

Abstract
In-frame premature termination codons (PTCs) account for ∼11% of all disease-associated mutations. PTC suppression therapy utilizes small molecules that suppress translation termination at a PTC to restore synthesis of a full-length protein. PTC suppression is mediated by the base pairing of a near-cognate aminoacyl-tRNA with a PTC and subsequently, the amino acid becomes incorporated into the nascent polypeptide at the site of the PTC. However, little is known about the identity of the amino acid(s) inserted at a PTC during this process in mammalian cells, or how the surrounding sequence context influences amino acid incorporation. Here, we determined the amino acids inserted at the cystic fibrosis transmembrane conductance regulator (CFTR) W1282X PTC (a UGA codon) in the context of its three upstream and downstream CFTR codons during G418-mediated suppression. We found that leucine, cysteine and tryptophan are inserted during W1282X suppression. Interestingly, these amino acids (and their proportions) are significantly different from those recently identified following G418-mediated suppression of the CFTR G542X UGA mutation. These results demonstrate for the first time that local mRNA sequence context plays a key role in near-cognate aminoacyl-tRNA selection during PTC suppression. We also found that some variant CFTR proteins generated by PTC suppression exhibit reduced maturation and activity, indicating the complexity of nonsense suppression therapy. However, both a CFTR corrector and potentiator enhanced activity of protein variants generated by G418-mediated suppression. These results suggest that PTC suppression in combination with CFTR modulators may be beneficial for the treatment of CF patients with PTCs.
Introduction
Nonsense mutations, which generate in-frame premature termination codons (PTCs) within an open reading frame, represent ∼11% of all disease-associated gene lesions (1). When a ribosome encounters a PTC in its acceptor (A) site, translation will terminate prior to the synthesis of a full-length protein, which often leads to loss of protein function. Unlike decoding during translation elongation that involves base pairing between mRNA codons and aminoacyl-tRNAs, termination codons (UAA, UAG, UGA) are decoded by a release factor complex composed of eRF1 and eRF3. eRF1 recognizes and binds to all three termination codons, while eRF3 is a GTPase that enhances polypeptide release by eRF1. Thus, no aminoacyl-tRNAs exist that are cognate to termination codons. However, near-cognate aminoacyl-tRNAs occasionally become accommodated at a PTC by base pairing with two of the three nucleotides of the termination codon, leading to the incorporation of an amino acid into the nascent polypeptide chain. This event, known as PTC suppression or readthrough, allows translation elongation to continue in the correct reading frame to produce a full-length protein. Certain low-molecular weight compounds have been identified, including aminoglycosides and PTC124 (also known as ataluren or TranslarnaTM), that enhance readthrough frequency at PTCs (2–6). Nonsense suppression therapy aims to utilize readthrough agents with the goal of restoring enough full-length, functional protein to alleviate diseases caused by PTCs (7).
However, since the amino acids inserted at a PTC during readthrough may differ from the amino acid normally found in the wild-type protein, the resulting full-length protein may not mature or function as effectively as the wild-type protein. From the perspectives of both the basic mechanism of PTC suppression and the potential therapeutic benefit afforded by PTC suppression therapy, it is important to determine both the identities and functional consequences of the amino acids inserted at a PTC during readthrough. Currently, only limited information is available regarding the amino acids inserted during PTC suppression in eukaryotes. In some retroviruses such as the Maloney murine leukemia virus (MuLV), a programmed stop codon suppression event is used to generate the full-length Gag-Pol protein. Glutamine (Gln) is inserted during suppression at UAA and UAG codons in this MuLV programmed readthrough system, while arginine (Arg), cysteine (Cys) and tryptophan (Trp) are incorporated at a UGA (8). Another study found that tyrosine, lysine and Trp are inserted during UAG suppression in yeast (9). More recently, it was found that glutamine, tyrosine and lysine are incorporated at both UAA and UAG codons, whereas Arg, Cys and Trp are inserted at a UGA codon in both yeast and human cells (10,11). While the same general sets of amino acids are inserted during both basal or stimulated readthrough, the proportions of the different amino acids inserted vary as a function of the readthrough conditions (11,12), consistent with a mechanism mediated via the accommodation of a subset of aminoacyl-tRNAs that are near-cognate to the PTC.
Cystic fibrosis (CF) is an autosomal-recessive disorder caused by mutations in the gene encoding the CF transmembrane conductance regulator (CFTR). The CFTR protein is an anion channel primarily localized to the apical membranes of secretory epithelial cells lining the airways and multiple organs (13). Our current study addresses other important questions related to the mechanism and consequences of PTC suppression for the treatment of not only CF, but also other diseases caused by nonsense mutations. For example, previous studies have shown that the mRNA sequence context near a PTC influences its susceptibility to suppression (14–17). While one study that examined PTC suppression in three different contexts did not observe significant context effects on the amino acids inserted (10), further investigation is clearly needed. Second, the relative function of full-length proteins generated by PTC readthrough that differ from the wild-type protein (referred to hereafter as protein variants) is unclear. To address these questions, we designed reporter constructs that carry the clinically relevant CFTR W1282X PTC and its surrounding sequence context. Following the induction of PTC suppression with the aminoglycoside G418, we found that leucine (Leu) (58%), Cys (38%) and Trp (4%) are inserted at the CFTR W1282X UGA termination codon during suppression. Surprisingly, these amino acids (and proportions) are significantly different from those that we recently identified following suppression of the CFTR G542X UGA mutation, which included Cys (44%), Trp (36%) and Arg (20%) (11). These results provide compelling evidence that sequence context has the potential to influence both the identity and frequency of the amino acids inserted during PTC suppression. We also examined the functionality of variant CFTR proteins generated by PTC suppression. We found that both the processing and function of many CFTR variants was reduced, indicating the complexity of nonsense suppression therapy. However, both parameters could be enhanced by the CFTR modulators VX-809 (lumacaftor) and VX-770 (ivacaftor). These results suggest that combining nonsense suppression drugs with CFTR modulators is a promising therapeutic approach to maximize CFTR function in CF patients that harbor nonsense mutations.
Results
Identification of amino acids incorporated during suppression of the CFTR W1282X nonsense mutation
CFTR is a large, polytopic, integral membrane protein that is expressed at low levels in epithelial cells. In addition, PTC suppression occurs at relatively low frequencies (often 5% or less). Together, these factors make it exceedingly difficult to analyze the amino acid(s) inserted during PTC suppression in the context of the intact CFTR protein. To overcome these obstacles, we recently developed a TurboGFP reporter to determine the amino acids inserted during readthrough of the clinically relevant CFTR G542X mutation using tandem mass spectrometry (MS–MS) (11) (Fig. 1A). Because previous studies have shown that the local mRNA sequence context flanking a PTC acts as a primary determinant of readthrough (15,16,18), we introduced the G542X (UGA) codon, as well as the surrounding local CFTR context (three codons upstream and downstream of the CFTR G542 position), into the TurboGFP reporter. Suppression of the PTC was induced using the aminoglycoside G418. The full-length protein products resulting from PTC suppression were then purified by Ni-NTA chromatography and subjected to MS–MS analysis. Three amino acids were found to be inserted at the CFTR G542X UGA codon upon readthrough: Arg, Cys and Trp (Fig. 1D).
Figure 1.
