

Abstract
A major challenge in human genetics is the validation of pathogenicity of heterozygous missense variants. This problem is well-illustrated by PROKR2 variants associated with Isolated GnRH Deficiency (IGD). Homozygous, loss of function variants in PROKR2 was initially implicated in autosomal recessive IGD; however, most IGD-associated PROKR2 variants are heterozygous. Moreover, while IGD patient cohorts are enriched for PROKR2 missense variants similar rare variants are also found in normal individuals. To elucidate the pathogenic mechanisms distinguishing IGD-associated PROKR2 variants from rare variants in controls, we assessed 59 variants using three approaches: (i) in silico prediction, (ii) traditional in vitro functional assays across three signaling pathways with mutant-alone transfections, and (iii) modified in vitro assays with mutant and wild-type expression constructs co-transfected to model in vivo heterozygosity. We found that neither in silico analyses nor traditional in vitro assessments of mutants transfected alone could distinguish IGD variants from control variants. However, in vitro co-transfections revealed that 15/34 IGD variants caused loss-of-function (LoF), including 3 novel dominant-negatives, while only 4/25 control variants caused LoF. Surprisingly, 19 IGD-associated variants were benign or exhibited LoF that could be rescued by WT co-transfection. Overall, variants that were LoF in ≥ 2 signaling assays under co-transfection conditions were more likely to be disease-associated than benign or ‘rescuable’ variants. Our findings suggest that in vitro modeling of WT/Mutant interactions increases the resolution for identifying causal variants, uncovers novel dominant negative mutations, and provides new insights into the pathogenic mechanisms underlying heterozygous PROKR2 variants.
Introduction
In the current era of high-throughput genome sequencing, investigators are continually refining and streamlining their analytical methodologies (1–9), increasing their access to normative and pathological datasets (10–13), and establishing new standards for interpreting and reporting genetic variants that occur in disease states (14–17). However, a remaining obstacle is how best to assess the functional consequences of rare, heterozygous missense mutations that will help distinguish disease-associated mutations from benign variation (15,18–20). This problem is becoming unwieldy as some genetic variants, including those associated with disease states, are increasingly found in control sequencing cohorts (21,22). Access to biologically relevant tissues and tractable assays have made it possible to develop high-throughput functional testing methods for some diseases (23); however, for most genetic disorders, such robust functional assays are unavailable.
To address these challenges, in silico algorithms incorporating evolutionary conservation, underlying protein structure, and/or functional information have been widely used to assist in the interpretation of missense variants (19,24–27). Nonetheless, each of these bioinformatics approaches has inherent limitations that constrain their broader utility to assert pathogenicity (6,17,20,28–30). Thus, to affirm causality, it is critical that appropriate functional assays are selected to assess the biological consequences of each genetic variant and that appropriate attention is paid to gene dosing, particularly in evaluating heterozygosity.
Isolated GnRH deficiency (IGD) is a rare Mendelian disorder with known genetic heterogeneity that is clinically characterized by absent pubertal development and infertility (31–35). One gene that causes IGD, PROKR2 [MIM: 607123], was discovered when homozygous ablation of its murine orthologue produced a precise IGD phenocopy (36). While bi-allelic loss-of-function (LoF) mutations were initially associated with autosomal recessive IGD in patients (37–40), the vast majority (>90%) of PROKR2 variants associated with IGD are heterozygous with no evident mutation on the second allele (38,41–43). Moreover, IGD cohorts are significantly enriched for heterozygous PROKR2 variants when compared with controls (37,44) (R.B., unpublished data).
Despite strong human genetic data and biological plausibility indicting PROKR2 as an IGD gene (36,43,45), there is less certainty about individual PROKR2 variants in some IGD patients for several reasons. First, most variants are missense rather than truncating frame-shift or stop-gain mutations. Second, these variants often display incomplete penetrance and variable expressivity within IGD families (38,39,44). Third, some of these variants occur in concert with variants in other IGD genes, i.e. in IGD subjects demonstrating a digenic mode of inheritance, suggesting that some variants may not be sufficiently penetrant by themselves to cause disease (37,46–48). Fourth, data from large exome-sequencing cohorts (13,49) have increasingly revealed many comparably rare (minor allele frequency [MAF] <0.01) heterozygous missense variants in PROKR2 in apparently reproductively normal controls. Finally, our group has shown that two variants in PROKR2 are founder alleles which, although enriched in IGD (44,50), are also seen in controls.
These features, along with the fact that the PROKR2 gene is not under evolutionary constraint for rare, non-synonymous mutations (1), suggests that neither rarity nor the protein-altering nature of these variants serve as sufficiently reliable surrogates for interpreting disease-causality. However, systematic functional testing is necessary for the discrimination of disease-causing variants from benign variants. Due to the complex ontogeny of GnRH neurons and neuroanatomic organization of the hypothalamus, no appropriate human cellular systems with which functional assay methods currently exist that can thoroughly model the in vivo functions of PROKR2. In vitro studies have indicated that PROKR2, a transmembrane G-protein-coupled receptor, dimerizes on the cellular surface (51,52), and activates several intracellular signaling pathways (38,53–57). PROKR2 variants can cause LoF in one (or more) of these pathways (38,56,57), and at least one mutant has been shown to have a dominant-negative effect on WT protein expression (53). Despite these findings, no studies have systematically investigated how defects in PROKR2 dimerization in the presence of both a mutant and a functional wild-type (WT) allele might impact upon pathogenicity. Previous studies have also failed to address the issue of their incomplete penetrance and systematically investigate the functionality of rare heterozygous variants seen exclusively in controls. Therefore, we hypothesized that assessing WT/Mutant interactions across multiple signaling pathways in parallel would allow us to address several remaining puzzles about the genetics of PROKR2 heterozygous missense mutations.
Results
In silico analyses fails to distinguish between IGD-associated and control PROKR2 variants
At the onset of this study in 2013, a total of 59 rare (MAF < 0.01) missense variants represented all known human PROKR2 variants reported (Supplementary Material, Table S1) from the following sources: 1) Massachusetts General Hospital (MGH) IGD patient cohort of >1, 500 patients, 2) the IGD literature, and/or 3) the 1000 Genomes (1 KG) (11) or the National Heart, Lung, and Blood Institute Exome Sequencing Project (ESP) (Exome Variant Server, Seattle, WA [12/2013]) databases. These rare variants alter amino acids that are widely dispersed throughout the body of the PROKR2 protein, thus representing a spectrum of mutations that could potentially alter the protein’s structure. During the course of this study, additional normative sequencing data were released from the ExAC consortium (13,49) that allowed us to further refine our groupings. Overall, seven variants remained exclusive to our IGD cohort at MGH (Supplementary Material, Table S1, Figs S1 and S2) and/or in the IGD literature (35,37) while an additional 27 variants were associated with IGD but were also present in the ExAC database (37–41,46,56,58–60); thus, these 34 variants were categorized as “IGD Variants”. The remaining 25 variants were found only in the ExAC database and not associated with IGD, hence their designation as “Control Variants” (13,49).
A priori, one might expect that variants in these 2 categories could be distinguished by in silico approaches of assessing rarity, conservation of amino acids, etc. We first tested this hypothesis by examining minor allele frequencies (MAFs) for each variant from the ExAC consortium database (13,49). There were several IGD Variants (A51T, V331M, and R268C) which the ExAC database showed are likely ethnic SNPs and one variant is a ∼9000-year-old founder allele, L173R, which we previously reported (44,50) (Supplementary Material, Table S1). With these variants included in the analysis, the average MAF for the IGD Variants was actually higher than the average MAF for the Control Variants, counter to our hypothesis. Moreover, even when these more common variants were removed, there was no difference between the two groups in the average MAF of variants (Supplementary Material, Table S1, P = 0.0579), suggesting that, collectively, Control Variants were as rare as IGD Variants.
Next, we assessed whether the IGD Variants would be more likely to disrupt conserved amino acids. To determine the genetic conservation of these residues, GERP scores (61) were retrieved from the UCSC Table Browser. As expected, the IGD Variants had slightly higher average GERP scores than the Control Variants (Supplementary Material, Table S1, *P < 0.041), suggesting that they were more likely to occur in conserved residues. However, there was a great deal of variation in scores across and within groups and the number of variants that were in conserved residues (GERP score >2) did not differ between groups (Supplementary Material, Table S1, P = 0.2649).
