumptive hypothalamus and midbrain cerebellum, indicated by an open arrowhead and asterisk, respectively (Fig. 3A and B). This data suggested that the perturbation of thap11 expression in early embryogenesis can lead to structural brain abnormalities in the zebrafish.
Figure 3.

Thap11 regulates neural stem cell proliferation and differentiation. (A,B) 2 day post fertilization (dpf) zebrafish embryos analysed for anti-acetylated tubulin expression using antibody immunohistochemistry. Arrowhead indicates hypothalamus and asterisk labels the presumptive cerebellum. (A) Non-injected (NI), (B) thap11 morpholino (thap11 Mo). (C–H) Immunohistochemistry with anti-Sox2 (green) and anti-HuC/D (red) antibodies at 2 day post fertilization (dpf) from (C) non-injected, (D) thap11 morphants (thap11 Mo), (E) THAP11 mRNA injected (THAP11), (F) thap11 morphants co-injected with THAP11 mRNA, (G)THAP11 c. 240C>G mRNA injected, and (H) thap11 morphants co-injected with the THAP11 c240C>G. Arrowheads demonstrate regions of HuC/D expression and arrows indicate Sox2 expression. HuC/D expression is consistently observed in the optic tectum and tegmentum, regions indicated by the arrowheads in (C). A thin lining of neural precursors are present surrounding the ventricle region and indicated by the arrow. Anatomically comparable regions from serial sections are demonstrated in the treated groups. n = 9 per group. I. Bar graph showing the cell counts of Sox2+ cells in sections from C-H. NI= non-injected, MO = thap11 morpholino injected, THAP11 = wild type THAP11 mRNA injected, c240C>G = mutant THAP11 mRNA injected, MO + THAP11 = co-injected as in (F), MO + c240C>G = co-injected as in (H). The counts were carried out in 9 sections in each category (n = 9) and error bars are shown. Asterisks denote results that were statistically significant.
Precise control of THAP11 function is necessary for neural progenitor lineage commitment
One possible explanation for the reduction in the number of axons observed in thap11 morphants could be a decrease in the number of differentiated neurons. To test this possibility we used immunohistochemistry to evaluate the expression of the pan neuronal marker HuC/D (Elavl3) (18) and Sox2, a marker of neural progenitors (19). At 2 dpf we observed a thin layer of cells lining the ventricles of the optic tectum and midbrain tegmentum, and in the center of the hypothalamus expressing Sox2 in non-injected embryos (n = 9) (Fig. 3C). We counted the Sox2+ cells in sections from the forebrain, midbrain, and hindbrain from representative non-injected embryos. There were an average of 180 Sox2+ cells in the non-injected embryos which were adjacent to the HuC/D+ neurons within the tissue of the midbrain and hypothalamus (Fig. 3C). In contrast, the knockdown of thap11 resulted in an increase in the number of Sox2 + cells in the forebrain and midbrain (n = 9) relative to non-injected control embryos (Fig. 3C, D, G). The increased number of Sox2+ cells in representative morphant embryos represented a 1.8 fold increase in neural precursors (Fig. 3I, P < 0.01). Concomitantly, there was a decrease in the number of HuC/D+ cells and an expanded fourth ventricle in the morphants (n = 9) (Fig. 3C and D). The small number of cells expressing HuC/D in the morphant animals is consistent with the possibility that Thap11 is required for neuronal differentiation.
Based on the results from knockdown experiments, we wanted to determine if overexpression of thap11 would result in an aberrant phenotype. In order to test this, we injected wildtype embryos with human THAP11 mRNA. Embryos injected with THAP11 mRNA (n = 9) appeared normal in size and overall appearance, however, analysis of brain development in THAP11 injected embryos demonstrated a statistically significant increase, approximately 2.1 fold more, in the number of Sox2+ cells lining the fourth ventricle and the tectal ventricle region proximal to the hypothalamus (Fig. 3E and I, P = 0.006), relative to the non-injected control. However, THAP11 injected embryos maintained the ability to produce differentiated neurons in the hypothalamus and midbrain tegmentum, but not in the presumptive optic tectum, which had fewer HuC/D+ cells (Fig. 3E). Since the overexpression of THAP11 mRNA resulted in more Sox2+ cells than the knockdown of thap11 (Fig. 3I), we did not expect the THAP11 mRNA to rescue the morphant phenotype which also results in increased Sox2+ cells. As expected, the co-injection of THAP11 mRNA and thap11 morpholino resulted in embryos which maintained a significant number of Sox2+ cells, which was approximately 1.4 fold more than the non-injected controls (Fig. 3I). However, the number of Sox2+ cells in the co-injected embryos was 1.3 fold lower than the thap11 morphant suggesting some antagonistic effect, although this difference was not statistically significant (Fig. 3F and I). These data suggested that any changes in the level of thap11 expression, either by down-regulation or overexpression, during brain development can have a dramatic effect on the fate of neural precursors.
Having established that perturbations in THAP11 expression alter neuronal differentiation, we next wanted to test the effect of the c.240C > G (p.Phe80Leu) mutation in THAP11, on neuronal differentiation. To test this, we injected a mutated version of THAP11 mRNA containing the c.240C > G mutation into wildtype embryos (n = 9). Expression of the c.240C > G mRNA increased the population of cells expressing HuC/D and decreased the number of Sox2+ cells by approximately 3 fold (P < 0.001) (Fig. 3G and I). Sox2 positive cells were not present in the ventricular zone and we could not differentiate the optic tectum from the midbrain tegmentum (Fig. 3G). These results dramatically contrasted from the knockdown of thap11, which increased the number of Sox2+ cells, as well as from the overexpression of THAP11 mRNA, which increased the number of Sox2 positive cells and reduced the HuC/D expression in the optic tectum (Fig. 3C–I). The co-injection of c.240C > G variant and thap11 morpholino resulted in more Sox2+ cells relative to control non-injected embryos (Fig. 3H and I), but also had more HuC/D+ cells than the thap11 morphant (Fig. 3D). Thus, these data suggest that c.240C > G THAP11 mutation promotes a neuronal fate at the expense of neural precursors, a completely opposite phenotype when compared to the knockdown of thap11 mRNA. Furthermore, these data also demonstrate that the altered function of the c.240C > G variant is not sufficient to completely reverse the effect of knockdown by the thap11 morpholino.