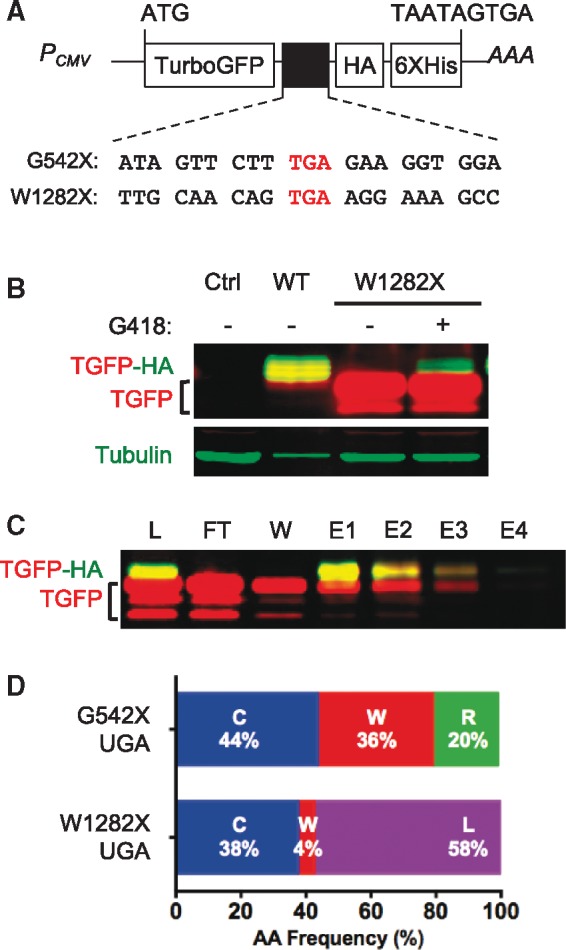
Determination of the amino acids inserted during G418-mediated suppression of the CFTR W1282X PTC in HEK293 cells. (A) TurboGFP reporters used to identify amino acids inserted at CFTR PTCs during readthrough. CFTR W1282X and G542X reporters (and the CFTR context included) are shown. (B) Representative western blot showing an increased amount of full-length protein (containing both TurboGFP and HA tag) after 24 h of G418 treatment in HEK293 cells transiently transfected with the W1282X reporter. Tubulin was used as a loading control. (C) Western blot showing protein purification using Ni-NTA resin. L, crude extract; FT, flow-through; W, wash; E, elution. (D) Comparison of the amino acids inserted (and their relative proportions) during G418 suppression of the G542X and W1282X alleles. Raw spectra of the W1282 construct can be seen in Supplementary Material, Figure S1.
In the current study, we modified the TurboGFP reporter system to identify the amino acids incorporated during PTC suppression of the CFTR W1282X nonsense mutation. To do this, the W1282X UGA PTC was introduced into the TurboGFP reporter within the context of its three upstream and three downstream codons of CFTR sequence. Since both the G542X and W1282X CFTR mutations introduced UGA codons into the TurboGFP reporter and both reporters were expressed in HEK293 cells, the only difference between the G542X and W1282X reporters was the identity of the three upstream and downstream CFTR codons flanking the UGA stop codon (Fig. 1A).
As in our previous study (11), readthrough was induced in HEK293 cells expressing the W1282X TurboGFP construct with G418 for 48 h, full-length readthrough products were purified using Ni-NTA chromatography (Fig. 1B and C), and MS–MS was carried out (Supplementary Material, Fig. S1). Similar to the CFTR G542X allele, Cys and Trp were also incorporated at CFTR W1282X following G418-mediated suppression. However, Arg was not detected (Fig. 1D) and a new amino acid, Leu, was identified. Our previous study of the amino acids inserted during G542X suppression found that Cys was incorporated at the highest frequency (44%), followed by Trp (36%) and Arg (20%) (Fig. 1D). In contrast, we found that Leu was the most abundant amino acid inserted at W1282X (58%), followed by Cys (38%) and Trp (4%). Since the same PTC (UGA) is found in both the G542X and W1282X constructs and they differ only by the three codons upstream and three codons downstream of the PTC, these results indicate that the local sequence context surrounding a PTC can influence both the amino acid(s) inserted during readthrough and their relative proportions.
Analysis of the amino acids incorporated at different PTC codons within a common context
Many studies have shown that the local sequence context flanking a PTC influences its readthrough efficiency (14–17). In particular, the presence of a CAA codon (encoding glutamine) on either side of a PTC has been shown to induce particularly robust readthrough in both yeast and mammalian systems (9,15,16,18). To further investigate the amino acids inserted at different PTC codons during readthrough, we utilized TurboGFP reporters containing either UGA, UAG or UAA termination codons flanked by upstream and downstream CAA codons (encoding Gln, or ‘Q’ in the single letter code). This set of reporters (collectively referred to as QXQ constructs) allowed us to monitor the amino acids inserted at different PTCs flanked by identical sequence contexts (Fig. 2A). G418 was again used to induce readthrough of each PTC in HEK293 cells.
Figure 2.
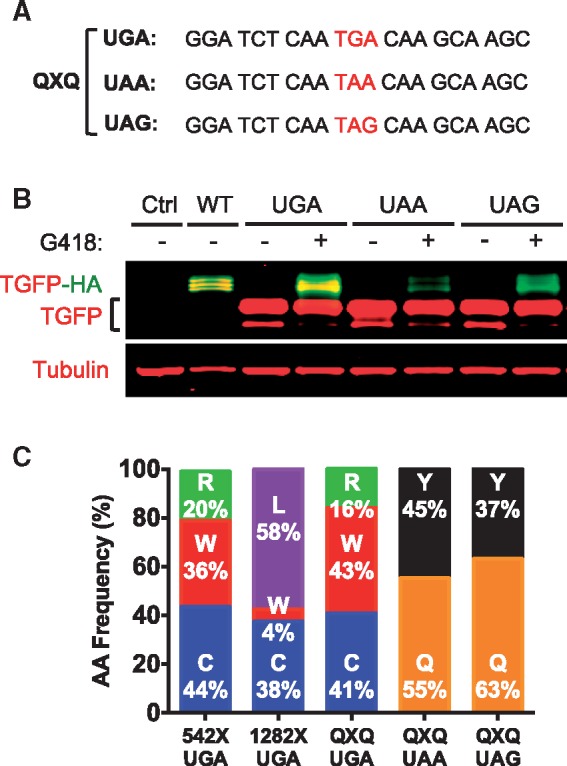
Determination of the amino acids inserted during G418-mediated suppression of UGA, UAA and UAG codons in the same context in HEK293 cells. (A) PTCs and context used to determine amino acids inserted (and their relative proportions). (B) Representative western blot showing full-length proteins produced after 24 h of G418 treatment in HEK293 cells transiently transfected with the QXQ constructs. (C) Comparison of amino acids inserted (and relative frequencies of each) as determined from by MS–MS of samples purified from HEK293 cells. Raw spectra of the QXQ constructs can be seen in Supplementary Material, Figure S1.
Consistent with previous results (15), the highest level of G418-mediated suppression was observed in the QXQ UGA construct, followed by QXQ UAG and QXQ UAA (Fig. 2B). MS–MS analysis of the purified QXQ readthrough peptides revealed that the identity and proportions of the three amino acids inserted at the QXQ UGA codon following G418-mediated suppression (Cys, Arg and Trp) were very similar to those observed following the suppression of the CFTR G542X UGA codon (Fig. 2C, compare columns one and three; also see Supplementary Material, Fig. S2). MS–MS analysis also showed that Gln and Tyr were inserted at both QXQ UAA and QXQ UAG with similar proportions following G418-mediated suppression (Fig. 2C, columns four and five), which is also consistent with results from previous studies (11). When taken together, these results suggest that at stop codons within multiple contexts, a common set of amino acids are often, but not always, inserted during readthrough.
