

Abstract
Usher syndrome type 1C (USH1C/harmonin) is associated with profound retinal, auditory and vestibular dysfunction. We have previously reported on an antisense oligonucleotide (ASO-29) that dramatically improves auditory function and balance behavior in mice homozygous for the harmonin mutation Ush1c c.216G > A following a single systemic administration. The findings were suggestive of improved vestibular function; however, no direct vestibular assessment was made. Here, we measured vestibular sensory evoked potentials (VsEPs) to directly assess vestibular function in Usher mice. We report that VsEPs are absent or abnormal in Usher mice, indicating profound loss of vestibular function. Strikingly, Usher mice receiving ASO-29 treatment have normal or elevated vestibular response thresholds when treated during a critical period between postnatal day 1 and 5, respectively. In contrast, treatment of mice with ASO-29 treatment at P15 was minimally effective at rescuing vestibular function. Interestingly, ASO-29 treatment at P1, P5 or P15 resulted in sufficient vestibular recovery to support normal balance behaviors, suggesting a therapeutic benefit to balance with ASO-29 treatment at P15 despite the profound vestibular functional deficits that persist with treatment at this later time. These findings provide the first direct evidence of an effective treatment of peripheral vestibular function in a mouse model of USH1C and reveal the potential for using antisense technology to treat vestibular dysfunction.
Introduction
Dizziness, vertigo and balance disorders may result from peripheral vestibular impairment. Peripheral vestibular dysfunction affects an estimated 35% of adults aged 40 and older (69 million in the USA) (1) and another 1.8 million develop severe to profound bilateral vestibular hypofunction worldwide (2). Treatment options for vestibular dysfunction can be limited, in part, because specific underlying mechanisms are often poorly understood and effective animal models may not be available for the evaluation and development of effective interventions. Genetic models are ideal for studying the isolated causes of vestibular dysfunction, for determining the requirements for treatment and for developing therapeutics. One genetic disorder affecting the vestibular system is Usher syndrome, which at present, apart from rehabilitation, has no effective clinical treatment option. In its most severe form (type I Usher, USH1), Usher is characterized by profound vestibular dysfunction, sensorineural hearing loss and progressive blindness.
There are six genes associated with USH1, including USH1C, which encodes harmonin, a scaffolding protein that is essential for proper hearing, vision and balance (3,4). In the inner ear, harmonin is found near the tips of stereocilia where it appears to have a structural role that is critical for sensory transduction in auditory and vestibular hair cells (5–8). One common mutation that causes Usher syndrome type 1C is a single nucleotide change within exon 3, USH1C c.216G > A, which results in the creation of a de novo 5’ splice site, 35 nucleotides upstream of the authentic 5’ splice site (4,9). This cryptic 5’ splice site created by the 216G > A substitution is used preferentially over the authentic site. This aberrant splicing causes a frame-shift in the mRNA and the production of a truncated, non-functional protein (4,9). We have previously shown that an antisense oligonucleotide (ASO) designed to basepair at the 216A position can block the aberrant 5’ splice site and redirect splicing to the correct site (10). This ASO, ASO-29, effectively rescues hearing, as well as behavioral abnormalities such as circling and swimming deficits, features commonly associated with vestibular dysfunction, in mice homozygous for the Ush1c c.216AG > A mutation (216AA mice). More generally, ASO technology is a rapidly emerging therapeutic platform that is being developed to treat a number of different conditions and diseases (11). Thus, an ASO drug has promise for the treatment of Usher syndrome and all of its symptoms in humans.
Though much has been learned about hereditary hearing loss by studying mouse models of human genetic disease (e.g.12), less is known about the role that genes play in peripheral vestibular impairment. Reasons for this include the fact that it can be difficult to recognize vestibular deficits in animal models by behavior alone, thus leading to false negative findings. Moreover, measurements of abnormal behaviors may be falsely linked to peripheral vestibular pathology, causing false positive findings. Finally, research aims, in many cases, are restricted to studying auditory or other sensors rather than vestibular, and thus vestibular data associated with hearing loss, for example, are simply not available. Recently, however, outcomes for specific genetic mutations have been revealed by direct assessment of peripheral vestibular function using electrophysiological methods such as vestibular sensory evoked potentials (VsEPs) (13–16). VsEPs are neural electrical responses initiated within the labyrinth of the inner ear by the utricular and saccular vestibular maculae (gravity receptors) in response to mechanical stimuli such as linear acceleration of the head (17,18). Linear head translation or gravity itself introduces shearing forces to the dense otoconial membrane overlying the epithelial hair cell mechanoreceptors of the maculae (e.g.19). These forces cause a shearing motion that displaces stereociliary bundles of macular receptors and initiates mechanoelectric transduction in receptor hair cells, which in turn activates vestibular primary afferent neurons. Ultimately, these events lead to the perception of head tilt or motion. VsEPs measure the collective neural activity generated by the peripheral vestibular system in response to such stimulation. The study of VsEP responses in animal models has led to new insights regarding vestibular dysfunction in human patients (16,20). The approach is well established and provides a meaningful direct measurement of vestibular function.
Abnormal spatiotemporal behaviors such as circling, head bobbing, hyperactivity, abnormal posturing and swimming difficulties are some examples of classic phenotypes that have been associated with genetic mutations and inner ear morphological pathologies (21–28), and as such, have been adopted and applied widely to define peripheral vestibular functional deficits in mutant mice. 216AA mice modeling human USH1C exhibit all of these classical behaviors including circling, head tossing and tumbling during swim tests (10,29). Systemic treatment of 216AA mice with ASO-29 at any age between P5 and P13, but not at P16 partially corrected Ush1c mRNA splicing, which resulted in an increase in the encoded harmonin protein, elimination of circling behavior and rescue of swimming ability for more than nine months (10). Despite a normalization of balance behaviors, hearing rescue was incomplete, with improvement of response to low frequency but not high frequency sound stimuli. These results reveal abnormal balance behaviors as a defect that responds exceptionally well to ASO-29 treatment. The complete elimination of disordered balance behaviors with ASO treatment conditions that were less effective at rescuing hearing raises the question of whether harmonin may play a selective critical role in vestibular function; a role that imparts some specific advantage for rescue by ASO treatment. Abnormal balance behaviors, can be readily produced by many other neural deficits including for example, central pathologies in the brain and spinal cord (e.g.30) and it may be the case that such central abnormalities and their correction by ASOs mediate behavioral changes in 216AA mice. Moreover, the absence of disordered behaviors after treatment does not guarantee normal peripheral vestibular function (13–15,20).
The relationship between peripheral vestibular impairment and balance behavior in 216AA Usher mice is not clear and little is known about the potential for treating these two phenotypes. To definitively address whether 216AA mice have a deficit in peripheral vestibular function that could account for the circling behavior, and whether such a defect can be corrected by treatment with ASO-29, we measured vestibular sensory evoked potentials (VsEPs), which provides a neurophysiological assessment of the otolithic organs of the inner ear. Our findings provide direct definitive evidence of a profound peripheral vestibular deficit in 216AA mice and a complete rescue of vestibular function in 216AA mice following treatment with ASO-29 shortly after birth. Our results also reveal a remarkable relationship between the day of treatment and the extent of vestibular rescue. This study shows for the first time, the rescue of vestibular function in an animal with the profoundly impaired vestibular system and suggests that the opportunity for treating animals occurs during a short window of time during the final stages of inner ear vestibular development in the neonatal mouse. Finally, our results demonstrate the potential of ASOs as a therapeutic approach to treating vestibular dysfunction and behavior.
Results
ASO-29 corrects abnormal spatiotemporal behavior in Usher mice
We have previously reported that a single intraperitoneal (IP) injection of ASO-29, targeting the USH1C c.216A mutation, administered before postnatal day 16 (P16) eliminates abnormal spatiotemporal behaviors such as circling and swimming in 216AA mutant Usher mice (10). In order to further determine the therapeutic window for treating these abnormal behaviors, we treated 216AA mice with either a single IP dose of ASO-29 (300 mg/kg) at P1, P5, or P15 or with a total of four doses administered at P1, 3, 5 and 7. Circling behavior and swimming ability of treated mice were assessed between P21-P30. 216AA mice treated with ASO-29 at P1 or P5 did not circle and displayed normal swimming and thus, were behaviorally indistinguishable from heterozygous, Ush1c216GA mice, which have normal hearing and balance behavior (216GA; Fig. 1). 216AA mice that were either untreated or treated with an untargeted, control ASO (ASO-C) at P5 had dramatically abnormal spatiotemporal behavior as seen from their inability to swim and their significantly higher number of rotations compared to heterozygous or ASO-29 treated Usher mice. 216AA mice treated with ASO-29 at P15 had improved swimming behavior and circled significantly less than untreated or ASO-C-treated Usher mice. Some of the mice treated with ASO-29 at P15 had abnormal swimming, and overall circling behavior was significantly higher than heterozygote (216GA) mice, though not significantly different than ASO-29 treatments at P1, P5 or P1,3,5,7 (Fig. 1). These results suggest that spatiotemporal behavioral deficits in these mice can be completely corrected by treatment with ASO-29 by P5 and largely corrected by treatment as late as P15.
