

Abstract
Myosin storage myopathy (MSM) is a congenital skeletal muscle disorder caused by missense mutations in the β-cardiac/slow skeletal muscle myosin heavy chain rod. It is characterized by subsarcolemmal accumulations of myosin that have a hyaline appearance. MSM mutations map near or within the assembly competence domain known to be crucial for thick filament formation. Drosophila MSM models were generated for comprehensive physiological, structural, and biochemical assessment of the mutations’ consequences on muscle and myosin structure and function. L1793P, R1845W, and E1883K MSM mutant myosins were expressed in an indirect flight (IFM) and jump muscle myosin null background to study the effects of these variants without confounding influences from wild-type myosin. Mutant animals displayed highly compromised jump and flight ability, disrupted muscle proteostasis, and severely perturbed IFM structure. Electron microscopy revealed myofibrillar disarray and degeneration with hyaline-like inclusions. In vitro assembly assays demonstrated a decreased ability of mutant myosin to polymerize, with L1793P filaments exhibiting shorter lengths. In addition, limited proteolysis experiments showed a reduced stability of L1793P and E1883K filaments. We conclude that the disrupted hydropathy or charge of residues in the heptad repeat of the mutant myosin rods likely alters interactions that stabilize coiled-coil dimers and thick filaments, causing disruption in ordered myofibrillogenesis and/or myofibrillar integrity, and the consequent myosin aggregation. Our Drosophila models are the first to recapitulate the human MSM phenotype with ultrastructural inclusions, suggesting that the diminished ability of the mutant myosin to form stable thick filaments contributes to the dystrophic phenotype observed in afflicted subjects.
Introduction
Myosin is a highly conserved, ubiquitous protein found in all eukaryotic cells. It acts as a motor protein powered by ATP hydrolysis to drive diverse cellular movements such as muscle contraction, cytokinesis, or translocation of vesicles or cargo. Type II myosin is a hexameric molecule (Fig. 1A) composed of two myosin heavy chains (MHC), two essential light chains (MLC-1 or ELC) and two regulatory light chains (MLC-2 or RLC) (1) and is the major component of thick filaments. The MHC amino terminal region forms a globular head structure containing the actin and nucleotide-binding sites (S1 domain), while the carboxy-terminal α-helices of two heavy chains intertwine to form a coiled-coil rod structure that assembles with other rods to form filaments (Fig. 1A).
Figure 1.

Schematic of the structure of myosin and multiple sequence alignment of C-terminal β-MHC. (A) Illustration of the myosin structure. The N-terminal portion of two MHCs (gray) fold into the globular head structure containing the actin and nucleotide-binding site (S1 domain), while the coiled C-terminal helices intertwine to form the α-helical coiled-coil rod structure. The location of the assembly competence domain (ACD) and the loci of the MSM mutations with their positions within the heptad coiled-coil are displayed. (B) Schematic of the heptad repeat pattern of amino acids within the coiled-coil. The position of the residues in the heptad is denoted a–g. (C) Clustal Omega multiple sequence alignment of full-length β-MHC LMMs from various species with the IFM MHC isoform of Drosophila melanogaster. Hs- Homo sapiens (RefSeq P12883.5); Bt- Bos taurus (RefSeq NP_777152.1); Ss- Sus scrofa (RefSeq NP_999020.1); Cl- Canis lupus familiaris (RefSeq NP_001107183.1); Oa- Ovis aries (RefSeq XP_004010374.1); Rn- Rattus norvegicus (RefSeq NP_058936.1); Mm- Mus musculus (RefSeq NP_542766.1); Gg- Gallus gallus (RefSeq NP_001001302.1); Dm- Drosophila melanogaster (RefSeq NP_724008.1). The distal rod region, corresponding to exons 37 through 40, of the human β-MHC is displayed. Residues are shaded based on degree of conservation. An (*) indicates positions that have identical residues, a (:) indicates conservation with strongly similar residues and a (.) indicates weakly similar residues. Residues in human β-MHC known to cause MSM when mutated are highlighted (red).
In striated muscle, myosins associate to form a bipolar thick filament with the S1 heads projecting away from the backbone. Thick filament assembly is facilitated by a specific pattern of amino-acid sequences and intramolecular charge dependent interactions among myosin rods and myosin binding proteins such as M-protein, myosin-binding protein C, and myomesin. Ordered assembly of MHC dimers and thick filaments is highly reliant on the sequence periodicity of the rod region (2). A heptameric repeat (denoted a–g) pattern is evident in the rod region (Fig. 1B) with hydrophobic residues predominating at internal positions a and d, forming the interface ‘seam’ between the superhelices, which is stabilized by salt bridges between residues e and g. Residues in the outer b, c and f positions are typically charged and mediate interactions between the myosin rods to pack them into filaments (2). These heptad repeats are known to be part of a larger 28 amino acid repeat composed of 14 positively charged followed by 14 negatively charged residues (2). The alternating positively and negatively charged zones along the molecule facilitate interactions with oppositely charged amino acids of other myosin rods resulting in the ordered formation of the thick filaments (3,4).
Sohn et al. (5) identified a region in the MHC rod that is necessary, but not sufficient, for filament assembly. This region spans amino acids 1871-1899 (human β-MHC) and is termed the assembly competence domain (ACD) (Fig. 1A); it has an evolutionarily conserved and unique charge ‘fingerprint’ with four central negative residues flanked by two distinct positive blocks (5,6). Basic residues in the ACD are required for tail–tail interactions, while the positively charged regions are critical for filament assembly and stabilize the tail-tail interactions (7).
There are eleven members of the human sarcomeric MHC family (8), with seven isoforms expressed in human skeletal muscles. β-MHC, encoded by MYH7, is expressed in the adult heart ventricles and slow skeletal muscle fibers. The ∼23 kb MYH7 gene contains 40 exons that direct the synthesis of the 1935 amino acid β-MHC isoform. The gene is important clinically as it is associated with over 500 disease-causing mutations (9). Myosin storage myopathy (MSM) was the first skeletal muscle myopathy identified as being caused by mutation in MYH7 (10) (OMIM #608358).
MSM is a protein aggregate myopathy with fewer than 75 cases reported in the literature to date. It was initially described by Cancilla et al. (11) as ‘familial myopathy with probable lysis of myofibrils of type I fibers’. Tajsharghi et al. (10) introduced the unifying term ‘myosin storage myopathy’ after molecular nosologic identification of the mutation in the rod region of MYH7. Since then, genotypic analyses have identified eight MSM mutations- R1845W (10), H1901L (12), L1793P (13), E1883K (14), L1779P (15), X1936W (16), K1784del (17) and R1820W (18) mapping to the distal C-terminal region of the myosin rod (Fig. 1A).
MSM patients exhibit heterogeneity in presentation and patterns of inheritance. There are young-onset (10–12,19,20) and adult forms (10,12,14,18–26), which can be sporadic (10,17) or familial (10,12,13,21,22,24–26) with an autosomal dominant- R1845W, H1901L, L1793P, L1779P, X1936W and K1784del (10,12,21,22,24–26) or recessive- E1883K and R1820W (14,18) phenotype. The clinical findings are equally diverse and commonly include hypotonia and delayed motor milestones with proximal muscle weakness that could be non-progressive or rapidly progressive with variations in severity and progression within the family (12,26). The pattern of muscle weakness is symmetrical, with weakness accentuated in the proximal muscles of both the upper and lower limbs (19,25). Muscle from the affected individuals show subsarcolemmal hyaline bodies (HBs) in the type 1 fibers, which is the hallmark of the disease (10,15,17,19–26). Type 2 fibers remain unaffected (10). Type I fiber predominance is noted in several patients (10,18,19,22,23,25). Immunohistochemical analyses have shown that the inclusions in the HBs are immunoreactive with anti β-MHC monoclonal antibodies, with retained ATPase reactivity. HBs lack activity for glycogen and oxidative enzymes but are found to be rimmed with desmin (21–25,27). Electron microscopy shows accumulations of amorphous and proteinaceous granulo-filamentous material, which is not membrane bound but found in continuity with the myofibrils (10,17,19–22,24,26,28). Despite pathogenetic characterization of MSM, the mechanism of formation and functional consequences of hyaline inclusions in muscle remain to be discovered. Mutations in the rod domain are hypothesized to result in altered stability of MHC coiled-coils with defective assembly and/or instability of thick filaments (10,12,14,29).
For this study, we generated Drosophila melanogaster transgenic MSM models for comprehensive physiological, structural, biochemical, and biophysical analysis of the human L1793P, R1845W and E1883K mutations to examine the mechanistic basis of the disease. Drosophila is a powerful model system for studying skeletal muscle myopathies as the thoracic indirect flight muscles (IFMs) have a well-characterized sarcomeric structure analogous to that of vertebrates (30). Drosophila possesses a single muscle Mhc gene and relies solely on alternative RNA splicing for different isoform expression in distinct muscles (31,32). The MSM mutant myosins were expressed in IFM and jump muscle MHC-null organisms (33–35) to obviate confounding effects of non-mutated isoforms. We found that flies expressing exclusively the L1793P, R1845W, or E1883K mutant myosin exhibit severe impairment of flight and jump ability concomitant with progressively disrupted muscle ultrastructure and myosin aggregates, as found in human biopsies. In vitro studies revealed reduced myosin filament assembly with all three mutant myosins and decreased proteolytic stability of the L1793P and E1883K filaments. We propose that altered thick filament assembly and integrity primarily contribute to the muscle pathology in MSM patients.