Characterization of CFTR W1282 readthrough variants
We recently reported that the CFTR G542R, G542C and G542W variants found to be produced by G418-mediated suppression of the CFTR G542X allele exhibited different levels of CFTR activity (11). To determine how incorporation of the variant amino acids produced by suppression of the CFTR W1282X mutation affects CFTR protein maturation and function, we mutated a WT CFTR cDNA at codon W1282 to create the W1282C (Cys) and the W1282L (Leu) CFTR variants. HEK293 cells that encoded these variant CFTR proteins were also examined for CFTR protein expression and maturation from band B [the immature, endoplasmic reticulum (ER) form that is present in CF patients with the common F508del mutation, a deletion of codon 508] to band C (the mature, fully glycosylated form) by western blotting (Fig. 3A). While cells transfected with each construct expressed similar levels of CFTR protein, the Band C/B ratio differed among the different CFTR variants. The majority of WT CFTR was present as Band C (C/B ratio = 1.7), indicating that most CFTR had exited the ER and moved through the Golgi to the cell surface. Cells expressing the W1282L CFTR variant showed only a slightly lower ratio of Band C/B relative to WT CFTR (C/B ratio = 1.2). In contrast, a much lower C/B ratio was observed in cells expressing the W1282C variant (C/B = 0.6), suggesting a more severe maturation and/or protein stability defect. We next made stable cell lines expressing W1282 wild-type and variant forms in Fischer rat thyroid (FRT) cells in order to carry out electrophysiological assessments of CFTR channel function. In these experiments, a cAMP agonist, forskolin, was used to activate CFTR channel opening, followed by the addition of the CFTR inhibitor, Inh172. This provided further confidence that currents observed had resulted from CFTR function. In each assay, CFTR function was corrected for modest differences in steady-state CFTR mRNA abundance. Using short circuit current (Isc) assays to monitor CFTR-mediated chloride movement, we found that the W1282L CFTR variant displayed similar activity as WT CFTR, while the W1282C variant exhibited only 36% of WT CFTR function (Fig. 3B and C). Independent assays measuring transcellular conductance (Gt) showed similar results (Fig. 3D and E) (see Supplementary Material, Fig. S3A and B for representative tracings). When taken together, we conclude that each of the CFTR W1282 variants, like the G542 variants (11), exhibited varying maturation defects and corresponding CFTR functional defects due to the nature of the amino acid inserted during the suppression event.
Figure 3.

Steady-state abundance and function of CFTR W1282L and W1282C variants. (A) Representative western blot showing CFTR W1282C and W1282L abundance. (B) Representative tracings of Isc (left) and (C) mRNA normalized ΔIsc (right) in FRT monolayers expressing WT CFTR or W1282 variants. N = 8 per variant, ****P < 0.0001. (D) mRNA normalized ΔGt in FRT cell monolayers expressing WT CFTR or W1282 variants. Fsk, forskolin (20 µM). Inh172, CFTRInh172 (10 μM). See Supplementary Material, Figure S3 for representative tracings of Gt measurements. (E) Correlation between ΔIsc and ΔGt.
CFTR modulators further stimulate CFTR W1282 variant function
A number of drugs, known as CFTR modulators, have recently been developed to treat CF patients. These drugs target specific CFTR defects that arise from different mutation classes. One of these, VX-809 (lumacaftor), is a CFTR corrector thought to bind CFTR protein and improve its processing in the ER (19). It has also been reported that VX-809 stabilizes CFTR F508del on the cell surface, thus augmenting CFTR half-life (20). Another CFTR modulator, VX-770 (ivacaftor), is an FDA-approved, orally bioavailable potentiator originally developed to treat CF patients with the G551D mutation (21–23). VX-770 is thought to bind CFTR at the cell surface in a manner that enhances the activity of mutant proteins with channel gating defects (24,25). Phase 3 clinical trials recently demonstrated that co-administration of VX-770 and VX-809 provides a therapeutic benefit in CF patients homozygous for F508del, the most common CF mutation (26), leading to FDA approval of this combination therapy (marketed as Orkambi) (27,28).
We next asked whether these CFTR modulators could be used to analyze the maturation and/or functional defects of CFTR readthrough variants. First, HEK293 cells were transfected with plasmids expressing W1282 CFTR variants and treated with VX-809 for 24 h to examine the effects on CFTR maturation. Western blots show an increase in the ratio of band C/B in HEK293 cells expressing either the W1282C or W1282L CFTR variants or WT CFTR upon VX-809 treatment (Fig. 4A and B), indicating an enhanced escape of CFTR from the ER. We also observed an increase in the absolute amount of band B for both the WT and variant forms of CFTR, indicating that VX-809 also stabilizes immature CFTR in the ER. In FRT cells stably expressing W1282 CFTR variants, we found that CFTR function was enhanced significantly when cells were treated with VX-809, and to a lesser extent, with VX-770. Moreover, the combined treatment of cells with these compounds produced greater chloride conductance via Isc measurements than treatment with either drug alone (Fig. 4C–E). Importantly, we found forskolin-dependent Isc of W1282 variants to strongly correlate with the band C/B ratio (Fig. 4F), suggesting that the level of CFTR function in this subset of CFTR variants is determined largely by their maturation and overall abundance on the cell surface. A similar strong correlation was observed between W1282 CFTR variant epithelial conductance (Gt) and the variant band C/B ratio (Fig. 4G and H;Supplementary Material, Fig. S3A and B). When taken together, these results indicate that VX-809 and VX-770 can significantly enhance W1282 CFTR variant function following readthrough.
Figure 4.

The effect of VX-809 and VX-770 on CFTR abundance and function in W1282 variants. (A) Representative western blot showing effects of VX-809 treatment on the abundance of CFTR bands B and C of WT CFTR and W1282 variants. HEK293 cells were transfected with each of the W1282 variants. Three hours later VX-809 (3 μM) was added to the culture medium. Cells were harvested 24 h later. *P < 0.05, ****P < 0.0001 versus untreated. (B) Quantitation of band C/B ratio. (C and D) Representative tracings of ΔIsc in FRT monolayers expressing W1282 variants. Cells were treated as described above. (E) Summary of ΔIsc data of WT CFTR and W1282 variants after mRNA normalization. N = 8 per condition. *P < 0.0001 versus untreated. (F) Correlation between ΔIsc and CFTR band C/B ratio. (G) Summary of ΔGt in of WT CFTR and W1282 variants after mRNA normalization. N = 6 per condition. *P < 0.0001 versus vehicle. See Supplementary Material, Figure S3 for representative tracings of ΔGt experiments. (H) Correlation between ΔGt and CFTR band C/B ratio.
Enhancement of CFTR G542 variant function by CFTR modulators
We next tested how CFTR modifiers affect G542 CFTR variant maturation and function. The G542 variants previously identified from G418-mediated readthrough (G542R, G542C and G542W) (11) were constructed and HEK293 and FRT cell lines expressing each variant were prepared. First, HEK293 cells were cultured +/− VX-809 treatment for 24 h and western blotting was carried out. VX-809 induced a significant increase in the band C/B ratio for WT CFTR, as well as for each G542 variant protein (Fig. 5A and B). Chloride conductance in FRTs was also enhanced significantly, as indicated by increased forskolin-dependent Isc (see Fig. 5C–F for representative Isc traces and Fig. 5G for a summary of all Isc data). Similarly, VX-809 also increased Gt in FRT cells expressing the G542X variants (Fig. 5I;Supplementary Material, Fig. S3C). Acute addition of VX-770 resulted in a further small increase in function of all G542 CFTR variants in both assays. When VX-809 and VX-770 treatments were combined, CFTR function was enhanced more than with either drug alone. Finally, we observed a strong correlation between G542 CFTR variants as determined by both ΔIsc and ΔGt functional measurements and the variant band C/B ratio (Fig. 5H and J). We conclude that CFTR modulators, particularly the corrector VX-809, can also enhance the function of the G542 CFTR variants produced as a result of G418-mediated readthrough.
Figure 5.

The effect of VX-809 and/or VX-770 on CFTR abundance and function in G542 variants. (A) Representative western blot showing effects of VX-809 treatment on CFTR bands B and C of WT CFTR and G542 variants. HEK293 cells were transfected with each of the G542 variants. VX-809 (3 μM) was added to the culture medium 3 h later, and cells were harvested 24 h later. **P < 0.01, ****P < 0.0001 versus untreated. (B) Quantitation of band C/B ratio. (C–F) Representative Isc tracings from FRT monolayers expressing WT CFTR or the indicated G542 variants treated with VX-809 (3 μM) for 24 h and/or VX-770 for 10 min (10 μM). (G) Summary of ΔIsc data of WT CFTR and G542 variants after mRNA normalization. N = 8 per condition. *P < 0.0001 versus untreated. (H) Correlation between ΔIsc and CFTR band C/B ratio. (I) Summary of ΔGt data of WT CFTR and G542 variants after mRNA normalization. N = 6 per condition. *P < 0.0001 versus vehicle. See Supplementary Material, Figure S3 for representative tracings of Gt experiments. (J) Correlation between ΔGt and CFTR band C/B ratio.