Figure 1.

Circling and swim behavior of Usher mice treated with ASO-29 at different ages. (A) Circling activity in an open field box of mice with indicated genotype and treatment with total number of rotations quantitated. Mutant mice (216AA) were treated at postnatal day P1 (n = 21), P5 (n = 24), P15 (n = 23) with ASO-29 or on multiple days at P1, 3, 5 and 7 (n = 9). 216AA mice also were either untreated (-; n = 14) or treated with a non-specific control ASO–C at P5 (n = 16). Heterozygotes (Het) were untreated (-; n = 14) or treated with ASO-29 at P5 (GA; n = 14). Error bars represent S.E.M. All ASO-29 treatment groups including heterozygotes had significantly fewer rotations than ASO-C treated 216AA mice, which had a similar, non-significant difference in rotations compared to untreated 216AA mice (F(6,114)=23.00, P < 0.0001; ****P < 0.0001, Dunnett’s multiple comparison test). ASO-29-treated 216AA mice were not significantly different than ASO-29 treated heterozygote 216GA mice except 216AA mice treated at P15 which had a significantly higher number of rotations compared to control-treated heterozygote mice, (F(5,101)=17.05, P < 0.0001; #P < 0.0471, Dunnett’s multiple comparison test). (B) Swim test scores of 216AA and 216GA mice described in (A) above, demonstrating normal swimming behavior of all 216AA mutant mice treated with ASO-29 compared to untreated and ASO-C treated mutant Usher mice. Swim test scores were classified as: 2, normal swimming; 1, irregular/circular swimming; 0, underwater tumbling. Error bars represent S.E.M.; ****P < 0.0001, Fisher’s exact test.
Rescue of vestibular function in Usher mice with ASO-29 treatment
In order to directly assess vestibular function and correlate it with behavioral deficits in 216AA Usher mice from the different treatment groups, we measured VsEPs. The VsEP gives three measures of vestibular function: vestibular response threshold, latencies and amplitude (31). VsEP threshold provides an estimate of the minimum stimulus level required to activate gravity receptors, thus yielding a metric that represents how sensitive the macular vestibular organs are to a transient head motion. Low thresholds reflect more sensitive receptors. VsEP latency represents the time required to activate vestibular neurons following a stimulus. This measurement includes time associated with sensory transduction, synaptic transmission, subsequent activation of the primary afferent neuron and conduction of the action potential to the vestibular nuclei in the brainstem. Response latencies are measured as the time between the onset of the stimulus and the appearance of the first positive (p1) and negative (n1) response peaks (Fig. 2). The response, recorded as the VsEP, is a compound action potential normally generated by hundreds of vestibular primary afferent neurons, thus reflecting the functional status of large regions of the sensory epithelium (19,32–36). The amplitudes of the response reflect the number of cells activated as well as how well their activation is synchronized. The larger the amplitude, the more robust the response, reflecting either more cells responding, more precise collective discharge synchronization or both. Amplitude is measured as the difference in voltage between p1 and n1 in microvolts.
Figure 2.

Vestibular sensory evoked potential (VsEP) waveforms for individual representative treated and untreated Usher mice. Response tracings for each mouse are represented by two separate VsEP recordings that are superposed to show replicability of response components. There are four of such response pairs shown for example in group I representing four individual heterozygote mice. Response trace pairs are shown for representative animals for all groups I through VI. Treatments of ASO-29 and ASO-C were 300 mg/kg doses injected intraperitoneally on days indicated. The Ush1c heterozygotes (216GA, group I) had robust VsEPs. Response peaks p1 and n1 are marked with a vertical line in the first pair of response waveforms of treatment group I. Ush1c 216AA mice not receiving treatment (group II) and those receiving ASO-C (group III) lacked discernable normal vestibular responses. Responses were present in Ush1c homozygotes 216AA receiving ASO-29 at P4 or P5 (P4/5, group IV). Robust responses were present in 216AA mice treated ASO-29 at P1 (group V). Responses were poor or absent in 216AA mice treated with ASO-29 at P15 (group VI). Mean values for threshold, latency and amplitudes are given in Table 1 for each group. Stimulus level for all responses was +6 dB re:1g/ms. Scale bar indicates amplitude and time. The stimulus is initiated at time = 0 at the beginning (left) of response traces. Although not shown, vestibular responses in 216AA mice treated with multiple doses of ASO-29 were comparable to those shown for groups I and V.
We first examined vestibular function in heterozygote 216GA mice treated with ASO-29 or ASO-C to confirm that they had the expected normal vestibular responses. For this, VsEP testing was performed on heterozygote mice injected at P4 with ASO-C or at P1, P4 or P1,3,5,7 with ASO-29 (ages at the time of VsEP testing: mean 3.64 months, range 2.3 to 5.1). There were no significant differences in VsEP responses among the heterozygote treatments and thus, results for all 216GA mice were combined to form one control group for statistical analysis. All VsEP responses in these 216GA control mice had relatively short onset latencies (< 2 ms, p1 and n1 response peaks are shown marked in Fig. 2-I) and peak-to-peak amplitudes (p1–n1) generally between 0.8 and 2.0 microvolts (Figs 2-I, 4 and 5, and Table 1). The mean threshold for heterozygotes was −12.0 ± 2.0 (n = 33) dB re:1g/ms (Fig. 3 and Table 1). Vestibular response thresholds, latencies and amplitudes in these heterozygote mice were comparable to age-matched C57BL/6J laboratory control mice (37) with the exception of slightly larger response amplitudes on average at the highest stimulus level (p1–n1: ANOVA F(1,49)=9.218, P = 0.004; see Table 1). These results indicate that treatment with ASO-29 and ASO-C had no adverse effects on vestibular function in the 216GA heterozygotes.
Figure 4.

ASO-29 rescues vestibular responses in Usher mice over a wide range of stimulus levels. Shown are input/output (IO) functions, where VsEP latencies (upper panels A and B, p1, n1) and amplitudes (lower panels A and B, p1–n1) are plotted as a function of stimulus level in dB SL. Usher mice rescued with ASO-29 treatment (open triangles, 216AA) at P1 (A, n = 20) and P4/5 (B, n = 19) are shown in contrast to normal heterozygotes (216GA, filled circles, n = 33). Threshold for each animal is taken into consideration by specifying the stimulus level as the level in dB above each individual’s own threshold (dB SL). Latencies (upper panels) and amplitudes (lower panels) for heterozygotes (216GA, filled circles) and rescued Ush1c homozygotes (216AA, open triangles) virtually superpose for both P1 and P4/5-treated animals, although the amplitudes for P1 treated animals were slightly larger than the heterozygotes (rmANOVA F(1,40) = 4.135, P = 0.049). Quantitative evaluation was completed for levels 4.5, 7.5 and 10.5 dB SL (levels bracketed by vertical dashed lines, rmMANOVA). These levels were chosen to preserve the largest sample sizes for the different levels. There were no significant differences in latencies or amplitudes for the P4/5 treatment group.
Figure 5.
