

Abstract
ABCA4 is a member of the superfamily of ATP-binding cassette (ABC) proteins that transports N-retinylidene-phosphatidylethanolamine (N-Ret-PE) across outer segment disc membranes thereby facilitating the removal of potentially toxic retinoid compounds from photoreceptor cells. Mutations in the gene encoding ABCA4 are responsible for Stargardt disease (STGD1), an autosomal recessive retinal degenerative disease that causes severe vision loss. To define the molecular basis for STGD1 associated with the p.Asn965Ser (N965S) mutation in the Walker A motif of nucleotide binding domain 1 (NBD1), we generated a p.Asn965Ser knockin mouse and compared the subcellular localization and molecular properties of the disease variant with wild-type (WT) ABCA4. Here, we show that the p.Asn965Ser ABCA4 variant expresses at half the level of WT ABCA4, partially mislocalizes to the endoplasmic reticulum (ER) of photoreceptors, is devoid of N-Ret-PE activated ATPase activity, and causes an increase in autofluorescence and the bisretinoid A2E associated with lipofuscin deposits in retinal pigment epithelial cells as found in Stargardt patients and Abca4 knockout mice. We also show for the first time that a significant fraction of WT ABCA4 is retained in the inner segment of photoreceptors. On the basis of these studies we conclude that loss in substrate-dependent ATPase activity and protein misfolding are mechanisms underlying STGD1 associated with the p.Asn965Ser mutation in ABCA4. Functional and molecular modeling studies further suggest that similar pathogenic mechanisms are responsible for Tangiers disease associated with the p.Asn935Ser (N935S) mutation in the NBD1 Walker A motif of ABCA1.
Introduction
Stargardt disease (STGD1: MIM 248200) is an autosomal recessive retinal degenerative disease caused by mutations in the gene encoding the ATP-binding cassette (ABC) transporter ABCA4, also known as ABCR or the Rim protein (1–4). Affected individuals typically show bilateral loss in central vision, atrophy of the macula, defects in color vision, delayed dark adaptation, and accumulation of yellow-white flecks at the level of the retinal pigment epithelium (RPE) (5–8). The age of onset and disease severity varies widely, but in most cases, STGD1 patients experience a significant reduction in visual acuity in their first or second decade of life and progressive loss in vision throughout life with visual acuity reaching 20/200 or greater in the advanced stages of the disease (7,9). Mutations in ABCA4 also cause the related retinopathies, cone-rod dystrophy and a subset of retinitis pigmentosa (10–12). Over 1000 mutations in the ABCA4 gene are known to cause ABCA4-associated diseases (13–15). These include missense and nonsense mutations, frameshifts, truncations, small deletions, and splicing mutations with the majority of the mutations being missense mutations that cause single amino acid substitutions at residues at sites throughout the protein.
ABCA4 is highly expressed in rod and cone photoreceptor cells where it localizes to the rim region of outer segment disc membranes (4,16–18). ABCA4 functions as a retinoid transporter flipping its substrate N-retinylidene-phosphatidylethanolamine (N-Ret-PE), the Schiff-base adduct of retinal and phosphatidylethanolamine (PE), from the lumen to the cytoplasmic side of outer segment disc membranes (19,20). This facilitates the clearance of all-trans and 11-cis retinal from photoreceptors via the visual cycle thereby preventing the accumulation of potentially toxic retinoid compounds in photoreceptors and retinal pigment epithelial (RPE) cells following phagocytosis of photoreceptor outer segments (21–25).
Several studies have examined the effect of various missense mutations and deletions on the expression and functional properties of ABCA4 expressed and purified from culture cells (20,26–28). Most mutations were found to cause a reduction in the functional activity and in some instances mislocalization of ABCA4 in cells (27,29,30). More recently, the effect of two disease-causing missense mutations in ABCA4 has been reported in a knockin mouse model for STGD1 (28). For these studies the wild-type (WT) Abca4 allele was replaced with a complex Abca4 allele encoding the disease associated variants p.Leu541Pro/p.Ala1038Val frequently found in the German STGD1 patient population. In mice homozygous for this double mutation, the ABCA4 variant expressed at only trace amounts. The phenotype of these mice was essentially identical to that of Abca4 knockout mice (23) leaving one to question whether disease-linked missense mutations are more deleterious than ABCA4 null mutations in STGD1 patients (31).