CFTR function is enhanced when cells expressing CFTR G542X or W1282X nonsense mutations are treated with a readthrough drug and CFTR modulators
We next determined the level of CFTR activity that can be restored in FRT cells expressing either the CFTR G542X or CFTR W1282X nonsense alleles when treated with a readthrough drug alone, CFTR modulators alone, or a combination of readthrough drug and CFTR modulators. First, the FRT line stably expressing CFTR G542X was treated with G418 for 48 h before assaying CFTR function. G418 treatment alone increased CFTR function in cells expressing CFTR G542X by ∼7-fold as measured by an increase in forskolin-dependent Isc (Fig. 6A and B) or Gt (Fig. 6E;Supplementary Material, Fig. S3C). Treatment of those cells with VX-809 alone or the combination of VX-809 and VX-770 without readthrough did not enhance CFTR function, suggesting that the CFTR protein fragment terminated at position 542 was not functional even when treated with both a corrector and a potentiator. However, a further increase in CFTR function from the G542X allele was observed when G418 and VX-809 were combined as compared to G418 alone, consistent with enhanced processing of the full-length variant proteins produced by readthrough of the G542X mutation by the corrector. Further addition of VX-770 enhanced the current slightly in G418-treated cells in either the presence or absence of VX-809, suggesting that VX-770 can increase gating of the full-length G542 CFTR variants on the cell surface. When taken together, these results show that the triple combination treatment of cells expressing the CFTR G542X allele increased CFTR function almost 15-fold (resulting in 4–5% of the activity observed in cells expressing WT CFTR), a level more than double the amount of CFTR activity produced with G418 alone.
Figure 6.

Effects of nonsense suppression, corrector, and potentiator compounds on CFTR function restored in cells expressing CFTR G542X and W1282X alleles. (A, C) Representative tracings of Isc in FRT monolayers expressing CFTR G542X or W1282X alleles after treatment with various combinations of G418 (250 µg/ml) and VX-809 (3 µM) for 24 h and VX-770 (10 µM) for 10 min. (B, D) Summaries of Isc data following mRNA normalization. N = 12 per condition. (E, F) Summaries of ΔGt data following mRNA normalization. N = 6 per condition. See Supplementary Material, Figure S3C for representative Gt tracings. (G) Representative equivalent current (Ieq) tracings of G542X/G542X primary HBE cells treated with DMSO vehicle, G418 (100 µM) or VX-809 (3 µM), or a combination of G418 and VX-809 for 48 h. (H) Summary data expressed as AUC between activation of CFTR-dependent Ieq with forskolin (10 µM) and ivacaftor (VX-770, 1 µM) for 30 min prior to inhibition of anion transport with bumetanide (10 µM). N = 4/condition. *P < 0.05, **P < 0.01, ***P < 0.001,****P < 0.0001 versus untreated or vehicle (unless otherwise indicated).
Interestingly, the response observed in FRT cells expressing the CFTR W1282X allele was quite different than that observed with CFTR G542X (Fig. 6C and D). G418 treatment alone increased CFTR function only two to three-fold, indicating that the W1282X allele is less responsive to readthrough induction by this compound. In contrast, treatment of these cells with VX-770 in the absence of G418 increased CFTR function by 4- to 5-fold, while treatment with the combination of VX-770 and VX-809 in the absence of G418 enhanced Isc by ∼10-fold. This finding suggests that the truncated CFTR protein produced from the W1282X allele, which contains much more of the CFTR protein than the truncated protein produced from the G542X allele, may be able to escape from the ER following VX-809 treatment, transit to the cell surface, and undergo channel opening when exposed to the potentiator VX-770. Remarkably, this occurs even though much of second NBD of CFTR is missing. Similar responses of the truncated protein expressed from the W1282X allele to correctors was also reported in another recent study (29). Treatment of these cells with the triple combination of G418, VX-809 and VX-770 increased forskolin-dependent Isc (Fig. 6C and D) and Gt (Fig. 6F) to a level ∼13-fold above background (3–4% of cells expressing WT CFTR), again suggesting that combining a readthrough drug with CFTR modulators enhances total CFTR activity obtained from the W1282X allele.
The results described above suggest that the function of CFTR variant proteins produced by suppression of the G542X and W1282X alleles can be significantly enhanced by co-treatment with CFTR modulators such as the potentiator VX-770 or the corrector VX-809. To confirm this conclusion, we turned to a more physiologically relevant cell type. It was previously suggested that modulator compounds that showed a positive response in primary human bronchial epithelial (HBE) cells would have the highest likelihood of showing clinical efficacy (30). Accordingly, we next examined the effect of combination therapy on CFTR activity in primary HBE cells homozygous for the CFTR G542X mutation using an equivalent current (Ieq) assay, which is functionally similar to the Isc and Gt assays (Fig. 6G and H). We found that growth for 48 h in the presence of 50 µM G418 led to an ∼3-fold increase in CFTR activity following activation with forskolin and VX-770. In addition, growth in the presence of both G418 and VX-809 effectively doubled the amount of CFTR activity observed (∼6-fold increase). A higher (100 µM) G418 dose stimulated CFTR activity ∼4.5-fold, while the co-treatment with VX-809 further enhanced the increase to ∼7.5-fold. Consistent with the results obtained using FRT cells expressing CFTR cDNAs, these results indicate that the co-administration of CFTR modulators with readthrough agents can enhance the amount of CFTR activity obtained by as much as 2-fold over the activity obtained by treatment with a readthrough drug alone.
Discussion
For this study, MS–MS was used to identify the amino acids incorporated at CFTR PTCs (and other PTCs) during readthrough. To avoid the difficulties of isolating adequate quantities of full-length CFTR protein generated by PTC suppression for MS–MS, we utilized a previously characterized TurboGFP reporter system that allows us to rapidly purify large quantities of full-length protein generated by readthrough of a PTC in the same flanking sequence context as the intact CFTR protein (11). Using this system, we determined: (i) the amino acids inserted at CFTR PTCs in human cells during G418-mediated readthrough, (ii) the effects of proximal mRNA sequence context upon the identity and proportions of amino acids inserted at PTCs, and (iii) the functional response of individual variant CFTR proteins produced by PTC suppression.
We found that inducing readthrough with G418 promoted insertion of Arg, Cys and Trp at UGA in both the CFTR G542X and QXQ contexts (Fig. 2A and Table 1). This subset of amino acids was previously found to be incorporated at UGA during basal readthrough, as well as readthrough induced by either G418 or ataluren in both yeast and human cells (10,12). We further found that Tyr and Gln were inserted at both UAA and UAG codons in the QXQ context (Tables 2 and 3), a finding also consistent with previous results (11). While previous studies found Lys to be incorporated at low levels during UAA and UAG readthrough (11,12), incorporation of Lys was not observed using our reporters. All of these near-cognate amino acid incorporations require a mismatch between the anticodon and stop codon at either the first or third nucleotide positions (Tables 1–3). In the CFTR W1282X context, we found that Cys and Trp were also inserted via mismatches with the 3rd position of the UGA stop codon. However, a new amino acid, Leu, was also inserted at the W1282X UGA codon (Table 1), which would occur through a G:A mismatch at the second nucleotide position of the UGA codon. It should be noted that Leu and Ile have identical masses, preventing us from unambiguously distinguishing between these two amino acids when examining the CFTR W1282X MS–MS data. However, we favor the interpretation that Leu was incorporated at the W1282X UGA codon, since a Leu codon is near-cognate to UGA (with a single mismatch), while all Ile codons are non-cognate to UGA (with mismatches at two or more positions).
Table 1.