Results
Localization and conservation of MSM alleles
MSM-inducing mutations are located at the distal end of the α-helical coiled-coil rod region of the β-cardiac/slow muscle myosin heavy chain (MHC) (Fig. 1A), encoded by the MYH7 gene. The myosin rod has an extremely ordered and evolutionarily conserved heptapeptide repeat pattern of amino acids (Fig. 1B). The distal rod domain, corresponding to exons 37–40 of the human MYH7 gene, is the locus of all MSM-inducing mutations (10,12–18). Clustal omega multiple sequence alignments of full-length β-MHC of various species with the IFM MHC sequence of Drosophila corroborate a high degree of conservation of the distal rod residues, including some residues that, when mutated, induce MSM (Fig. 1C).
Generation of transgenic MSM flies
Three MSM mutations were selected for investigation, L1793P, R1845W and E1883K, based on their prevalence reported in literature (10,13,14,20,24–26) and their conservation (Fig. 1C). Drosophila genomic Mhc encoding the human L1793P, R1845W or the E1883K mutation was cloned into a P element pCaSpeR 4 vector (36) and used to create transgenic lines by germline transformation (37). Transformed lines with transgene cytolocations mapped to the X, third, and fourth chromosome were crossed into the Mhc10 genetic background, where Mhc10 is a mutation that prevents endogenous myosin heavy chain mRNA production in the indirect flight muscles (IFM) and the tergal depressor of trochanter (TDT) jump muscles (33). Transgenic MHC abundance in IFMs was measured using polyacrylamide gel electrophoresis and densitometry. Transgenic lines with near wild-type MHC expression levels were selected for further analyses. Three L1793P lines (L1793P-1, L1793P-4 and L1793P-6), two R1845W lines (R1845W-3 and R1845W-6) and two E1883K lines (E1883K-2 and E1883K-5) with transgenic MHC levels equivalent to yw ‘wild-type’ control were identified (Table 1). To assuage concerns of chromosomal position effects on phenotype, we elected to study three lines per genotype. To generate a third line for the R1845W mutant, we crossed two lines (R1845W-1 and R1845W-2) with transgenes on the third chromosome that were each homozygous lethal to create a trans-heterozygote (R1845W-1/2) that had an MHC content similar to control (Table 1). Two additional lines for the E1883K mutant (E1883K-3 and E1883K-4) were generated by recombining the transgene on the second chromosome onto the Mhc10 chromosome. Both showed MHC abundance in IFMs comparable to yw (Table 1). Female PwMhc2 flies, which express two inserted copies of the normal Mhc gene in the Mhc10 background, were used as the transgenic wild-type control in all studies (38). RT-PCR using RNA isolated from IFMs confirmed that the mutant Mhc transcript substituted for the wild-type transcript in all lines generated. In addition, we verified appropriate alternative splicing of the transgenic Mhc transcripts in the IFMs (see Materials and Methods).
Table 1.
Transgene chromosomal locations, MHC abundance, flight indices and jump distances of transgenic lines
| Transgenic line | Transgene cytolocation | %MHC abundancea Mean ± SEM (n) | Flight Index Mean ± SEM (n) | Jump distance (cm) Mean ± SEM (n) |
|---|---|---|---|---|
| PwMhc2 | X | 99.8 ± 2.2 (6) | 5.20 ± 0.10 (241) | 5.53 ± 0.06 (50) |
| L1793P- 1 | 4 | 97.1 ± 6.4 (6) | 0.33 ± 0.06 (204) | 3.02 ± 0.04 (50) |
| L1793P-4 | 4 | 94.9 ± 3.8 (6) | 0.45 ± 0.07 (205) | 2.95 ± 0.04 (50) |
| L1793P-6 | X | 94.2 ± 5.2 (6) | 0.38 ± 0.06 (219) | 3.00 ± 0.04 (50) |
| R1845W-3 | 3 | 91.5 ± 3.5 (6) | 0.38 ± 0.06 (213) | 2.97 ± 0.05 (50) |
| R1845W-6 | 3 | 98.6 ± 6.3 (6) | 0.42 ± 0.06 (224) | 3.04 ± 0.05 (50) |
| R1845W-1/2 | 3 | 100.9 ± 7.0 (6) | 0.43 ± 0.06 (210) | 2.96 ± 0.04 (50) |
| E1883K-2 | 3 | 98.6 ± 4.8 (6) | 0.32 ± 0.05 (235) | 2.97 ± 0.04 (50) |
| E1883K-3 | 2 | 95.8 ± 6.2 (6) | 0.28 ± 0.05 (233) | 2.93 ± 0.04 (50) |
| E1883K-4 | 2 | 93.7 ± 4.1 (6) | 0.27 ± 0.05 (242) | 2.92 ± 0.04 (50) |
| E1883K-5 | 3 | 92.8 ± 3.7 (6) | 0.31 ± 0.06 (246) | 2.98 ± 0.04 (50) |
Relative to yw control.
The L1793P, R1845W and E1883KMhc mutations depress flight and jump ability
To determine if the mutant myosins can support muscle function, flight and jump tests were performed on one-day-old flies homozygous for the MSM-inducing mutations. Female flies were used for all studies, as some transgenic lines have the Mhc transgene located on the X chromosome, with the males expressing only a single copy. PwMhc2 transgenic controls displayed normal flight behavior with the majority flying upward [flight index (FI) of 5.20 ± 0.10]. On the contrary, all mutant lines demonstrated a lack of flight ability (FI = 0.27–0.45, P < 0.0001) (Table 1). In addition, expression of the L1793P, R1845W or E1883K MHC resulted in a wings-up phenotype that is frequently associated with damaged and/or ‘hypercontracted’ IFM fibers (39–41). The average jump distance of one-day-old PwMhc2 controls was 5.53 ± 0.06 cm (Table 1). In contrast, Drosophila expressing mutant MHC had severely diminished jump ability (2.92–3.04 cm, P <0.0001) (Table 1). Thus, the skeletal muscle function in Drosophila parallels the muscle weakness symptom documented in afflicted patients.
Loss of flight in MSM flies is associated with abnormal IFM fibers, disrupted sarcomeric structure and altered muscle proteostasis
To investigate if IFM morphology is altered in the MSM mutants relative to controls, we employed fluorescent microscopy imaging of bisected thoraces. One-day-old PwMhc2 flies demonstrated an ordered dorsal-longitudinal IFM (DLM) structure with the six myofibers spanning the length of the thorax (Fig. 2A). All three MSM mutants, however, displayed strikingly altered IFM histopathology, with torn fibers that were bunched at the attachment sites (Fig. 2A). We used confocal microscopy to study sarcomeric structure in the transgenic lines. Whole mount confocal micrographs of PwMhc2 DLMs revealed a regular sarcomeric pattern (Fig. 2B and C). The wild-type transgenic MHC had incorporated uniformly into sarcomeres as evident by the distinct A-bands with their central M-lines (Fig. 2C). In opposition, thoraces of all three MSM mutant flies showed no obvious sarcomeric structures (Fig. 2B). Notably, abundant aggregates composed of MHC were identified within the mutant flies’ musculature, reminiscent of the hyaline bodies (HBs) in afflicted patient muscles (10,17,19–22,24,26,28) (Fig. 2C).
Figure 2.

Expression of MSM mutations in flies is characterized by disruption of IFM fibers and sarcomeric integrity, along with accumulation of myosin and poly-ubi-quitin aggregates. (A) Fluorescent micrographs of one-day-old transgenic control (PwMhc2) and mutant dorsal longitudinal IFMs. PwMhc2 Drosophila show normal IFM morphology, while all three mutants display torn and separated fibers. (B–E) Whole mount confocal images from one-day-old PwMhc2 control and mutant hemithoraces. PwMhc2 flies show ordered sarcomeres with regular I-bands (TRITC-Phalloidin labeled actin) (B) and A-bands with prominent M-lines (anti-MHC) (C). All three mutants, however, show no evident sarcomeres and have large MHC aggregates within their thoracic musculature (C). In addition, all three mutant IFMs contain several poly-ubiquitinated protein inclusions, indicative of altered proteostasis (D). Scale bar = 5 µm. (F) Fluorescent micrographs of bisected P9 pupae depict the presence of all six DLMs in the PwMhc2 and MSM mutant Drosophila. Arrows delineate the enveloping pupal case. (G,H) Whole mount confocal micrographs of the bisected pupae. PwMhc2 pupal muscles show assembled myofibrils with organized sarcomeres with apparent Z-lines (G) and M-lines (H). All three MSM mutants show myofibrils, but lack an ordered sarcomeric pattern. However, as opposed to 1-day-old flies (C), pupal muscles showed MHC localization throughout the length of the myofibrils with no distinct aggregates (H). Scale bar = 5 µm. Arrows in B, G denote Z-lines and arrows in C, H demarcate M-lines.
Protein surplus myopathies, characterized by molecular aggregates are often accompanied by altered proteostasis. Damaged and/or aggregated proteins are post-translationally targeted for degradation by ubiquitination with subsequent degradation via the proteasome or macroautophagy (42–44). To test the hypothesis that muscle dysfunction during MSM could be associated with reduced protein homeostasis, we immunohistochemically assessed aggregation of poly-ubiquitinated substrates. IFMs of young, one-day-old PwMhc2 flies revealed no discernible aggregates (Fig. 2D). Strikingly, IFMs of all three MSM mutants contained numerous distinct poly-ubiquitinated aggregates (Fig. 2D). Interestingly, none of the abundant myosin aggregates included ubiquitinated protein (Fig. 2E), which is reminiscent of the immunohistochemical analyses of human skeletal muscle biopsies from MSM patients, where HBs similarly did not display ubiquitin staining (10). This potentially indicates that the mutant MHC in the aggregates, which failed to stably integrate into sarcomeres, may not be destined for degradation.