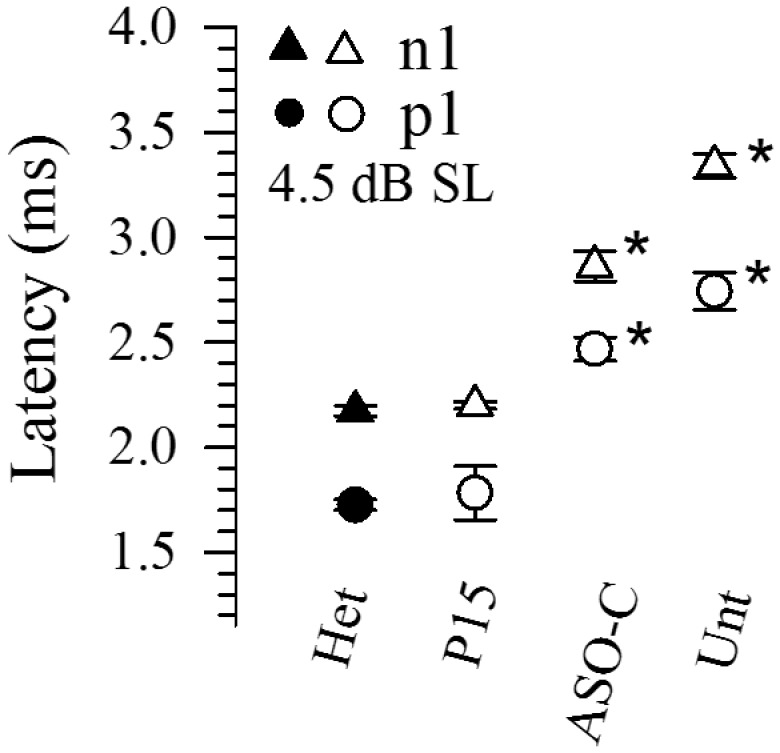
Latency of remnant vestibular responses in Usher mice. Onset latencies (p1, n1) of remnant vestibular responses in Usher mice (216AA, open symbols) treated with ASO-29 at P15 (P15, n = 3), ASO-C at P4/5 (ASO-C, n = 4) or untreated (Unt, n = 5) in contrast to those of heterozygotes (216GA, filled symbols, n = 16). Sample numbers were very small for remnant responses since responses were absent in most animals of these groups. A stimulus level of 4.5 dB SL was chosen to maximize sample sizes for all groups. One striking common feature for the ASO-C and untreated groups was a substantial delay in activation timing as reflected in response onset latency (*MANOVA F(6,64) = 12.25, P = 3.97 × 10−9; Post Hoc Bonf: Het vs ASO-C: p1 & n1: P < 1.6 × 10−10; Het vs Untreated, p1 & n1: P < 3.0 × 10−15). In contrast, activation timing was normal in remnant responses of Usher mice treated at P15 with ASO-29 (compare Het and P15 latencies).
Table 1.
Summary data for all genotypes and treatments. Vestibular sensory evoked potential (VsEP) latencies (p1, n1 in ms) and amplitudes (p1-n1 in µV) obtained at the highest stimulus level of 6 dB re:1g/ms. VsEP thresholds (V-threshold) are shown in dB re:1g/ms). Numbers are expressed as mean ±standard deviation. The number of mice is indicated in parentheses. The fraction of animals with no vestibular response for each treatment group is listed in the column labeled: ‘NR Fraction V-threshold’. Genotypes include C57BL/6J laboratory standard (216GG), Ush1c heterozygote (216GA) and Ush1c homozygote (216AA). Treatments are indicated as ASO-29, ASO-C or untreated along with the postnatal day(s) of ASO administration
| Genotype | Treatment | Age at test (months) | V-Threshold (dB re:1g/ms) | NR Fraction V-threshold | p1 (ms) | n1 (ms) | p1–n1 (µV) | |
|---|---|---|---|---|---|---|---|---|
| 216GG | 1 | None | 3.73 ± 0.82 (21) | −11.83 ± 2.95 (18) | 0/18 | 1.364 ± 0.171 (18) | 1.645 ± 0.171 (18) | 0.73 ± 0.25 (18) |
| 216GA | 2 | Het, ASO-29 or -C | 3.52 ± 0.98 (34) | −12.05 ± 2.00 (33) | 0/33 | 1.339 ± 0.060 (33) | 1.689 ± 0.077 (33) | 1.00 ± 0.33 (33) |
| 216AA | 3 | P1, ASO-29 | 2.29 ± 0.28 (20) | −9.90 ± 2.50(20) | 0/20 | 1.408 ± 0.073 (20) | 1.760 ± 0.093 (20) | 0.99 ± 0.25 (20) |
| 4 | P4/5, ASO-29 | 3.47 ± 1.04 (20) | −6.55 ± 4.13 (19) | 0/19 | 1.577 ± 0.136 (19) | 1.964 ± 0.120 (19) | 0.61 ± 0.28 (19) | |
| 5 | P1,3,5,7, ASO-29 | 2.49 ± 0.10 (10) | −10.2 ± 2.21 (10) | 0/10 | 1.386 ± 0.063 (10) | 1.720 ± 0.049 (10) | 0.74 ± 0.12 (10) | |
| 6 | P4/5, ASO-C | 3.88 ± 0.30 (15) | −4.50 ± 2.45 (4) | 10/14 | 2.195 ± 0.123 (4) | 2.596 ± 0.118 (4) | 0.65 ± 0.24 (4) | |
| 7 | P15, ASO-29 | 2.54 ± 0.16 (10) | −5.25 ± 2.87 (4) | 6/10 | 1.521 ± 0.050 (3) | 2.016 ± 0.137 (4) | 0.46 ± 0.13 (3) | |
| 8 | Unt (Untreated) | 2.11 ± 0.08 (10) | 0.50 ± 2.45 (6) | 4/10 | 2.745 ± 0.201 (6) | 3.348 ± 0.143 (6) | 0.42 ± 0.06 (6) | |
Post-hoc pair-wise comparisons of differences between treatment groups (Bonferroni):.
VsEP Thresholds.
3 vs 2: P1 vs Het: P = 0.046.
4 vs 2: P4/5 vs Het: P = 7.70 × 10−9.
4 vs 3: P4/5 vs P1: P = 0.002.
4 vs 5: P4/5 vs P1,3,5,7: P = 0.007.
6 vs 2: ASO-C vs Het: P = 9.0 × 10−6.
7 vs 2: P15 vs Het: P = 5.0 × 10−5.
8 vs 4: Unt vs P4/5: P = 0.002.
8 vs 6: Unt vs ASO-C: P = 0.035.
8 vs 7: Unt vs P15: P = 0.016.
Latencies (p1, n1, +6 dB re:1g/ms).
3 vs 2: P1 vs Het: p1: P = 0.041, n1: P = 0.044.
4 vs 2: P4/5 vs Het: p1: P = 8.3 × 10−14, n1: P = 7.4 × 10−16.
8 vs 2: Unt vs Het::p1: P = 3.39 × 10−10, n1: P = 5.0 × 10−35.
7 vs 2: P15 vs Het: p1: P = 0.016, n1: P = 1.26 × 10−4.
6 vs 2: ASO-C vs Het: p1: P = 8.7 × 10 − 20, n1: P = 1.5 × 10−21.
Amplitudes (p1−n1, +6 dB re:1g/ms).
4 vs 2:P4/5 vs Het: P = 6.2 × 10−5.
8 vs 2: Unt vs Het: P = 0.001.
7 vs 2: P15 vs Het: P = 0.028.
Figure 3.

ASO-29 treatment during a critical time period rescues VsEP thresholds in Usher mice. Graphical representation of the mean thresholds (horizontal dashed bars) and distributions of individual thresholds for control heterozygous (216GA, ‘Het’, n = 33) and homozygous Ush1c mice (216AA, ‘Mutant’) treated with ASO-29 at days indicated (P1,3,5,7, n = 10; P1, n = 20; P4/P5, n = 19; P15, n = 10). Individual animals of adjacent treatment groups are distinguished by alternating filled and open triangles. Threshold is expressed in dB relative to 1.0 g/ms, where g = 9.81 m/s2. Treatment of 216AA mice with a single dose of ASO-29 at P1 or with multiple doses at P1,3,5,7 resulted in thresholds comparable to 216GA mice although mean thresholds for P1 (P = 0.046) but not P1,3,5,7 mice were slightly higher than heterozygotes (see Table 1). Both mean thresholds and distributions were within normal limits for the mouse. Thresholds for 216AA treated at P4/P5 were modestly elevated and had a wider threshold distribution. Responses were absent or thresholds were substantially elevated in 216AA that were untreated (‘Untreated’, n = 10) or received ASO-C (‘P4/P5, ASO-C’, n = 14) or were treated at P15 with ASO-29 (ANOVA F(3,63) = 63.8, P = 4.4 × 10−19;Post hoc Bonf: P < 7.7 × 10−12). Animals showing no response were assigned a threshold value of +10 dB (‘No Response’) and resulting mean thresholds for P15-treatment with ASO-29, ASO-C treatment and untreated animals are represented as horizontal dashed bars shown.