Finally, to assess the predicted pathogenicity of variants in silico, Condel (24) was used to generate consensus deleteriousness scores using a weighted average of five computational tools: SIFT (26), Polyphen2 (25), MAPP (62), LogR (63), and Pfam (64). Again, Chi-square analysis found no difference in the proportion of variants predicted to be deleterious vs. the number predicted to be benign between the two groups (P = 0.1134, Supplementary Material, Table S1; Fig. 1). Taken together, these data demonstrate that in silico approaches are not sufficiently sensitive to distinguish between IGD and Control Variants. Thus, functional testing of these variants is necessary to affirm pathogenicity and make distinctions that are crucial for clinical management and genetic counseling of IGD patients.
Figure 1.

Summary of findings for all variants from computational predictions, functional testing of mutants alone, and functional testing of mutant and WT interactions. Testing of mutants alone suggested no enrichment for deleterious variants in IGD compared with controls. However, Mutant + WT co-transfections revealed that IGD-associated variants were more likely to be deleterious, (i.e. LoF in 2 or 3 assays), than Control variants (*P = 0.0270). Red= deleterious, Gray= Benign.
In vitro testing of mutant PROKR2 alone also fails to differentiate between IGD-associated and control variants
To assess the functional effects of each variant in vitro, we followed an approach used previously (38,53–57) to test signaling activity of mutant PROKR2 across three different pathways: Gαi (MAPK), Gαq (IP-one), and Gαs (cAMP). Mutant vectors were transfected into HEK293 or GS-22A cells (chosen for low endogenous PROKR2 expression and high transfection efficiency) and signaling was compared with WT PROKR2. Complete signaling assay results, including calculated EC50 and Emax values for the MAPK pathway and relative Emax values for the IP-One and cAMP pathways, are shown in Supplementary Material, Table S2. Each of the 59 variants was first classified as either LoF or benign separately for each assay, with LoF mutants defined as those exhibiting significantly reduced EC50, Emax, or both, vs. WT. Somewhat surprisingly, 55/59 variants showed LoF in signaling in at least 1 assay when tested alone, including 16 Control Variants that were LoF in 2 or more assays (Supplementary Material, Table S2). Chi-square analysis found no difference between the two groups in the proportion of variants that were LoF in 2 or more assays (deleterious), versus variants that were benign or LoF in 1 assay (benign, P = 0.2960, Fig. 1). Therefore, similar to our in silico analysis, conventional functional testing of mutants transfected alone also failed to discriminate between IGD and Control variants in aggregate and did not allow for clear classification of pathogenic variants.
Co-transfections of WT and mutant PROKR2 suggest that PROKR2 variants discovered in control populations are less likely to cause IGD
More than 90% of IGD-associated PROKR2 variants and almost all of the variants seen in controls occur exclusively in the heterozygous state. Therefore, to more accurately model the heterozygous genetic context as they are actually observed in IGD patients, the signaling function of mutants that tested LoF in the mutant-alone experiments was subsequently assessed by co-transfection with WT in a series of DNA dosing experiments. Details of these co-transfection assay results, including their calculated EC50 and Emax values for the MAPK pathway and relative Emax values for the IP-One and cAMP pathways, are compared in Supplementary Material, Table S2. After functional testing with co-transfections, the proportion of IGD Variants that remained LoF in ≥ 2 assays (deleterious) was higher than the proportion of deleterious Control Variants, while the proportion of likely benign variants was lower (*P = 0.0270, Fig. 1).
In addition, while, a priori, the bioinformatics prediction programs were not useful for distinguishing control variants from those associated with IGD, post-hoc analysis on a variant-by-variant level revealed that functional testing using co-transfections substantially bolstered the predictions made by using ConDel alone and provided further validation for a classification of pathogenic variants based on LoF in ≥ 2 assays. With co-transfection, mutants that were LoF in ≥ 2 assays were more likely to be associated with IGD than those that were benign (Fig. 2, P < 0.006) and more likely predicted to be deleterious by ConDel (P < 0.0001). In contrast, mutants that tested benign were most likely predicted to be neutral, regardless of whether they were associated with IGD or seen in controls. These results suggest that, although the prediction programs were able to accurately predict many LoF variants when testing mutant signaling alone, they were less sensitive to pathway-specific reductions in signaling and largely unable to anticipate WT and mutant protein interactions found in Mutant + WT co-transfection experiments.
Figure 2.
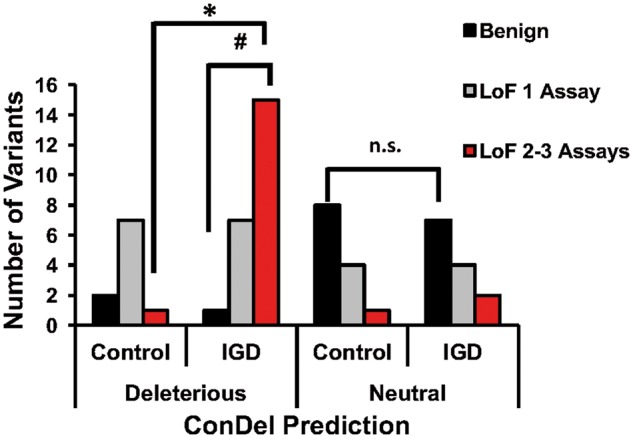
Direct comparison of ConDel predictions with results of functional testing. ConDel predictions were largely aligned with the results of functional testing for each mutant, except for mutants that caused LoF in only 1 signaling pathway. For those mutants, some were predicted to be neutral and others deleterious, and these were evenly split as to whether they were IGD-associated or seen only in Controls. Mutants that were LoF in 2–3 assays were more likely predicted to be deleterious by ConDel (#P < 0.0001), and more commonly associated with IGD (*P < 0.0006), while mutants that tested benign were more mostly predicted to be neutral, regardless of whether they were associated with IGD or seen in Controls (n.s.).
Despite this improved distinction between the two groups of variants, there were still a number of variants in each group that demonstrated LoF in one signaling pathway (typically cAMP) with co-transfection (Supplementary Material, Table S2), suggesting that these might represent milder variants that may rely upon the genetic context for their phenotypic penetrance. Alternatively, this lack of distinction may be due to differences in relative assay sensitivity across the three different signaling pathways. Mutations that were LoF in one signaling pathway were evenly split as to whether they were predicted in silico to be neutral or deleterious and in their occurrence in control and IGD cohorts (Fig. 2, n.s.). Thus, while this combination of co-transfection experiments to model heterozygosity significantly improved current predictions of likelihood of LoF, particularly for variants in control cohorts, further testing will be required to definitively determine if these mutants are preferentially affecting individual pathways (i.e. exhibit pathway specificity) and if these effects have downstream pathophysiologic consequences.
Co-transfection of WT and mutant PROKR2 reveals complex interactions between WT and mutant PROKR2 protein in heterozygous carriers
To elucidate the mechanisms underlying the results from co-transfection experiments, dose-response curves were generated from co-transfections of mutant PROKR2 with varying doses of WT in the MAPK signaling assay. Overall, these detailed dose-response curve results revealed a surprising complexity in the interactions between mutant and WT PROKR2, resulting in four distinct signaling signatures. Although there were many examples for each signature, for consistency, Figure 3 shows representative examples of variants found in both IGD patients and controls. Complete co-transfection signaling assay results, including calculated EC50 and Emax values for the MAPK pathway, are shown in Supplementary Material, Table S2.
Figure 3.