To ascertain if the abnormal structure of the IFMs and absence of myofibrils in the MSM adult flies were due to defects in the DLMs fiber formation, we examined the IFM morphology and myofibrillar structure of P9 pupae (45). Fluorescent microscopy of bisected pupae showed the presence of six DLMs in the PwMhc2 control and all three MSM mutants (Fig. 2F). As all DLM fibers appeared to be intact, we presume that the MSM mutations do not cause overt defects in fiber formation. High magnification confocal micrographs exhibited a regular sarcomeric pattern in the PwMhc2 controls with apparent Z- and M-lines with the phalloidin and anti-MHC antibody staining, respectively (Fig. 2G and H). All three MSM mutants showed distinct cylindrical myofibrils, albeit with no regular sarcomeric arrangement (Fig. 2G). Some sarcomere-like repeating units were noticeable in the L1793P pupal muscles, but none was distinguishable in the R1845W and E1883K mutants (Fig. 2H). In contrast to the staining observed in younger adults (Fig. 2C), the mutant pupal muscles showed myosin co-localized to the myofibrils with actin, with no evidence of distinct MHC aggregates (Fig. 2H). Thus, it appears that the MHC aggregates do not form during early myofibrillogenesis.
MSM mutations result in progressive ultrastructural defects in IFM myofibrils
Drosophila IFMs have a highly ordered myofilament lattice that is amenable to ultrastructural analysis. We used transmission electron microscopy (TEM) to ascertain how MSM-inducing myosin affected muscle development and maintenance. IFMs of late stage (P12-P13) pupae and young (<12 h old) adults were imaged. Longitudinal sections of homozygous PwMhc2 controls displayed normal sarcomeric structure with easily discernable Z-lines, A-bands, and M-lines both in the pupal stage and in young adults (Fig. 3A, I, M, U). Transverse sections showed round myofibrils with an organized double hexagonal array of myofilament packing (Fig. 3E and Q). However, sections through mutant thoraces revealed severe ultrastructural abnormalities, even in pupae. Longitudinal sections depicted sarcomeric disarray with misaligned myofilament packing and loss of integrity of the M-lines and to some extent the Z-lines (Fig. 3B–D). Transverse sections through pupal fibers revealed misaligned subsections of myofibrils with several areas showing disruption of the hexagonal arrangement of the thick and thin filaments (Fig. 3F–H). Remarkably, IFMs of the mutants contained areas composed largely of granular and filamentous material adjacent to myofibrils that appeared very similar to the HB inclusions identified in muscle biopsies of MSM patients (10,17,19–22,24,26,28) (Fig. 3J–L). These defects, observed in the contracting pupal muscle, could indicate disordered myofibrillar assembly or decreased stability of the myosin filaments causing myofibril degeneration.
Figure 3.

MSM mutant IFMs show progressive ultrastructural defects. TEM micrographs of P12-P13 late-stage pupal (A–L) and young adult (M–X) IFMs. Longitudinal sections of PwMhc2 DLMs show normal sarcomeric structure with evident Z- and M-lines (A, I, M, U). In contrast, longitudinal sections of all three mutants reveal sarcomeric disarray and myofilament mispacking in both pupal (B–D) and adult DLMs (N–P). Transverse sections of PwMhc2 display crystalline-like double hexagonal myofilamentous arrays (E, Q) that are altered by the MSM mutations (F–H, R–T). Inset in (E) shows the hexagonal arrangement of the thick filaments. Mutants show disruptions in regions of myofibrils and misaligned filament lattice structures (arrows in F–H). In addition, the MSM homozygotes contain large granular/fibrillar inclusions (J–L, V–X) between the myofibrils or adjacent to sarcomeres that appear similar in composition to the hyaline inclusions noted in affected humans. In all sections, the ultrastructural defects worsen with age in the mutants. Magnification is the same in all longitudinal sections (A–D, I–L, M–P, U–X). Magnification is the same in all transverse sections (E–H, Q–T). Scale bar = 1 µm. Z and M denote Z-lines and M-lines respectively.
Electron micrographs from muscles of young mutant adults showed a worsening myofibrillar structure relative to pupal muscles, with few intact sarcomeres. Longitudinal sections through thoraces of adult mutant flies displayed loss of myofilaments and M-lines from extant sarcomeres (Fig. 3N–P). Cross-sections demonstrated additional interruptions to the myofilament lattice with evidence of increasing gaps consistent with thick filament loss (Fig. 3R–T). Several large areas showed completely disrupted myofibrillar structures and are composed predominantly of hyaline-like inclusions (Fig. 3V–X), indicative of a progressive decline in the muscle architecture.
MSM mutations diminish myosin filament assembly
We performed in vitro assembly assays to examine potential differences in polymerization between wild-type and MSM mutant myosins by exploiting the ability of myosin to form synthetic filaments under low ionic salt conditions. Ten µg of purified myosin from IFMs were added to solutions at various salt concentrations (500 mM to 50 mM NaCl) and then incubated on ice for 30 min to permit filament assembly. Samples were centrifuged and the abundance of myosin in the fractions was quantified by gel electrophoresis and densitometry. Supernatants contained unpolymerized myosin molecules, while pellets contained myosin molecules that formed insoluble filaments. As anticipated, at high salt concentrations, most wild-type and mutant myosins remained in solution (Fig. 4A). However, starting at 275 mM NaCl, a differential assembly rate was observed (Fig. 4B), with the three mutant myosins displaying a reduced percent of myosin assembled into filaments relative to control. An EC50 value, indicative of the effective concentration at which 50% of myosin assembles into filaments was extrapolated. Wild-type myosin formed filaments with an EC50 of 201.90 ± 9.24 mM NaCl (Fig. 4C). In contrast, EC50 for L1793P, R1845W and E1883K myosins were 162.70 ± 7.77, 163.10 ± 5.89 and 166.20 ± 5.60 mM NaCl, respectively (Fig. 4C, P < 0.01). Importantly, at physiological salt concentration (150 mM NaCl) wild-type myosin almost completely assembled into filaments while a significant fraction of all three mutant myosins remained soluble (Fig. 4B and D, P < 0.01). Our results indicate that L1793P, R1845W and E1883K myosins do not polymerize into filaments as readily as wild-type myosin.
Figure 4.

MSM mutant myosins are characterized by altered in vitro filament forming abilities. (A) SDS-PAGE demonstrates that following incubation in various salt concentrations and sedimentation, more mutant myosins remain in the soluble fraction and less in the pellet fraction compared with wild-type. This is indicative of reduced myosin filament assembly in the mutant. (B) Densitometric analyses of gels plotted as a percentage of myosin filaments formed from high to low salt conditions. Mutant myosins require lower salt concentrations to drive filament formation compared with wild-type. (C) MSM mutant myosins have lower EC50 values (effective salt concentration at which 50% of myosin assembles into filaments, P < 0.01). (D) Under physiological salt conditions (150 mM NaCl), MSM mutant myosins form significantly fewer filaments than wild-type (P < 0.01). Values represented are mean ± SEM.
L1793P myosin assembles into short filaments
To investigate gross morphology of synthesized myosin filaments by TEM, wild-type and L1793P myosins were diluted from high salt storage buffer into 100 mM KCl buffer. The L1793P myosin was chosen for this characterization as proline residues in coiled-coils are predicted to introduce kinks in the α-helical strands resulting in distortions and destabilization (46). Thus, structural differences in assembled filaments, if any, would hypothetically be best appreciated in this mutant myosin. Electron microscopy of positively stained molecules revealed linear filaments with gradually tapered ends and no major morphological differences between the wild-type and L1793P filaments (Fig. 5A). However, measures of filament contour lengths demonstrated that the L1793P mutant formed significantly shorter filaments (495.0 ± 128.3 nm) compared with wild-type (713.7 ± 131.8 nm) (Fig. 5B, P < 0.0001).
Figure 5.

L1793P mutant myosin forms shorter filaments relative to wild-type. (A) TEM micrographs of synthesized wild-type myosin filaments show linear structures of relatively uniform size and shape. L1793P mutant myosin assembles into morphologically similar filaments. Scale bar = 500 nm. (B) Randomized contour measures of myosin filament lengths show that L1793P mutant myosin, on average, formed significantly shorter filaments than wild-type myosin (P < 0.0001). Error bars indicate SD.
L1793P and E1883K mutations result in reduced stability of synthetic filaments
To determine if myosin filament stability is influenced by the MSM mutations, in vitro proteolysis experiments were conducted with synthetic myosin filaments. Assembled filaments were treated with α-chymotrypsin for 2 h, with samples retrieved at various time points (0 to 120 min). Incubation with α-chymotrypsin yielded the anticipated 130 kD rod and 90 kD S1 fragments within a minute of digestion (47) (Fig. 6A). No major differences in the stability of full-length filaments were identified as manifested by complete digestion into the S1 and rod components within 10 min (Fig. 6A). Extension of the proteolysis reaction for 30–60 min resulted in generation of an additional fragment from wild-type myosin, originating from a single cleavage site in the rod fragment. Interestingly, three truncated fragments were observed to originate from the L1793P and E1883K rods (Fig. 6A). The R1845W myosin, however, formed just one intact rod fragment and one truncated product similar to wild-type myosin (Fig. 6A). Despite extension of proteolysis to 120 min, no other truncated rod fragments were detected in the wild-type and R1845W myosin.
Figure 6.