Hcfc1 and Thap11 regulate overlapping cellular behaviors during brain development
We have previously demonstrated that hcfc1b, an ortholog of HCFC1, plays an important role in craniofacial development (13), similar to thap11. Although zebrafish have two paralogs, hcfc1a and hcfc1b, we focused on the hcfc1b ortholog because we have previously demonstrated that loss of hcfc1b results in a phenotype consistent with mutations in HCFC1, however loss of hcfc1a alone is not sufficient to replicate all of the phenotypes associated with cblX (13). We next wanted to test if hcfc1b, like thap11, also plays a role in brain development. To begin to test this hypothesis, we injected validated (13) hcfc1b antisense morpholinos to knockdown the expression of hcfc1b in the developing zebrafish. Similar to our analysis in thap11 morphants, we analysed both the number and location of neural precursors and neurons at 2 dpf using immunohistochemistry. In non-injected animals (n = 12), HuC/D was localized to both the hypothalamus and the midbrain bordering the Sox2 expression in the 4th and tectal ventricles (Fig. 4A). The phenotypes in hcfc1b morphants (n = 12) mirrored those observed in thap11 morphants, with approximately 2.8 fold more Sox2+ cells (Fig. 4A–C,G, P <0.001) surrounding the ventricular zone with an enlarged fourth ventricle region (Fig. 4C). These effects were more pronounced in the forebrain/midbrain when compared with the hindbrain. As expected, the knockdown of hcfc1a, resulted in a similar, yet more moderate phenotype (data not shown).
Figure 4.

hcfc1b regulates neural stem cell differentiation. (A–F) Sox2 and HuC/D expression analysis using antibody immunohistochemistry in 2 days post fertilization (dpf) zebrafish embryos are shown. (A) non-injected embryos (NI), (B) embryos injected with HCFC1 mRNA, (C) hcfc1b morpholino-injected (hcfc1b Mo), (D) co-injected with hcfc1b Mo and wild-type HCFC1 mRNA, (E) co-injected with hcfc1b Mo and mutated HCFC1 c.217G>A (217), (F) co-injected with hcfc1b MO and mutated HCFC1 c.344C>T (344) mRNA. Arrowheads demonstrate regions of HuC/D expression and arrows indicate Sox2 expression. For A, B, C, and D, n = 12 per group. For E&F, n = 9 per group. G) Bar graph showing the cell counts of Sox2+ cells in sections from A-F. NI= non-injected, MO = hcfc1b morpholino injected, HCFC1 = wild type HCFC1 mRNA injected, MO + HCFC1 = co-injected as in (D), MO + c.217 = co-injected as in (E), MO + c.344 = co-injected as in (F). The counts were carried out in 9 sections in each category (n = 9) and error bars are shown. Asterisks denote results that were statistically significant. Cells were counted in 12 sections each of A–D, and 9 sections each of E–F. Error bars are shown * denotes significance relative non-injected control embryos and ** denotes significance relative to hcfc1b morphant embryos.
We next asked whether overexpression of HCFC1 would result in an increased number of Sox2+ cells. Injection of HCFC1 mRNA caused a more moderate, yet statistically significant 1.8 fold increase in the number of Sox2+ cells (P = 0.009) (Fig. 4B and G). These data are consistent with the increase in the number of Sox2+ cells observed when THAP11 expression was perturbed (Fig. 3). Embryos co-injected with hcfc1b morpholino and HCFC1 mRNA had approximately 227 Sox2+ cells per section, which was more than observed in the non-injected control embryos, but not statistically significant. However, co-injected embryos had significantly fewer Sox2+ cells than observed in hcfc1b morphants (P < 0.05) (Fig. 4A–D,G). Furthermore, co-injection of HCFC1 mRNA and hcfc1b morpholino (n = 12) led to a reduction in the overall size of the fourth ventricle and an increase in the expression of HuC/D in the midbrain (Fig. 4D). Despite this improvement in neural phenotype of co-injected embryos, the fact that HCFC1 mRNA injection increases Sox2+ cells limits the ability of this assay to demonstrate complete rescue of the morphant phenotype. Taken together, these data suggest that any perturbations in HCFC1 or THAP11 expression interfere with Sox2 expression and neural precursor development.
We have previously reported multiple HCFC1 mutations in cblX patients (1) and have shown that the different mutations have different effects on the expression of downstream targets (1) as well as craniofacial phenotypes (13). We next wanted to test if the same variability will be observed in the effect of various mutations on the brain phenotypes. To test this, we injected mRNAs encoding the HCFC1 mRNAs resulting in a) c.217G > A, p.Ala73Thr or b) c.344C > T, p.Ala115Val mutations observed in our cblX patients (1). We focused on these two mutations, because we have previously demonstrated that the c.217G > A mutation has a milder effect on the expression of downstream targets than the c.344C > T (1). However, both mutations result in severe neurological impairment (1). The overexpression of these HCFC1 versions did not disrupt the number of neural precursors or lead to abnormalities in size and overall development (Supplementary Material, Fig. S1).
Next, we wanted to determine the effect of the expression of these variants on the neural developmental phenotype associated with knockdown of hcfc1b. To test this we co-injected hcfc1b morpholino with the two mutant versions of HCFC1, either with the a) c.217G > A, p.Ala73Thr (n = 9) or b) c.344C > T, p.Ala115Val mutations (n = 9) (1). We analysed the co-injected embryos at 2 dpf using anti-Sox2 and anti-HuC/D immunohistochemistry. Co-injection of either the c.217G > A HCFC1, or c.344C > T HCFC1, did not correct the expression of the Sox2 expressing cells (Fig. 4E and F). Embryos co-injected with the c.217G > A variant had an average of 238 Sox2+ cells per section (Fig. 4G), which was approximately 1.3 fold lower than the morphant, however these changes are not statistically significant (P = 0.12). Interestingly, co-injection of the c.217G > A HCFC1 did not improve the expression of HuC/D in any of the brain tissue. In contrast, embryos co-injected with c.344C > T HCFC1 mRNA resulted in a slightly larger region of HuC/D expressing cells than the c.217G > A HCFC1 co-injected ones, but they occupied significantly less tissue regions than those observed in the non-injected controls (Fig. 4A, E, F). Moreover, the c.344C > T co-injected embryos had an average 242 Sox2+ cells per section, a modest decrease from the hcfc1b morphant, but not a statistically significant difference (P = 0.14) (Fig. 4E–G). Thus, both of the cblX-associated mutations in HCFC1 did not improve the abnormal neuronal differentiation phenotype in hcfc1b morphants, suggesting impaired function of the mutant proteins.