Dose Response Curves of MAPK Signaling for Mutants Co-transfected with Varying Doses of WT PROKR2. (A–D) Representative data from the MAPK signaling assay are shown as log-dose-response curves from three separate experiments run in triplicate (± SEM) calculated relative to the maximum WT response. (A) Signature #1: When co-transfected with WT, the G234D variant had a dominant negative effect on WT signaling as shown by reduced signaling at an equal (50: 50, G234D/WT) ratio, and a further reduction with increased mutant (75: 25, G234D/WT), both < 50 WT < 100 WT (P < 0.05). (B) Signature #2: Co-transfection of L173R with WT revealed the mutant was unable to contribute to MAPK signaling, but did not impact WT signaling (50: 50 L173R/WT different from 100 WT; P < 0.05). (C) Signature #3: Co-transfection of P290S with WT seemed to partially rescue P290S signaling (50 WT < 50: 50 P290S/WT < 100 WT; P < 0.05). (D) Signature #4: When co-transfected with WT, MAPK signaling by the R268C mutation was completely rescued (50: 50 R268C/WT = 100 WT). (E) Western blots were probed with antibodies to V5 (tagging PROKR2) and β-Actin. Graphed data are presented as mean (± SEM) from 3 separate experiments. For representative blots, some lanes from gels have been removed and reordered, but all lanes from the same blot are shown together with the WT they were compared with and were unaltered. Relative protein expression and representative blots of mutants alone and mutants + WT as compared with WT alone. Red bars: mutant G234D showed potential dominant negative interactions in the MAPK signaling pathway and reduced protein expression as compared with WT, *P < 0.05. (F) Confocal images of WT-mCherry-PROKR2, mutant G234D GFP-tagged PROKR2, and co-transfection of WT and mutant. In contrast to other mutants rescued by WT, the G234D mutation appeared to sequester WT-PROKR2 intracellularly (yellow labeling).
Representing Signature #1, variant G234D decreased WT signaling when co-transfected at a 50: 50 ratio with WT (Fig. 3A). However, signaling was decreased further with an increased (75: 25) MT/WT ratio revealing a dose-response relationship and a previously unrecognized dominant negative effect of this mutant. Two additional IGD Variants, W178S and N325K, showed similar indications of previously unappreciated dominant negative effects on WT signaling in the MAPK assay (Supplementary Material, Table S2). Representing Signature #2, the L173R variant had no effect on WT signaling when co-transfected at a 50: 50 ratio but also could not signal by itself (Fig. 3B), making the case for its functional haploinsufficiency. In Signature #3, variant P290S, had signaling deficits capable of being partially rescued in the presence of WT PROKR2 (Fig. 3C). Finally, representing Signature #4, variant R268C was completely rescued by co-transfection with WT (Fig. 3D).
To determine whether altered protein trafficking underlies the rescue of mutants and/or dominant negative interactions, PROKR2 protein expression was measured in cells transfected either with mutants alone or co-transfected with WT. Average relative protein expression data for each mutant are shown in Supplementary Material, Table S2. While none of the IGD Variants had reduced protein expression, several of the IGD Variants demonstrated reduced expression compared with WT when transfected alone. Curiously, 2 IGD-associated LoF mutants (R85L and V274D) exhibited increased expression compared with WT (Supplementary Material, Table S2). Interestingly, when mutants with reduced protein expression were co-transfected with WT, several appeared to have rescued or partially rescued protein expression (Supplementary Material, Table S2) while others, such as the dominant negative G234D, appeared to reduce expression of WT protein (Fig. 3E).
To further determine whether altered intracellular trafficking could underlie the dominant negative interactions of G234D, transfected proteins in fixed cells were visualized with a confocal microscope using GFP-tagged mutant and mCherry-tagged WT PROKR2. GFP-tagged G234D not only reduced its membrane expression but also appeared to reduce the membrane expression of WT PROKR2 with co-transfection (Fig. 3F). Although these results warrant further investigation using more sensitive assay methods, they nonetheless suggest that some of the PROKR2 mutants are capable of reducing WT PROKR2 expression a dominant-negative manner in the heterozygous state.
Classification of IGD-associated PROKR2 variants based on ACMG criteria
Taken together, our co-transfection results indicated that a number of variants previously considered LoF by previously published studies are, in fact, misclassified due to the fact that their interactions with WT PROKR2 were never modeled. Therefore, we next sought to formally classify the 34 IGD Variants based on ACMG criteria (17) using a combination of our assay results (Supplementary Material, Table S2), as well as published data on PROKR2 variants and pedigrees, data from prediction algorithms, and MAF data from the ExAC database (Supplementary Material, Table S1). As shown in Table 1, there were 8 variants classified as benign or likely benign and 10 variants classified as likely pathogenic. Five of these variants are unique to the IGD cohort at Massachusetts General Hospital (Supplementary Material, Table S1, Figs S1 and S2). While the N325K variant (found in one proband) is likely pathogenic variants A51T, D112Y, V297I, and V317L are categorized as benign or likely benign. Thus, there are a number of IGD pedigrees that remain “unsolved” in their genetic diagnoses and offer opportunities for novel genetic discovery.
Table 1.
Quick-reference for variant classification based on ACMG criteria
| IGD variant | ACMG criteria met | ACMG classification |
|---|---|---|
| A51T | BS1, BS3, BS4, BP4, BP5 | Benign |
| R85C | PM1, PP3, BS3, BS4, BP5 | Benign |
| D112Y | PM2, PP3, BS3, BS4 | Benign |
| S202G | BS3, BS4, BP4, BP5 | Benign |
| V331M | BS1, BS3, BS4, BP4, BP5 | Benign |
| M64V | PM2, BS3, BP4 | Likely Benign |
| V317L | PM2, BS3, BP4, BP5 | Likely Benign |
| R357W | BS3, BP4 | Likely Benign |
| S188L | PS3 (hom), BS3 (het), BS4, BP4 | Het Benign, Hom Uncertain Significance |
| V297I | PS3 (hom), BS 3 (het), BS4, BP4, BP5 | Het Benign, Hom Uncertain Significance |
| V158I | PS3 (hom), BS3 (het), BP4 | Het Likely Benign, Hom Uncertain Significance |
| R248Q | PS3 (hom), BS3 (het), BP4, BP5 | Het Likely Benign, Hom Uncertain Significance |
| R85L | PS3 (hom), BS3 (het) PM1, PP3, BP5 | Hom Likely Pathogenic, Het Likely Benign |
| Q210R | PS3 (hom), BS3 (het), PM2, PP3, BP2 | Hom Likely Pathogenic, Het Likely Benign |
| M323I | PS3 (hom), BS3 (het), PM2, PM3, BP4 | Hom Likely Pathogenic, Het Likely Benign |
| T260M | PS3 (hom), BS3 (het), PM3, PP3 | Hom Likely Pathogenic, Het Uncertain Significance |
| V274D | PS3 (hom), BS3 (het), PM2, PP3 | Hom Likely Pathogenic, Het Uncertain Significance |
| M111R | PS3, PM2, PM3, PP3, BP2 | Likely Pathogenic |
| Y113H | PS3, PM3, PP3 | Likely Pathogenic |
| R80C | PS3, PM2, PP3 | Likely Pathogenic |
| R85G | PS3, PM1, PP3 | Likely Pathogenic |
| R85H | PS3, PM1, PP3, BP5 | Likely Pathogenic |
| R164Q | PS3, PM2, PM3, PP3 | Likely Pathogenic |
| G234D | PS3, PM3, PP3 | Likely Pathogenic |
| W251L | PS3, PM2, PP3 | Likely Pathogenic |
| R270C | PS3, PM2, PP3 | Likely Pathogenic |
| N325K | PS3, PM2, PP3, BS4 | Likely Pathogenic |
| V115M | PS3, PP3 | Uncertain Significance |
| R117W | PS3, PP3 | Uncertain Significance |
| W178S | PS3, BP4 | Uncertain Significance |
| P290S | PS3, PP3, BS4 | Uncertain Significance |
| L173R | PS3, PS4, PP3, BS4, BP5 | Uncertain Significance |
| R268C | PS3, PM3, PP3, BS1, BP2, BP5 | Uncertain Significance |
| V334M | PS3 (hom), BS3 (het), PP3 | Uncertain Significance |
| KEY: | ||
|---|---|---|
| Term | Description | Evidence of impact |
| BS1 | Allele frequency greater than expected for disorder | Strong |
| BS3 | Well-established functional studies show no damaging effect | |
| BS4 | Lack of segregation in affected family members | |
| BP2 | In cis with a pathogenic variant in any inheritance pattern | Supporting |
| BP4 | Computational evidence suggesting no impact on gene or gene product | |
| BP5 | Found in a case with alternate molecular basis for disease | |
| Benign | 2 or more Strong | |
| Likely Benign | 1 Strong and 1 Supporting OR 2 or more Supporting | |
| PS3 | Well-established functional studies supportive of damaging effect | Strong |
| PS4 | Prevalence of variant in affected individuals significantly increased compared with controls | |
| PM1 | Located in a mutational hot spot and/or critical functional domain | Moderate |
| PM2 | Absent from controls (or extremely low frequency) | |
| PM3 | In trans with a pathogenic variant | |
| PP3 | Computational evidence supporting a deleterious effect | Supporting |
| Likely Pathogenic | 2 or more Strong OR 1 Strong and 1-2 Moderate OR 1 Strong 2 or more Supporting | |
| Uncertain Significance | Criteria not met OR contradictory criteria for Benign vs. Pathogenic | |
NOTE: Unused ACMG criteria (i.e. those not met by any variant) have been excluded from this table.