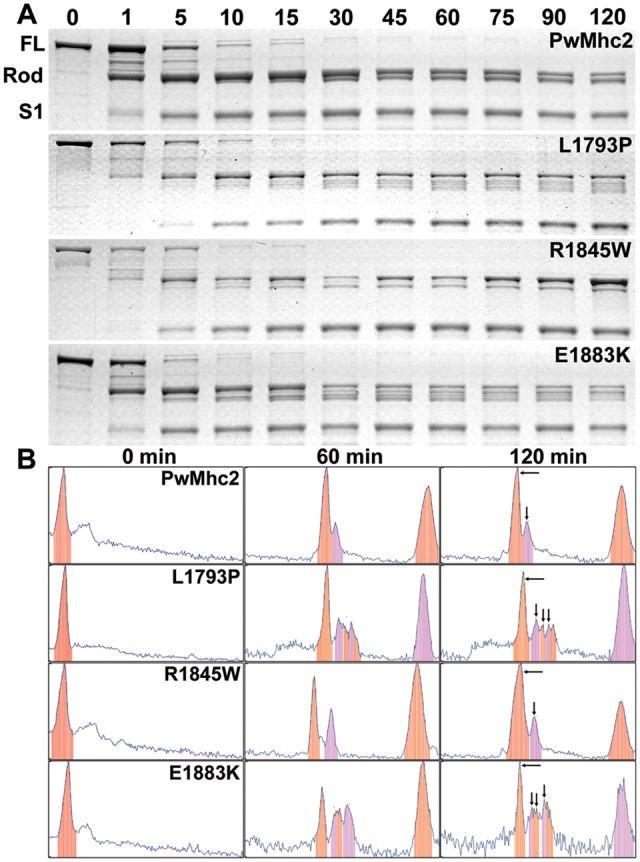
L1793P and E1883K mutant myosin filaments show altered stability compared with wild-type. (A) SDS-PAGE separation of wild-type and mutant filaments subjected to α-chymotrypsin proteolysis with samples collected at various time points during the reaction. Proteolysis of full-length (FL) myosin filaments yields ∼130 kD sized rod fragments and ∼90 kD S1 head fragments within 5–10 min. After 30-60 min of digestion, additional cleaved products originating from the rod fragments are apparent. Wild-type and R1845W myosins show one truncated rod fragment while the L1793P and E1883K myosins show three, indicative of reduced stability. Extended proteolysis of 120 min does not yield any new truncated products for wild-type or R1845W myosins. (B) Un-Scan-It peak profiles of protein bands detected in the electrophoresed gels at baseline (0 min), 60 and 120 min after proteolysis. Peak analyses of digested wild-type and R1845W mutant myosin filaments corroborated presence of one truncated rod peak, while those of the L1793P and E1883K mutants confirmed the presence of three truncated fragments in addition to the intact rod fragment (designated by arrows in the right panel).
Corroborative evidence of myosin digestion products was obtained with additional analyses using Un-Scan-It gel analysis software. Gel bands signal analyses confirmed the presence of two rod peaks 60 min post-digestion with wild-type and R1845W (Fig. 6B). In contrast, E1883K and L1793P showed four prominent rod peaks (Fig. 6B). In all cases a substantial amount of intact rod fragment remained at 120 min post-digestion, suggesting that the MSM mutations did not make myosin filament rods completely susceptible to proteolytic degradation. Our proteolysis data indicate that differences in stability of myosin filament rods exist in the E1883K and L1793P mutant filaments compared with wild-type, but not in the R1845W filaments.
MSM mutant myosins in the Mhc1 background induce lethality
To determine whether any of the MSM alleles that result in impaired filament assembly and integrity would support viability, we expressed the transgenic Mhc in the absence of wild-type myosin. To this end we crossed each MSM allele into the Mhc1 mutant background. The Mhc1 mutation deletes most of constitutive exon 5 and part of the preceding intron, resulting in a complete MHC null phenotype that is homozygous lethal (35). Two copies of PwMhc2 transgenic wild-type myosin rescued the homozygous lethal phenotype of the Mhc1 mutant. Notably, we found that L1793P, R1845W and E1883K transgenes crossed into the Mhc1 background yield no surviving MSM homozygotes, indicating that the mutant myosins alone are not sufficient to sustain organismal survival.
Discussion
The assembly of myosin into thick filaments is a multistep process that requires proper folding of the α-helices into coiled-coils and ordered assembly of these coiled-coils in correct register to form thick filaments during myofibrillogenesis. Disease-causing mutations in the distal rod domain of MYH7 primarily result in dilated cardiomyopathy, myosin storage myopathy or Laing early onset distal myopathy (9). Few in vitro molecular characterization studies of such mutations have reported defects in filament assembly or stability. Armel and Leinwand (48) expressed rod fragments to examine biochemical characteristics resulting from mutating the R1500 residue to a proline or tryptophan. R1500P causes Laing early onset distal myopathy (49) while R1500W myosin is reported to cause dilated cardiomyopathy (50). Both mutations showed differences in paracrystal assembly rates and stability, albeit to varying degrees, indicating that even subtle differences in the molecular phenotypes could result in diverse clinical phenotypes. Similarly, the E1356K mutation (51) causes hypertrophic cardiomyopathy with no skeletal muscle defects while the E1883K mutation causes both hypertrophic cardiomyopathy and MSM (14). Despite identical charge changes in surface residues of the heptad repeat, different pathological phenotypes were observed, which could be attributed to variance in localized alterations to the protein (29,52). Another fascinating LMM mutation is Drosophila Mhc6, which affects flight ability and IFM ultrastructure (39). It substitutes an invariant arginine with a histidine (R1559H). Despite charge conservation, the muscles showed structural and functional defects and the mutant myosin was prone to cleavage in vivo. This indicates that in addition to charge conservation, steric properties of LMM residues are vital, shedding light on the importance of the conservation of specific residues in precise positions of myosin rods through evolution. Thus, individual characterization of LMM mutations is crucial to identify the combination of biophysical and biochemical effects that form the molecular basis of disease etiology for each myosin allele.
MSM patients, all with mutations in the distal portion of LMM, exhibit a wide spectrum of clinical heterogeneity, with variability in disease severity within families (12,26). Genetic modifiers, lifestyle, and environmental factors influence genotype-phenotype correlations, making predictions of the molecular basis of the disease perplexing. This is further complicated by the fact that humans express several sarcomeric MHC isoforms that may partly compensate for the mutant myosin, both in the skeletal muscles and in the heart. Thus, models that permit analyses of the mutant myosin without the influence of wild-type protein would greatly assist with determining pathogenic mechanisms. The presence of the single Drosophila melanogaster Mhc gene coupled with availability of myosin-null lines (34,53) facilitates relatively straightforward generation of isogenized populations of transgenic flies with muscle-specific expression of exclusively mutant myosin.
Here, we generated several Drosophila MSM models to resolve the basis by which mutations in MHC cause skeletal muscle myopathy from the tissue to the molecular level. Our fly models mimic the phenotypes of MSM patients, who show progressive proximal muscle weakness with characteristic sub-sarcolemmal accumulation of MHC in skeletal muscles. We demonstrated that expression of the L1793P, R1845W and E1883K MSM myosins severely compromises function of the flight and jump muscles (Table 1) and disrupts muscle ultrastructure (Fig. 3) with evidence of altered protein homeostasis (Fig. 2D). The mutants display myosin aggregates akin to hyaline bodies (HBs) (Fig. 2C) and these aggregates are apparently stable, since they are not marked for degradation by ubiquitination (Fig. 2E). The myosin aggregates appeared scattered throughout the thorax, as opposed to the compact HBs in the skeletal muscles of MSM patients (Fig. 2C). This could be due to a lack of desmin in flies, which may serve to organize the aggregates in humans, where HBs are found to be rimmed by this intermediate filament protein (21–25,27), as well as the high contraction frequency and power output of the indirect flight muscles, which could disrupt formation of such structures. In human patients, filamentous inclusions between myofibrils have been described as abortive filaments (54), suggesting that the inclusions may be primarily composed of thick filaments that failed to assemble in an ordered fashion for inclusion within the myofibrils. Presence of inclusions that reduce the effective functioning sarcomeric area could be a contributing factor to the weakness in patients’ muscles. However, the specificity of proximal muscle weakness, and the scapuloperoneal muscle in particular, is perplexing as MYH7 is expressed in all slow twitch muscles. Clearly, the mutant myosin yields muscles that differentially fail to maintain muscle function in the human disease. Levels of expression of additional myosin isoforms as well as muscle architecture and usage likely play roles in the degree of dysfunction of each muscle type. Animal models of the disease may not be particularly useful in unraveling the pathology of specific muscle involvement due to these factors.
We observed that Drosophila muscle fibers and myofibril-like structures are present at the pupal stage in MSM homozygotes (Figs 2F–H and 3B–D). Since thick filaments are evident in TEMs of late-stage P12-P13 pupae (Fig. 3), rod dimerization and filament assembly can transpire in vivo. Our in vitro analysis using isolated pure populations of full-length mutant protein shows that MSM myosin is constrained in its ability to assemble into filaments relative to wild-type myosin at physiological salt concentrations (Fig. 4) and that assembled synthetic L1793P and E1883K myosin filaments are more susceptible to proteolytic degradation compared with wild-type controls (Fig. 6). The progressive severity of myofibrillar disarray observed in MSM adult flies (Fig. 3N–P, R–T) suggests that, like synthetic myosin filaments in vitro, thick filament stability is compromised in vivo as well. In addition, distinct myosin aggregates are not evident in the P9 pupal muscles (Fig. 2H), but appear in young MSM adults (Fig. 2C), as do foci of polyubiquitin (Fig. 2D), indicating a degenerative process. Altered assembly in the contracting muscles could explain the deteriorating myofibrillar architecture and identification of HBs in the in vitro proteolytically stable R1845W myosin filaments.
These results together facilitate insights into the mechanistic basis of the disease model. Assembly of myofibril-like structures in vivo occurs in the early stages of development, with intact DLMs that lack distinct myosin aggregates. The bulk of the myosin aggregates appears to form from unassembled or disassembled thick filaments as the pupal muscle contraction continues and metamorphosis transpires. Our in vitro proteolysis studies using pure myosin may mimic the behavior of in vivo thick filaments, which could also exhibit decreased stability leading to increasingly abnormal myofibrils, altered proteostasis and accumulation of hyaline-like inclusions. Overall, there are both assembly defects and degenerative components to the disease model.