ABCA4, a single polypeptide consisting of 2273 amino acids, is organized into two non-identical tandem halves with each half containing three core domains – nucleotide binding domain (NBD), exocytoplasmic domain (ECD) and transmembrane domain (TMD) (32). The p.Asn965Ser (N965S) variant in the first nucleotide binding domain (NBD1) of ABCA4 is the most common STGD1 mutation found in the Danish population and is frequent in STGD1 patients of Chinese descent (33,34). Patients homozygous for this variant experience a reduction in visual acuity in their second decade of life, progressive deterioration of vision throughout life, peripheral dystrophy, color vision defects, delayed dark adaptation, and reduced ERG amplitudes (33). To define the molecular basis for STGD1 associated with the p.Asn965Ser mutation, we generated a p.Asn965Ser knockin mouse and compared the expression, localization, and functional properties of this disease variant with WT ABCA4. Here, we show that the p.Asn965Ser ABCA4 variant expresses, but at a lower level than WT ABCA4, partially mislocalizes to the ER of photoreceptors, lacks N-Ret-PE activated ATPase activity, and exhibits an increase in autofluorescence and A2E characteristic of lipofuscin accumulation in RPE cells. On the basis of the high degree of similarity in the structure of ABCA4 and ABCA1, our results on the p.Asn965Ser mutation in ABCA4 are relevant to understanding the molecular basis for Tangiers disease associated with the p.Asn935Ser mutation in the Walker A motif of ABCA1. Finally, we also show for the first time that a significant fraction of WT ABCA4 is retained in the inner segment of photoreceptors.
Results
ABCA4 is expressed at reduced levels in photoreceptors of p.Asn965Ser mutant mice
To determine the level of expression of the p.Asn965Ser ABCA4 variant, retinal membranes and enriched photoreceptor outer segment preparations were isolated from the retina of 2-month-old WT and p.Asn965Ser STGD1 mice. Equal amounts of protein were resolved on SDS polyacrylamide gels, and western blots were labeled with monoclonal antibodies to ABCA4 and the cyclic nucleotide-gated channel A1 subunit (CNGA1) as a control. As shown in Figure 1A and quantified in Figure 1B, an equal amount of the 63 kDa CNGA1 was detected in photoreceptor outer segments (POS) from WT and p.Asn965Ser mouse retina whereas the amount of ABCA4 in POS of p.Asn965Ser mice was half that of WT. A similar reduction in ABCA4 was observed in retinal membranes containing inner segments as revealed by Na/K ATPase labeling. These results indicate that the p.Asn965Ser ABCA4 variant is expressed in retinal photoreceptors, but at half the level observed for WT ABCA4.
Figure 1.

Quantification of ABCA4 in WT and p.Asn965Ser photoreceptor outer segments and retina membranes by Western blotting. Enriched photoreceptor outer segments (POS) and retinal membranes (Retina) were prepared from WT and p.Asn965Ser ABCA4 mice. (A) Equal amounts of protein from POS (20 µg) and Retina (50 µg) preparations isolated from WT and homozygous p.Asn965Ser (N965S) mice were applied to the lanes of a SDS polyacrylamide gel and resulting western blots were labeled with the Rim3F4 antibody to ABCA4, the PMc1D1 antibody to the CNGA1 channel, and an antibody to the Na/K ATPase (α subunit) as a marker for inner segments. (B) Quantification of ABCA4 and CNGA1 in POS and Retina from p.Asn965Ser and WT mice. The amount of CNGA1 in the p.Asn965Ser preparations was identical to the amount in WT preparations, whereas quantity of p.Asn965Ser (N965S) ABCA4 was about half that of WT ABCA4 in both POS and Retina preparations. Data are presented as the mean ± SD for three independent preparations.
WT and p.Asn965Ser ABCA4 are localized in photoreceptor outer and inner segments
In previous studies ABCA4 was reported to localize in rod and cone outer segments and more specifically to the rim region of discs by immunofluorescence and immunoelectron microscopy (4,16–18). Figure 2A and D confirms the preferential localization of ABCA4 to the photoreceptor outer segment layer of WT mouse retina by immunofluorescence microscopy using monoclonal antibodies (Rim 3F4 and Rim 5B4) to ABCA4. p.Asn965Ser ABCA4 also preferentially localized to the photoreceptor outer segment layer of mice homozygous for this mutation (Fig. 2B and E). In control experiments, neither the Rim 3F4 nor the Rim 5B4 antibodies labeled photoreceptors of the Abca4 knockout mouse further confirming the specificity of these antibodies (Fig. 2C and F).
Figure 2.