Despite strong experimental evidence, 7 IGD Variants remained of uncertain significance, either due to the fact that there was missing genetic evidence or contradictory criteria suggesting they might be benign. Among these, the variant L173R, previously shown to be a founder allele, is significantly enriched in IGD cases (P < 0.0001; Supplementary Material, Table S3), a feature that strongly supports its pathogenicity despite its lack of complete segregation in pedigrees. Importantly, the classification of the remaining nine variants differed based on genetic context (ie. homozygous or compound heterozygous vs. heterozygous status). Four of these variants (S188L, V297I, V158I, and R248Q) would have remained categorized as unknown significance without modeling of mutant/WT interactions, and the other 5 (R85L, Q210R, M323I, T260M, and V274D) would previously have been classified as pathogenic, even in heterozygotes. Therefore, our combined approach of functional testing with in silico prediction has clarified the pathogenic status of many PROKR2 missense variants.
Discussion
Rare, heterozygous missense variants in PROKR2 are enriched in IGD, but many similar variants are also seen in controls. Therefore, we directly and systematically compared the functional consequences of variants found in IGD with those found in controls using in silico analyses and in vitro functional assays across multiple pathways and varying ratios of WT/Mutant PROKR2. Importantly, the 59 variants tested could not be distinguished from each other using traditional in silico approaches at the inception of these studies. Moreover, traditional in vitro testing examining mutations alone across several signaling pathways revealed a large number of LoF mutants in the Control Variant group. These findings suggested that either: 1) heterozygous controls bearing these variants might exert a subtle reproductive defect such that their phenotype cannot be easily ascertained from publically available genetic data; or 2) heterozygous LoF variants in PROKR2 alone are insufficient to cause IGD in the absence of additional genetic hits or environmental effects; or 3) that in vitro modeling of the mutant proteins in isolation was resulting in erroneously classifying some control variants as LoF. To directly test this third hypothesis, we adapted the traditional transfection assay methods to reflect the “heterozygous” context by co-transfecting mutant PROKR2 with WT. Our co-transfection experiments indicated that modeling of Mutant/WT protein interactions discerns true LoF alleles from those rescued by WT PROKR2, thereby permitting a reclassification of many of the variants and resulting in improved genotype–phenotype correlations.
The decision to use co-transfections for functional testing was based on several observations. First, most PROKR2 variants in IGD patients are found in the heterozygous state (38,41–43). Thus, most individuals with mutant PROKR2 also have a normal functioning WT copy of the PROKR2 protein. Second, PROKR2 has been shown to dimerize in vivo (51). While the complete functional role of its dimerization in downstream signaling is not yet known, one study using deletion mutants found that the transmembrane domains 4–5 (TM4–5) are most critical for PROKR2 dimerization and may modulate second messenger signaling (52). Finally, co-transfections were necessary to assess the contributions of dominant negative interactions. Only one variant, R80C, was previously shown to have a dominant negative effect on WT protein expression (53), a finding that we replicate with our protein expression data (Supplementary Material, Table S2). However, prior to this study no other PROKR2 missense variants had been shown to exert such effects (55).
There are 10 PROKR2 variants found in both the homozygous and heterozygous states within families that appear to exhibit incomplete penetrance (38,39,44), making it possible to assess genotype-phenotype correlations. IGD patients homozygous for the PROKR2 mutations R85C, R85H, L173R, or P290S have a severe phenotype consisting of cryptorchidism and anosmia (60) and 3 other homozygous mutations, R268C, Y113H, and V274D, are also associated with the more severe anosmic form of IGD (Kallmann Syndrome [KS]) (40,58,65). In this study, except for R85C, which was benign in 2/3 signaling assays, these variants all display severe LoF when tested alone, consistent with their severe IGD phenotype of KS. Interestingly, of the variants that were LoF across all three signaling pathways when transfected alone, most were completely or partially rescued in co-transfection experiments. This observation offers a potential explanation as to why they are inherited from reproductively normal heterozygous parents and are also seen in heterozygous controls (i.e. their incomplete penetrance). Two of the above variants, L173R and R268C, are also found in the homozygous state in controls (13), suggesting that they may not be sufficient by themselves to cause disease despite their ability to cause LoF in vitro. While previous studies have suggested pathway-specific effects of variants, these results demonstrate more clearly how these effects could translate into subtle phenotypes that might still be seen in heterozygous carriers (56).
Two other variants, A51T and V331M, are seen in the homozygous state in controls yet reported only in the heterozygous state in IGD. These variants tested benign in 2 assays and were rescued by WT in the third assay, suggesting that they are unlikely to cause IGD. Thus, prior reports of their pathogenicity (37,38,41) likely represent false positive associations. Within the IGD patient cohort at MGH, there are 6 probands from varied genetic backgrounds (both European and South Asian) with the A51T mutation (Supplementary Material, Fig. S1). While 4/6 are sporadic in their genetic presentation, in pedigrees where segregation analysis was feasible, 2 probands inherited the A51T mutation from unaffected parents. Importantly, 2 probands harboring the A51T mutation also have mutations in another IGD gene that is likely to be pathogenic [Supplementary Material, Fig. S1, FGFR1 and POLR3B (66)]. Taken together with the results of our functional tests, we conclude that both A51T and V331M are either completely benign variants or have only mild functional defects, and that the cases with these variants lacking other known IGD gene variants should be categorized as “unsolved” in their genetic diagnoses.
Our co-transfection experiments also suggest that some of the functions of PROKR2 may require dimerization and that, in some cases, Mutant/WT dimers can function normally. To this point, four of these tested variants lie within TM4–5, the region of PROKR2 previously shown to be critical for dimerization (52). All of these were IGD Variants and tested LoF in all 3 signaling assays when transfected alone. One of these variants, S188L, was rescued by co-transfection with WT suggesting that it could form functional dimers in contrast to the other 3 variants, L173R, W178S, and G234D that were not rescuable (Supplementary Material, Table S2). Both W178S and G234D also had reduced protein expression when transfected alone (Supplementary Material, Table S2), as has been shown previously (40,55,67), again supporting the notion that some of these variants may cause defects in protein folding and/or intracellular trafficking. Interestingly, two mutations (Q210R and R80C) with normal membrane trafficking were previously shown to have reduced ligand binding or altered recruitment of beta-arrestins (56). Hence, it may be important for future experiments to investigate whether dimerization with WT alters either of these additional functions of mutant PROKR2.
Importantly, co-transfection experiments also uncovered three novel dominant negative mutations in PROKR2. The IGD Variant N325K (found in a patient with KS, Supplementary Material, Fig. S2) was LoF in all three assays with or without WT co-transfection and caused a dose-responsive decrease in WT signaling in the MAPK assay (Supplementary Material, Table S2). Although this variant did not fully segregate within the pedigree (Supplementary Material, Fig. S2), the genetic evidence is in favor of its pathogenicity, as the GNRHR variant (A50V) carried by the proband is likely benign (48). Interestingly, mutant N325K protein was expressed at normal WT levels and had no apparent effect on WT protein expression in co-transfection experiments (Supplementary Material, Table S2). In contrast, two other IGD Variants, G234D and W178S, were both expressed at lower levels than WT and G234D (Fig. 3E) and may cause WT PROKR2 to also be expressed at lower levels (Fig. 3F), offering a potential explanation for its dominant negative actions. More detailed analyses of these mutant/WT interactions are warranted to confirm the mechanism and further investigate the trafficking of the mutant G234D. While G234D was categorized as likely pathogenic, W178S remains of uncertain significance because it currently fails to meet other criteria supporting its pathogenicity. Although our protein expression results replicated a previous finding that suggested a dominant negative effect of R80C (53), our signaling results did not support that finding (Supplementary Material, Table S2). Nonetheless, R80C remains categorized as likely pathogenic, even if the mechanism of its pathogenicity is unclear. These findings highlight the importance of using a composite of biological assays and integrating genetic information when investigating dominant negative mutations.