Accumulation of poly-ubiquitinated protein aggregates in the young MSM flies suggests altered proteostasis. It is interesting that poly-ubiquitin is not detected in the myosin aggregates, since the ubiquitin-proteasome pathway is responsible for the bulk of intracellular protein degradation (55). In a human patient, MuRF1 E3 ubiquitin ligase involvement with normal thick filament degradation was found, in that mutations in MuRF1 and MuRF3 yielded MSM-like skeletal myopathy and cardiomyopathy (56). Histopathological analyses of a skeletal muscle biopsy revealed subsarcolemmal β-MHC inclusions in slow skeletal muscle fibers that were non-ubiqutinated with ultrastructural sarcomeric disorganization. This myopathology is strikingly similar to human MSM and to our transgenic Drosophila. MuRF1 and MuRF3 double mutant knock-out mice with muscle impairment and myosin accumulation both in the heart and skeletal muscle has also been reported (57). Thus, MSM mutations and altered UPS-mediated proteostasis can result in similar protein aggregation myopathy.
A previous effort to characterize the biochemical basis of MSM (29) used purified wild-type and mutant LMM protein fragments in vitro. This study identified unique molecular phenotypes in each mutant, as opposed to a unifying mechanism resulting in MSM. L1793P myosin showed the most severe defects with a slow nucleation of filament assembly, highly reduced thermal stability, and moderately reduced resistance to proteolysis. E1883K myosin displayed truncated filament assembly, lower thermal stability, and high susceptibility to proteolysis. The location of the E1883K mutation in the assembly competence domain potentially contributes to the elevated instability of this protein. Similar molecular phenotypes were observed with the R1845W myosin that showed altered assembly properties and reduced thermal stability, but with resistance to proteolysis. This is reminiscent of our finding for full-length R1845W myosin molecules when assembled into filaments.
Both the R1845W and the E1883K mutations affect charged surface residues of the alpha-helical coiled coil (Fig. 1A) and the mutations therefore are expected to disrupt the charged interactions necessary for thick filament formation and stability. Despite the similarity in locations, the former causes dominant human disease while the latter is reported to be recessive. In contrast, the L1793P mutation affects a residue that is buried in the hydrophobic seam of the coiled-coil. L1793P is likely to have its effect due to introduction of a kink in the secondary structure that disrupts coiled-coil formation. This could reduce dimerization at physiological salt concentrations and cause long-range effects on the ability to assemble thick filaments. No obvious property that sets apart L1793P myosin from the other two forms was observed in our study, nor was there an apparent reason for the reported recessive nature of E1883K. It was interesting that the L1793P and E1883K mutations uncovered proteolytic sites in synthetic thick filaments that are distant from the mutations. The abnormal coiled-coil structure and/or thick filament assembly that can arise due to the incorrect charge or helicity introduced by each mutation could engender propagated effects which reveal proteolytic sites that are typically buried in wild-type myosin filaments. Proteolysis at a distant site in the rod was previously observed both in vivo and in vitro in a Drosophila myosin rod mutant (39).
Very recent attempts to study MSM mutant myosin function in vivo provided some insights into disease causation, but did not recapitulate the disease phenotypes as completely as our Drosophila models. Dahl-Halvarsson et al. (58) expressed MSM myosins in developing human myoblasts in vitro and found that R1845W and H1901L myosins yielded aggregates and disrupted myofibrillogenesis. However, this was not the case for L1793P myosin, which appeared to incorporate normally in developing myofibrils. They also expressed wild-type, R1845W, E1883K and H1901L myosins in C. elegans, but found that motility of all organisms, including wild-type was compromised, likely due to myosin overexpression. They did observe partial rescue of paralyzed unc-54 (MHC B null) worms by MSM myosins, demonstrating some functionality of the mutant proteins.
In summary, our Drosophila models are the first to demonstrate explicitly the effects of MSM mutations on skeletal muscle. We provide the first evidence of aggregates formed by the mutant myosins within a model organism and reproduce filamentous inclusions between myofibrils. Our data suggest that the mutations in MHC engender the skeletal muscle phenotype by altering their filament assembly properties and/or by causing instability of the formed filaments. Our in vitro results using purified full-length myosin substantiate and augment some previous results using rod fragments (29). Our Drosophila models should serve as an effective tool for testing potential therapeutic approaches by exploiting the connection between thick filament turnover and the UPS system to alleviate accumulation of the myosin aggregates that contribute to the myopathic phenotype. While the E1883K mutation is recessive in humans and therefore is faithfully mimicked by our Drosophila homozygote model, future characterization of the effects of the dominant L1793P and R1845W MSM myosins in the heterozygous state on skeletal and heart muscle will provide insights into their confounding effects on wild-type myosin function.
Materials and Methods
Multiple sequence alignments
Sequence comparison of full-length β-MHC (encoded by MYH7) from several species and Drosophila IFM MHC isoform K were performed using Clustal Omega multiple sequence alignment program. Residues are shaded based on degree of conservation.
Construction of the MSM Mhc transgenes
Genomic DNA from the single Drosophila melanogaster Mhc gene was used to generate analogues of the human L1793P, R1845W and E1883K MSM mutations using standard cloning techniques (59). Production of the mutant Mhc constructs was initiated by making subclones of the 12.6 kb 3’ Mhc end cloned in a pBluescript KS vector. A 9 kb fragment was isolated using the Nco I restriction endonuclease and cloned into pLitmus 28i vector. A 3.1 kb Stu I enzyme-generated subfragment was made from the Nco I fragment and cloned into pLitmus 28i vector. A 1.8 kb Bgl II endonuclease cleaved subfragment was then made, which included exon 17 (the locus of the L1793, R1845 and E1883 codons) and was used for in vitro site directed mutagenesis (QuikChange Site-directed mutagenesis kit, Agilent Technologies) using the following specific oligonucleotide primer pairs (Integrated DNA technologies).
L1793P: 5’ ATCAAGGAGCCGCAGGTCCGT 3’ and 5’ ACGGACCTGCGGCTCCTTGAT 3’
R1845W: 5’ AAGTCCGAGTGGCGCGTCAAG 3’ and 5’CTTGACGCGCCACTCGGACTT 3’
E1883K: 5’ AGGCAGATCAAGGAGGCTGAG 3’ and 5’ CTCAGCCTCCTTGATCTGCCT 3’
Upon sequence confirmation of the mutation in the site directed mutagenesis product, each subfragment was sequentially cloned back into the bigger fragment from which it originated to regenerate the complete 3’ Mhc clone. The 12.6 kb 3’ Mhc construct with the mutation was removed from its pBluescript KS vector by Eag I mediated restriction digestion and ligated to the 19.1 kb 5’ Mhc clone to generate the full length genomic Mhc mutant construct. The final construct was in the P element vector, pCaSpeR 4 (36) with a Drosophila miniwhite gene w+ as a selectable eye-color marker (38). The mutant constructs were amplified and purified using QIAfilter Plasmid Maxi Kit (Qiagen Inc.) and full-length clones verified by DNA sequencing prior to microinjections into Drosophila embryos.
Generation of transgenic Drosophila
Drosophila germline microinjections were performed at BestGene Inc. as described (37). The mutant genomic DNA along with the Δ 2-3 helper plasmid (60) expressing transposase was injected into the yw strain Drosophila embryos (61). Surviving adults from the injections (G0 generation) were crossed back to yw flies and the progeny (G1 generation) screened for pigmented eye color, an indicator of successful transformation reflecting the presence of both the miniwhite (w+) marker and the Mhc transgene. A single transformed progeny from each G1 was then back-crossed into the yw background and self-crossed to generate stable homozygous transgenic lines. One thousand embryos were injected for each construct and 6 viable lines for the R1845W, 7 lines for the L1793P and 5 lines for the E1883K constructs were generated.
The chromosomal location of each transgene insertion for viable transformed lines was mapped by standard genetic crosses using multiple balancer chromosomes. Transgenic lines with the Mhc transgene on the X, third or fourth chromosome were crossed into the IFM and jump muscle MHC null Mhc10 background (33) and were used for all investigative studies. The yw strain and the transgenic wild-type PwMhc2 (38) line in the Mhc10 background were used as controls.
For recombination of a transgene on the second chromosome onto the Mhc10 chromosome, male flies from homozygous viable lines was crossed with female Mhc10/Mhc10; MKRS/TM2 flies. Mhc10/Mhc+, MhcE1883K; TM2/+ female progeny were selected and crossed with Mhc10/Mhc10; MKRS/TM2 males to permit potential recombination of the transgene with the Mhc10 chromosome during meiosis. Approximately 30–40 individual male offspring from this cross, with a possibly recombined Mhc10 and MhcE1883K(Mhc10, MhcE1883K), were back-crossed individually with female Mhc10/Mhc10; MKRS/TM2 flies. Males were used for crosses at this step, as recombination is nearly non-existent in Drosophila males. Progeny with pigmented eyes and only the TM2 marker from each individual cross were picked. The Mhc10/Mhc+, MhcE1883K genotype was differentiated from the Mhc10/Mhc10, MhcE1883K genotype based on jump ability. Flies with the Mhc10/Mhc+, MhcE1883K genotype were able to jump better in comparison to the Mhc10/Mhc10, MhcE1883K flies due to the presence of a wild-type copy of the Mhc gene. Mhc10/Mhc10, MhcE1883K flies were selected and self-crossed to create Mhc10, MhcE1883K homozygotes. Expression of the transgene and absence of wild-type Mhc transcripts were verified by RT-PCR (see below) before further analyses.
To attempt to generate flies expressing exclusively mutant myosin in all tissues, MSM transgenic lines L1793P-1, R1845-6 and E1883K-2 were crossed into the Mhc1 background (35) by standard mating crosses using multiple balancer chromosomes.