Immunofluorescence micrographs of ABCA4, peripherin-2, and KDEL ER proteins in WT, homozygous p.Asn965Ser (N965S) and homozygous ABCA4 KO mouse photoreceptors. Retinal cryosections were stained with monoclonal antibodies to ABCA4 (Rim 3F4 and Rim 5B4), monoclonal antibody to peripherin-2 (Per-5H2), and monoclonal antibody to KDEL ER retention sequence followed by a secondary fluorescent-labeled goat anti-mouse antibody (red) and counterstained with DAPI nuclear stain (blue). Right side of each panel is a line scan showing the relative fluorescence intensity profile across the inner and outer segments arising from antibody labeling. No staining of ABCA4 was observed for the Abca4 KO retina as expected. OS, outer segment; IS, inner segment; ONL, outer nuclear layer. Bar = 20 µm.
Closer inspection of photoreceptors labeled with the ABCA4 monoclonal antibodies showed detectable immunofluorescence staining in the inner segment layers of both the WT and homozygous p.Asn965Ser mice (Fig. 2A, B, D and E). This is most evident in the line scans that traverse the inner and outer segment layers in the right panel of these figures. In WT mice, a gradual increase in fluorescence intensity was observed from the proximal part of the inner segment [adjacent to the outer nuclear layer (ONL)] to the distal end near the junction with the connecting cilium with little labeling above the ONL. This inner segment staining profile was not observed for other outer segment proteins such as peripherin-2 (Fig. 2G), cyclic nucleotide-gated channel subunit A1 (CNGA1), guanylate cyclase-1 (RetGC1), and glutamic acid rich proteins (GARP) (Fig. 3). Instead, very low level of continuous staining was observed for these proteins within the inner segment of WT and p.Asn965Ser mice as exemplified in the line scan for peripherin-2 in Figure 2G.
Figure 3.

Immunofluorescence microscopy of outer segment proteins in WT and homozygous p.Asn965Ser (N965S) mouse photoreceptors. Retinal cryosections were stained with antibodies to the CNGA1 channel (PMc1D1), RetGC1 (GC-8A5), GARP (Garp 4B1), rhodopsin (Rho 1D4), red/green cones (R/G opsin), and GFAP and counterstained with DAPI (blue). Similar labeling was observed for these proteins in WT and p.Asn965Ser retina. Bar = 20 µm.
The photoreceptor inner segment staining of p.Asn965Ser ABCA4 was more intense than that observed for WT ABCA4, and interestingly, displayed a different fluorescent intensity profile. As shown in the line scan of Figure 2B and E, a distinct peak of p.Asn965Ser ABCA4 staining intensity was evident just above the ONL layer within the inner segment layer. To determine if this peak intensity coincides with the location of the endoplasmic reticulum (ER) in the inner segment of photoreceptors, we stained retinal sections with a monoclonal antibody against KDEL, an established ER retention signal. As shown in Figure 2J–L, a peak of KDEL immunofluorescence staining was evident just above the ONL layer similar to that observed for ABCA4 staining in the p.Asn965Ser mouse. This suggests that a significant fraction of p.Asn965Ser ABCA4 is retained in the ER of photoreceptor cells. This was further confirmed in a double labeling experiment in which a retinal section of a p.Asn965Ser mouse was labeled with the Rim 5B4 monoclonal antibody to ABCA4 and a polyclonal antibody to KDEL for immunofluorescence microscopy (Supplementary Material, Fig. S1). Co-staining of these antibodies was observed just above the ONL layer in the merged image supporting the localization of a fraction of p.Asn965Ser ABCA4 in the ER.
The localization of other photoreceptor proteins in the retina of the p.Asn965Ser mouse was also investigated by immunofluorescence microscopy to determine if this mutation affected the trafficking of these proteins in photoreceptors. Figures 2H and 3 show that peripherin-2, CNGA1, RetGC1, rhodopsin and GARP primarily localized to the outer segments in the retina of the p.Asn965Ser mouse similar to that for the WT mouse. Likewise, the distribution of M/L cone opsin in cone photoreceptors of the p.Asn965Ser mouse was similar to the distribution in WT mice indicating that the trafficking of M/L cone opsin to the outer segment was not affected by the p.Asn965Ser mutation in ABCA4 (Fig. 3). We also labeled retina sections with an antibody to glial fibrillary acidic protein (GFAP) to determine if the p.Asn965Ser ABCA4 variant induced stress in Mueller cells. Figure 3 shows that the labeling pattern of the p.Asn965Ser retina is similar to that of WT retina, suggesting that retinal cells in the p.Asn965Ser mouse do not exhibit significant stress. Finally, analysis of the outer nuclear layer (ONL) thickness of the p.Asn965Ser mice indicated that no significant photoreceptor degeneration occurred over 2 months, a finding that is consistent with the Abca4 KO mouse and the p.Leu541Pro/p.Ala1038Val knockin mouse (23,28).