Hcfc1 regulates the differentiation of neural precursors in the developing brain
We had observed that the Sox2 expressing cells in our hcfc1b morphants maintained an elongated shape, consistent with a proliferative population. In order to test if these cells were actively proliferating, we next evaluated whether morphant animals had higher numbers of proliferating cells in the developing forebrain using a 5-ethynyl-2'-deoxyuridine (EdU) uptake assay. EdU is a thymidine analog that is incorporated into the DNA of dividing cells. We pulsed 2 day old non-injected (n = 9) and morpholino injected embryos (n = 9) with EdU and then measured incorporation using EdU Click-it technology (Life Technologies). We observed an increase of EdU positive cells throughout the forebrain/midbrain, with significant EdU uptake in the cells surrounding the enlarged fourth ventricle in morphant animals (Fig. 5A and B). In addition, we noted a high number of EdU positive cells in the cells surrounding the developing eye. In the developing hypothalamus, an increase in EdU uptake was observed in close proximity to the tectal ventricle (Fig. 5A’ and B’). The number of EdU positive cells was higher in the midbrain, with proliferative cells occupying presumptive cerebellar tissue that would normally be occupied by post-mitotic neurons. These data are consistent with the expansion of Sox2 expression, as these cells are highly proliferative, and provide evidence that hcfc1b regulates neural precursor differentiation in the developing brain.
Figure 5.
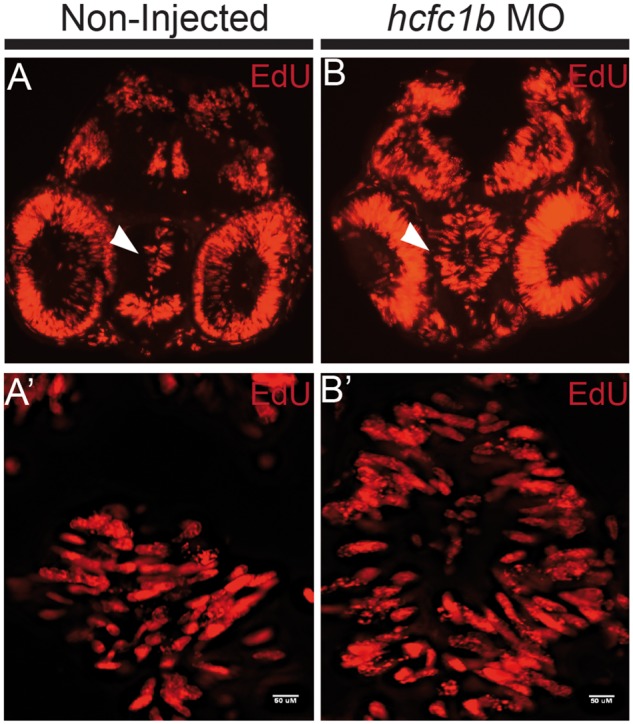
hcfc1b regulates neural stem cell proliferation. (A,B) Non-injected or hcfc1b morpholino (hcfc1b Mo) injected embryos were pulsed with 10mM 5-ethynyl-2’-deoxyuridine (EdU) and analysed using the EdU Click-It technology. (A’-B’). Higher magnification images of the area indicated by the arrowhead in A and B. n = 9 per group.
RNA-seq analysis of patient-derived fibroblasts
The overlap in the phenotypes observed between cblX patients and the patient with the THAP11 mutation, combined with our observations in the zebrafish experiments, suggested that HCFC1 and THAP11 regulate an overlapping set of genes involved in early vertebrate development. In order to test this possibility, we carried out a transcriptome analysis using RNA-seq on fibroblast lines derived from patient samples. RNA was isolated from seven control fibroblast lines, 12 cblX fibroblast lines, the proband with homozygous p.Phe80Leu mutation in THAP11, and 17 additional patients with inborn errors of cobalamin metabolism (2 cblD, 3 cblG, 3 cblE, 3 cblF, 3 cblC, and 3 patients with MTHFR deficiency) (Supplementary Material, Table S1). Given that we had only one subject with the THAP11 mutation, we first assessed by unsupervised clustering if there were similarities of this patients’ transcriptome as compared to fibroblasts derived from patients with mutations in other complementation groups, and control fibroblasts. The THAP11 patient transcriptome clustered in the same clade with majority of cblX patients; this was also seen in principal-component analyses (Supplementary Material, Fig. S2). We observed 46 genes that were differentially expressed in all cblX patients and the patient with the THAP11 mutation (Table 1). Furthermore, all these differentially expressed genes showed the same pattern of expression in the fibroblasts of cblX patients and the patient with the THAP11 mutation (Supplementary Material, Fig. S3). The most downregulated gene was TMOD2, followed by ZNF883, ZNF717, SEC11C, and MMACHC.
Table 1.
Top differentially expressed genes in CblX and THAP11 patients, selected as Bejamini-Hochberg corrected P-value < 0.05
| Gene | DESeq adjusted P-value | Gene | DESeq adjusted P-value |
|---|---|---|---|
| TMOD2 | 2.8x10−87 | ZNF132 | 0.00031 |
| ZNF883 | 1.1x10−27 | KBTBD8 | 0.00034 |
| ZNF717 | 3.5x10−25 | ANKRD13B | 0.00039 |
| SEC11C | 2.7X10−23 | CCDC163P | 0.0004 |
| MMACHC | 5.4X10−23 | FHIT | 0.00045 |
| NAT1 | 1.5X10−21 | ZNF154 | 0.00078 |
| LOC100134868 | 2.7X10−20 | ZNF74 | 0.0013 |
| ZNF454 | 1.3X10−19 | LGMN | 0.0015 |
| PPAPDC3 | 6X10−15 | DPF1 | 0.0016 |
| FBXL19−AS1 | 2.3X10−13 | MUTYH | 0.0016 |
| NEIL1 | 2.2X10−12 | MGC70870 | 0.0025 |
| ALG14 | 1.9X10−09 | PIEZO2 | 0.0036 |
| C16orf5 | 1.1X10−08 | TPTEP1 | 0.0055 |
| ZNF229 | 1.4X10−08 | SCN2A | 0.0075 |
| ADAM21 | 1X10−07 | ZNF140 | 0.011 |
| FSD1 | 8.4X10−07 | TMEM150A | 0.013 |
| PAFAH2 | 8.5X10−07 | ASB13 | 0.014 |
| RG9MTD2 | 1.2x10−06 | ARL10 | 0.014 |
| C3orf39 | 9.8x10−06 | ZNF334 | 0.019 |
| ZNF233 | 0.000018 | CDH13 | 0.028 |
| NPL | 0.000044 | ZIK1 | 0.031 |
| UBXN8 | 0.00015 | COQ10A | 0.038 |
| WDR46 | 0.00017 | VANGL2 | 0.043 |
Besides MMACHC, no other genes known to be involved in the cobalamin pathway were affected, confirming our hypothesis that, similar to cblX patients, the aberrant cobalamin metabolism phenotype in the fibroblasts of the patient with THAP11 mutation is caused by a lack of MMACHC expression. This is further supported by our observation that retroviral transduction of MMACHC into patient-derived fibroblasts correct the biochemical phenotype measured by [14C]propionate incorporation (Supplementary Material, Fig. S4). Furthermore, there is a reduction in mRNA expression of MMACHC and TMOD2 in both the THAP11 patient and cblX patients, while the expression of HCFC1 and THAP11 in both remains similar to control levels (Fig. 6). Our data support the idea that HCFC1 and THAP11 co-regulate an overlapping set of genes which may play a role in the phenotypes observed in patients with cblX and related disorders.