Quantitation of transgenic MHC abundance
Transgenic MHC expression levels were estimated by one-dimensional SDS-PAGE (62). yw strain flies were used as controls. Dorsal hemi-thoraces from young females were individually homogenized in 1x Laemmli sample buffer and electrophoresed in 10% polyacrylamide tris-glycine gels (BioRad). Gels were stained with Coomassie Brilliant Blue, destained, scanned on an Epson flatbed scanner and images analysed using the NIH ImageJ program gel plotting macros (https://imagej.nih.gov/ij/download.html). The myosin to actin ratio was determined for each experimental sample and compared with the ratio calculated from the yw control. Protein quantification was performed on six independent biological samples with three to four technical replicates per sample. The normalized transgenic MHC expression levels measured against control are represented as mean ± SEM.
Verification of transgene expression
Expression of mutant Mhc was verified by isolating total RNA from the IFMs of 50 transgenic flies. RT-PCR was carried out using the Protoscript M-MuLV First Strand cDNA Synthesis Kit (New England BioLabs) and 1 µg total RNA. The cDNA from the exon 17 region was amplified using specific oligonucleotide primers (5’ GAACAGCTGGGTATCTCCGAG 3’ and 5’ TTCTTATAAGCTATAATTCTC 3’) and the L1793P, R1845W and E1883K mutations verified by Sanger sequencing.
To ensure correct alternative splicing of the IFM-specific myosin transcript (that includes exon 3 b, exon 7d, exon 9a, exon 11e, exon 15a and exon 18), the following primer pairs were used for cDNA amplification and PCR products were verified by the Sanger sequencing method.
Exon 3 b: 5’ TGGATCCCCGACGAGAAGGA 3’ and 5’ GACATGATCTGGTAGTAGATGTGG 3’
Exon 7d: 5’ GGCTGGTGCTGATATTGAGA 3’ and 5’ TGTAGTGCCAACCAACCTCTTCT 3’
Exon 9a: 5’ GTTCCCCAAGGCCTCCGATCA 3’ and 5’ GCGGTCTTCTCAGCCAAAAGC 3’
Exon 11e: 5’ CTGCATGCCGCTGAAGTGAAG 3’ and 5’ GGGTGACAGACGCTGCTTGGT 3’
Exon 15a: 5’ GAACAGCTGGGTATCTCCGAGC 3’ and 5’ GGTCGAATCTTGGTGGGAAGGC 3’
Exon 18: 5’ GGCCGCAAGAGCGCGCTGC 3’ and 5’ GGTCGAATCTTGGTGGGAAGGCC 3’
Flight and jump tests
Flight tests were performed as described (63) at room temperature. Newly eclosed female flies were aged for a day at room temperature. For flight tests, each fly was released into the center of a Plexiglas chamber with a light source positioned at the top. Each fly was assigned a score of six for upward flight, four for horizontal, two for downward, or zero for no flight. The average flight index from 204-246 flies was calculated for each genotype. Flight indices are represented as mean ± SEM. Significance was assessed using Kruskal-Wallis one-way ANOVA using Dunn’s post-hoc tests. Large sample populations tested mitigated concerns due to heterogeneity in variance within the sample sets.
For jump tests, newly eclosed female flies were collected. Their wings were clipped and they were aged one day at room temperature. Individual flies were placed atop an inverted vial and coaxed with a paint brush to jump onto a paper printed with concentric circles spaced 0.5 cm apart. The three farthest jump distances out of ten trials per fly were recorded and averaged for 50 flies per genotype. Values are represented as mean ± SEM. Measured values were logarithmically transformed to ensure normal distribution of all sample sets and significance was assessed via one-way ANOVA with Bonferroni’s multiple comparison tests.
Fluorescent microscopy
Fluorescent microscopy of stained hemi-thoraces for examination of gross morphology of 1-day-old Drosophila IFMs was performed as described previously (64,65). Briefly, flies were anesthetized, and heads and abdomens were removed. Thoraces were fixed overnight in 4% formaldehyde at 4 °C and rinsed several times in 1x PBS. Fixed thoraces were arranged on a glass slide, snap frozen in liquid nitrogen and immediately bisected along the midsagittal plane using a razor blade and stained with Alexa-568 Phalloidin (1: 100 in 1x PBST, Invitrogen) overnight at 4 °C. Samples were rinsed with 1x PBS before imaging with the EVOS® FL cell imaging system (Life Technologies) at 4x magnification.
For imaging IFM morphology of pupae, P9 pupae were arranged dorsal side up on a glass slide, snap frozen in liquid nitrogen and bisected along the midsagittal plane using a razor blade. Bisected pupae were fixed in 4% formaldehyde for 30 min at room temperature and stained with Alexa-568 Phalloidin (1: 100 in 1x PBST, Invitrogen) overnight at 4 °C before imaging with the EVOS® FL cell imaging system (Life Technologies) at 4x magnification.
Confocal microscopy
For whole-mount immunostaining of the IFMs, one-day-old fly thoraces and P9 pupae were prepared and bisected as described above. Hemi-thoraces were stained with rabbit anti-Drosophila muscle myosin (1: 500, kind gift from Dr. Dan Kiehart, Duke University) and mouse anti-ubiquitinylated protein (clone FK2, 1: 250, EMD Millipore) primary antibodies. Secondary stainings were done with Alexa-488 goat anti-rabbit, Cy5-goat anti-mouse antibodies and Alexa-Fluor 568 phalloidin (Invitrogen). Bisected pupae were stained with rabbit anti-Drosophila muscle myosin (1: 500) with secondary Alexa-488 goat anti-rabbit antibody and Alexa-568 phalloidin. Samples were rinsed in 1x PBS, mounted with Vectashield (Vector Laboratories) and visualized using a Leica TCS SPE RGBV confocal microscope (Leica Microsystems) at 100x magnification.
Transmission electron microscopy
TEM of IFMs was carried out as previously described (35). Late-stage female pupae and thoraces from females <12h old were fixed in 3% paraformaldehyde and 2% gluteraldehyde in 100 mM sodium phosphate pH 7.2 overnight at 4 °C. Samples were then rinsed in 100 mM sodium phosphate buffer (pH 7.2) and fixed secondarily with 1% osmium tetroxide in 100 mM sodium phosphate (pH 7.2) for 2 h at 4 °C. Samples were then rinsed with distilled water, dehydrated using increasing concentrations of acetone, embedded in Spurr’s resin, with increasing concentrations of resin from 75 to 100%, before placing into molds. The molds were cured at 60 °C overnight. Thin sections were cut using a glass or diamond knife and stained with 2% uranyl acetate followed by lead citrate. Images were acquired on a FEI Tecnai 12 transmission electron microscope using a TVIPS 214 high-resolution camera.
For TEM of synthetic myosin filaments, formed filaments were attached to a copper grid and positively stained with 1% uranyl acetate for 10 s. Grids were washed with distilled water and dried using filter paper. Images were acquired as described above at 15 000X to 110 000X magnification.
Myosin isolation and purification from Drosophila indirect flight muscles
Thoraces of one-day-old adult Drosophila were bisected to expose the flight musculature. Dorsal longitudinal muscles (DLMs) were isolated and then skinned in a YMG [York modified glycerol: 20 mM KPi, 1 mM EGTA, 2 mM MgCl2, 10 mM DTT, 50% glycerol] and 2% Triton X-100 solution (66). Myosin was extracted and purified following a series of high-salt suspensions and low-salt precipitations (66). Protein concentrations were determined from the 280 nm peak absorbance measures on a Beckman DU-640B spectrophotometer (Beckman and Coulter).
Myosin solubility assays
Myosin from 1-day-old adult female flies (150 PwMhc2 control and 180 each of MSM homozygous mutants) was purified as described (66) and resuspended in myosin storage buffer [MSB: 20 mM MOPS, 2 mM MgCl2, and 10 mM DTT] with 500 mM NaCl instead of KCl. Ten µg of myosin were diluted in 100 µl NaCl with concentrations ranging from 500 mM to 50 mM. Samples were incubated on ice for 30 min to permit assembly of myosin filaments and then centrifuged (on a Beckman Coulter X-103 using a TLA 100.3 rotor) at 50,000 rpm (∼104,000 g) for 30 min at 4 °C. Monomeric myosins that do not form filaments remain in the supernatant (soluble) fraction, whereas formed filaments are present in the pellet fraction. One hundred µl of supernatant were collected from each sample, diluted in 2X Laemmli buffer with 50 mM DTT and heat denatured at 95 °C for 5 min. Pellets were re-suspended in the same volume of Laemmli buffer and denatured. Ten µl from the two protein fractions were electrophoresed in 10% polyacrylamide tris-glycine gels (BioRad) and stained overnight with Gel Code Blue (Thermo Scientific) at a 1: 5 dilution. Gels were scanned on an Epson Perfection 1640SU scanner, using VueScan software (Hamrick, v9.5.35). Myosin band densities in both the supernatant and pellet fractions were analysed using Un-Scan-It gel software (Silk Scientific, version 6.1). Percent myosin filaments formed at each NaCl concentration was extrapolated as the ratio of insoluble myosin to total myosin. Solubility assays were performed on three to four independent myosin preparations (150–180 flies/myosin preparation). Significance was assessed by nonlinear regression analysis and one-way ANOVA with Bonferroni multiple comparison tests.
In vitro synthetic myosin filament assembly and analysis
Synthetic myosin filaments were formed using the fast dilution method by lowering the salt concentration of myosin storage buffer (MSB). Myosin purified from IFMs of 80 PwMhc2 control and 100 MSM homozygous mutants was re-suspended in MSB with 500 mM KCl. Fifty µg of purified myosin from each genotype were diluted with MSB buffer without KCl to a final concentration of 100 mM KCl in 100 µl (0.5 µg/µl myosin concentrations). The samples were gently mixed and incubated on ice for 30 min. Synthetic myosin filaments formed were stained and visualized using TEM as described above. Images were acquired from filaments formed from three independent biological sample preparations. Filament contour length measures from individually resolved filaments from the TEM images were performed using NIH ImageJ software (n = 300; 100 measures per genotype per biological sample). Filaments were chosen for measurement using a random number generator after numbering all resolved filaments on each micrograph. Significance was assessed using Mann–Whitney t-test (two-tailed with 95% confidence intervals).