Similar point mutations in other G-protein Coupled Receptors that have been shown to alter folding and trafficking can sometimes be rescued by chemical chaperones (67,68). The results presented herein suggest that heterozygosity may be sufficient for the WT allele to rescue some mutants in these cases by correcting abnormal trafficking or folding of the mutant protein. However, the data from the protein expression assays suggest that most variants do not cause an obvious trafficking defect. Therefore, there may be other as-yet-unknown mechanisms by which dimerization with WT can rescue signaling. For example, it is possible that forming a functional dimer with WT is necessary for a mutant protein to interact with another co-receptor or surface binding protein, to trans-activate other receptor(s) (69), and/or to bind PROK2 ligand with full affinity (70,71). Future studies are necessary to investigate these mechanisms for variants that lie in domains known to affect either of these aspects of PROKR2 signaling.
While cell-based assays are tractable and useful in interpreting the function of individual mutants, they do have limitations. Intrinsic differences in assay sensitivities, even in a seemingly thorough combined analysis, may generate some false positives and negatives. It is not known which (if any) of the three signaling pathways measured in this study is preferred by endogenous PROKR2 signaling or what cellular functions they perform. Although PROKR2 is involved in the development and migration of GnRH neurons as well as the neuroendocrine regulation of GnRH release (34,42), it is not clear which pathways or cell types expressing PROKR2 are critical for the normal development and function of GnRH neurons. In addition, none of the current methods allow for adequate assessment of neomorphic functions aquired by mutations that may, in turn, produce some of the diverse phenotypes associated with IGD. This last point is especially important, as several variants appeared to increase signaling above and beyond WT levels (Supplementary Material, Table S2). If the expression of PROKR2 is tightly controlled or time-sensitive across development as our current understanding of its biological roles suggest, it is certainly possible that some of these variants could move from a benign categorization to a gain-of-function. In fact, a recent study found a frameshift mutation in PROKR2 that increased WT PROKR2 signaling in a heterozygous context and was associated with precocious, rather than delayed, puberty (72). To assert causality to any variant, future studies will need to assess the effects of altered signaling in a context that reflects the true biology of PROKR2 in vivo and across development. In particular, although these methods are not currently feasible, future studies would benefit greatly from the use of isogenic olfactory, hypothalamic, and/or neural precursor cell lines to assess the endogenous downstream targets of PROKR2 signaling. Alternatively, it may be possible to perform transcriptomics analysis on PROKR2-expressing cells from mouse models to give insights to downstream signaling in vivo.
In summary, modeling of heterozygous PROKR2 variants in vitro reveals pathogenic variants with new mutant-WT interactions and calls into question variants that were previously only tested in a mutant-alone context. We have provided an up-to-date resource (Table 1) for clinicians and genetic counselors to interpret IGD-associated PROKR2 variants that will be built upon as more sequencing and experimental data are available. This study also provides a new and more complete framework for developing methodical, composite (multiple-pathway) functional assay paradigms that, going forward, will aid in the biological interpretation of heterozygous genetic variants associated with other genetic diseases, particularly when combined with bioinformatics approaches.
Materials and Methods
General methods and reagents
HEK293 cells (acquired from ATCC, USA) were chosen based on their lack of endogenous PROKR2 expression and used for all assays except for cAMP signaling assays, which employed the GS-22A1 cell line (73), a HEK-293-derived cell line stably transfected with the luciferase-based cAMP reporter plasmid, pGlosensor-22F (Promega) (74). HEK293 cells were cultured in EMEM with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin, while GS-22A1 cells were cultured in DMEM supplemented with 10% (v/v) FBS, both at 37 °C in a humidified atmosphere with 5% CO2.
WT PROKR2 coding transcript was cloned into a pcDNA3.1/V5HisD TOPO expression vector (Invitrogen, USA) and this V5-tagged WT construct was used to generate mutant PROKR2 vectors using a Quickchange site-directed mutagenesis kit (Agilent) as previously described (38). All mutant constructs were purified and verified by Sanger sequencing performed by the CCIB DNA Core Facility at MGH (Boston, MA). A monster GFP (mGFP) vector (Promega) on a pcDNA backbone was used as an empty vector (EV) control. For confocal imaging, GFP-tagged WT and mutant PROKR2 vectors were created using the mGFP backbone and site-directed mutagenesis. An mCherry tagged WT PROKR2 vector was generously given to us by Dr. Ursula Kaiser. Transfections were performed using Fugene HD transfection reagent (Promega) in Optimem reduced-serum media (Life Technologies) mixed with EMEM following product recommendations, and mGFP was used to ensure transfection efficiency was >80%. Prior to each transfection, vector DNAs were quantified with a nanodrop (Thermoscientific) to ensure correct ratios were added. PROK2 ligand (Bachem) was serially diluted and used to assess PROKR2 signaling.
PROKR2 encodes a G-protein-coupled receptor known to signal through multiple intracellular cascades, including Gαi-, Gαq-, and Gαs-mediated pathways (38,53,56,57). Prior studies have attempted to assess the functional consequences of some of the IGD-associated PROKR2 variants using the three in vitro assays employed here (38,55–57,75). All mutants were first transfected alone and screened independently for an impact on each of the signaling assays (see below for details). If a mutant demonstrated loss-of-function (LoF) in a particular assay, then it was subsequently tested for Mutant/WT interactions in co-transfection experiments in that assay. Several benign variants were initially also tested in co-transfection experiments but, as expected, did not differ from WT (data not shown). To save on costs, dose-response curves were only run for the MAPK (Gαi) signaling pathway, whereas signaling in the IP-One (Gαq) and cAMP (Gαs) assays were tested at the dose that elicited a maximum WT response as determined in preliminary dose-response experiments (see details below). All assays were performed with 3 biological replicates and repeated in 2–3 independent experiments.
Egr1-luciferase reporter (MAPK) assay to assess Gαi signaling
Cells were plated in 24-well plates (Corning) at ∼50% confluence (100 000 cells/well). The next day, WT and mutant PROKR2 vectors were transiently transfected alone or together in three different ratios (1: 1, 3: 1, 1: 3), for a total of 8 ng of PROKR2 vector/well, along with 5 ng Egr1-Luc reporter vector and 187 ng of mGFP vector. The mGFP vector (200 ng) was transfected alone as an EV control. After 24 h, transfected cells were treated with increasing doses of PROK2 ligand. After 16–18 h, cells were lysed in passive lysis buffer (Promega) and 10 µl of lysate was transferred to a 96-well plate (Corning-Costar). Luciferase activity was measured using a dual-luciferase reporter assay system (Promega) and Novostar plate-reader (BMG Labtech) injecting 50 µl of luciferase assay buffer/well. Four-parameter logistical dose-response curves were fitted to each dataset based on the calculated relative response to WT for each assay. Curve fits were analysed by non-linear regression and an EC50 was calculated for each curve. Emax was calculated relative to the maximum WT PROKR2 response.
IP-one assay to assess Gαq signaling
Signaling via the PLC/IP3 pathway was assessed using the IP-One Tb assay (CisBio), which is an antibody-based, homogenous time-resolved fluorescence method for quantifying cellular levels of inositol monophosphate (IP1), a stable intermediate in the PLC/IP3 signaling pathway. PROK2 ligand at a pre-determined concentration eliciting the maximum WT response (30 nM) was diluted in assay buffer [Hank’s Balanced Salt solution containing 10 mM HEPES pH 7.4 and supplemented with 0.1% bovine serum albumin (BSA) and LiCl (20 mM) and added to GS-22A cells in 96-well plates]. The cells were incubated at 37 °C for 1 h. IP1 detection solution [IP1 conjugate and lysis buffer + 2.5% anti-IP1 cryptate-Tb conjugate + 2.5% D-myo-inositol monophosphate (IP1)-d2 conjugate] was then added to the plate (10 µl/well) and the plate was incubated in the dark for 1 h at room temperature. The plate was then read on an EnVision Multilabel Reader (PerkinElmer Life and Analytical Sciences); excitation at 340 nm and measurements of emission at 615 nm and 665 nm. The fluorescence resonance energy transfer ratios (665 nm/615 nm) were converted to IP1 by interpolating values from an IP1 standard curve.
cAMP accumulation assay to assess Gαs signaling
GS-22A cells were plated in 96-well plates (Corning) and, 24 h later, transfected with WT (80 ng), mutant (80 ng), or WT + mutant PROKR2 together (1: 1, 160 ng total). After 24 h, transfected cells were pre-loaded with luciferin in CO2-independent DMEM supplemented with 0.1% bovine serum albumin (BSA) for 20 min at room temperature then treated with PROK2 ligand at a pre-determined concentration eliciting the maximum WT response (300 nM) and placed into a PerkinElmer Envision plate reader for an additional 60 min, during which time the development of cAMP-dependent luminescence was measured at 2-min intervals. The peak luminescence signal, typically occurring 10–15 min after ligand addition, was used for assessment. Curves were fit to the data using a four-parameter sigmoidal dose-response equation.