Quantitative analysis of ABCA4 and other outer segment proteins in photoreceptors of WT and homozygous p.Asn965Ser mice
To better define the relative amount of ABCA4 present in the inner segment compared with the outer segment, we quantified the pixel densities in a defined area of the inner and outer segment layers using Image J software. The percent fluorescence staining intensity of the inner segment relative to the total staining (inner and outer segment staining) is summarized in Figure 4 for ABCA4 and other outer segment proteins in photoreceptors of WT and p.Asn965Ser mice. In WT photoreceptors, ABCA4 inner segment staining was 13% for the C-terminal antibody Rim 3F4 and 19.5% for the internal antibody Rim 5B4. In comparison, CNGA1, GARP, peripherin-2 and RetGC1 exhibited very low inner segment staining of WT photoreceptors in the range of 3–6%. Rhodopsin staining was somewhat higher at ∼8%. In the p.Asn965Ser mouse, inner segment staining of the ABCA4 variant was markedly higher reaching a mean of 31% for Rim 3F4 and 33% for Rim5B4 antibody (Fig. 4). No significant change in intensity for CNGA1, GARP, peripherin-2, RetGC1 or rhodopsin was observed in the photoreceptor inner segment layer of the p.Asn965Ser mouse relative to WT mice.
Figure 4.
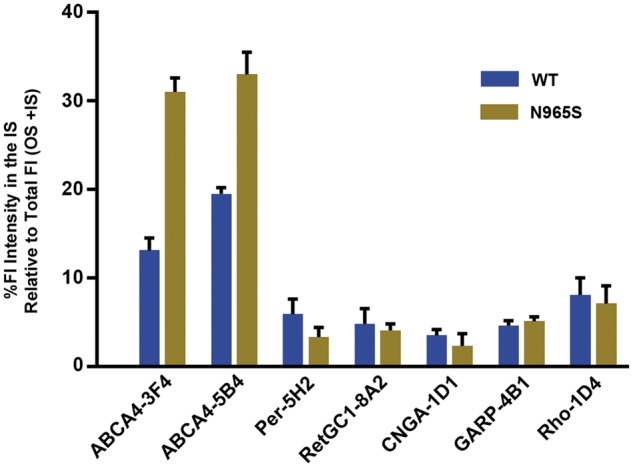
Percent immunofluorescence staining intensity of ABCA4 and other outer segment proteins in the inner segment (IS). Data obtained from immunofluorescence microscopy of WT and p.Asn965Ser (N965S) cryosections as per Figures 2 and 3. Ten squares (6 × 6 µm) were selected in each the inner and outer segment layers and the mean fluorescence pixel intensities were obtained using Image J software. The data are expressed as the % unit area of fluorescence (Fl) intensity in the IS relative to the total intensity (IS + OS). The data provide a quantitative assessment of the staining of antibodies in the inner vs. the outer segment. ABCA4 shows significant staining in the IS for WT and p.Asn965Ser (N965S) photoreceptors with increased staining in the IS for the p.Asn965Ser photoreceptors relative to WT photoreceptors. Data from two sets of mice were similar.
Localization of WT and p.Asn965Ser ABCA4 in transfected culture cells
Transiently transfected culture cells have been used to examine the effect of disease-causing mutations on the cellular localization of ABCA4 (27,28,30). The distribution of ABCA4 in these cells has not been compared with the distribution in photoreceptors. We have examined the subcellular distribution of WT ABCA4 and the p.Asn965Ser mutant in transfected COS7 cells by immunofluorescence microscopy. Figure 5 shows that WT ABCA4 preferentially co-localized with calnexin in large intracellular vesicle-like structures as previously reported (27). The p.Asn965Ser mutant also localized to vesicle-like structures, but a significant fraction also distributed in a reticular pattern characteristic of ER. The reticular distribution was similar to that observed for the highly misfolded p.Thr983Ala ABCA4 STGD1 mutant which was devoid of vesicular structures (Fig. 5). These studies indicate that the p.Asn965Ser mutation has a similar effect on ABCA4 localization in both culture cells and photoreceptors with a substantial fraction of the mutant protein being retained in the ER as misfolded protein.
Figure 5.