In vitro proteolysis of synthetic myosin filaments
Myosins from one-day-old adult female flies (80 PwMhc2 control and 100 MSM homozygous mutants) were purified as described above and re-suspended in MSB with 500 mM NaCl. Myosin filaments were synthesized in vitro in MSB with 100 mM NaCl, and centrifuged at 50,000 rpm (104, 000 g) for 30 min at 4 °C. Supernatants were collected and heat denatured in 5X sample buffer [5X SB: 250 mM Tris-Cl pH 6.8, 10% SDS, 30% glycerol, 5% β-mercaptoethanol, 0.02% bromophenol blue]. The pellets were gently re-suspended to a final protein concentration of 1 mg/ml in 100 mM NaCl Digestion Buffer [DB: 20 mM NaPi pH 7.0, 1 mM EDTA pH 7.0, 0.1 M NaCl, 4 mM DTT] and incubated in a 20 °C water bath. Myosin filament samples were digested with α-chymotrypsin (0.05 mg/ml; Worthington Biochemical) for 0 to 120 min. A 5 µl sample volume of the digestion mixture was taken out at specific time points, diluted in 5X SB buffer and denatured at 95 °C for 5 min. Samples were electrophoresed in 10% polyacrylamide tris-glycine gels (Bio-Rad) and stained overnight with Gel Code Blue (Thermo Scientific) at a 1: 5 dilution. Gels were scanned using on an Epson Perfection 1640SU scanner, using VueScan scanner software (Hamrick, v9.5.35). Proteolysis assays with mutant MHC were run in parallel with control PwMhc2 wild-type MHC. Twelve biological replicates of PwMhc2 and four biological replicates each of the mutant MHC were performed from independent myosin purification preparations. Band densities were analysed using Un-Scan-It gel software (Silk Scientific, v6.1).
Acknowledgements
The authors thank Dr. Nigel Laing for initial discussions and insights about MSM that led to this project and Dr. Dan Kiehart for providing the rabbit anti-Drosophila skeletal muscle myosin antibody. We would also like to thank Jennifer Suggs and Anju Melkani (San Diego State University) for helpful advice and assistance with fly genetics.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health [R01HL124091 to AC and R21OD010561 and R37GM032443 to SIB]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. AST is a Fellow of the Rees-Stealy Research Foundation and the San Diego State University Heart Institute.
References
- 1. Emerson C.P. Jr., Bernstein S.I. (1987) Molecular genetics of myosin. Annu. Rev. Biochem., 56, 695–726. [DOI] [PubMed] [Google Scholar]
- 2. McLachlan A.D., Karn J. (1982) Periodic charge distributions in the myosin rod amino acid sequence match cross-bridge spacings in muscle. Nature, 299, 226–231. [DOI] [PubMed] [Google Scholar]
- 3. Atkinson S., Stewart M. (1991) Molecular basis of myosin assembly: coiled-coil interactions and the role of charge periodicities. J. Cell. Sci. Suppl., 14, 7–10. [DOI] [PubMed] [Google Scholar]
- 4. Atkinson S.J., Stewart M. (1992) Molecular interactions in myosin assembly:: Role of the 28-residue charge repeat in the rod. J. Mol. Biol., 226, 7–13. [DOI] [PubMed] [Google Scholar]
- 5. Sohn R.L., Vikstrom K.L., Strauss M., Cohen C., Szent-Gyorgyi A.G., Leinwand L.A. (1997) A 29 residue region of the sarcomeric myosin rod is necessary for filament formation. J. Mol. Biol., 266, 317–330. [DOI] [PubMed] [Google Scholar]
- 6. Cohen C., Parry D.A. (1998) A conserved C-terminal assembly region in paramyosin and myosin rods. J. Struct. Biol., 122, 180–187. [DOI] [PubMed] [Google Scholar]
- 7. Ricketson D., Johnston C.A., Prehoda K.E. (2010) Multiple tail domain interactions stabilize nonmuscle myosin II bipolar filaments. Proc. Natl. Acad. Sci. U S A, 107, 20964–20969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oldfors A. (2007) Hereditary myosin myopathies. Neuromuscul. Disord., 17, 355–367. [DOI] [PubMed] [Google Scholar]
- 9. Colegrave M., Peckham M. (2014) Structural implications of beta-cardiac myosin heavy chain mutations in human disease. Anat. Rec. (Hoboken, N.J.), 297, 1670–1680. [DOI] [PubMed] [Google Scholar]
- 10. Tajsharghi H., Thornell L.E., Lindberg C., Lindvall B., Henriksson K.G., Oldfors A. (2003) Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann. Neurol., 54, 494–500. [DOI] [PubMed] [Google Scholar]
- 11. Cancilla P., Kalyanaraman K., Verity M., Munsat T., Pearson C. (1971) Familial myopathy with probable lysis of myofibrils in type I fibers. Neurology, 21, 579–585. [DOI] [PubMed] [Google Scholar]
- 12. Bohlega S., Abu-Amero S., Wakil S., Carroll P., Al-Amr R., Lach B., Al-Sayed Y., Cupler E., Meyer B. (2004) Mutation of the slow myosin heavy chain rod domain underlies hyaline body myopathy. Neurology, 62, 1518–1521. [DOI] [PubMed] [Google Scholar]
- 13. Dye D.E., Azzarelli B., Goebel H.H., Laing N.G. (2006) Novel slow-skeletal myosin (MYH7) mutation in the original myosin storage myopathy kindred. Neuromuscul. Disord., 16, 357–360. [DOI] [PubMed] [Google Scholar]
- 14. Tajsharghi H., Oldfors A., Macleod D.P., Swash M. (2007) Homozygous mutation in MYH7 in myosin storage myopathy and cardiomyopathy. Neurology, 68, 962.. [DOI] [PubMed] [Google Scholar]
- 15. Chai J., Liu C., Lai P., Yee W. (2007) CP 1.15 Myosin storage myopathy with a novel slow-skeletal myosin (MYH7) mutation in a Chinese patient. Neuromuscul. Disord., 17, 838. [Google Scholar]
- 16. Ortolano S., Tarrio R., Blanco-Arias P., Teijeira S., Rodriguez F., Garcia.-Murias M., Delague V., Levy N., Fernandez J.M., Quintans B.. et al. A novel MYH7 mutation links congenital fiber type disproportion and myosin storage myopathy. Neuromuscul. Disord., 21, 254–262. [DOI] [PubMed] [Google Scholar]
- 17. Stalpers X., Verrips A., Braakhekke J., Lammens M., van den Wijngaard A., Mostert A. (2011) Scoliosis surgery in a patient with “de novo” myosin storage myopathy. Neuromuscul. Disord., 21, 812–815. [DOI] [PubMed] [Google Scholar]
- 18. Yuceyar N., Ayhan O., Karasoy H., Tolun A. (2015) Homozygous MYH7 R1820W mutation results in recessive myosin storage myopathy: scapuloperoneal and respiratory weakness with dilated cardiomyopathy. Neuromuscul. Disord., 25, 340–344. [DOI] [PubMed] [Google Scholar]
- 19. Barohn R.J., Brumback R.A., Mendell J.R. (1994) Hyaline body myopathy. Neuromuscul. Disord., 4, 257–262. [DOI] [PubMed] [Google Scholar]
- 20. Laing N., Ceuterick-de Groote C., Dye D., Liyanage K., Duff R., Dubois B., Robberecht W., Sciot R., Martin J., Goebel H. (2005) Myosin storage myopathy: slow skeletal myosin (MYH7) mutation in two isolated cases. Neurology, 64, 527–529. [DOI] [PubMed] [Google Scholar]
- 21. Masuzugawa S., Kuzuhara S., Narita Y., Naito Y., Taniguchi A., Ibi T. (1997) Autosomal dominant hyaline body myopathy presenting as scapuloperoneal syndrome: clinical features and muscle pathology. Neurology, 48, 253–257. [DOI] [PubMed] [Google Scholar]
- 22. Bohlega S., Lach B., Meyer B., Al Said Y., Kambouris M., Al Homsi M., Cupler E. (2003) Autosomal dominant hyaline body myopathy: clinical variability and pathologic findings. Neurology, 61, 1519–1523. [DOI] [PubMed] [Google Scholar]
- 23. Rafay M.F., Bril V., Halliday W. (2005) Hyaline body myopathy: Adulthood manifestations. Can. J. Neurol. Sci., 32, 253–256. [DOI] [PubMed] [Google Scholar]
- 24. Shingde M.V., Spring P.J., Maxwell A., Wills E.J., Harper C.G., Dye D.E., Laing N.G., North K.N. (2006) Myosin storage (hyaline body) myopathy: a case report. Neuromuscul. Disord., 16, 882–886. [DOI] [PubMed] [Google Scholar]
- 25. Pegoraro E., Gavassini B.F., Borsato C., Melacini P., Vianello A., Stramare R., Cenacchi G., Angelini C. (2007) MYH7 gene mutation in myosin storage myopathy and scapulo-peroneal myopathy. Neuromuscul. Disord., 17, 321–329. [DOI] [PubMed] [Google Scholar]
- 26. Uro-Coste E., Arne-Bes M.C., Pellissier J.F., Richard P., Levade T., Heitz F., Figarella-Branger D., Delisle M.B. (2009) Striking phenotypic variability in two familial cases of myosin storage myopathy with a MYH7 Leu1793pro mutation. Neuromuscul. Disord., 19, 163–166. [DOI] [PubMed] [Google Scholar]
- 27. Ceuterick C., Martin J.J., Martens C. (1993) Hyaline bodies in skeletal muscle of a patient with a mild chronic nonprogressive congenital myopathy. Clin. Neuropathol., 12, 79–83. [PubMed] [Google Scholar]
- 28. Supala-Berger A., Fine E., Heffner R., Young-McLain E. (2009) Hyaline inclusion myopathy: Unmasked by statin therapy. Muscle Nerve, 40, 657–661. [DOI] [PubMed] [Google Scholar]