PROKR2 protein extraction and Western blotting
Cells were plated in 6-well plates (Corning) at ∼50% confluency (600 000 cells/well). The next day, WT and mutant PROKR2 vectors were transiently transfected alone or together in two different ratios (1: 1, 3: 1), for a total of 600 ng PROKR2 vector/well along with 400 ng of mGFP Empty Vector. The mGFP vector (1000 ng) was transfected alone as an EV control. After 24 h, medium was changed to fresh EMEM. After 48 h, cells were washed with PBS and lysed with M-PER Lysis Buffer (Thermoscientific) with protease inhibitors (1: 1000) and protein was quantified using a BCA assay (Thermoscientific).
Total proteins were diluted in 5X Laemmli sample buffer (Boston Bioproducts) with 10% beta-mercaptethanol. 5 µg of protein was loaded into 10% Tris gels (BioRad) along with Precision Plus Protein Kaleidoscope Standard (BioRad) and separated using SDS-PAGE, then transferred overnight to PVDF membranes and immunoblotted to detect V5-tagged PROKR2 protein expression. Supplementary Material, Table S4 gives information about the antibodies used. Blocking buffer was milk for anti-V5 and BSA for anti-β-actin. PROKR2-V5 tagged protein was visualized with HRP-conjugated anti-mouse secondary antibody as a ∼45 KD band using Supersignal West Pico chemiluminescent substrate kit (Thermoscientific), then membranes were stripped using Restore Western Blot Stripping buffer (Thermoscientific) and similarly probed for β-actin (HRP-tagged, also ∼45 KD). All blots were developed on x-ray film (Kodak), films were scanned using an Epson Perfection 3200 photo scanner, and images quantified using ImageJ software (76) for densitometry relative to β-actin intensity.
Confocal imaging
Cells were plated in EMEM in 24-well plates with a 12 mm Poly-L-lysine coated glass coverslip (both from Corning) in each well. After 24 h, cells were transfected with 50 ng of either WT-mCherry -PROKR2, mutant-GFP-PROKR2, or a 1: 1 combination of both. The next day, medium was removed and adherent cells were washed once with PBS, fixed for 10 min at 37 °C in PBS containing 4% paraformaldehyde (Electron Microscopy Sciences) and 5% sucrose, and washed three times in PBS. Coverslips were then mounted onto slides with Vectashield hardset mounting medium with 4, 6-diamidino-2-phenylindole (DAPI; Vector Laboratories). Cells were visualized using a Nikon A1R confocal microscope. Images were taken with a Hamamatsu ORCA CCD camera, deconvolved using Nikon NIS-Elements software, and processed for publication using FIJI analysis software (77).
Statistics
Average MAF and GERP scores were analysed by Kruskal-Wallis tests with Dunn’s multiple-comparisons post-test. Contingencies for GERP scores and ConDel predictions were analysed using Fisher’s Exact tests. For MAPK Assays, in mutant alone experiments activity of each mutant at the EC50 dose was calculated and compared with WT EC50 from the same experiment via one-way ANOVA with Bonferroni’s post-test or student’s t-test with Welch’s correction. Emax for each mutant was calculated relative to WT from the same experiment and compared with WT via Kruskall-Wallis tests with Dunn’s multiple-comparisons post-test or Student’s t-tests with Welch’s correction. In experiments testing WT and mutant interactions, activity of each mutant and 50: 50 mutant/WT combination at the EC50 dose was calculated along with the Emax and compared with the maximum dose of WT (100 WT), the minimum dose of WT (50 WT), and the 75: 25 mutant/WT combination via one-way ANOVA with Dunn's multiple-comparisons post-test. For IP-One and cAMP assays, mutant responses were compared with WT via one-way ANOVA or student's T-test.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
The authors would like to thank Dr. Jim Gusella and Dr. Joel Hirschorn for invaluable input on this manuscript, as well as Jenny Jing, Caroline Copacino, Dr. Richard Bouley, Thomas Dean, Dr. Rona Carroll, and Dr. Ana Paula Abreu for technical assistance in completing this work.
Conflict of Interest statement. None declared.
Funding
Eunice Kennedy Shriver National Institute of Child Health and Development (R01 HD015788), Harvard Reproductive Sciences Center (P50 HD028138). MGH Endocrine Division Training Grant (T32 DK007028) and the Training Program in Reproductive and Developmental Biology (T32 HD007396) to KH Cox, NIH Career Development Award (K23 HD077043) to R.B, MGH Center for Skeletal Research (CSR), which is supported by NIAMS (P30 AR066261), Microscopy Core facility of the MGH Program in Membrane Biology, which receives support from the Boston Area Diabetes and Endocrinology Research Center (DK 57521) and the MGH Center for the Study of Inflammatory Bowel Disease (DK 43351) and whose Nikon A1R confocal microscope was purchased using an NIH Shared Instrumentation Grant S10 RR031563–01.
Supplementary Material
Supplementary Material is available at HMG online.