List of potential amino acids incorporated at UGA
| Codons with one mismatch | Anticodon | Mismatch base pairing | Potential AA inserted | AA identified in sample |
|---|---|---|---|---|
| AGA | UCU | U–U | Arg | G542X, QXQ UGA |
| CGA | UCG | U–G | Arg | G542X, QXQ UGA |
| GGA | UCC | U–C | Gly | |
| UAA | UUA | U–G | STOP | |
| UCA | UGA | G–G | Ser | |
| UUA | UAA | G–A | Leu | W1282X |
| UGC | GCA | G–A | Cys | G542X, QXQ UGA |
| UGU | ACA | A–A | Cysa | G542X, QXQ UGA |
| UGG | CCA | A–C | Trp | All three |
tRNA is absent in Homo sapiens.
Table 2.
List of potential amino acids incorporated at UAA
| Codons with one mismatch | Anticodon | Mismatch base pairing | Potential AA inserted | AA identified in sample |
|---|---|---|---|---|
| AAA | UUU | U–U | Lys | |
| CAA | UUG | U–G | Gln | QXQ UAA |
| GAA | UUC | U–C | Glu | |
| UGA | UCA | A–C | SelCys | |
| UCA | UGA | G–A | Ser | |
| UUA | UAA | A–A | Leu | |
| UAC | GUA | G–A | Tyr | QXQ UAA |
| UAU | AUA | A–A | Tyr | QXQ UAA |
| UAG | CUA | A–C | STOP |
Table 3.
List of potential amino acids incorporated at UAG
| Codons with one mismatch | Anticodon | Mismatch base pairing | Potential AA inserted | AA identified in sample |
|---|---|---|---|---|
| AAG | CUU | S | Lys | |
| CAG | CUG | U–G | Gln | QXQ UAG |
| GAG | CUC | U–C | Glu | |
| UGG | CCA | A–C | Trp | |
| UCG | CGA | G–A | Ser | |
| UUG | CAA | A–A | Leu | |
| UAA | UUA | U–G | STOP | |
| UAC | GUA | G–G | Tyr | QXQ UAG |
| UAU | AUA | G–A | Tyr | QXQ UAG |
Recent structural studies have revealed significant new insights into mechanisms underlying near-cognate misreading during translation elongation (31). Codon:anticodon complementarity is monitored via the ribosomal decoding center, where several universally conserved nucleotides in the ribosomal RNA probe the geometry of base pairings. Rather than examining base complementarity through the formation of hydrogen bonds, the ribosome appears to probe codon:anticodon pairings primarily using steric restraints. Since similar codon:anticodon interactions occur in the ribosomal decoding center during PTC suppression, it is likely that these geometrical constraints also limit the possible interactions between near-cognate tRNAs and PTCs.
The accuracy of codon selection between cognate, wobble, and near-cognate pairings during elongation can vary by 400-fold, and these differences in incorporation depend on the identity and modifications of the misreading tRNA, the position of the mismatch within the codon:anticodon interaction, and the specific type of basepairing mismatch (32). Our identification of the insertion of Leu during suppression of the W1282X mutation would require a G:A mismatch in the middle position of the codon:anticodon pairing. Interestingly, this mismatch has been observed in a previous nonsense suppression study that examined how a pseudouridylated termination codon (ΨAG) could be decoded by a serine tRNA (33). It was shown that the adenosine in the middle position adopted a syn conformation that led to base-pairing with guanosine in a conformation similar to canonical Watson-Crick purine-pyrimidine base pairing. Additional insights into the key features of near-cognate mispairing will greatly enhance our ability to predict the amino acids most likely to be inserted during nonsense suppression.
Even though the G542X and W1282X mutations are both UGA stop codons, different amino acids (and different proportions of shared amino acids) are inserted at the PTCs upon readthrough with G418 (Fig. 2C). Since the only difference between the G542X and W1282X constructs resides in the three upstream and three downstream codons flanking the UGA termination codons, the reason for the incorporation of different amino acids, as well as the difference in the proportions of the same amino acids, must lie in the differences in the sequence context. This is the first study to show that the proximal sequence surrounding a PTC can influence the amino acids incorporated at a PTC during readthrough.
The importance of sequence context in the translation termination efficiency, as well as the susceptibility to readthrough, is well known (34,35). However, the mechanism by which this occurs is less well understood. Previous studies have shown that the most C-terminal residues of the nascent polypeptide chain can interact with the ribosome, and thus potentially influence decoding events (36,37). The first residue following the termination codon can also strongly influence the decoding event, and has led to the stop codon and the following nucleotide being referred to as a ‘tetranucleotide termination sequence’ (16,38). We are now beginning to understand how this effect occurs. A recent study reported that the N-terminal domain of eRF1 reaches into the decoding center of the ribosome to form a pocket that accommodates not only the stop codon, but also the following nucleotide in a novel ‘U-turn’ RNA configuration (39). This four-base recognition event could explain how the first nucleotide following the stop codon influences the efficiency of stop codon recognition (15,16). Whether the tetranucleotide termination sequence can also influence the selection of near-cognate tRNAs during nonsense suppression will be examined in future studies.
The results discussed above indicate that the amino acids inserted during readthrough may often be different from the amino acids present at that position in the wild-type protein. Therefore, the variant proteins produced by readthrough may not fully recapitulate wild-type protein stability or function. Few studies have investigated the relative protein activity restored by PTC suppression as compared to the WT protein. To address this question, we examined how the amino acids incorporated during readthrough of the CFTR W1282X PTC affect CFTR protein maturation and function. HEK293 cells expressing the W1282C and W1282L proteins showed variable defects in CFTR maturation and stability compared to WT CFTR, with W1282C having more severe defects. Evaluation of W1282C and W1282L CFTR variant channel function by Isc measurements and by transcellular conductance (Gt) assays in FRT cells showed that while both W1282C and W1282L CFTR variants were functional, the W1282L variant produced significantly more channel activity. These results are similar to those recently published for the CFTR G542X variants (11). These data demonstrate that the amino acid inserted at a PTC can often have significant effects on the function of the full-length protein generated. Future research on nonsense suppression therapies should explore whether different readthrough agents can bias the amino acids inserted in distinct ways so that optimal protein function can be obtained.
Our results demonstrate that the resulting full-length, variant CFTR proteins resulting from PTC suppression may still be defective due to defects in CFTR maturation, gating or stability. Recently, a number of CFTR modulator drugs have been developed to address these defects. For example, the corrector VX-809 stabilizes the immature form of CFTR in the ER and facilitates its trafficking to the cell surface (19), while the potentiator VX-770 increases the gating of CFTR protein at the cell surface (21–23). Aside from their therapeutic potential, CFTR modulators can be used to better understand the defects in variant CFTR proteins generated by PTC suppression. We found that the corrector VX-809 significantly increased total variant CFTR protein accumulation from both the G542X and W1282X alleles. It also increased their band C/B ratio, suggesting that it enhanced CFTR maturation and/or stability at the cell surface. VX-770 treatment also provided modest enhancements in variant CFTR function. When both compounds were used, CFTR channel function was enhanced more than with either compound alone. These results indicate that the combination of a CFTR corrector with a CFTR potentiator can significantly enhance the function of CFTR variant proteins generated by PTC suppression.
We also found that FRT cells expressing either CFTR G542X or W1282X alleles treated with the readthrough compound G418 showed increased CFTR channel activity. However, a greater level of activity was restored in cells expressing the CFTR G542X cDNA, indicating that a more robust level of G418-mediated readthrough of the CFTR G542X allele was achieved. No increase in CFTR activity was observed when cells expressing CFTR G542X were treated with VX-809 alone, VX-770 alone, or a combination of both compounds, indicating that the truncated CFTR protein produced by the G542X allele does not possess any residual CFTR activity. Most importantly, these results obtained in FRT cells were confirmed in primary, homozygous G542X HBE cells, thus showing the utility of combining readthrough compounds with CFTR modulators in these more clinically relevant cells. In contrast, an increase in CFTR channel activity was detected when cells expressing CFTR W1282X were treated with VX-809 alone. Co-treatment of these cells with VX-809 and VX-770 stimulated CFTR activity to a much greater extent, as previously reported (17,40). We interpret these results to mean that the truncated CFTR protein produced by the W1282X allele retains residual activity, but most is unable to escape the ER. VX-809 can facilitate the movement of more truncated CFTR protein to the cell surface, but the channel is largely unable to open due to the C-terminal truncation within NBD2. However, the addition of VX-770 overcomes this gating defect, resulting in a significant stimulation of CFTR activity. The rescue of latent CFTR activity from the CFTR W1282X allele is completely consistent with another recent report (29,41), and suggests that a partial restoration of CFTR activity may be obtained in patients carrying this allele through the action of CFTR modulators in the absence of nonsense suppression. Most CFTR nonsense mutations, like G542X, are expected to lack the latent activity associated with the W1282X allele. Thus, this study provides a proof-of-concept that the combination of CFTR modulators and nonsense suppression drugs may be the most efficient way to restore maximal CFTR function in CF patients that harbor non-sense mutations.