Figure 6.
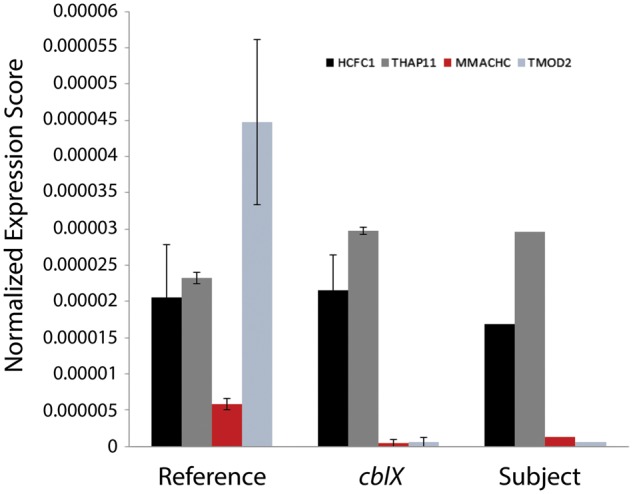
Expression of target genes in patient samples. Normalized expression of MMACHC, TMOD2, HCFC1, and THAP11 in control, cblX patients, and THAP11 patients are shown. The Reference includes average expression from 7 control individuals, cblX includes average expression from 12 patients with cblX, and Subject includes expression data from the single patient with the THAP11 c.240C > G mutation.
Discussion
We have identified an individual presenting with a cblX-like syndrome who did not have mutations in the known candidate gene, HCFC1. Instead, this individual was found to carry a homozygous missense mutation in THAP11. HCFC1 is known to interact with THAP11, a transcription factor, in the regulation of the expression of downstream targets including MMACHC, an enzyme in the cobalamin pathway (1,11,15). THAP11 belongs to a family of THAP proteins, all of which have been demonstrated to contain a conserved HCFC1 binding motif (HBM) (7). The missense mutation we have identified in THAP11, c.240C > G, alters a highly conserved phenylalanine residue at amino acid 80 to a leucine, within the DNA-binding THAP domain (Fig. 1 (20)). Homozygous mutation of c.240C > G suggested that cblX-like syndrome is a recessive condition caused by the mutation in THAP11. It is supported by the fact that c.240C > G is novel and there is no homozygote with this mutation reported in any publicly available database, such as Exome Variant Server (EVS) or Exome Aggregation Consortium (ExAC). This alteration likely affects the ability of THAP11 to efficiently bind the promoters of its downstream targets. Alternatively, the mutation we have uncovered may affect the conformation of THAP11 and its ability to interact with HCFC1 or other protein interactors. The THAP11 HBM begins at amino acid 254, which is 4–9 amino acids upstream the conserved coiled-coil domain (Fig. 1), which is involved in dimerization of transcription factors at gene promoters (21). It is noteworthy that the mutations identified in HCFC1 affect the kelch domain, a domain that is also important for protein-protein interactions (1,22). Our finding strongly suggests that mutations in either HCFC1 or THAP11 can lead to dysregulation of MMACHC and other downstream targets, subsequently resulting in the phenotypes associated with cblX.
We have previously shown that individuals with cblX, carrying mutations that affect the kelch domain of HCFC1, often have dysmorphic features, which was further supported by our knockdown studies of hcfc1b in zebrafish (1,3,13). Furthermore, this role of HCFC1 in craniofacial development is modulated, at least in part by one of its downstream targets, MMACHC (13). We have demonstrated here that even THAP11 is important in craniofacial development as knockdown of thap11 in zebrafish led to craniofacial abnormalities. We observed a complete loss of the viscerocranium in the thap11 morphants, which is consistent with results obtained in both hcfc1b and mmachc morphants (13). These data strongly support the role of HCFC1 and THAP11 in the regulation of facial development. Although, the exact mechanisms and the downstream effectors, other than MMACHC, of facial development regulated by HCFC1 and THAP11 remain to be further elucidated, our previous data support an impairment in the differentiation of cranial neural crest cells (CNCCs) which give rise to the cartilaginous structures of the viscerocranium (13).
In addition to craniofacial abnormalities and deficits in cobalamin metabolism, individuals with cblX have significant neurological impairment (1). Recent evidence from in vitro assays in murine neural stem cells have demonstrated that aberrant expression of HCFC1 results in defects in both neural precursor proliferation and neuronal differentiation (2,12). Here, we tested the role of both Hcfc1 and Thap11 in neuronal differentiation using morpholino mediated knockdown in the developing zebrafish embryo. We show that the reduction in expression of either gene results in an increase in the number of proliferating neural precursors at the expense of neuronal differentiation. Our Hcfc1 knockdown is consistent with similar observation in the developing mouse brain with loss-of-function mutations in Hcfc1 (2,12). The function of THAP11 in brain development appears to be very complex relying on precise control of the expression of THAP11, where either the overexpression or knockdown can result in neural precursor dysfunction. However, the observation that the expression of the mutated THAP11 results in a unique phenotype, biasing differentiation towards a neuronal phenotype, was quite intriguing. This phenotype is the opposite of what is observed when the level of wildtype THAP11 expression is aberrant, suggesting that the mechanism by which THAP11 regulates downstream genes may be more complicated. THAP11 is known to regulate a significant fraction of the genome including genes involved in metabolism (4), the electron transport chain (23), and cell proliferation (15). Presumably, mutations in THAP11 modify its ability to interact with protein partners, which can subsequently lead to qualitative differences in the expression of target genes, thus resulting in unique phenotypes. Qualitative differences in gene expression have been noted for other transcription factors, namely those of the MYB family (24,25). In contrast, direct perturbation of wildtype THAP11 expression may not lead to qualitative differences, but rather quantitative differences in gene expression, leading to a significantly different phenotype.