In contrast to the normal vestibular function in heterozygous 216GA mice, 216AA mice receiving no treatment (n =10) or ASO-C (n =14) at P4 or P5 (P4/5) had no vestibular responses (14 of 24 tested) or displayed only remnants of responses, which bore little resemblance to normal VsEPs and are generally indicative of profound vestibular deficits (Fig. 2-II and -III). The lowest threshold for the abnormal response remnants was −7.5 dB re:1g/ms, which was found in only one ASO-C-treated 216AA animal (Fig. 3). Mean thresholds for remnant responses of 216AA mice treated with ASO-C (+5.9 ± 6.9 (n = 14)) and untreated 216AA mice (+4.3 ± 5.2 (n = 10)) were significantly and substantially higher than heterozygotes (mice with no response were assigned a value of +10 dB here, Fig. 3). The findings of normal vestibular function in heterozygotes and profound vestibular deficits in animals harboring the homozygous 216AA mutation are consistent with the observed phenotype for this Usher mouse model.
We next tested whether treatment of 216AA mice with ASO-29 at P4 or P5 could rescue peripheral vestibular function. Remarkably, vestibular responses were present in all 216AA mice treated with a single dose of ASO-29 at P4/5 (Fig. 2-IV, Table 1). These rescued response waveforms are comparable to those from normal heterozygotes (Fig. 2-I) and are a dramatic improvement compared to mutant 216AA mice that were either untreated (Fig. 2-II) or treated at P4/5 with ASO-C (Fig. 2-III), most of which had no response (Figs 2-II and III, 3, Table 1). The mean threshold value for the P4/5-ASO-treated 216AA mice was −6.5 ± 4.1 (n = 19) dB re:1g/ms, and was modestly higher than those of normal heterozygotes (−12.0 ± 2.0 (n = 33) dB re:1g/ms, P = 7.7 × 10−9, Fig. 3, Table 1). In addition to having an elevated mean threshold, the distribution of individual thresholds for 216AA mice treated with ASO-29 at P4/5 was much broader than that of control heterozygotes (Fig. 3), with a range of −13.5 to +1.5 dB re:1g/ms (Fig. 3). These results reveal some overlap with heterozygote 216GA mice (normal range is −16.5 to −4.5 dB re:1g/ms). The wide variation of VsEP thresholds in 216AA mice treated with ASO-29 at P4/5 corresponds functionally to a range from normal to moderately severe (+1.5 dB re:1g/ms) loss of vestibular function. This distribution presumably indicates that there is some variability in the nature and degree of successful rescue with systemic administration of ASO-29 at P4/5.
Vestibular sensitivity in 216AA mice treated with ASO-29 on P4/5 was significantly better (Table 1, P = 0.002) than that observed for untreated 216AA mice, many of which had no response (Fig. 2-II and IV, 3). In contrast, most of the 216AA mice treated with ASO-29 at P15 had profound vestibular deficits (Fig. 2VI), with responses absent in 6 of the 10 animals tested (Fig. 2VI and 3, Table 1). The mean threshold for Usher mice treated at P15 with ASO-29 was 3.9 ± 8.05 dB re:1g/ms (n = 10), with mice exhibiting no response assigned a threshold of 10 dB (Fig. 3). Thresholds in the few P15-treated mice with discernable remnant responses (4 of 10) ranged from −7.5 to −1.5 dB re:1g/ms (Fig. 3). There was no difference in remnant mean thresholds between ASO-C and P15 ASO-29 treatments in 216AA mice (Table 1). Mean threshold values for the few remnant responses were slightly lower in mice treated with ASO-C (n = 4) and with ASO-29 at P15 (n = 4) compared to untreated mutants (ASO-C: P = 0.035, P15: P = 0.016; Table 1). Threshold values for remnant responses of 216AA mice treated with ASO-C or ASO-29 at P15 were significantly higher than heterozygotes (P = 5.0 × 10−5; Table 1) and were within the middle to upper distribution of the mice treated with ASO-29 at P4/5. These results provide definitive evidence of a profound peripheral vestibular deficit in 216AA mice and substantial improvement of vestibular function in all animals following ASO-29 treatment at P4/5 but not at P15. A major vestibular deficit in 216AA mice treated with ASO-29 at P15 was revealed only by VsEP measurements, inasmuch as spatiotemporal behavior in these mice was much improved such that most animals were not visibly different from heterozygote mice (Fig. 1). Together, these results suggest a critical treatment window prior to two weeks-of-age, which defines an important intervention time period for complete rescue of peripheral vestibular function but not for rescue of spatiotemporal behavior.
Rescue of vestibular function in Usher mice depends on treatment time
Because many of the final steps in the gravity receptor development in mice occur between P2 and P10 (38–40), we hypothesized that the wide variance in thresholds and lack of a complete rescue of vestibular function in the ASO-29 mice treated at P4/5 was due to the time of injection and that correction of splicing earlier in the development of the stereociliary bundle of vestibular hair cells would result in a more complete rescue. To test this idea, ASO-29 was administered to neonates at P1 and VsEP responses were measured as in the previous groups. Treatment of 216AA mice at P1 resulted in mean thresholds that were comparable to, though significantly different than, heterozygotes (−9.90 ± 2.50 (n = 20) dB re:1g/ms, P = 0.046; Table 1). The distribution of thresholds fell within the normal range from −13.5 to −4.5 dB re:1g/ms (Fig. 3) and closely matched that of heterozygotes of the present study (Fig. 2V, 3, range −16.5 to −7.5 dB re:1g/ms) as well as age-matched C57BL/6J controls (−16.5 to −4.5 dB re:1g/ms) (37).
In order to determine whether vestibular function could be improved further by increasing the dose regimen during early neonatal periods, we administered ASO-29 in multiple single injections at postnatal days 1, 3, 5 and 7 (P1,3,5,7). Similar to the single P1 ASO-29 dose, multiple early doses produced a normal range of vestibular thresholds (−13.5 to −7.5 dB re:1g/ms; Fig. 3). The mean threshold for P1,3,5,7 animals was not significantly different from that of heterozygote controls (−10.2 ±2.2 (n = 10) dB re:1g/ms, Table 1). There was no significant difference in mean thresholds between a single dose at P1 and multiple doses of ASO-29. Thresholds for these early and multiple ASO-29 treatments were within 3 dB of the mean for heterozygote control animals (differences: 2.15, 1.85 dB respectively) and were consistent with normal vestibular function and rescue for both the single P1 and multiple dose treatment groups (for comparisons with other studies see Honaker et al., 2015 (41)). The mean threshold for 216AA mice treated with ASO-29 at P4/5 mutants was significantly higher than both the P1 (P = 0.002) and P1,3,5,7 (P = 0.007) treatment groups (Table 1). It should be noted that in some cases, the dose of ASO-29 at P4/5 could produce a full rescue given that some individual animals had thresholds well within normal limits, though the probability of doing so was lower than that of a single dose at P1 or a series of doses between P1 and P7 (compare to P1 and P1,3,5,7: Fig. 3, Table 1).
ASO-29 treatment results in normal encoding of vestibular stimulus levels by sensory epithelia in Usher mice
Differences in response latency (p1, n1) at the highest stimulus level between the control groups and Usher mice treated with ASO-29 at P1 or at P1,3,5,7 were quite small and within the normal variation of mean values for the VsEP in the C57BL/6 strain (Table 1). However, for the P4/5-treated group, the latency was longer, on average, than heterozygote controls (P < 9.0 × 10−14, Table 1). Similarly, the response amplitudes of the P4/5 ASO-29 group were less than heterozygotes at +6 dB re:1g/ms (Table 1, P = 6.2 × 10−5). These differences are likely due to the modestly higher thresholds, on average, for the 216AA mice treated with ASO-29 at P4/5.
To more clearly understand this rescue of vestibular function by ASO-29, we examined whether the rescued vestibular system encodes different stimulus levels in a normal manner. For this, we evaluated response characteristics at seven stimulus levels between response threshold and the maximum stimulus level available (+6 dB re:1g/ms), and measured the input/output (I/O) functions of Usher mice treated with single injections of ASO-29 at P1 and P4/5 (Fig. 4). Rescued animals exhibited a direct relationship between stimulus and response levels in the form of an increase in amplitude and decrease in latency with increasing stimulus level, similar to that seen in vestibular responses from normal heterozygotes (Fig. 4). Similar results were obtained from 216AA Usher mice treated with multiple doses of ASO-29 on days P1,3,5,7 (data not shown). Differences in threshold among the mice were taken into account in these plots, which are represented in relation to the threshold of each mouse in dB SL. Remarkably, the curves virtually superpose for the rescued 216AA Usher and 216GA heterozygote mice, indicating comparable response properties across the entire sensory dynamic range. Analysis of latencies at the three stimulus levels containing the largest sample sizes (4.5, 7.5, 10.5 dB SL) revealed no difference between normal heterozygotes and ASO-29-treated 216AA mice for both P1-treated and P4/5-treated mutants. P1-n1 amplitudes were marginally larger across stimulus levels for rescued 216AA mice treated at P1, although they were within the normal range (Fig. 4A, P = 0.049). There were no differences in I/O amplitudes for mutants treated at P4/5 and heterozygotes when the stimulus level was expressed in dB SL. These findings, suggest that the improvement in vestibular function in Usher mice treated at P1 with ASO-29 includes the rescue of the normal processes associated with encoding different stimulus levels as well as the mechanisms underlying activation timing and response onset. This result indicates that even in mice with thresholds that remained somewhat elevated after rescue with a single dose (e.g. P4/5 treatment, Figs 3 and 4B), a normal onset timing and input/output function was present when threshold was taken into consideration. This normalization was found even in those P4/5 animals with high thresholds above −7.5 dB re 1g/ms.