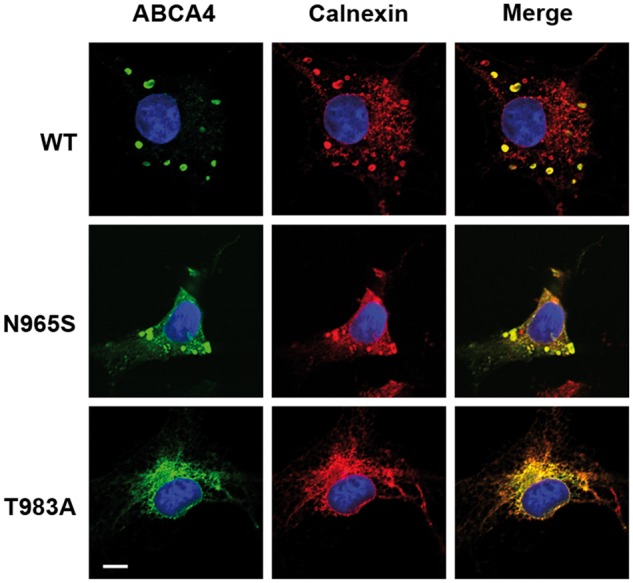
Localization of WT ABCA4 and disease-associated mutants expressed in culture cells. WT ABCA4 and p.Asn965Ser (N965S) and p.Thr983Ala (T983A) STGD1 ABCA4 mutants were expressed in COS7 cells and double labeled with the ABCA4 monoclonal antibody Rim3F4 (green) and polyclonal antibody to calnexin (red) as an ER marker. WT ABCA4 primarily localizes in large calnexin-containing intracellular vesicle-like structures (Merge image). The p.Thr983Ala mutant localizes in a reticular pattern characteristic of ER. The p.Asn965Ser mutant is found in both with the vesicular structures and in a reticular pattern characteristic of ER. Bar = 10 µm.
Functional characterization of the p.Asn965Ser ABCA4 variant
Previous studies have shown that the basal ATPase activity of immunoaffinity purified ABCA4 reconstituted into phosphatidylethanolamine (PE)-containing liposomes is stimulated by all-trans and 11-cis retinal (24,26,35). To determine the effect of the p.Asn965Ser mutation on the functional activity of ABCA4, we purified ABCA4 from WT and p.Asn965Ser mice by immunoaffinity chromatography on a Rim 3F4 antibody matrix. Figure 6A shows that the one-step immunoaffinity procedure effectively purifies ABCA4 and the p.Asn965Ser variant from detergent-solubilized retinal membranes as analyzed on SDS gels stained with Coomassie blue and Western blots labeled with an anti-ABCA4 antibody. The basal and retinal stimulated ATPase activity of p.Asn965Ser variant was compared with WT ABCA4 after reconstitution of the purified protein into PE-containing liposomes. As shown in Figure 6B, the basal ATPase activity of the p.Asn965Ser variant was about 50% that of WT ABCA4 when measured at the same protein concentration. Importantly, the addition of all-trans retinal used to generate N-Ret-PE stimulated the ATPase activity of WT ABCA4, but failed to increase the activity of p.Asn965Ser ABCA4. Similar results were obtained for p.Asn965Ser ABCA4 expressed and purified from transfected HEK293 cells as shown in Figure 6C and as previously reported (26).
Figure 6.

Purification and functional analysis of p.Asn965Ser (N965S) ABCA4. (A) WT and p.Asn965Ser (N965S) ABCA4 were purified from detergent-solubilized retina membranes by immunoaffinity chromatography on a Rim 3F4 matrix and analyzed on SDS gels stained with Coomassie blue (CB) and western blot labeled with the anti-ABCA4 antibody (Rim 3F4). Approximately, 0.2 µg of WT and 0.1 µg of p.Asn965Ser ABCA4 was applied to the lanes for CB staining and 0.08 µg was applied to the lanes for Western blotting. (B, C) ATPase activity of WT (black) and p.Asn965Ser ABCA4 (grey) purified and reconstituted into phosphatidylethanolamine-containing liposomes in the presence and absence of all-trans retinal. (B). ATPase activity of ABCA4 proteins purified from mouse retinal membranes. Mean ± SD for n = 3. (C) ATPase activity of ABCA4 proteins expressed and purified from HEK293 cells. Mean ± SD for n = 4. ATPase assays were carried out using the same concentration of WT and p.Asn965Ser ABCA4. Data are plotted as a percentage of WT ABCA4 ATPase activity in the absence of retinal.