References
- 1. Samocha K.E., Robinson E.B., Sanders S.J., Stevens C., Sabo A., McGrath L.M., Kosmicki J.A., Rehnstrom K., Mallick S., Kirby A.. et al. (2014) A framework for the interpretation of de novo mutation in human disease. Nat. Genet., 46, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nielsen R., Korneliussen T., Albrechtsen A., Li Y., Wang J., Awadalla P. (2012) SNP calling, genotype calling, and sample allele frequency estimation from New-Generation Sequencing data. PLoS One, 7, e37558.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nielsen R., Paul J.S., Albrechtsen A., Song Y.S. (2011) Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet., 12, 443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fischer M., Snajder R., Pabinger S., Dander A., Schossig A., Zschocke J., Trajanoski Z., Stocker G., Raghava G.P.S. (2012) SIMPLEX: cloud-enabled pipeline for the comprehensive analysis of exome sequencing data. PLoS One, 7, e41948.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M.. et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet., 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pabinger S., Dander A., Fischer M., Snajder R., Sperk M., Efremova M., Krabichler B., Speicher M.R., Zschocke J., Trajanoski Z. (2014) A survey of tools for variant analysis of next-generation genome sequencing data. Brief Bioinform., 15, 256–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Genome Project Data Processing S. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang K., Li M., Hakonarson H. (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res., 38, e164.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ware J.S., Samocha K.E., Homsy J., Daly M.J. (2015) Interpreting de novo Variation in Human Disease Using denovolyzeR. Curr. Protoc.Hum. Genet., 87, 7 25 21-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bamshad M.J., Shendure J.A., Valle D., Hamosh A., Lupski J.R., Gibbs R.A., Boerwinkle E., Lifton R.P., Gerstein M., Gunel M., Mane S., Nickerson D.A. (2012) The Centers for Mendelian Genomics: a new large-scale initiative to identify the genes underlying rare Mendelian conditions. Am. J. Med. Genet. A, 158A, 1523–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Genomes Project C., Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A. (2012) An integrated map of genetic variation from 1, 092 human genomes. Nature, 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Amberger J., Bocchini C.A., Scott A.F., Hamosh A. (2009) McKusick's Online Mendelian Inheritance in Man (OMIM). Nucleic Acids Res., 37, D793–D796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B.. et al. (2016) Analysis of protein-coding genetic variation in 60, 706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Daneshjou R., Zappala Z., Kukurba K., Boyle S.M., Ormond K.E., Klein T.E., Snyder M., Bustamante C.D., Altman R.B., Montgomery S.B. (2014) PATH-SCAN: a reporting tool for identifying clinically actionable variants. Pac. Symp. Biocomput., 229–240. [PMC free article] [PubMed] [Google Scholar]
- 15. Kircher M., Witten D.M., Jain P., O'Roak B.J., Cooper G.M., Shendure J. (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet., 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. MacArthur D.G., Manolio T.A., Dimmock D.P., Rehm H.L., Shendure J., Abecasis G.R., Adams D.R., Altman R.B., Antonarakis S.E., Ashley E.A.. et al. (2014) Guidelines for investigating causality of sequence variants in human disease. Nature, 508, 469–476. 10.1038/nature13127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E.. et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med., 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lindblom A., Robinson P.N. (2011) Bioinformatics for human genetics: promises and challenges. Hum. Mutat., 32, 495–500. [DOI] [PubMed] [Google Scholar]
- 19. Cooper G.M., Shendure J. (2011) Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat. Rev. Genet., 12, 628–640. [DOI] [PubMed] [Google Scholar]
- 20. Grimm D.G., Azencott C.A., Aicheler F., Gieraths U., MacArthur D.G., Samocha K.E., Cooper D.N., Stenson P.D., Daly M.J., Smoller J.W.. et al. (2015) The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum. Mutat., 36, 513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Minikel E.V., Vallabh S.M., Lek M., Estrada K., Samocha K.E., Sathirapongsasuti J.F., McLean C.Y., Tung J.Y., Yu L.P., Gambetti P.. et al. (2016) Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med., 8, 322ra329.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Akinrinade O., Koskenvuo J.W., Alastalo T.P. (2015) Prevalence of Titin Truncating Variants in General Population. PLoS One, 10, e0145284.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Majithia A.R., Flannick J., Shahinian P., Guo M., Bray M.-A., Fontanillas P., Gabriel S.B., Rosen E.D., Altshuler D., Flannick J.. et al. (2014) Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc. Natl. Acad. Sci. U S A, 111, 13127–13132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gonzalez-Perez A., Lopez-Bigas N. (2011) Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet., 88, 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kumar P., Henikoff S., Ng P.C. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc., 4, 1073–1081. [DOI] [PubMed] [Google Scholar]
- 27. Schwarz J.M., Rodelsperger C., Schuelke M., Seelow D. (2010) MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods, 7, 575–576. [DOI] [PubMed] [Google Scholar]
- 28. Thusberg J., Olatubosun A., Vihinen M. (2011) Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat., 32, 358–368. [DOI] [PubMed] [Google Scholar]
- 29. Stitziel N.O., Kiezun A., Sunyaev S. (2011) Computational and statistical approaches to analyzing variants identified by exome sequencing. Genome Biol., 12, 227.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hicks S., Wheeler D.A., Plon S.E., Kimmel M. (2011) Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum. Mutat., 32, 661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Balasubramanian R., Crowley W.F. Jr (2007) Isolated Gonadotropin-Releasing Hormone (GnRH) Deficiency, Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J.H., Bird T.D., Ledbetter N., Mefford H.C., Smith R.J.H., Stephens K. (eds.), Gene Reviews, Seattle (WA): University of Washington, Seattle; 1993-2017. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1334/. [Google Scholar]
- 32. Balasubramanian R., Crowley W.F. Jr. (2011) Isolated GnRH deficiency: a disease model serving as a unique prism into the systems biology of the GnRH neuronal network. Mol. Cell. Endocrinol., 346, 4–12. 10.1016/j.mce.2011.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Balasubramanian R., Dwyer A., Seminara S.B., Pitteloud N., Kaiser U.B., Crowley W.F. Jr. (2010) Human GnRH deficiency: a unique disease model to unravel the ontogeny of GnRH neurons. Neuroendocrinol., 92, 81–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stamou M.I., Cox K.H., Crowley W.F. Jr. (2016) Discovering Genes essential to the hypothalamic regulation of human reproduction using a human disease model: adjusting to life in the “-Omics” era. Endocr. Rev., 2016, 4–22. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35. Sykiotis G.P., Pitteloud N., Seminara S.B., Kaiser U.B., Crowley W.F. Jr. (2010) Deciphering genetic disease in the genomic era: the model of GnRH deficiency. Sci. Transl. Med., 2, 32rv32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matsumoto S., Yamazaki C., Masumoto K.H., Nagano M., Naito M., Soga T., Hiyama H., Matsumoto M., Takasaki J., Kamohara M.. et al. (2006) Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc. Natl. Acad. Sci. U S A, 103, 4140–4145. 10.1073/pnas.0508881103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dode C., Teixeira L., Levilliers J., Fouveaut C., Bouchard P., Kottler M.L., Lespinasse J., Lienhardt-Roussie A., Mathieu M., Moerman A.. et al. (2006) Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet., 2, e175.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cole L.W., Sidis Y., Zhang C., Quinton R., Plummer L., Pignatelli D., Hughes V.A., Dwyer A.A., Raivio T., Hayes F.J.. et al. (2008) Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin-releasing hormone deficiency: molecular genetics and clinical spectrum. J. Clin. Endocrinol. Metab., 93, 3551–3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Abreu A.P., Trarbach E.B., de Castro M., Frade Costa E.M., Versiani B., Matias Baptista M.T., Garmes H.M., Mendonca B.B., Latronico A.C. (2008) Loss-of-function mutations in the genes encoding prokineticin-2 or prokineticin receptor-2 cause autosomal recessive Kallmann syndrome. J. Clin. Endocrinol. Metab., 93, 4113–4118. [DOI] [PubMed] [Google Scholar]
- 40. Tommiska J., Toppari J., Vaaralahti K., Kansakoski J., Laitinen E.M., Noisa P., Kinnala A., Niinikoski H., Raivio T. (2013) PROKR2 mutations in autosomal recessive Kallmann syndrome. Fertil. Steril., 99, 815–818. [DOI] [PubMed] [Google Scholar]
- 41. Sarfati J., Guiochon-Mantel A., Rondard P., Arnulf I., Garcia-Pinero A., Wolczynski S., Brailly-Tabard S., Bidet M., Ramos-Arroyo M., Mathieu M.. et al. (2010) A comparative phenotypic study of kallmann syndrome patients carrying monoallelic and biallelic mutations in the prokineticin 2 or prokineticin receptor 2 genes. J. Clin. Endocrinol. Metab., 95, 659–669. [DOI] [PubMed] [Google Scholar]