Incomplete or flawed repair of vestibular function with late correction of Harmonin expression
Although 70% (24/34) of 216AA animals receiving no treatment, ASO-C or ASO-29 at P15, had no vestibular responses, there was a small proportion of animals that had remnant responses in all groups (Table 1). These responses, for the most part, were abnormal in waveform and showed interesting differences in I/O activation timing. A comparison between treatment groups of remnant response latencies was made for the stimulus level of 4 dB SL in order to preserve the largest sample sizes possible (Fig. 5). A key common feature of remnant responses for ASO-C-treated and untreated 216AA mice was a striking delay in the onset activation latencies for p1 & n1 (Fig. 5). In contrast, p1 and n1 latencies for 216AA mice treated at P15 were normal despite significantly reduced amplitudes (P = 0.045). Indeed, the remnant I/O functions for the P15 treatment group had a normal appearance over the three to five stimulus levels available for each animal (data not shown). The dramatic difference in activation time for the P15 group versus the ASO-C and untreated treatment groups may provide hints about the role of harmonin in the processes linking mechanoelectric transduction and spike generation at the primary afferent trigger zone. Although it is not possible to be certain, the latency I/O pattern shown by the 216AA Usher mice treated with ASO-29 at P15 may represent a very modest, incomplete repair of activation timing. All remnant responses of the P15 group (n = 4) showed this pattern. All remnant responses for the ASO-C (n = 4) and untreated (n = 6) groups showed the highly abnormal prolonged onset latencies.
Vestibular function in 216AA mice treated with ASO-29 correlates with corrected splicing
Our finding that rescue of vestibular function depends on the time of treatment raises the question of whether the amount of correctly spliced Ush1c c.216A mRNA was different between the different dose regimens. To test this possibility, 216AA mice were treated at different post-natal days of life in order to assess the dependence of ASO-29-mediated Ush1c c.216A splicing correction on the time of treatment. RNA was isolated from the inner ear tissue of the treated mice at P21–30 in order to assess splicing at a time-point as close as possible to the time of treatment. Ush1c c.216A mRNA was quantitated by RT-PCR analysis using primers that specifically amplify only correctly spliced mRNA. We found that the amount of correctly spliced Ush1c c.216A mRNA was significantly higher in samples from mice treated with ASO-29 at P0–1, P3–5 or P1,3,5,7 compared to those treated at P15 (Fig. 6). However, despite differences in vestibular function, little difference was observed in the amount of correct splicing in samples from mice treated at P0–1 compared to those treated later at P3–5. These findings suggest that the timing of treatment is likely a key factor responsible for the differences in the rescue of vestibular function between mice receiving ASO at P1 compared to those treated at P4/5, whereas less effective splicing correction could account for the lack of functional rescue in P15-treated mice.
Figure 6.

ASO-29 treatment corrects splicing of Ush1c c.216A mRNA. (A) Ush1c c.216A and GAPDH RT-PCR products amplified from cDNA generated from RNA isolated from the inner ear of 216AA mice treated at the indicated post-natal day with ASO-C or ASO-29. (B) Quantitation of RT-PCR products in A. There is a significant increase in correctly spliced Ush1c c.216A mRNA in 216AA mice treated with ASO-29, compared to those treated with ASO-C, at all treatment time-points except the P15 treatment group, which had a significantly lower level of correctly splice Ush1c c.216A mRNA compared to the other ASO-29 treatments. Error bars are S.E.M. One-way ANOVA, (F(4,16) =13.58, P < 0.0001.) ***P < 0.001; ****P < 0.0001 relative to ASO-C treatment. #P < 0.05, ##P < 0.01 relative to P15 treatment with ASO-29 using Tukey’s multiple comparison test.
Discussion
This study provides direct definitive evidence of the profound loss of peripheral vestibular sensory function in 216AA mutant mice and shows that the strategically designed ASO, ASO-29, rescues peripheral vestibular function when administered to the 216AA neonatal mouse as a single dose or as a series of doses over several days during the early postnatal period. An ASO-specific effect was evident based on the findings of normal vestibular response thresholds, latencies and amplitudes in ASO-29-treated mutants compared to the absence of normal vestibular responses in animals that were untreated or were treated with ASO-C. The most effective treatment ages were restricted to the neonatal period from P1 to P5. Collectively, treatments with ASO-29 at P4/5 were less effective at rescuing vestibular function than treatment at P1, and P15 treatment failed to substantially improve vestibular function of mutants, a finding that suggests the existence of a critical period for the robust rescue of peripheral vestibular function that diminishes over time.
Harmonin and development of the hair cell bundle
There are three major isoforms of harmonin (4). The Ush1c c.216G > A mutation is considered a functional null mutation that creates an aberrant splice site which is used exclusively over the natural splice, resulting in the production of an mRNA with a reading frame shift and the incorporation of a stop codon in exon 4. This mRNA isoform encodes a truncated isoform of harmonin, the formation of which precludes production of all other forms of the protein. Mice homozygous for this mutation have severely disorganized cochlear hair cell stereocilia and compromised mechanotransduction characteristics (10,42,43). Mice harboring a different harmonin null mutation, one lacking the first exon of harmonin, which contains the translational initiation site, were shown to have severely fragmented and disorganized cochlear hair cell bundles early in development as well as altered planar cell polarity (6). These structural abnormalities were attributed to pathological developmental processes associated with the absence of harmonin. Reportedly, vestibular hair cells showed less severe bundle anomalies than cochlear hair cells but, nonetheless, had notable staircase differential growth defects. It is not clear that structural defects such as these contribute to the profound loss of vestibular function observed here. The extent to which this is the case remains to be seen inasmuch as there is little detailed information regarding vestibular bundle morphology in any harmonin null mutant.
Harmonin is critical for setting normal sensitivity and timing of neural activation in the vestibular epithelium
Harmonin is localized at the upper tip link density (UTLD) in stereocilia of mature mice (5,6) and has been proposed to bind and link the upper tip link segment (cadherin 23, Cdh23) to cytoplasmic actin filaments of the stereocilium (5,7). This assembly is thought to be critical for normal mechanotransduction and adaptation to hair bundle shear during the mechanoelectric transduction process. Consistent with this hypothesis is the recent observation of reduced or absent mechanotransduction currents as well as reduced rates and extent of adaptation in utricular hair cells of neonatal 216AA Usher mice (43). Others have also reported changes in adaptation in non-null harmonin mutations such as Ush1cdfcr-2j (7), and Ush1cdfcr (5). The profound loss of function in the functional null mutants reported here can be explained by altered transduction and defective adaptation mechanisms in macular hair cells. This hypothesis is particularly attractive because the most severe changes were seen in hair cells of the striola (43), an epithelial region thought to be a major contributor to the generation of the VsEP (19,31).
VsEP threshold as a physiological metric reflects the minimum stimulus level required to produce a discernable collective activation of vestibular neurons and thus it reflects the overall sensitivity of the neuroepithelium to transient stimulation. A key effect of the harmonin 216A mutation was to decrease the sensitivity of the macular sensory epithelium and in most animals it was reduced to the extent that no response could be obtained at the highest stimulus level. Thus, the present results confirm a critical role for harmonin in establishing macular neuroepithelial sensitivity to head translation.