HCFC1 and THAP11 are known to interact with other transcription factors including additional THAP family members, YY1, and ZNF143 to regulate the expression of over 5000 mammalian genes (6,7,9,16,26,27). Furthermore, mutations in ZNF143 were recently also shown to have an effect on cobalamin metabolism (28). The THAP11:HCFC1 protein complex has been shown to be important for the growth of embryonic stem cells (11,15). Thus, it is likely that both HCFC1 and THAP11 play a bigger role as master regulators of development and disease. However, our data suggest that the mutations observed in HCFC1 and THAP11 in individuals with cblX, or disorders with overlapping phenotypes, lead to the dysregulation of only a subset of all the genes that are potentially regulated by these two transcription regulators. This is further supported by the observation that mutations in HCFC1, which are outside the kelch domain or in regulatory sequences, have a phenotypic spectrum that may not necessarily include defects in cobalamin metabolism (2,3). Therefore, it is possible that specific mutations in HCFC1 and THAP11 will have distinct phenotypic outcomes resulting from the dysregulation of subsets of downstream effectors that are either partially or completely non-overlapping. This is further supported by our current and previous observations that distinct cblX-associated mutations have variable effects on the expression of MMACHC (1), craniofacial development (13) and neurological impairment.
Since HCFC1 and THAP11 co-regulate multiple genes, we would expect to see an overlapping set of downstream genes to be affected in cblX patients and the patient with THAP11 mutation. We used high-throughput RNA sequencing in patient derived cell lines in order to identify these common effector genes. We identified a small number of differentially expressed genes in both cblX and THAP11 patient fibroblasts as shown in Table 1. We verified that either mutation of HCFC1 or THAP11 results in reduced MMACHC expression. Our RNA sequencing data also demonstrated significant differential expression of TMOD2 which encodes a neuronal specific tropomodulin that regulates actin elongation and depolymerization via interaction with tropomysin (29). Tropomodulins are negative regulators of neurite growth and mutations in tropomodulin affect its affinity for tropomysin altering neurite formation and extension (30–32). Furthermore, a loss of TMOD2 in mice causes defects in learning and memory, and may suggest a mechanistic role in the intellectual disability/developmental delay observed in cblX (1,33). Interestingly, TMOD2 RNA was also strongly downregulated in the patient with ZNF143 mutations (data available in European Genome-phenome Archive). We are aware that our expression analysis is very preliminary and available information on many of the differentially expressed genes is not sufficient to predict a role in the clinical phenotype of the patient. However, these preliminary data provide a glimpse into the types of genes regulated by HCFC1 and THAP11 whose perturbation may lead to the phenotypes observed in individuals with cblX. A more comprehensive and detailed analysis of the transcriptional dysregulation in patient samples is needed to further elucidate the pathophysiology of HCFC1 and THAP11 mutations in cblX and related disorders.
In summary, we have discovered that a mutation in THAP11 can also result in a cblX-like disorder characterized by defects in cobalamin metabolism, intractable epilepsy and intellectual disability. Loss of both thap11 and hcfc1b leads to structural brain deficits in zebrafish, apparently due to an increase in neural precursor proliferation and a concomitant decrease in neuronal differentiation. Further, we have used patient derived fibroblasts to identify several candidate genes that may contribute to the phenotypes associated with cblX. Our in vivo functional analysis demonstrates that knockdown of hcfc1 or thap11 results in an overlapping craniofacial and brain phenotypes suggesting that the HCFC1 and THAP11 function as a unit to promote the development of these tissues. It is likely that other transcription factors interact with HCFC1 and/or THAP11, and that mutations in these could also lead to cblX-like disorders. Thus, we suggest that patients presenting with cblX-like phenotypes of unknown genetic etiology should be tested for mutations in the known HCFC1:THAP11 protein interacting partners.
Materials and Methods
Human samples
Patient fibroblast cell lines from the skin tissue biopsy were obtained from the Repository for Human Mutant Cells at the Montreal Children’s Hospital. Cells were grown in minimum essential medium plus non-essential amino acids (Wisent Bioproducts, St Bruno QC) with additional 5% fetal bovine serum (Wisent) and 5% iron enriched calf serum (Wisent).
Clinical summary of the THAP11 patient
The patient was the first born child to unrelated Moroccan parents living in Denmark (Supplementary Material, Fig. S5). He was born in 1992 at 34 weeks gestation with a birth weight of 1820 g. Head circumference at birth was not available. He had no congenital heart defects or other structural anomalies. At two months of age, the boy presented with myoclonic seizures, and at four months his overall development was delayed. At twelve months, a metabolic work-up showed mild methylmalonic aciduria in the range of 191–383 µmol MMA/mmol creatinine (reference range for children >6 months is n.d. – 6.4 µmol MMA/mmol creatinine); methylcitrate was also present in the urine, but homocystine could not be detected. Plasma methionine was in the low-normal range. Treatment was initiated with a protein-restricted diet, intramuscular hydroxocobalamin injections, pyridoxine, folic acid, and betaine, none of which had any effect on his methylmalonic aciduria or on the course of his disease. He gradually developed encephalopathy, tetraplegia and profound mental retardation. In the late 90s, when the measurement of total plasma homocysteine became available, his levels were within the range of 9.4–13.1 µM (reference range <15 µM). The patient had an eye exam in 2001, which showed esotropia and +4/+5 hyperopia. CNS imaging was never performed. The boy died from pneumonia at age ten, and the parents did not consent to postmortem examination. The family declined subsequent prenatal diagnosis and has since had four healthy children. They did not respond to an offer of carrier testing and genetic counseling based on the new genetic findings.
Studies of cultured patient fibroblasts showed decreased [14C]propionate and [14C]methyltetrahydrofolate incorporation (measurements of the function of the cobalamin-dependent enzymes methylmalonyl-CoA mutase and methionine synthase, respectively). Propionate incorporation was decreased to a smaller extent than usually observed in cblC patients. Patient fibroblasts accumulated less cobalamin than did control cells when incubated with [57Co]cyanocobalamin, and synthesis of both adenosylcobalamin and methylcobalamin was decreased, although to a smaller extent than usually seen in cblC patients. The results of the complementation were consistent with a diagnosis of cblC.
Retroviral transduction
Fibroblast lines were immortalized by transducing the E7 gene from the human papilloma virus and the human telomerase using the protocol described in Yao and Shoubridge (34). The THAP11 construct was purchased in pDONR vector (GeneCopoeia, Rockville, USA) and was cloned into pLXSH mammalian retroviral expression vector using the Gateway cloning system (Invitrogen, Carlsbad, USA). The MMACHC-GFP-pLXSH vector was cloned as previously described (35). Retroviral transduction was performed on immortalized patient and control fibroblasts. Phoenix packaging cells were plated in 10 cm tissue culture plates to be at 50–75% confluence on the first day of the procedure. They were treated with 3.75 µl of 50 mM chloroquine in 7.5 ml of media, incubated for 5 min at 37 °C to prepare the cells for transient transfection. The following was then added to the cells: 5–10 µg of DNA construct in pBABE or pLXSH vector, 2.5 µl salmon sperm DNA, 152 µl 2M CaCl2 in 2500 µl of HBS buffer pH 7.05. After 24 h, the media was changed and target cells were plated to be 50–60% confluent the following day. 24 h later the Phoenix supernatant containing the retroviral particles were passed through a 0.45 µm filter. 4 mL of the supernatant was mixed with 4 µl of 4 mg/µl of polybrene (Merck Millipore, Billerica, USA) and added to the target cells. They were incubated at 37 °C for 4 h then another 6 mL of supernatant and 6 µl of polybrene were added and the cells were infected overnight. The following day the media was detoxified and cells were split if confluent. Antibiotic selection began 2 to 3 days later with 100 µg/mL of hygromycin for pLXSH vectors on 50% confluent cells. Cells transfected with pBABE and pLXSH vectors were maintained in medium with added 1 µg/µl puromycin and 100 µg/µl of hygromycin respectively.