Materials and Methods
Plasmid construction
The following reporters were constructed that contain a readthrough cassette with CFTR G542WT, G542X, W1282WT, W1282X, QXQ UGA, QXQ UAA, QXQ UAG or QWQ along with three codons of upstream and downstream sequence (Fig. 1A). The first five cassettes listed above were fused downstream of TurboGFP through three steps of PCR reactions using a template pT7CFE1-CGFP-HA-His (ThermoFisher). The forward primer is the same in all three steps and the reverse primer of the third step is the same for all constructs. QXQ UAA, QXQ UAG and the corresponding WT constructs were obtained through site-directed mutagenesis based on QXQ UGA. The oligonucleotides used to construct the reporter cassettes are shown in Supplementary Material, Table S1. The resulting fragment was cut with NheI and XhoI and inserted into the pcDNA3.1Zeo(+) expression vector (Invitrogen). All constructs were confirmed by DNA sequencing. The CFTR G542X, W1282X and the WT fragment was cloned between NheI and XhoI in the pcDNA3.1Zeo(+) expression vector, respectively. G542R, G542C, G542W, W1282C and W1282L constructs were obtained through site-directed mutagenesis and confirmed by sequencing. The oligonucleotides used to build CFTR constructs are shown in Supplementary Material, Table S2.
Mammalian cell culture and transfection
Human embryonic kidney 293 (HEK293) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS) and 200 nM of l-glutamine (Invitrogen). HEK293s were transfected using Lipofectamine LTX (Invitrogen) according to the manufacturer’s instructions and then placed in selection medium containing 200 μg/ml of zeocin for 2 weeks. FRT cells were cultured in Ham's F-12 medium (Sigma) supplemented with 5% FBS and 2.68 g/l sodium bicarbonate (Sigma). FRTs were transfected with CFTR constructs using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions and selected with 800 μg/ml of zeocin for 2 weeks. In order to stimulate readthrough, cells were treated with G418 (0.3 mg/ml) for 24–48 h before harvesting.
Quantitative real-time PCR
Total RNA was extracted using the RNeasy Mini Kit (Qiagen) as per the manufacturer’s directions. cDNA was obtained through reverse transcription using iScript™ Reverse Transcription Supermix (Bio-Rad). Quantitative real-time PCR was performed using the CFX96 Real-Time PCR Detection System (Bio-Rad). Primer efficiencies for human CFTR (5’-CCTATGACCC GGATAACAAG GA-3’ and 5’-GAACACGGCT TGACAGCTTT A-3’) and tubulin (5'-CAACACCTTC TTCAGTGAGA CAGG-3' and 5'-TCAATGATCT CCTTGCCAAT GGT-3’) were 93.4 and 95.8%, respectively.
Western blot
HEK293 cells were washed with 1× PBS, harvested, and lysed with lysis buffer (pH 8.0, 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole) containing protease inhibitor cocktail tablet (Roche) using three freeze (on dry ice) and thaw (37 °C) cycles. Lysates were centrifuged at 14 000g for 10 min at 4 °C. Lysates were boiled for 5 min, centrifuged, loaded onto SDS-PAGE gels, separated by electrophoresis and transferred to Immobilon-FL PVDF membrane (EMD Millipore). Membranes were blocked in 1× PBS with 5% milk (w/v) and 0.3% Tween 20 (v/v) overnight. The next day membranes were incubated with TurboGFP antibody (Pierce, 1:2000), anti-HA antibody (Covance Research, 1:2000) and anti-tubulin (Abcam, 1:2000, or DSHB, 1:2000) for 2 h and washed followed by secondary antibodies (IRDye 680RD and IRDye 800CW, Li-Cor, 1:20 000) incubation for 2 h and washed. Then blots were imaged using Li-Cor Odyssey® CLx Infrared Imaging System.
For CFTR western blotting using FRT cells, the cells were washed with 1× PBS, harvested and lysed with RIPA buffer (50 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris–HCl) containing protease inhibitor cocktail tablet (Roche). Proteins were separated using 4–15% gradient SDS-Page gels (Biorad). Transfer, block, antibody incubation and imaging were the same as HEK293 samples. CFTR antibodies used here: 1:1 mixture of 570 and 596 monoclonal CFTR antibodies (UNC, 1:20 000).
Protein purification
HEK293 cells were harvested and lysed with lysis buffer using three freeze and thaw cycles. Supernatant was transferred to a new tube after centrifuge and incubated with Ni-NTA Superflow resin (Qiagen) overnight at 4 °C. The next day, resin was washed three times with lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0) and eluted with elution buffer (50 mM NaH2PO4, 300 mM NaCl, 500 mM imidazole, pH 8.0).
Sample preparation for mass spectrometry
The enriched protein fractions were concentrated, and the buffer exchanged using 3 kDa MW cut-off columns (Millipore). The sample was then diluted in LDS PAGE buffer (Invitrogen) followed by reducing, denatured and separation on an SDS Bis–Tris gel (4–12%, Invitrogen). The gel was stained overnight with colloidal blue (Invitrogen). Based on western blot analysis, the region of interest (based on molecular weight) was cut out and equilibrated in 100 mM ammonium bicarbonate (AmBc). Gel slices were reduced, carbidomethylated, dehydrated and digested with Trypsin Gold (Promega) as per the manufacturers’ instructions. Following digestion, peptides were extracted, concentrated under vacuum and resolubilized in 0.1% formic acid prior to analysis by 1D reverse phase LC-ESI-MS2 as outlined below.
Mass spectrometry
Peptide digests (8 µl each) were injected onto a 1260 Infinity nHPLC stack (Agilent) and separated using a 100 μm I.D. ×13 cm pulled tip C-18 column (Jupiter C-18 300 Å, 5 μm, Phenomenex). This system runs in-line with a Thermo Orbitrap Velos Pro hybrid mass spectrometer, equipped with a nano-electrospray source (Thermo Fisher Scientific), and all data were collected in CID mode. The nHPLC was configured with binary mobile phases that included solvent A (0.1% FA in ddH2O), and solvent B (0.1% FA in 15% ddH2O/85% ACN), programmed as follows: 10 min at 0% B (2 µl/min, load), 90 min at 0–40% B (0.5 nl/min, analyze), 15 min at 0% B (2 µl/min, equilibrate). Following each parent ion scan (350–1200 m/z at 60k resolution), fragmentation data (MS2) was collected on the top most intense 15 ions. For data dependent scans, charge state screening and dynamic exclusion were enabled with a repeat count of 2, repeat duration of 30 s and exclusion duration of 90 s.
MS data conversion and searches
The XCalibur RAW files were collected in profile mode, centroided and converted to MzXML using ReAdW v. 3.5.1. The mgf files were then created using MzXML2Search (included in TPP v. 3.5) for all scans with a precursor mass between 350 and 1200 Da. These data were searched using SEQUEST, which was set for three maximum missed cleavages, a precursor mass window of 20 ppm, trypsin digestion, variable modification C at 57.0293, and M at 15.9949. For the fragment-ion mass tolerance, 0.0 Da was used. Searches were performed with a human subset of the UniRef100 database, which included common contaminants such as digestion enzymes and human keratin, in addition to sequences specific to these experiments.