Autofluorescence and A2E accumulation in the p.Asn965Ser mouse
A major consequence arising from the loss in ABCA4 expression and function in mice and STGD1 patients is the accumulation of the bis-retinoid A2E and an increase in autofluorescence resulting from the accumulation of lipofuscin deposits in RPE cells (28,36,37). To determine if the p.Asn965Ser mouse exhibits similar properties, we compared A2E levels and autofluorescence intensity in the p.Asn965Ser mice with that for WT mice. There was a 10-fold increase in A2E levels in 6-month-old p.Asn965Ser homozygous mice relative to age-matched WT mice (Fig. 7A). Little, if any increase in A2E, was observed in the p.Asn965Ser heterozygous mice. A significant increase in autofluorescence was also observed for the p.Asn965Ser homozygous mice relative to WT mice as measured by scanning laser ophthalmoscopy (SLO) (Fig. 7B and C).
Figure 7.

Analysis of A2E and lipofuscin autofluorescence in WT and p.Asn965Ser (N965S) 6-month-old mice. (A) Quantification of A2E in the eyes of WT mice (WT/WT), heterozygous (WT/N965S) and homozygous (N965S/N965S) mice as measured by HPLC. Error bars indicate mean ± SD for n = 3. P < 0.001 between homozygous and WT or heterozygous mice and WT. No significant difference was found between WT and heterozygous p.Asn965Ser mice. (B) Fundus images of autofluorescence from WT (WT/WT) and homozygous (p.Asn965Ser/p.Asn965Ser) mice as measured by SLO. C. Quantification of the autofluorescence images of WT and p.Asn965Ser (N965S/N965S) homozygous mice. Mean fluorescence intensity ± SD for n = 6. P < 0.001 as measured by two-tailed Student’s t-test.
Discussion
Mice deficient in ABCA4 have been widely used as an animal model for STGD1 since they exhibit many of the characteristic features found in STGD1 patients including the accumulation of lipofuscin and A2E in RPE cells, delayed dark adaptation, oxidative stress, and in some cases photoreceptor degeneration (23,28,38–40). However, the effect of disease-causing missense mutations on the molecular properties and subcellular localization of ABCA4 cannot be evaluated in the Abca4 knockout mouse and are impeded by the extremely low level of expression in the p.Leu541Pro/p.Ala1038Val dual knockin mouse (28). In contrast, the homozygous p.Asn965Ser STGD1 mouse expresses the ABCA4 variant in photoreceptors at significant levels (∼50% WT ABCA4) enabling us to determine the effect of this mutation on the cellular distribution and functional properties of ABCA4. Immunofluorescence microscopy showed that a major fraction of the p.Asn965Ser variant (∼65% of the total) is able to exit the ER and traffic properly to the photoreceptor outer segments. The remaining fraction, however, accumulates in the inner segment with a significant portion retained in the ER apparently as misfolded protein as depicted in Figure 8. Interestingly, the p.Asn965Ser variant expressed in COS7 cells is also distributed between vesicular structures characteristic of WT ABCA4 and reticular pattern indicative of misfolded protein sequestered in the ER.
Figure 8.
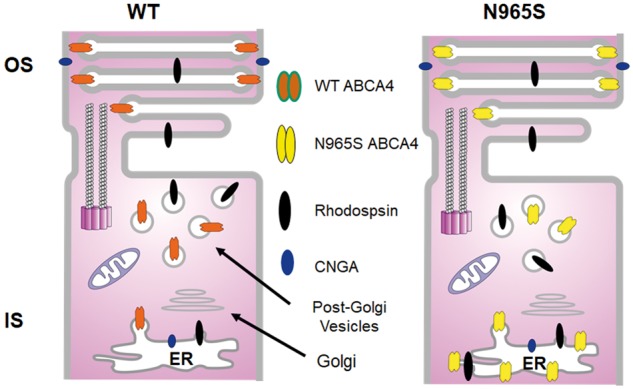
Schematic diagram showing the distribution of ABCA4, CNGA1 and rhodopsin in WT and p.Asn965Ser (N965S) photoreceptors. In WT photoreceptors a significant portion of ABCA4 is present in the post-Golgi vesicles of the IS, where it can function with retinol dehydrogenase 12 (Rdh12) and other dehydrogenases to reduce retinal to retinol which has diffused from OS thereby protecting subcellular organelles from retinoid toxicity. In contrast, a significant fraction of ABCA4- p.Asn965Ser is partially misfolded and a significant fraction is retained in the ER. Rhodopsin is also detected in the inner segment and presumably in post-Golgi vesicles.