[14C]propionate incorporation
[14C]propionate incorporation indirectly measures the function of the mitochondrial MCM (Methylmalonyl CoA Mutase) enzyme. 400,000 cells were plated from each line in triplicates in 35 mm tissue culture dishes and allowed to grow for 48 h. The cultures were then incubated in medium containing 0.1 µM [14C]propionate for 18 h. The labelled cells were washed with PBS three times, following which the cellular macromolecules were precipitated in 3 treatments of 1.5 ml 5% trichloroacetic acid for 15 min at 4 °C. The precipitate was subsequently resuspended in 1 mL 0.2N NaOH and incubated for 4 h at 37 °C. The amount of radioactivity was determined by liquid scintillation counting, taking 700 µl of the NaOH mixture. The values were normalized to protein levels, determined by the Lowry assay done on 100 µl of the NaOH mixture.
Sanger sequencing
Genomic DNA from the subject (100 ng) were amplified by using two sets of primers specific to THAP11 (NM_020457.2). Forward primer 1: 5’-GAAAGAATCTGCTCTTCGGAGGCTATCGCA-3’, reverse primer 1: 5’- AAACTCCACTTGCACTGTGAGATCGATGGG-3’, forward primer 2: 5’- CCAGCTGCAGCCGAACCTGGTATCT-3’, and reverse primer 2: 5’- GCTGAGGCCTATTTCCCCATGTTTCAAGGA-3’. PCR reaction and conditions were as follows: Promega (Madison, WI) GoTaq Hot Start kit with 1X Master Mix and 400nM of each primer. PCR began with an initial cycle at 95 °C for 3 min, followed by 30 cycles of 94 °C for 30 s, 60 °C for 30 s and 72 °C for 1 min, finishing with extension at 72 °C for 5 min. Amplified PCR products were sequenced using the PCR primers as sequencing primers on an ABI (Carlsbad, CA) PRISM 3730xl at a commercial sequencing facility.
Zebrafish maintenance
For all experiments, embryos [Tupfel long fin (TL) or (AB)] were maintained in embryo medium at 28.5° C. Zebrafish (Danio rerio) were maintained at the University of Colorado Anschutz Medical Campus or The University of Texas at El Paso according to the Institutional Animal Care and Use Committee (IACUC) guidelines (protocols #B-85411(08)1D or 811689-5).
Morpholino injections
hcfc1b morpholino injections and HCFC1 mRNA rescue experiments were performed as previously described (13). Co-injection with a morpholino targeting the tp53 gene was performed as a control for off target effects. For thap11 knockdown experiments, embryos were injected with 2nL of 0.5 mM thap11 translational inhibiting morpholino (GeneTools LLC, Philomath, OR) (ACGCAGCAAGTGAAGCCGGGCATGA) at the single cell stage. For THAP11 experiments, in vitro synthesized mRNA was produced from a THAP11 cDNA clone MGC. 20469 (GE Dharmacon, Lafayette, CO) using Sp6 mMessage Machine kit (Life Technologies, Grand Island, NY) according to manufacturer’s instructions. Plasmid DNA was linearized either with HpaI (New England Biolabs, Ipswitch, MA) or SalI (ThermoFisher, Grand Island, NY). Approximately, 800pg of THAP11 mRNA was injected per embryo.
Sequential staining of bone and cartilage
Embryos were harvested at 4 days post fertilization and fixed for 1 h in 2% paraformaldehyde (Boston BioProducts, Ashland, MA) at room temperature. They were washed in wash buffer [100mM Tris pH 7.5 (Boston BioProducts, Ashland, MA), 10mMgCl2 (Boston BioProducts, Ashland, MA), 80% ethanol]. Embryos were then sequentially rehydrated in 80%, 50%, and 25% ethanol buffered in 100mM Tris pH 7.5 (Boston BioProducts, Ashland, MA). Stained larvae were bleached in 3% hydrogen peroxide (Sigma, St. Louis, MO) and 0.5% potassium hydroxide (KOH) (Boston BioProducts, Ashland, MA) at room temperature for 10 min to remove pigment and then were washed in 25% glycerol (Sigma, St. Louis, MO) containing 100 mM Tris pH 7.5 (Boston BioProducts, Ashland, MA). Subsequent bone staining was performed by incubation of alizarin red stain (0.01%) (Sigma, St. Louis, MO) for 30 min at room temperature. Embryos were destained in 50% glycerol (Sigma, St. Louis, MO) for 10 min at room temperature and stored at 4°C for future analysis. Statistical analysis of the craniofacial phenotype was performed using a Fisher’s exact T-test comparing wildtype embryos with thap11 morpholino injected embryos or thap11 injected embryos with thap11 morpholino co-injected with THAP11 mRNA. Abnormal embryos were classified as those with an abnormal ceratohyal angle, truncated Meckel’s cartilage, and/or missing ceratobranchial cartilages.
Immunohistochemistry
Embryos were fixed in 4% paraformaldehyde (Boston BioProducts, Ashland, MA) for 2 h at room temperature, then washed in PBS and embedded in 1.5% agar and 5% sucrose. Embedded blocks were incubated in 30% sucrose overnight, dried, frozen on dry ice, and sectioned with at cryostat microtome (20 µM). Slides were incubated with appropriate antibody; anti-acetylated tubulin (1:1000 dilution) (Sigma, St. Louis, MO), anti-sox2 (1:250 dilution) (Abcam, Cambridge, MA), anti-HuC/D (1:500 dilution) (Life Technologies, Grand Island, NY) overnight at 4° C. Samples were washed in PBS and incubated with an appropriate Alexa Fluor secondary antibody (Life Technologies, Grand Island, NY). Stained samples were washed and cover slipped with VectaShield (Vector Laboratories, Burlingame, CA) and imaged with appropriate filter. Images were captured using an LSM 700 confocal microscope fitted with Zen 2009 software or a Ziess Axiovert 200 including Velocity Software from PerkinElmer. Image manipulations were limited to levels and contrast adjustments. The total number of Sox2+ cells were counted from serial sections obtained from injected embryos. Statistical analysis was performed using a student’s T-test.