- 42. Balasubramanian R., Plummer L., Sidis Y., Pitteloud N., Martin C., Zhou Q.Y., Crowley W.F. Jr. (2011) The puzzles of the prokineticin 2 pathway in human reproduction. Mol. Cell. Endocrinol., 346, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martin C., Balasubramanian R., Dwyer A.A., Au M.G., Sidis Y., Kaiser U.B., Seminara S.B., Pitteloud N., Zhou Q.Y., Crowley W.F. Jr. (2011) The role of the prokineticin 2 pathway in human reproduction: evidence from the study of human and murine gene mutations. Endocr. Rev., 32, 225–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Avbelj Stefanija M., Jeanpierre M., Sykiotis G.P., Young J., Quinton R., Abreu A.P., Plummer L., Au M.G., Balasubramanian R., Dwyer A.A.. et al. (2012) An ancient founder mutation in PROKR2 impairs human reproduction. Hum. Mol. Genet., 21, 4314–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Prosser H.M., Bradley A., Caldwell M.A. (2007) Olfactory bulb hypoplasia in Prokr2 null mice stems from defective neuronal progenitor migration and differentiation. Eur. J. Neurosci., 26, 3339–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sykiotis G.P., Plummer L., Hughes V.A., Au M., Durrani S., Nayak-Young S., Dwyer A.A., Quinton R., Hall J.E., Gusella J.F.. et al. (2010) Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc. Natl. Acad. Sci. U S A, 107, 15140–15144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Canto P., Munguia P., Soderlund D., Castro J.J., Mendez J.P. (2009) Genetic analysis in patients with Kallmann syndrome: coexistence of mutations in prokineticin receptor 2 and KAL1. J. Androl., 30, 41–45. [DOI] [PubMed] [Google Scholar]
- 48. Gianetti E., Hall J.E., Au M.G., Kaiser U.B., Quinton R., Stewart J.A., Metzger D.L., Pitteloud N., Mericq V., Merino P.M.. et al. (2012) When genetic load does not correlate with phenotypic spectrum: lessons from the GnRH receptor (GNRHR). J. Clin. Endocrinol. Metab., 97, E1798–E1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Karczewski K.J., Weisburd B., Thomas B., Solomonson M., Ruderfer D.M., Kavanagh D., Hamamsy T., Lek M., Samocha K.E., Cummings B.B.. et al. (2017) The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res., 45, D840–D845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Choi J.-H., Balasubramanian R., Lee P.H., Shaw N.D., Hall J.E., Plummer L., Buck C.L., Kottler M.-L., Jarzabek K., Wołczynski S.. et al. (2015) Expanding the spectrum of founder mutations causing isolated gonadotropin-releasing hormone deficiency. J. Clin. Endocrinol. Metab., 100, E1378–E1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Marsango S., Bonaccorsi di Patti M.C., Barra D., Miele R. (2011) Evidence that prokineticin receptor 2 exists as a dimer in vivo. Cell. Mol. Life Sci., 68, 2919–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sposini S., Caltabiano G., Hanyaloglu A.C., Miele R. (2015) Identification of transmembrane domains that regulate spatial arrangements and activity of prokineticin receptor 2 dimers. Mol. Cell. Endocrinol., 399, 362–372. [DOI] [PubMed] [Google Scholar]
- 53. Abreu A.P., Noel S.D., Xu S., Carroll R.S., Latronico A.C., Kaiser U.B. (2012) Evidence of the importance of the first intracellular loop of prokineticin receptor 2 in receptor function. Mol. Endocrinol, 26, 1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lin D.C., Bullock C.M., Ehlert F.J., Chen J.L., Tian H., Zhou Q.Y. (2002) Identification and molecular characterization of two closely related G protein-coupled receptors activated by prokineticins/endocrine gland vascular endothelial growth factor. J. Biol. Chem., 277, 19276–19280. [DOI] [PubMed] [Google Scholar]
- 55. Monnier C., Dode C., Fabre L., Teixeira L., Labesse G., Pin J.P., Hardelin J.P., Rondard P. (2009) PROKR2 missense mutations associated with Kallmann syndrome impair receptor signalling activity. Hum. Mol. Genet., 18, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sbai O., Monnier C., Dode C., Pin J.P., Hardelin J.P., Rondard P. (2014) Biased signaling through G-protein-coupled PROKR2 receptors harboring missense mutations. faseb J., 28, 3734–3744. [DOI] [PubMed] [Google Scholar]
- 57. Libri D.V., Kleinau G., Vezzoli V., Busnelli M., Guizzardi F., Sinisi A.A., Pincelli A.I., Mancini A., Russo G., Beck-Peccoz P.. et al. (2014) Germline prokineticin receptor 2 (PROKR2) variants associated with central hypogonadism cause differental modulation of distinct intracellular pathways. J. Clin. Endocrinol. Metab., 99, E458–E463. [DOI] [PubMed] [Google Scholar]
- 58. Sinisi A.A., Asci R., Bellastella G., Maione L., Esposito D., Elefante A., De Bellis A., Bellastella A., Iolascon A. (2008) Homozygous mutation in the prokineticin-receptor2 gene (Val274Asp) presenting as reversible Kallmann syndrome and persistent oligozoospermia: case report. Hum. Reprod., 23, 2380–2384. [DOI] [PubMed] [Google Scholar]
- 59. Chan Y.M., de Guillebon A., Lang-Muritano M., Plummer L., Cerrato F., Tsiaras S., Gaspert A., Lavoie H.B., Wu C.H., Crowley W.F. Jr. et al. (2009) GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. U S A, 106, 11703–11708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sarfati J., Fouveaut C., Leroy C., Jeanpierre M., Hardelin J.P., Dode C. (2013) Greater prevalence of PROKR2 mutations in Kallmann syndrome patients from the Maghreb than in European patients. Eur. J. Endocrinol., 169, 805–809. [DOI] [PubMed] [Google Scholar]
- 61. Davydov E.V., Goode D.L., Sirota M., Cooper G.M., Sidow A., Batzoglou S., Wasserman W.W. (2010) Identifying a high fraction of the human genome to be under selective constraint using GERP ++. PLoS Comput. Biol., 6, e1001025.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stone E.A., Sidow A. (2005) Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res., 15, 978–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Clifford R.J., Edmonson M.N., Nguyen C., Buetow K.H. (2004) Large-scale analysis of non-synonymous coding region single nucleotide polymorphisms. Bioinformatics, 20, 1006–1014. [DOI] [PubMed] [Google Scholar]
- 64. Finn R.D., Coggill P., Eberhardt R.Y., Eddy S.R., Mistry J., Mitchell A.L., Potter S.C., Punta M., Qureshi M., Sangrador-Vegas A.. et al. (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res., 44, D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gu W.J., Zhang Q., Wang Y.Q., Yang G.Q., Hong T.P., Zhu D.L., Yang J.K., Ning G., Jin N., Chen K.. et al. (2015) Mutation analyses in pedigrees and sporadic cases of ethnic Han Chinese Kallmann syndrome patients. Exp. Biol. Med. (Maywood), 240, 1480–1489. 10.1177/1535370215587531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Richards M.R., Plummer L., Chan Y.M., Lippincott M.F., Quinton R., Kumanov P., Seminara S.B. (2017) Phenotypic spectrum of POLR3B mutations: isolated hypogonadotropic hypogonadism without neurological or dental anomalies. J. Med. Genet., 54, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen D.N., Ma Y.T., Liu H., Zhou Q.Y., Li J.D. (2014) Functional rescue of Kallmann syndrome-associated prokineticin receptor 2 (PKR2) mutants deficient in trafficking. J Biol. Chem., 289, 15518–15526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tao Y.X., Conn P.M. (2014) Chaperoning G protein-coupled receptors: from cell biology to therapeutics. Endocr. Rev., 35, 602–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ji I., Lee C., Song Y., Conn P.M., Ji T.H. (2002) Cis- and trans-activation of hormone receptors: the LH receptor. Mol. Endocrinol., 16, 1299–1308. [DOI] [PubMed] [Google Scholar]
- 70. Song J., Li J., Liu H.D., Liu W., Feng Y., Zhou X.T., Li J.D. (2016) Snapin interacts with G-protein-coupled receptor PKR2. Biochem. Biophys. Res. Commun., 469, 501–506. [DOI] [PubMed] [Google Scholar]
- 71. Yin W., Liu H., Peng Z., Chen D., Li J., Li J.D. (2014) Mechanisms that underlie the internalization and extracellular signal regulated kinase 1/2 activation by PKR2 receptor. Cell Signal, 26, 1118–1124. 10.1016/j.cellsig.2014.01.031 [DOI] [PubMed] [Google Scholar]
- 72. Fukami M., Suzuki E., Izumi Y., Torii T., Narumi S., Igarashi M., Miyado M., Katsumi M., Fujisawa Y., Nakabayashi K.. et al. (2017) Paradoxical gain-of-function mutant of the G-protein-coupled receptor PROKR2 promotes early puberty. J. Cell. Mol. Med., 21, 2623–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Carter P.H., Dean T., Bhayana B., Khatri A., Rajur R., Gardella T.J. (2015) Actions of the small molecule ligands SW106 and AH-3960 on the type-1 parathyroid hormone receptor. Mol. Endocrinol., 29, 307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Binkowski B.F., Butler B.L., Stecha P.F., Eggers C.T., Otto P., Zimmerman K., Vidugiris G., Wood M.G., Encell L.P., Fan F.. et al. (2011) A luminescent biosensor with increased dynamic range for intracellular cAMP. ACS Chem. Biol., 6, 1193–1197. [DOI] [PubMed] [Google Scholar]
- 75. Peng Z., Tang Y., Luo H., Jiang F., Yang J., Sun L., Li J.D. (2011) Disease-causing mutation in PKR2 receptor reveals a critical role of positive charges in the second intracellular loop for G-protein coupling and receptor trafficking. J. Biol. Chem, 286, 16615–16622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schneider C.A., Rasband W.S., Eliceiri K.W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B.. et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods., 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.