Although a significant fraction of the p.Asn965Ser protein escapes the ER and localizes to the photoreceptor outer segments, the mutant protein has low basal ATPase activity, and unlike WT ABCA4, its basal ATPase activity is not activated by its substrate N-Ret-PE. The low basal ATPase activity detected for the p.Asn965Ser mutant may arise from residual activity of NBD1 uncoupled to N-Ret-PE transport or from NBD2 that has been shown previously to bind ATP (26,41). Collectively, our results suggest that substitution of the asparagine for a serine at position 965 in NBD1 results in partial misfolding of ABCA4 with some retention in the ER and complete loss in substrate-dependent ATPase activity. The inability of p.Asn965Ser ABCA4 to hydrolyze ATP required for the transport of N-Ret-PE across disc membranes leads to the accumulation of retinal and N-Ret-PE in photoreceptor outer segments and the production of bis-retinoid compounds which accumulate in RPE cells upon phagocytosis as observed by an increase in autofluorescence measured by SLO and A2E accumulation quantified by HPLC. The loss in ABCA4 functional activity for the p.Asn965Ser variant is broadly consistent with the phenotypes displayed by STGD1 patients homozygous for this mutation including early onset and severe reduction in vision leading to blindness and an increase in autofluorescence (33). Since the phenotype of the p.Asn965Ser mouse is similar to the Abca4 knockout mouse and the p.Leu541Pro/p.Ala1038Val dual knockin mouse in relation to the increase in autofluorescence and A2E in RPE cells, the pathogenic mechanism of the p.Asn965Ser mouse appears to be primarily, if not solely, due to the loss in functional activity of ABCA4 and not the presence of misfolded protein.
The p.Asn965Ser mutation resides in the Walker A motif, also known as the P-loop, of NBD1. The Walker A motif characterized by the consensus sequence GXXXXGKT/S where X can be any amino acid is found in many ATP and GTP binding proteins and functions in the binding of the phosphate groups of ATP or GTP (42). Sequence analysis of members of the ABCA subfamily of ABC transporters indicates that the GXNGAGKT/S sequence is conserved in both NBD1 and NBD2 of the ABCA subgroup consisting of ABCA1, 2, 3, 4, 7, 12, 13, and is conserved in the NBD2 of the ABCA6-like subgroup which include ABCA5, 6, 8, 9, 10 (43). The latter group represents a gene cluster that maps to chromosome 17q24.2–3 and is suggested to arise through gene duplication. The importance of the conserved asparagine (N) in this consensus sequence is highlighted by the finding that substitution of asparagine (N) with serine (S) in ABCA4 causes STGD1 (33) and the same substitution in the NBD1 Walker A motif of ABCA1 (p.Asn935Ser) causes Tangiers disease, a disorder associated with defective phospholipid and cholesterol efflux from cells and high density lipoprotein deficiency (44). Recent studies indicate that these ABCA4 and ABCA1 Walker A mutants display similar properties when expressed in culture cells including a reduction in expression of detergent-solubilized protein (∼50% WT protein), reduced ATPase and phospholipid flippase activity, and subcellular mislocalization (30). This suggests that the Asn to Ser substitution in the Walker A motif of NBD1 has a similar effect on the structure and function of both ABCA transporters and perhaps other members of the ABCA subfamily. Another amino acid substitution within the Walker A motif has also been examined. Replacement of lysine (K) for a methionine (M) in the Walker A motifs of NBD1 and NBD2 abolishes the ATPase activity of ABCA4 and ABCA1 as well as other ABC transporters (26,30).
A low-resolution structure of ABCA4 has been obtained by negative stained, single particle electron microscopy (45). This study has provided some information on the global structure of the protein and conformational changes associated with nucleotide binding. Recently, the structure of ABCA1 in the absence of bound substrate or nucleotides has been determined at a resolution of 4.1 Å by cryoelectron microscopy revealing important insight into the structural features of this ABCA lipid exporter (46). ABCA4 shares an overall amino acid sequence identity of 52% with ABCA1. This identity increases to 64% in NBD1 and 71% in NBD2 with essentially identical Walker A motifs in both proteins. We have generated a homology model of ABCA4 using ABCA1 as a template (Fig. 9A). As in the case of ABCA1, the ABCA4 model has an overall shape of a torch. The structure of NBD1 in the ABCA4 model overlaps with that of ABCA1 as expected on the basis of their high sequence identity (Fig. 9B). The asparagine (N) in the Walker A motif of NBD1 is within the P loop flanked by a β-strand and an α-helix as observed in other ATP binding proteins (Fig. 9B). The Walker A motif of NBD1 is situated at the interface between the two NBDs of the ABCA transporter. Replacement of the conserved asparagine for serine most likely hinders the efficient folding of ABCA4 possibly due to the absence of stabilizing H-bonding. Currently, the structure of the ABCA1 with a bound ATP analogue is not available, but it is possible that this asparagine in the Walker A motif makes key contacts with ATP to promote stable nucleotide binding. This awaits a higher resolution structure of ABCA1 and ultimately ABCA4 with its bound ligands. The similarity in the structure and function of ABCA1 and ABCA4 with respect to the p.Asn965Ser mutation suggests that pharmacological treatments for ABCA4 may also have application in the treatment of Tangiers disease associated with the Asn935Ser mutation.