RNA-sequencing
RNA was isolated from seven control fibroblast lines, 12 cblX fibroblast lines (12/14 cblX patients described in Yu et al. (1)), the THAP11 patient, and 17 additional patients with inborn errors of cobalamin metabolism (2 cblD, 3 cblG, 3 cblE, 3 cblF, 3 cblC, and 3 patients with MTHFR deficiency) (Supplementary Material, Table S1). Cell lines for RNA sequencing studies were grown in T75 tissue culture flasks until confluent. To ensure uniform cell treatment in order to minimize colluding effects on gene expression, cells were plated at a density of 9 million cells per flask and allowed to grow for five days. Cells were visually inspected at this point, ensuring confluency, regular cell shape and no abnormalities. If any significant abnormalities were noted, the cultures were replated. If the cultures looked well, they were harvested, pelleted and dissolved in 2 mL of Tri-Reagent (Sigma) and immediately frozen at −80 °C.
Total RNA from the suspended cell pellets was extracted using the miRNAeasy Mini Kit (Qiagen). Quality was assessed using the Agilent 2100 Bioanalyzer and quantified on the Nanodrop (Thermo Scientific). The RNA seq library was prepared using the TruSeq Stranded Total RNA Sample Preparation Kit (Illumina RS-122–2301) including Ribo-Zero Gold depletion to remove ribosomal RNA. The library quality was tested using Agilent 2100 Bioanalyser on the DNA-1000 chip.
The samples were indexed and four samples per lane were run on the Illumina Hi-Seq2000 sequencer, using 100bp paired end reads. The raw sequencing reads were trimmed for quality with a phred quality score between 33 ≥ 30 and a length greater than 32bp. The Illumina adapters were removed using Trimmomatic v0.22. The filtered reads were aligned to the reference hg19 human genome using TopHat v1.4.1 and Bowtie v0.12.8. Duplicate reads were removed using Picard’s MarkDuplicates.jar v1.80 (http://picard.sourceforge.net). Fragment per Kilobase of exon model per Million mapped fragments (FPKM) values for the UCSC and Ensembl genes and transcripts were obtained using Cufflinks v2.1.1. FPKM normalizes the abundance of reads from a given gene for the transcript length and library size. The reads were counted using HTSeq-Count. DESeq was used to determine differential gene expression between patients and controls. Differences in gene expression between patients and age and sex-matched controls were identified in normalized RNA-seq using ANOVA. The RNA-seq data is in the process of being deposited to the European Genome-phenome Archive (EGA).
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
BA was supported in part by grants from the National Institutes of Health (NIH) and the Gates Frontiers Fund. AMQ was supported by institutional training grant T32MH015442 and 2G12MD007592 and by an institutional mentored award M-14-166 from the Colorado Clinical and Translational Science Institute. The University of Colorado, School of Medicine Zebrafish Core Facility was supported by National Institutes of Health (NIH). AB was supported by a Frederick Banting and Charles Best Canada Graduate Scholarship.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health (NIH) (GM081519, NS062717, P30 NSO48154), Institutional training grant T32MH015442 and 2G12MD007592, Gates Frontiers Fund, Colorado Clinical and Translational Science Institute and Frederick Banting and Charles Best Canada Graduate Scholarship.
References
- 1. Yu H.-C., Sloan J.L., Scharer G., Brebner A., Quintana A.M., Achilly N.P., Manoli I., Coughlin C.R. 2nd, Geiger E.A., Schneck U.. et al. (2013) An X-Linked cobalamin disorder caused by mutations in transcriptional coregulator HCFC1. Am. J. Hum. Genet., 93, 506–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang L., Jolly L.A., Willis-Owen S., Gardner A., Kumar R., Douglas E., Shoubridge C., Wieczorek D., Tzschach A., Cohen M.. et al. (2012) A noncoding, regulatory mutation implicates HCFC1 in Nonsyndromic Intellectual Disability. Am. J. Hum. Genet., 91, 694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gérard M., Morin G., Bourillon A., Colson C., Mathieu S., Rabier D., Billette de Villemeur T., Ogier de Baulny H., Benoist J.F. (2015) Multiple congenital anomalies in two boys with mutation in HCFC1 and cobalamin disorder. Eur. J. Med. Genet., 58, 148–153. [DOI] [PubMed] [Google Scholar]
- 4. Tyagi S., Herr W. (2009) E2F1 mediates DNA damage and apoptosis through HCF-1 and the MLL family of histone methyltransferases. embo J., 28, 3185–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Knez J., Piluso D., Bilan P., Capone J.P. (2006) Host cell factor-1 and E2F4 interact via multiple determinants in each protein. Mol. Cell. Biochem., 288, 79–90. [DOI] [PubMed] [Google Scholar]
- 6. Machida Y.J., Machida Y., Vashisht A.A., Wohlschlegel J.A., Dutta A. (2009) The deubiquitinating enzyme BAP1 regulates cell growth via interaction with HCF-1. J. Biol. Chem., 284, 34179–34188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mazars R., Gonzalez-de-Peredo A., Cayrol C., Lavigne A.-C., Vogel J.L., Ortega N., Lacroix C., Gautier V., Huet G., Ray A.. et al. (2010) The THAP-zinc finger protein THAP1 associates with coactivator HCF-1 and O-GlcNAc transferase: a link between DYT6 and DYT3 dystonias. J. Biol. Chem., 285, 13364–13371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wysocka J., Myers M.P., Laherty C.D., Eisenman R.N., Herr W. (2003) Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev., 17, 896–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Michaud J., Praz V., Faresse N.J., Jnbaptiste C.K., Tyagi S., Schütz F., Herr W. (2013) HCFC1 is a common component of active human CpG-island promoters and coincides with ZNF143, THAP11, YY1, and GABP transcription factor occupancy. Genome Res., 23, 907–916. 10.1101/gr.150078.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Adams J., Kelso R., Cooley L. (2000) The kelch repeat superfamily of proteins: propellers of cell function. Trends Cell Biol., 10, 17–24. [DOI] [PubMed] [Google Scholar]