- 29. Armel T.Z., Leinwand L.A. (2009) Mutations in the beta-myosin rod cause myosin storage myopathy via multiple mechanisms. Proc. Natl. Acad. Sci. U S A, 106, 6291–6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reedy M.C., Beall C. (1993) Ultrastructure of developing flight muscle in Drosophila. I. Assembly of myofibrils. Dev. Biol., 160, 443–465. [DOI] [PubMed] [Google Scholar]
- 31. Bernstein S.I., Mogami K., Donady J.J., Emerson C.P. Jr. (1983) Drosophila muscle myosin heavy chain encoded by a single gene in a cluster of muscle mutations. Nature, 302, 393–397. [DOI] [PubMed] [Google Scholar]
- 32. George E.L., Ober M.B., Emerson C.P. Jr. (1989) Functional domains of the Drosophila melanogaster muscle myosin heavy-chain gene are encoded by alternatively spliced exons. Mol. Cell. Biol., 9, 2957–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. O'Donnell P.T., Collier V.L., Mogami K., Bernstein S.I. (1989) Ultrastructural and molecular analyses of homozygous-viable Drosophila melanogaster muscle mutants indicate there is a complex pattern of myosin heavy-chain isoform distribution. Genes Dev., 3, 1233–1246. [DOI] [PubMed] [Google Scholar]
- 34. Collier V., Kronert W., O'Donnell P., Edwards K., Bernstein S. (1990) Alternative myosin hinge regions are utilized in a tissue-specific fashion that correlates with muscle contraction speed. Genes Dev., 4, 885–895. [DOI] [PubMed] [Google Scholar]
- 35. O'Donnell P.T., Bernstein S.I. (1988) Molecular and ultrastructural defects in a Drosophila myosin heavy chain mutant: differential effects on muscle function produced by similar thick filament abnormalities. J. Cell Biol., 107, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thummel C., Pirrotta V. (1992) New pCaSpeR P element vectors. Dros. Inf. Serv., 71, 150. [Google Scholar]
- 37. Spradling A.C., Rubin G.M. (1982) Transposition of cloned P elements into Drosophila germ line chromosomes. Science, 218, 341–347. [DOI] [PubMed] [Google Scholar]
- 38. Swank D.M., Wells L., Kronert W.A., Morrill G.E., Bernstein S.I. (2000) Determining structure/function relationships for sarcomeric myosin heavy chain by genetic and transgenic manipulation of Drosophila. Microsc. Res. Tech., 50, 430–442. [DOI] [PubMed] [Google Scholar]
- 39. Kronert W.A., O'Donnell P.T., Fieck A., Lawn A., Vigoreaux J.O., Sparrow J.C., Bernstein S.I. (1995) Defects in the Drosophila myosin rod permit sarcomere assembly but cause flight muscle degeneration. J. Mol. Biol., 249, 111–125. [DOI] [PubMed] [Google Scholar]
- 40. An H.S., Mogami K. (1996) Isolation of 88F actin mutants of Drosophila melanogaster and possible alterations in the mutant actin structures. J. Mol. Biol., 260, 492–505. [DOI] [PubMed] [Google Scholar]
- 41. Nongthomba U., Cummins M., Clark S., Vigoreaux J.O., Sparrow J.C. (2003) Suppression of muscle hypercontraction by mutations in the myosin heavy chain gene of Drosophila melanogaster. Genetics, 164, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Powell S.R. (2006) The ubiquitin-proteasome system in cardiac physiology and pathology. Am. J. Physiol. Heart Circ. Physiol., 291, H1–H19. [DOI] [PubMed] [Google Scholar]
- 43. Willis M.S., Patterson C. (2010) Hold me tight: Role of the heat shock protein family of chaperones in cardiac disease. Circulation, 122, 1740–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang X., Robbins J. (2014) Proteasomal and lysosomal protein degradation and heart disease. J. Mol. Cell Cardiol., 71, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bainbridge S.P., Bownes M. (1981) Staging the metamorphosis of Drosophila melanogaster. Development, 66, 57–80. [PubMed] [Google Scholar]
- 46. Barlow D., Thornton J. (1988) Helix geometry in proteins. J. Mol. Biol., 201, 601–619. [DOI] [PubMed] [Google Scholar]
- 47. Miller B.M., Nyitrai M., Bernstein S.I., Geeves M.A. (2003) Kinetic analysis of Drosophila muscle myosin isoforms suggests a novel mode of mechanochemical coupling. J. Biol. Chem., 278, 50293–50300. [DOI] [PubMed] [Google Scholar]
- 48. Armel T.Z., Leinwand L.A. (2010) Mutations at the same amino acid in myosin that cause either skeletal or cardiac myopathy have distinct molecular phenotypes. J. Mol. Cell Cardiol., 48, 1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Meredith C., Herrmann R., Parry C., Liyanage K., Dye D.E., Durling H.J., Duff R.M., Beckman K., de Visser M., van der Graaff M.M.. et al. (2004) Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause Laing early-onset distal myopathy (MPD1). Am. J. Hum. Genet., 75, 703–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kärkkäinen S., Heliö T., Jääskeläinen P., Miettinen R., Tuomainen P., Ylitalo K., Kaartinen M., Reissell E., Toivonen L., Nieminen M.S.. et al. (2004) Two novel mutations in the β‐myosin heavy chain gene associated with dilated cardiomyopathy. Eur. J. Heart Fail., 6, 861–868. [DOI] [PubMed] [Google Scholar]
- 51. Van Driest S.L., Jaeger M.A., Ommen S.R., Will M.L., Gersh B.J., Tajik A.J., Ackerman M.J. (2004) Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol., 44, 602–610. [DOI] [PubMed] [Google Scholar]
- 52. Armel T.Z., Leinwand L.A. (2010) A mutation in the beta-myosin rod associated with hypertrophic cardiomyopathy has an unexpected molecular phenotype. Biochem. Biophys. Res. Commun., 391, 352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mogami K., O'Donnell P.T., Bernstein S.I., Wright T.R., Emerson C.P. Jr. (1986) Mutations of the Drosophila myosin heavy-chain gene: effects on transcription, myosin accumulation, and muscle function. Proc. Natl. Acad. Sci. U S A, 83, 1393–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Oldfors A., Lamont P.J. (2008), In The Sarcomere and Skeletal Muscle Disease. Springer, New York: 78-91. [Google Scholar]
- 55. Rock K.L., Gramm C., Rothstein L., Clark K., Stein R., Dick L., Hwang D., Goldberg A.L. (1994) Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell, 78, 761–771. [DOI] [PubMed] [Google Scholar]
- 56. Olive M., Abdul-Hussein S., Oldfors A., Gonzalez-Costello J., van der Ven P.F., Furst D.O., Gonzalez L., Moreno D., Torrejon-Escribano B., Alio J.. et al. (2015) New cardiac and skeletal protein aggregate myopathy associated with combined MuRF1 and MuRF3 mutations. Hum. Mol. Genet., 24, 3638–3650. [DOI] [PubMed] [Google Scholar]
- 57. Fielitz J., Kim M.S., Shelton J.M., Latif S., Spencer J.A., Glass D.J., Richardson J.A., Bassel-Duby R., Olson E.N. (2007) Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J. Clin. Invest., 117, 2486–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dahl-Halvarsson M., Pokrzywa M., Rauthan M., Pilon M., Tajsharghi H. (2017) Myosin storage myopathy in C. elegans and human cultured muscle cells. PLoS One, 12, e0170613.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sambrook J., Fritsch E.F., Maniatis T. and others. (1989) Molecular cloning. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. [Google Scholar]
- 60. Robertson H.M., Preston C.R., Phillis R.W., Johnson-Schlitz D.M., Benz W.K., Engels W.R. (1988) A stable genomic source of P element transposase in Drosophila melanogaster. Genetics, 118, 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cripps R., Bernstein S. (2000) Generation of transgenic Drosophila melanogaster by P element-mediated germline transformation Introducing DNA into Living Cells and Organisms. PA Norton and LF Steel, editors. BioTechniques Books, Eaton Publishing, Natick, MA, 93–125. [Google Scholar]
- 62. Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 63. Drummond D.R., Hennessey E.S., Sparrow J.C. (1991) Characterisation of missense mutations in the Act88F gene of Drosophila melanogaster. Mol. Gen. Genet., 226, 70–80. [DOI] [PubMed] [Google Scholar]
- 64. Nongthomba U., Ramachandra N.B. (1999) A direct screen identifies new flight muscle mutants on the Drosophila second chromosome. Genetics, 153, 261–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Viswanathan M.C., Blice-Baum A.C., Schmidt W., Foster D.B., Cammarato A. (2015) Pseudo-acetylation of K326 and K328 of actin disrupts Drosophila melanogaster indirect flight muscle structure and performance. Front. Physiol., 6, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Swank D.M., Bartoo M.L., Knowles A.F., Iliffe C., Bernstein S.I., Molloy J.E., Sparrow J.C. (2001) Alternative exon-encoded regions of Drosophila myosin heavy chain modulate ATPase rates and actin sliding velocity. J. Biol. Chem., 276, 15117–15124. [DOI] [PubMed] [Google Scholar]