Peptide filtering, grouping and quantification
A list of peptide IDs were generated based on SEQUEST search results, which were filtered using Scaffold (Protein Sciences). Scaffold was applied in order to filter and group all of the matching peptides to generate and retain only high confidence IDs while also generating normalized spectral counts (SCs) across all samples for the purpose of relative quantification. The filter cut-off values were set with peptide length (>5 amino acids), no peptides with a MH+1 charge state were included, peptide probabilities were calculated and set to >90% CI, with the number of peptides per protein set at 2 or more, and protein probabilities were set to >97% CI, which all combined results in a list of protein IDs with >99% confidence. Scaffold incorporates the two most common methods for statistical validation of large proteome datasets, the false discovery rate and protein probability (42–44). Relative quantification across experiments are performed via spectral counting (45), and SC abundances are then normalized between samples.
Short-circuit current (Isc) measurements of FRT cell monolayers
Cells were seeded 4 days before assay on Costar 24 well 0.4 µM permeable supports (Corning) and maintained in medium at 37 °C with 5% CO2. Cells were grown to confluence and tight monolayer formation was determined by measuring transepithelial resistance using an epithelial voltohmmeter (WPI). VX-809 (Lumacaftor, 3 μM, Selleckchem) was added to medium 24 h before experiment. Isc was measured under voltage clamp conditions in Ussing Chambers (Physiologic Instruments), as previously described (17,46). Briefly, after equipment offset, cell monolayers were mounted onto chambers filled with a high chloride ringer solution with (millimolar units) 120 NaCl, 25 NaHCO3, 3.33 KH2PO4, 1.2 CaCl2, 0.83 K2HPO4, 1.2 MgCl2 and 10 mannitol. Solutions were vigorously stirred and gassed with 95% O2 and 5% CO2 continuously. When indicated, solutions in the basolateral side was changed to a low Cl− ringer which contained (millimolar units) 1.2 NaCl, 25 NaHCO3, 3.33 KH2PO4, 1.2 CaCl2, 0.83 K2HPO4, 1.2 MgCl2, 141 Na Gluconate and 10.8 mannitol. Amiloride (100 μM) was added to basolateral side to block residual ENaC current. Forskolin (10–20 μM, depending on experiment) was added to both sides to elevate cAMP and Isc was monitored for >10 min. The stimulated Isc induced by the forskolin was consistent with a cAMP-stimulated chloride current mediated by the CFTR channel. VX-770 (Ivacaftor, 10 μM, Seleckchem) was added to both sides. In addition, CFTR inhibitor-172 (CFTRInh-172) (10 μM, Sigma) was added to the basolateral side to further confirm specificity.
Trans-Gt measurements of FRT cell monolayers
Gt was measured using 24-channel voltage clamp, as previously described (17,47). After recording the baseline Gt, forskolin (20 μM) was added to both sides followed by CFTRInh-172 (10 μM) to the basolateral side.
Ieq on HBE cell monolayers
Primary HBE cells isolated from a homozygous G542X patient (graciously provided by Dr Scott Randell, at the University of North Carolina at Chapel Hill) were expanded and differentiated into cell monolayers grown at air liquid interface. Briefly, cells (Passage 3) were seeded onto HTS 24-well transwell filter inserts (Corning, #3378) at a density of ∼500 K/cm2 and grown until well-differentiated (30). HBE cell monolayers were then incubated for 48 h (37 °C, 5% CO2) in the presence of (basolateral) vehicle or readthrough agent G418 (50 µM or 100 µM) and/or the corrector molecule, VX-809 (3 µM) or the combination of G418 with VX-809. During Ieq assay, the incubation media is replaced with HEPES buffered (pH 7.4) F-12 Coon’s modification media (Sigma, F6636) and cells are equilibrated for 30 min at 36 °C. The Ieq measurements were obtained using a transepithelial current clamp (TECC-24, EP Design, Belgium) mounted on a robotic platform (P2300, Precise Automation, CA, USA), as previously described (17). After measuring the baseline Ieq, benzamil (6 µM) was added to block the epithelial sodium channel activity followed by the addition of CFTR agonist cocktail forskolin (10 µM) plus VX-770 (1 µM), and then anion transport was inhibited with bumetanide (10 µM). DMSO (0.03%) served as the vehicle control. The area under the curve (AUC) between additions of CFTR agonists and CFTR inhibitor are used as measures of functional CFTR Cl− transport activity.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
The authors would like to thank Ming Du for helpful discussions and Dr Scott Randell at the University of North Carolina at Chapel Hill for providing G542X/G542X primary HBE cells.
Conflict of Interest statement. D.M.B. is a consultant for PTC Therapeutics. S.M.R. receives grant support from PTC Therapeutics, Vertex Pharmaceuticals, Bayer Healthcare and Novartis to conduct clinical trials for the treatment of cystic fibrosis, and research support from Galapagos to evaluate potentiators and correctors to augment translational readthrough.
Funding
This work was supported by National Institutes of Health Grants R21 NS090928 (to D.M.B. and K.M.K.), R21 OD019922 (to K.M.K.), and P30 DK072482 (to S.M.R.); by the Cystic Fibrosis Foundation (D.M.B., S.M.R., R.J.B., and K.M.K.); and by the University of Alabama Institutional Core Support Program from National Cancer Institute Grant P30 CA13148-38 (to J.M.).
References
- 1. Mort M., Ivanov D., Cooper D.N., Chuzhanova N.A. (2008) A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat., 29, 1037–1047. [DOI] [PubMed] [Google Scholar]
- 2. Howard M., Frizzell R.A., Bedwell D.M. (1996) Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat. Med., 2, 467–469. [DOI] [PubMed] [Google Scholar]
- 3. Bedwell D.M., Kaenjak A., Benos D.J., Bebok Z., Bubien J.K., Hong J., Tousson A., Clancy J.P., Sorscher E.J. (1997) Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat. Med., 3, 1280–1284. [DOI] [PubMed] [Google Scholar]
- 4. Du M., Jones J.R., Lanier J., Keeling K.M., Lindsey J.R., Tousson A., Bebok Z., Whitsett J.A., Dey C.R., Colledge W.H.. et al. (2002) Aminoglycoside suppression of a premature stop mutation in a Cftr-/- mouse carrying a human CFTR-G542X transgene. J. Mol. Med., 80, 595–604. [DOI] [PubMed] [Google Scholar]
- 5. Du M., Liu X., Welch E.M., Hirawat S., Peltz S.W., Bedwell D.M. (2008) PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc. Natl. Acad. Sci. U.S.A., 105, 2064–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Welch E.M., Barton E.R., Zhuo J., Tomizawa Y., Friesen W.J., Trifillis P., Paushkin S., Patel M., Trotta C.R., Hwang S.. et al. (2007) PTC124 targets genetic disorders caused by nonsense mutations. Nature, 447, 87–91. [DOI] [PubMed] [Google Scholar]
- 7. Keeling K.M., Xue X., Gunn G., Bedwell D.M. (2014) Therapeutics based on stop codon readthrough. Annu. Rev. Genomics Hum. Genet., 15, 371–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feng Y.X., Copeland T.D., Oroszlan S., Rein A., Levin J.G. (1990) Identification of amino acids inserted during suppression of UAA and UGA termination codons at the gag-pol junction of Moloney murine leukemia virus. Proc. Natl. Acad. Sci. U.S.A., 87, 8860–8863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fearon K., McClendon V., Bonetti B., Bedwell D.M. (1994) Premature translation termination mutations are efficiently suppressed in a highly conserved region of yeast Ste6p, a member of the ATP-binding cassette (ABC) transporter family. J. Biol. Chem, 269, 17802–17808. [PubMed] [Google Scholar]
- 10. Blanchet S., Cornu D., Argentini M., Namy O. (2014) New insights into the incorporation of natural suppressor tRNAs at stop codons in Saccharomyces cerevisiae. Nucleic Acids Res., 42, 10061–10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roy B., Friesen W.J., Tomizawa Y., Leszyk J.D., Zhuo J., Johnson B., Dakka J., Trotta C.R., Xue X., Mutyam V.. et al. (2016) Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc. Natl. Acad. Sci. U.S.A., 113, 12508–12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roy B., Leszyk J.D., Mangus D.A., Jacobson A. (2015) Nonsense suppression by near-cognate tRNAs employs alternative base pairing at codon positions 1 and 3. Proc. Natl. Acad. Sci. U.S.A., 112, 3038–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rowe S.M., Miller S., Sorscher E.J. (2005) Cystic fibrosis. N. Engl. J. Med., 352, 1992–2001. [DOI] [PubMed] [Google Scholar]