- 11. Dejosez M., Levine S.S., Frampton G.M., Whyte W.A., Stratton S.A., Barton M.C., Gunaratne P.H., Young R.A., Zwaka T.P. (2010) Ronin/Hcf-1 binds to a hyperconserved enhancer element and regulates genes involved in the growth of embryonic stem cells. Genes Dev., 24, 1479–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jolly L.A., Nguyen L.S., Domingo D., Sun Y., Barry S., Hancarova M., Plevova P., Vlckova M., Havlovicova M., Kalscheuer V.M.. et al. (2015) HCFC1 loss-of-function mutations disrupt neuronal and neural progenitor cells of the developing brain. Hum. Mol. Genet., 24, 3335–3337. 10.1093/hmg/ddv083. [DOI] [PubMed] [Google Scholar]
- 13. Quintana A.M., Geiger E.A., Achilly N., Rosenblatt D.S., Maclean K.N., Stabler S.P., Artinger K.B., Appel B., Shaikh T.H. (2014) Hcfc1b, a zebrafish ortholog of HCFC1, regulates craniofacial development by modulating mmachc expression. Dev. Biol., 396, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Watkins D., Rosenblatt D.S. (2011) Inborn errors of cobalamin absorption and metabolism. Am. J. Med. Genet. C Semin. Med. Genet., 157C, 33–44. [DOI] [PubMed] [Google Scholar]
- 15. Dejosez M., Krumenacker J.S., Zitur L.J., Passeri M., Chu L.-F., Songyang Z., Thomson J.A., Zwaka T.P. (2008) Ronin is essential for embryogenesis and the pluripotency of mouse embryonic stem cells. Cell, 133, 1162–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gervais V., Campagne S., Durand J., Muller I., Milon A. (2013) NMR studies of a new family of DNA binding proteins: the THAP proteins. J. Biomol. NMR, 56, 3–15. [DOI] [PubMed] [Google Scholar]
- 17. Cambray-Deakin M.A., Burgoyne R.D. (1987) Posttranslational modifications of alpha-tubulin: acetylated and detyrosinated forms in axons of rat cerebellum. J. Cell Biol., 104, 1569–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marusich M.F., Furneaux H.M., Henion P.D., Weston J.A. (1994) Hu neuronal proteins are expressed in proliferating neurogenic cells. J. Neurobiol., 25, 143–155. [DOI] [PubMed] [Google Scholar]
- 19. Ellis P., Fagan B.M., Magness S.T., Hutton S., Taranova O., Hayashi S., McMahon A., Rao M., Pevny L. (2004) SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev. Neurosci., 26, 148–165. [DOI] [PubMed] [Google Scholar]
- 20. Cukier C.D., Maveyraud L., Saurel O., Guillet V., Milon A., Gervais V. (2016) The C-terminal region of the transcriptional regulator THAP11 forms a parallel coiled-coil domain involved in protein dimerization. J. Struct. Biol., 194, 337–346. [DOI] [PubMed] [Google Scholar]
- 21. Burkhard P., Stetefeld J., Strelkov S.V. (2001) Coiled coils: a highly versatile protein folding motif. Trends. Cell Biol., 2, 82–88. [DOI] [PubMed] [Google Scholar]
- 22. Li X., Zhang D., Hannink M., Beamer L.J. (2004) Crystal structure of the Kelch domain of human Keap1. J. Biol. Chem., 279, 54750–54758. [DOI] [PubMed] [Google Scholar]
- 23. Poché R.A., Zhang M., Rueda E.M., Tong X., McElwee M.L., Wong L., Hsu C.-W., Dejosez M., Burns A.R., Fox D.A.. et al. (2016) Ronin (Thap11) is an essential transcriptional regulator of genes required for mitochondrial function in the developing retina. Cell Rep., 14, 1684–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rushton J.J., Davis L.M., Lei W., Mo X., Leutz A., Ness S.A. (2003) Distinct changes in gene expression induced by A-Myb, B-Myb and c-Myb proteins. Oncogene, 22, 308–313. [DOI] [PubMed] [Google Scholar]
- 25. Liu F., Lei W., O’Rourke J.P., Ness S.A. (2006) Oncogenic mutations cause dramatic, qualitative changes in the transcriptional activity of c-Myb. Oncogene, 25, 795–805. [DOI] [PubMed] [Google Scholar]
- 26. Ajuh P.M., Browne G.J., Hawkes N.A., Cohen P.T., Roberts S.G., Lamond A.I. (2000) Association of a protein phosphatase 1 activity with the human factor C1 (HCF) complex. Nucleic Acids Res., 28, 678–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luciano R.L., Wilson A.C. (2003) HCF-1 functions as a coactivator for the zinc finger protein Krox20. J. Biol. Chem., 278, 51116–51124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pupavac M., Watkins D., Petrella F., Fahiminiya S., Janer A., Cheung W., Gingras A.-C., Pastinen T., Muenzer J., Majewski J.. et al. (2016) Inborn error of cobalamin metabolism associated with the intracellular accumulation of transcobalamin-bound cobalamin and mutations in ZNF143, which codes for a transcriptional activator. Hum. Mutat., 37, 976–982. [DOI] [PubMed] [Google Scholar]
- 29. Watakabe A., Kobayashi R., Helfman D.M. (1996) N-tropomodulin: a novel isoform of tropomodulin identified as the major binding protein to brain tropomyosin. J. Cell Sci., 109, 2299–2310. [DOI] [PubMed] [Google Scholar]
- 30. Guillaud L., Gray K.T., Moroz N., Pantazis C., Pate E., Kostyukova A.S. (2014) Role of tropomodulin’s leucine rich repeat domain in the formation of neurite-like processes. Biochemistry (Mosc.), 53, 2689–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moroz N., Guillaud L., Desai B., Kostyukova A.S. (2013) Mutations changing tropomodulin affinity for tropomyosin alter neurite formation and extension. PeerJ., 1, e7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fath T., Fischer R.S., Dehmelt L., Halpain S., Fowler V.M. (2011) Tropomodulins are negative regulators of neurite outgrowth. Eur. J. Cell Biol., 90, 291–300. 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cox P.R., Fowler V., Xu B., Sweatt J.D., Paylor R., Zoghbi H.Y. (2003) Mice lacking Tropomodulin-2 show enhanced long-term potentiation, hyperactivity, and deficits in learning and memory. Mol. Cell. Neurosci., 23, 1–12. [DOI] [PubMed] [Google Scholar]
- 34. Yao E., Shoubridge E.A. (1999) Expression and functional analysis of SURF1 in Leigh syndrome patients with cytochrome c oxidase deficiency. Hum. Mol. Genet., 8, 2541–2549. [DOI] [PubMed] [Google Scholar]
- 35. Mah W., Deme J.C., Watkins D., Fung S., Janer A., Shoubridge E.A., Rosenblatt D.S., Coulton J.W. (2013) Subcellular location of MMACHC and MMADHC, two human proteins central to intracellular vitamin B12 metabolism. Mol. Genet. Metab., 108, 112–118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.