Early treatment with ASO-29 at P1 rescued normal sensitivity. However, it was also possible to partially rescue sensitivity with treatment at older ages (P4/5). The fact that the levels of correctly spliced harmonin mRNA were the same for P1 and P4/5 single dose groups indicates a critical developmental difference in the state of the epithelium for the two periods (Figs 3 and 6). The reduced effectiveness of ASO-29 dosing at later times could reflect the presence of reduced numbers of HCs amenable to rescue or it could reflect a repair that was of poor or variable quality or both. In any case the probability of achieving full sensitivity would be diminished. Of interest is the finding that the relationship between stimulus level and response activation latency was preserved in partially rescued animals of the P4/5 group (Fig. 4B). Thus, although threshold remained high in many P4/5 mice, activation timing for the VsEP at levels above threshold was the same as normal animals. This outcome could be explained by residual diminished adaptation in the transduction apparatus. In this case, the transduction channel open probability versus bundle displacement curve (Po(x)) would be shifted to the right (thereby elevating threshold) with a normal Po(x) slope mediating normal activation timing at levels above threshold. Adaptation is widely regarded as essential for maintaining the sensitivity of the transduction apparatus by ensuring adequate tension on tip links for different static/dynamic shear levels (44–46). Adaptation thereby ensures tip link tension is adjusted to provide the maximum sensitivity to the smallest stimuli. In the context of the VsEP, inadequate/defective adaptation could be seen to increase the threshold of activation and hypothetically this could underlie the elevated thresholds in the partially rescued animals receiving P4/5 injections.
ASO-C administration and ASO-29 treatments outside the critical period
A few 216AA mutant mice treated with ASO-C or with ASO-29 at P15 had remnant response thresholds at the upper limits for the normal range of VsEP thresholds (n = 3 at −7.5 dB and n = 3 at −4.5 dB, respectively). These thresholds extended slightly below those in the untreated 216AA group (Fig. 3). For reasons outlined below, these few outliers might suggest subtle improvement with ASO-29 treatment at P15, but it is unlikely that remnant responses in ASO-C treated animals represent ASO induced improvements.
Two distinct profiles of activation timing are represented for a single stimulus level of 4.5 dB SL in the remnant response groups shown in Figure 5 (ASO-C, P15-ASO-29 and untreated). The few P15-treated animals evidencing responses demonstrated latencies that approached those of normal animals, whereas remnant responses in both ASO-C-treated and untreated animals, despite similar thresholds, showed extraordinary long onset latencies (p1 and n1) beyond 2 ms (Fig. 5). Such latencies are never seen in normal animals suggesting that there is a fundamental difference between the remnant responses of mice treated with ASO-29 at P15 and those treated with ASO-C or untreated animals. Remnant responses of ASO-C-treated mice are more likely vestiges of function left over from the pathological processes associated with the absence of harmonin. Indeed, that must be the case for remnant responses in the untreated mutants. In the case of the ASO-C-treated animals (P4/5), there was no convincing evidence of behavioral improvements (only 1 out of 25 was judged a normal swimmer), whereas 21 of 23 ASO-29-treated animals (P15) were scored normal. Given the absence of improved behavior, the idea of some meaningful improvement in vestibular function in ASO-C-treated Usher mice is, in our view, untenable. Thus, highly abnormal remnant vestibular responses in ASO-C-treated and untreated Usher mice are associated with no obvious behavioral benefit and thus provide no indication of effective vestibular function.
Our results provide evidence of improvements in circling and swimming behaviors in mutants with ASO-29 treatment as late as P15 despite persisting profound peripheral vestibular deficits. These findings suggest that ASO-29 treatment may 1) improve behavior through actions outside the gravity receptor epithelium (e.g. central nervous system (CNS) or elsewhere) and/or 2) there may be some benefit to vestibular function with ASO-29 treatment as late as P15. The demonstration of relatively normal latency functions in some (4/10) animals treated at P15 as noted above is consistent with the latter hypothesis. These outcomes together with possible vestibular improvements that are not readily detected by the VsEP may explain the improvement in behaviors. We have noted previously that the slightest amount of peripheral vestibular input can provide a sufficient cue to the central nervous system to support normal behaviors (16,20). ASO-29 treatment at P15 results in behavioral recovery and may do so in part by partially improving peripheral vestibular function.
Despite improved behaviors, severe to profound loss of peripheral vestibular function persisted in mice treated with ASO-29 at P15. Thus, as indicated here and previously (13–15,20), behavior alone may not reflect the severity of vestibular insult, presumably due to the remarkable capacity of the CNS to compensate for peripheral loss. Together, behavior and direct peripheral vestibular measures provide unique insight into these processes and can be very valuable in developing an understanding of the mechanisms underlying, as well as system levels involved in, vestibular functional rescue and recovery.
Conclusion
The present study demonstrates for the first time that properly timed systemic administration of ASO-29 to 216AA mutant mice restores normal gravity receptor function and behavior. Rescued vestibular response thresholds, latencies and amplitudes under these conditions are comparable to normal heterozygotes and laboratory controls. The results underscore the considerable potential for preventing catastrophic congenital sensory loss using ASO therapeutic strategies. The development of the vestibular system in humans occurs in utero (50) and thus, interventions aimed at treating vestibular deficits may require early administration to the developing fetus. We have recently demonstrated that ASOs can be effectively delivered to a mouse embryo via injection into the amniotic cavity, with effects on target gene expression lasting as long as a month after birth (51). These results suggest a potential treatment paradigm for ASOs as a therapeutic agent for disorders, such as Usher syndrome, that require intervention early in development.
Materials and Methods
Animals
All experimental procedures were carried out in accordance with the Institutional Animal Care and Use Committees (IACUCs) at Rosalind Franklin University of Medicine and Science (RFUMS) and the University of Nebraska-Lincoln (UNL) following NIH guidelines for The Care and Use of The Laboratory Animals. Ush1c216G>A mice on a mixed C57BL/6 and 129S6 background (42) were bred, treated and housed at RFUMS prior to being shipped to UNL. The temperature and humidity controlled room of RFUMS Animal Facility where animals were raised, has a noise ambiance level of about 60 dB as measured by a sound meter (RadioShack). Post weaning age mice were shipped overnight from RFUMS to UNL via ground-air-ground transportation using World Courier INc. (Bensville, IL) under ambient temperature and pressure controlled environment as certified by the Carrier.
Antisense oligonucleotides
2’-O-methoxyethyl-ASO (ASO-29: 5’-AGCTGATCATATTCTACC-3’) with phosphorothioate backbone was synthesized by Ionis Pharmaceuticals as previously described (10). ASOs were diluted in 0.9% saline sterile solution. Mouse neonates were injected intraperitoneally (IP) with 300 mg of ASOs per kg of body weight at postnatal day 1 (P1), once between Day 3 and 5 (P3–5), and at 15 (P15). Multiple day injections with 300 mg/kg of ASOs were performed four times; one dose at P1 and P3 and P5 and P7. Control mutant mice were treated with 300 mg/kg of body weight at P5 with a non-specific ASO, ASO-C (5’-TTAGTTTAATCACGCTCG-3’) as previously described (10).
RNA isolation and RT-PCR
Inner ear tissue was harvested from mice at P21–P30 and snap frozen in liquid nitrogen. RNA was isolated from the tissue using TRIzol reagent (Life Technologies, Carlsbad, CA) according to the manufacturer’s protocol. RNA was reverse transcribed using GoScript reverse transcription system (Promega, Madison, WI). Radiolabeled PCR was carried out using GoTaq Green Master Mix (Promega, Madison, WI) with α-32P-dCTP. A primer specific for the knocked-in human portion or of Ush1c c.216A, hUsh1c_Ex3WTF (5’-GAATATGATCAGCTGACC), and mUsh1c_Ex5R (5’-TCTCACTTTGATGGACACGGTCTTC-3’) were used. The forward primers only amplify cDNA from RNA transcribed from the Ush1c c.216A allele because the homozygous Ush1c 216AA mice have the human USH1C c.216A gene knocked in to exon 3 and 4, replacing the mouse sequence (Lentz et al., 2007, PMID: 17174357). Mouse-specific Gapdh primers were: mGapdh_Ex3F (5’-GTGAGGCCGGTGCTGAGTATG-3’) and mGapdh_Ex4R (5’-GCCAAAGTTGTCATGGATGAC-3’). Products were separated on a 6% nondenaturing polyacrylamide gel and quantified using a Typhoon 9400 phosphorimager (GE Healthcare).
Animal behavior
Swim test. Swimming was assessed in a transparent plastic container (Width 24 × High 20 × Length 35 cm) filled with 12–14 cm of 30°C water as previously described (10). The swimming capacity of each mouse was observed and recorded with a video camera for 15 s and then scored as follows: ‘Normal’ (score 2: straight swim without signs of distress), ‘Circling/twisting’ (score 1: difficulty swimming, circular swim motion, twisting), ‘Tumbling’ (score 0: inability to stay afloat, sinking into the water immediately or during the course of the test). When tumbling occurred, mice were immediately rescued and test was stopped.