- 14. Cassan M., Rousset J.P. (2001) UAG readthrough in mammalian cells: effect of upstream and downstream stop codon contexts reveal different signals. BMC Mol. Biol., 2, 3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Manuvakhova M., Keeling K., Bedwell D.M. (2000) Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. rna, 6, 1044–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bonetti B., Fu L., Moon J., Bedwell D.M. (1995) The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J. Mol. Biol., 251, 334–345. [DOI] [PubMed] [Google Scholar]
- 17. Xue X., Mutyam V., Tang L., Biswas S., Du M., Jackson L.A., Dai Y., Belakhov V., Shalev M., Chen F.. et al. (2014) Synthetic aminoglycosides efficiently suppress cystic fibrosis transmembrane conductance regulator nonsense mutations and are enhanced by ivacaftor. Am. J. Respir. Cell Mol. Biol., 50, 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Keeling K.M., Bedwell D.M. (2002) Clinically relevant aminoglycosides can suppress disease-associated premature stop mutations in the IDUA and P53 cDNAs in a mammalian translation system. J. Mol. Med., 80, 367–376. [DOI] [PubMed] [Google Scholar]
- 19. Van Goor F., Hadida S., Grootenhuis P.D., Burton B., Stack J.H., Straley K.S., Decker C.J., Miller M., McCartney J., Olson E.R.. et al. (2011) Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U.S.A., 108, 18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eckford P.D., Ramjeesingh M., Molinski S., Pasyk S., Dekkers J.F., Li C., Ahmadi S., Ip W., Chung T.E., Du K.. et al. (2014) VX-809 and related corrector compounds exhibit secondary activity stabilizing active F508del-CFTR after its partial rescue to the cell surface. Chem. Biol., 21, 666–678. [DOI] [PubMed] [Google Scholar]
- 21. Van Goor F., Hadida S., Grootenhuis P.D., Burton B., Cao D., Neuberger T., Turnbull A., Singh A., Joubran J., Hazlewood A.. et al. (2009) Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. U.S.A., 106, 18825–18830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Accurso F.J., Rowe S.M., Clancy J.P., Boyle M.P., Dunitz J.M., Durie P.R., Sagel S.D., Hornick D.B., Konstan M.W., Donaldson S.H.. et al. (2010) Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med., 363, 1991–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ramsey B.W., Davies J., McElvaney N.G., Tullis E., Bell S.C., Drevinek P., Griese M., McKone E.F., Wainwright C.E., Konstan M.W.. et al. (2011) A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med., 365, 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eckford P.D., Li C., Ramjeesingh M., Bear C.E. (2012) Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J. Biol. Chem., 287, 36639–36649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jih K.Y., Hwang T.C. (2013) VX-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc. Natl. Acad. Sci. U.S.A., 110, 4404–4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wainwright C.E., Elborn J.S., Ramsey B.W., Marigowda G., Huang X., Cipolli M., Colombo C., Davies J.C., De Boeck K., Flume P.A.. et al. (2015) Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med., 373, 1783–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gohil K. (2015) Pharmaceutical approval update. Pharm. Ther. Commun., 40, 567–568. [PMC free article] [PubMed] [Google Scholar]
- 28. Matthes E., Goepp J., Carlile G.W., Luo Y., Dejgaard K., Billet A., Robert R., Thomas D.Y., Hanrahan J.W. (2016) Low free drug concentration prevents inhibition of F508del CFTR functional expression by the potentiator VX-770 (ivacaftor). Br. J. Pharmacol., 173, 459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haggie P.M., Phuan P.W., Tan J.A., Xu H., Avramescu R.G., Perdomo D., Zlock L., Nielson D.W., Finkbeiner W.E., Lukacs G.L.. et al. (2017) Correctors and potentiators rescue function of the truncated W1282X-CFTR translation product. J. Biol. Chem., 292, 771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Neuberger T., Burton B., Clark H., Van Goor F. (2011) Use of primary cultures of human bronchial epithelial cells isolated from cystic fibrosis patients for the pre-clinical testing of CFTR modulators. Methods Mol. Biol., 741, 39–54. [DOI] [PubMed] [Google Scholar]
- 31. Rozov A., Demeshkina N., Westhof E., Yusupov M., Yusupova G. (2016) New Structural Insights into Translational Miscoding. Trends Biochem. Sci., 41, 798–814. [DOI] [PubMed] [Google Scholar]
- 32. Zhang J., Ieong K.W., Johansson M., Ehrenberg M. (2015) Accuracy of initial codon selection by aminoacyl-tRNAs on the mRNA-programmed bacterial ribosome. Proc. Natl. Acad. Sci. U.S.A., 112, 9602–9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fernandez I.S., Ng C.L., Kelley A.C., Wu G., Yu Y.T., Ramakrishnan V. (2013) Unusual base pairing during the decoding of a stop codon by the ribosome. Nature, 500, 107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Keeling K.M., Wang D., Conard S.E., Bedwell D.M. (2012) Suppression of premature termination codons as a therapeutic approach. Crit. Rev. Biochem. Mol. Biol., 47, 444–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brar G.A. (2016) Beyond the triplet code: context cues transform translation. Cell, 167, 1681–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Beringer M. (2008) Modulating the activity of the peptidyl transferase center of the ribosome. rna, 14, 795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shao S., Murray J., Brown A., Taunton J., Ramakrishnan V., Hegde R.S. (2016) Decoding mammalian ribosome-mRNA states by translational GTPase complexes. Cell, 167, 1229–1240, e1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brown C.M., Stockwell P.A., Trotman C.N., Tate W.P. (1990) Sequence analysis suggests that tetra-nucleotides signal the termination of protein synthesis in eukaryotes. Nucleic Acids Res., 18, 6339–6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Matheisl S., Berninghausen O., Becker T., Beckmann R. (2015) Structure of a human translation termination complex. Nucleic Acids Res., 43, 8615–8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rowe S.M., Varga K., Rab A., Bebok Z., Byram K., Li Y., Sorscher E.J., Clancy J.P. (2007) Restoration of W1282X CFTR activity by enhanced expression. Am. J. Respir. Cell Mol. Biol., 37, 347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mutyam V., Libby E.F., Peng N., Hadjiliadis D., Bonk M., Solomon G.M., Rowe S.M. (2017) Therapeutic benefit observed with the CFTR potentiator, ivacaftor, in a CF patient homozygous for the W1282X CFTR nonsense mutation. J. Cyst. Fibros, 16, 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Keller A., Nesvizhskii A.I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem., 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- 43. Nesvizhskii A.I., Keller A., Kolker E., Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem., 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- 44. Weatherly D.B., Atwood J.A. III, Minning T.A., Cavola C., Tarleton R.L., Orlando R. (2005) A Heuristic method for assigning a false-discovery rate for protein identifications from Mascot database search results. Mol Cell Proteomics, 4, 762–772. [DOI] [PubMed] [Google Scholar]
- 45. Liu H., Sadygov R.G., Yates J.R. III (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem., 76, 4193–4201. [DOI] [PubMed] [Google Scholar]
- 46. Rowe S.M., Sloane P., Tang L.P., Backer K., Mazur M., Buckley-Lanier J., Nudelman I., Belakhov V., Bebok Z., Schwiebert E.. et al. (2011) Suppression of CFTR premature termination codons and rescue of CFTR protein and function by the synthetic aminoglycoside NB54. J. Mol. Med. (Berl), 89, 1149–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mendoza J.L., Schmidt A., Li Q., Nuvaga E., Barrett T., Bridges R.J., Feranchak A.P., Brautigam C.A., Thomas P.J. (2012) Requirements for efficient correction of DeltaF508 CFTR revealed by analyses of evolved sequences. Cell, 148, 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.