Open field behavior. Mice behavioral testing was performed in a 55 (L) × 35 (W) × 38 cm (H) sound-proof ventilated box (MED Associates Inc., Georgia, Vermont) and filmed using a video camera (WideCam F100, KYE, New Taipei city, Taiwan) connected to HP E6500 laptop computer (Dell, Round Rock, TX). The open field test was done at weaning age (Day 21) over 2 min. Using Any-Maze software (Stoelting, Wood Dale, IL), the number of rotations, defined as number of times the mouse body completed an entire 360° angle continuous turn during the 2 min recording, was calculated. A one-way ANOVA and Tukey’s multiple comparison test was performed using Prism 6 software (GraphPad Software Inc. La Jolla, CA) to compare means of each mouse category.
Functional assessment of vestibular sensors
Animals and animal preparation. Animal preparation and functional testing for vestibular sensory evoked potentials (VsEPs) followed procedures comparable to those previously described (17,18,47,48). After arrival at UNL, animals were housed and maintained using standard husbandry methods until measurements were made. Ambient sound levels were monitored using a dosimeter (NoisePro DL, Quest Technologies, Oconomowoc, WI) and found to remain below 65 dB LAeq throughout the animal care work cycle. Animals used in VsEP testing were not evaluated for spatiotemporal behavioral deficits.
During VsEP testing, mice were anesthetized with ketamine (90–126 mg/kg) and xylazine (10–14 mg/kg) injected intraperitoneally. Body core temperature was maintained at 37.0 ± 0.2 °C using a homeothermic heating blanket and rectal thermocouple (FHC, Inc.). Subcutaneous stainless steel electrodes were placed at the nuchal crest (noninverting), behind the pinna (inverting) and at the hip (ground). VsEP recordings were performed in each mouse, after which the mouse was euthanized and tail snips were harvested for genotyping.
Vestibular sensory - evoked potentials (VsEPs). A linear jerk pulse was generated using a linear voltage ramp (2 ms duration) routed through a power amplifier, which drove a mechanical shaker (Labworks, Inc. Model E2-203). Linear jerk pulses (17 pulses/s) ranging in amplitude from +6 to −18 dB re:1.0 g/ms (where 1.0 g = 9.8 m/s2) adjusted in 3 dB steps were presented to the head in the naso-occipital axis. A broad band forward masker (50 to 50,000 Hz, 97 dB SPL) was presented during VsEP measurements (49). A noninvasive head clip was used to secure the head to the mechanical shaker for delivery of the vestibular stimuli. Mice were placed in a supine position with the nose up and stimuli were presented in the naso-occipital axis. Normal polarity began with upward movement (naso-occipital, +X). Inverted stimulus polarity began with downward movement (naso-occipital, -X). Responses were collected for both normal and inverted polarities and the resulting waveforms were averaged online to produce the final waveform for analysis.
The activity of the peripheral vestibular nerve is reflected by the first positive (p1) and negative (n1) response peaks of the VsEP (33). Amplitude and latencies for the first response peaks of the waveform were quantified. Peak-to-peak amplitude, measured in microvolts, represented the difference in voltage between p1 and n1, (p1–n1). Response peak latencies were defined as the time, in milliseconds, from the onset of the stimulus to the appearance of each peak, p1 and n1. VsEP response threshold was defined as the stimulus level midway between the minimum stimulus level that produced a discernible response and the maximum stimulus level that did not result in a visible response.
Recording overview and averaging. Single channel signal averaging was used to record VsEP response waveforms. Electroencephalographic activity reflected in surface electrodes was amplified (200,000X, Grass P511), band pass filtered (300–3000 Hz, −6 dB points, Grass P511) and digitized (10 µs per point for 1024 points, NI 6259) beginning at stimulus onset. 256 samples were averaged to produce the final VsEP waveforms. Offline analysis was used to determine response thresholds (in dB re: 1.0g/ms), response peak latencies (in ms), and peak-to-peak amplitudes (in µV).
Data a nalysis. In cases where animals evidenced no response to testing at any stimulus level, the animals were scored as ‘no response’ (‘NR’). Animals that had no response were distinguished from an animal where testing was not competed, in which case no data were entered.
Statistics
Univariate analysis of variance (ANOVA, Prism 6 v.6.0h) was used to evaluate circling and Ush1c splicing. Dunnett’s or Tukey’s multiple comparison tests were used for post-hoc analysis as noted. Fisher’s exact tests were used for the categorical swim test data. For VsEP data, univariate or multivariate analysis of variance (ANOVA, MANOVA, respectively) (SPSS, v22, IL) were used to evaluate gravity receptor function. Where appropriate, repeated measures analysis of variance was used (rmANOVA, rmMANOVA). Bonferroni (Bonf) tests were used as noted for post hoc analysis of VsEP data. Unless stated specifically, data are presented as mean ± standard deviation (sample size). The criterion for statistical significance was P < 0.05.
Acknowledgements
We thank Anthony Hinrich for technical assistance and members of the Jones and Hastings labs for comments on the manuscript.
Conflict of Interest statement. F.R. is an employee of Ionis Pharmaceuticals. M.L.H. receives funding from Ionis Pharmaceuticals.
Funding
National Institutes of Health R01DC012596 (MLH); Nebraska Tobacco Settlement Biomedical Research Foundation, and Department of Special Education and Communication Disorders, UNL (TAJ); National Institutes of Health U54GM104940 and P30GM103340 (JJL).
References
- 1. Agrawal Y., Carey J.P., Della Santina C.C., Schubert M.C., Minor L.B. (2009) Disorders of balance and vestibular function in US adults: data from the National Health and Nutrition Examination Survey, 2001-2004. Arch. Intern. Med., 169, 938–944. [DOI] [PubMed] [Google Scholar]
- 2. Ward B.K., Agrawal Y., Hoffman H.J., Carey J.P., Della Santina C.C. (2013) Prevalence and impact of bilateral vestibular hypofunction: results from the 2008 US National Health Interview Survey. JAMA Otolaryngol. Head Neck Surg., 139, 803–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bitner-Glindzicz M., Lindley K.J., Rutland P., Blaydon D., Smith V.V., Milla P.J., Hussain K., Furth-Lavi J., Cosgrove K.E., Shepherd R.M.. et al. (2000) A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nat. Genet., 26, 56–60. [DOI] [PubMed] [Google Scholar]
- 4. Verpy E., Leibovici M., Zwaenepoel I., Liu X.Z., Gal A., Salem N., Mansour A., Blanchard S., Kobayashi I., Keats B.J.. et al. (2000) A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat. Genet., 26, 51–55. [DOI] [PubMed] [Google Scholar]
- 5. Grillet N., Xiong W., Reynolds A., Kazmierczak P., Sato T., Lillo C., Dumont R.A., Hintermann E., Sczaniecka A., Schwander M.. et al. (2009) Harmonin mutations cause mechanotransduction defects in cochlear hair cells. Neuron, 62, 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lefevre G., Michel V., Weil D., Lepelletier L., Bizard E., Wolfrum U., Hardelin J.P., Petit C. (2008) A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development, 135, 1427–1437. [DOI] [PubMed] [Google Scholar]
- 7. Michalski N., Michel V., Caberlotto E., Lefevre G.M., van Aken A.F., Tinevez J.Y., Bizard E., Houbron C., Weil D., Hardelin J.P.. et al. (2009) Harmonin-b, an actin-binding scaffold protein, is involved in the adaptation of mechanoelectrical transduction by sensory hair cells. Pflugers. Arch., 459, 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boeda B., El-Amraoui A., Bahloul A., Goodyear R., Daviet L., Blanchard S., Perfettini I., Fath K.R., Shorte S., Reiners J.. et al. (2002) Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. embo J., 21, 6689–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lentz J., Savas S., Ng S.S., Athas G., Deininger P., Keats B. (2005) The USH1C 216G–>A splice-site mutation results in a 35-base-pair deletion. Hum. Genet., 116, 225–227. [DOI] [PubMed] [Google Scholar]
- 10. Lentz J.J., Jodelka F.M., Hinrich A.J., McCaffrey K.E., Farris H.E., Spalitta M.J., Bazan N.G., Duelli D.M., Rigo F., Hastings M.L. (2013) Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat. Med., 19, 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Havens M.A., Hastings M.L. (2016) Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res., 44, 6549–6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leibovici M., Safieddine S., Petit C. (2008) Mouse models for human hereditary deafness. Curr. Top. Dev. Biol., 84, 385–429. [DOI] [PubMed] [Google Scholar]
- 13. Goodyear R.J., Jones S.M., Sharifi L., Forge A., Richardson G.P. (2012) Hair bundle defects and loss of function in the vestibular end organs of mice lacking the receptor-like inositol lipid phosphatase PTPRQ. J Neurosci., 32, 2762–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jones S.M., Johnson K.R., Yu H., Erway L.C., Alagramam K.N., Pollak N., Jones T.A. (2005) A quantitative survey of gravity receptor function in mutant mouse strains. J. Assoc. Res. Otolaryngol., 6, 297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee S.I., Conrad T., Jones S.M., Lagziel A., Starost M.F., Belyantseva I.A., Friedman T.B., Morell R.J. (2013) A null mutation of mouse Kcna10 causes significant vestibular and mild hearing dysfunction. Hear. Res., 300, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jones S.M., Jones T.A. (2014) Genetics of peripheral vestibular dysfunction: lessons from mutant mouse strains. J. Am. Acad. Audiol., 25, 289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jones S.M., Erway L.C., Johnson K.R., Yu H., Jones T.A. (2004) Gravity receptor function in mice with graded otoconial deficiencies. Hear. Res., 191, 34–40. [DOI] [PubMed] [Google Scholar]
- 18. Jones S.M., Erway L.C., Bergstrom R.A., Schimenti J.C., Jones T.A. (1999) Vestibular responses to linear acceleration are absent in otoconia-deficient C57BL/6JEi-het mice. Hear. Res., 135, 56–60. [DOI] [PubMed] [Google Scholar]
- 19. Jones T.A., Lee C., Gaines G.C., Grant J.W. (2015) On the high frequency transfer of mechanical stimuli from the surface of the head to the macular neuroepithelium of the mouse. J. Assoc. Res. Otolaryngol., 16, 189–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mathur P.D., Vijayakumar S., Vashist D., Jones S.M., Jones T.A., Yang J. (2015) A study of whirlin isoforms in the mouse vestibular system suggests potential vestibular dysfunction in DFNB31-deficient patients. Hum. Mol. Genet., 24, 7017–7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deol M.S. (1956) The anatomy and development of the mutants pirouette, shaker-1 and waltzer in the mouse. Proc. R. Soc. Lond. B Biol. Sci., 145, 206–213. [DOI] [PubMed] [Google Scholar]
- 22. Deol M.S., Green M.C. (1966) Snell's waltzer, a new mutation affecting behaviour and the inner ear in the mouse. Genet. Res., 8, 339–345. [DOI] [PubMed] [Google Scholar]
- 23. Deol M.S. (1966) The probable mode of gene action in the circling mutants of the mouse. Genet. Res., 7, 363–371. [DOI] [PubMed] [Google Scholar]
- 24. Deol M.S. (1968) Inherited diseases of the inner ear in man in the light of studies on the mouse. J. Med. Genet., 5, 137–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deol M.S., Lane P.W. (1966) A new gene affecting the morphogenesis of the vestibular part of the inner ear in the mouse. J. Embryol. Exp. Morphol., 16, 543–558. [PubMed] [Google Scholar]
- 26. Erway L.C., Fraser A.S., Hurley L.S. (1971) Prevention of congenital otolith defect in pallid mutant mice by manganese supplementation. Genetics, 67, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lyon M.F. (1951) Hereditary absence of otoliths in the house mouse. J. Physiol., 114, 410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lyon M.F. (1958) Twirler: a mutant affecting the inner ear of the house mouse. J. Embryol. Exp. Morphol., 6, 105–116. [PubMed] [Google Scholar]
- 29. Lentz J., Pan F., Ng S.S., Deininger P., Keats B. (2007) Ush1c216A knock-in mouse survives Katrina. Mutat. Res., 616, 139–144. [DOI] [PubMed] [Google Scholar]
- 30. Loscher W. (2010) Abnormal circling behavior in rat mutants and its relevance to model specific brain dysfunctions. Neurosci. Biobehav. Rev., 34, 31–49. [DOI] [PubMed] [Google Scholar]
- 31. Jacobson G. P., Shepard Neil T. (ed.) (2016) Balance Function Assessment and Management. 2nd ed.Plural Publishing, San Diego. [Google Scholar]
- 32. Jones T.A. (1992) Vestibular short latency responses to pulsed linear acceleration in unanesthetized animals. Electroencephalogr. Clin. Neurophysiol., 82, 377–386. [DOI] [PubMed] [Google Scholar]
- 33. Nazareth A.M., Jones T.A. (1998) Central and peripheral components of short latency vestibular responses in the chicken. J. Vestib. Res., 8, 233–252. [PubMed] [Google Scholar]
- 34. Irons-Brown S.R., Jones T.A. (2004) Effects of selected pharmacological agents on avian auditory and vestibular compound action potentials. Hear. Res., 195, 54–66. [DOI] [PubMed] [Google Scholar]
- 35. Jones T.A., Jones S.M., Vijayakumar S., Brugeaud A., Bothwell M., Chabbert C. (2011) The adequate stimulus for mammalian linear vestibular evoked potentials (VsEPs). Hear. Res., 280, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jones T.A., Pedersen T.L. (1989) Short latency vestibular responses to pulsed linear acceleration. Am. J. Otolaryngol., 10, 327–335. [DOI] [PubMed] [Google Scholar]
- 37. Mock B.E., Vijayakumar S., Pierce J., Jones T.A., Jones S.M. (2016) Differential effects of Cdh23(753A) on auditory and vestibular functional aging in C57BL/6J mice. Neurobiol. Aging, 43, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Denman-Johnson K., Forge A. (1999) Establishment of hair bundle polarity and orientation in the developing vestibular system of the mouse. J. Neurocytol., 28, 821–835. [DOI] [PubMed] [Google Scholar]
- 39. Rusch A., Lysakowski A., Eatock R.A. (1998) Postnatal development of type I and type II hair cells in the mouse utricle: acquisition of voltage-gated conductances and differentiated morphology. J. Neurosci., 18, 7487–7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Curthoys I.S. (1983) In Romand R. (ed.), Development of Auditory and Vestibular Systems. Academic Press Inc, New York, pp. 425–461. [Google Scholar]
- 41. Honaker J.A., Lee C., Criter R.E., Jones T.A. (2015) Test-retest reliability of the vestibular sensory-evoked potential (VsEP) in C57BL/6J mice. J. Am. Acad. Audiol., 26, 59–67. [DOI] [PubMed] [Google Scholar]
- 42. Lentz J.J., Gordon W.C., Farris H.E., MacDonald G.H., Cunningham D.E., Robbins C.A., Tempel B.L., Bazan N.G., Rubel E.W., Oesterle E.C.. et al. (2010) Deafness and retinal degeneration in a novel USH1C knock-in mouse model. Dev. Neurobiol., 70, 253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pan B., Askew C., Galvin A., Heman-Ackah S., Asai Y., Indzhykulian A.A., Jodelka F.M., Hastings M.L., Lentz J.J., Vandenberghe L.H.. et al. (2017) Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat. Biotechnol., 35, 264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Eatock R.A., Corey D.P., Hudspeth A.J. (1987) Adaptation of mechanoelectrical transduction in hair cells of the bullfrog's sacculus. J. Neurosci., 7, 2821–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Eatock R.A. (2000) Adaptation in hair cells. Annu. Rev. Neurosci., 23, 285–314. [DOI] [PubMed] [Google Scholar]
- 46. Gillespie P.G., Muller U. (2009) Mechanotransduction by hair cells: models, molecules, and mechanisms. Cell, 139, 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mock B., Jones T.A., Jones S.M. (2011) Gravity receptor aging in the CBA/CaJ strain: a comparison to auditory aging. J. Assoc. Res. Otolaryngol., 12, 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vijayakumar S., Lever T.E., Pierce J., Zhao X., Bergstrom D., Lundberg Y.W., Jones T.A., Jones S.M. (2015) Vestibular dysfunction, altered macular structure and trait localization in A/J inbred mice. Mamm. Genome., 26, 154–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jones T.A., Jones S.M. (1999) Short latency compound action potentials from mammalian gravity receptor organs. Hear. Res., 136, 75–85. [DOI] [PubMed] [Google Scholar]
- 50. Jones T.A., Jones S.M., (2016) Ontogeny of the Vestibular System and Balance In: Balance Function Assessment and Management, Second Edition Eds: Jacobson G.P., Shepard N.T., Plural Publishing, San Diego. [Google Scholar]
- 51. Depreux F.F., Wang L., Jiang H., Jodelka F.M., Rosencrans R.F., Rigo F., Lentz J.J., Brigande J.V., Hastings M.L. (2016) Antisense oligonucleotides delivered to the amniotic cavity in utero modulate gene expression in the postnatal mouse. Nucleic Acids Res., 44, 9519–9529. [DOI] [PMC free article] [PubMed] [Google Scholar]