Figure 9.

Structural model of ABCA4 and the NBD1 domains of ABCA4 and ABCA1. (A) Model of ABCA4 derived from the structure of ABCA1 determined by cryo-EM (46). NBD1 is colored in blue and NBD2 is colored in yellow. The position of the Asn965 residue in NBD1 is shown in space-filling spheres. NBD – nucleotide binding domain; ECD – exocytoplasmic domain; TMD – transmembrane domain. (B) Comparative structural model of NBD1 domains of ABCA1 (tan) and ABCA4 (silver). The similarity in structure of these NBDs is evident. The Asn965 residue of ABCA4 is shown in spheres with red indicating an oxygen atom and blue indicating the nitrogen atom of the asparagine side chain.
As part of this study, we have used highly specific monoclonal antibodies together with refined quantitative confocal scanning microscopy to examine the distribution of ABCA4 and other key photoreceptor proteins in WT mouse retina. As previously reported ABCA4, rhodopsin, peripherin-2, CNG channel, GARP proteins, and RetGC1 all localized primarily to the photoreceptor outer segment layer. However, unlike the other outer segment proteins, ABCA4 was detected in significant amounts (∼ 17% based on fluorescence intensity measurements) in the inner segments of photoreceptors. Fluorescence intensity profiling further indicates that there was a higher density of ABCA4 in WT photoreceptors just below the connecting cilium, a region rich in post Golgi vesicles. This is in contrast to the other outer segment proteins which do not accumulate in photoreceptor inner segments apparently due to their rapid trafficking across the photoreceptor inner segment on route to the outer segment. The presence of ABCA4 in the inner segment (Fig. 8) could be due to inefficient trafficking of this high molecular weight glycoprotein within photoreceptors. However, it is also possible that ABCA4 retained in the inner segment plays an important role in maintaining the function and survival of photoreceptors under conditions of excess retinal production. Although most all-trans retinal produced from the photobleaching of rhodopsin and cone opsin is detoxified to all-trans retinol by retinol dehydrogenase 8 (RDH8) in concert with the N-Ret-PE transport activity of ABCA4 in photoreceptor outer segment disc membranes, some all-trans retinal can diffuse from the outer to the inner segments. Earlier in vitro studies indeed have shown that retinal can partition between membrane vesicles (20,47). We suggest that ABCA4 present in the inner segments functions in conjunction with retinol dehydrogenase 12 (RDH12) and possibly other retinol dehydrogenases to reduce residual retinal to retinol in photoreceptor inner segments thereby protecting mitochondria and other subcellular organelles from the potential toxic effects of retinal and its condensation products.
The p.Asn965Ser mouse is a useful animal model to investigate potential therapeutic strategies to prevent vision loss in Stargardt patients. To date most studies have used Abca4 knockout mice to examine the potential for gene delivery and drug therapy targeting the visual cycle or the production of bisretinoids (48–52). A knockin model for STGD1 such as the p.Asn965Ser mouse is important in further exploring the potential of gene delivery and drug therapy in an animal model which expresses the mutant protein. It is also useful in evaluating the effect of drugs that activate the functional activity and folding of ABCA4 variants associated with Stargardt disease.
Materials and Methods
Generation of p.Asn965Ser ABCA4 mouse
The p.Asn965Ser mutant mouse was generated by Ingenious Targeting Laboratories (Ronkonkoma, NY, USA). Briefly, the targeting vector (Supplementary Material, Fig. S2) was subcloned from a positively identified C57BL/6 BAC clone. The region was designed such that the long homology arm (LA) extends ∼6.08 kb 3’ to the site of the point mutation (A→G) in exon 19 and the FRT flanked Neo cassette is inserted 604 bp 5’ to the point mutation. The short homology arm (SA) extends 1.94 kb 5’ to the FRT flanked Neo cassette. The BAC was subcloned into a ∼2.4kb backbone vector (pSP72, Promega) containing an ampicillin section cassette for retransformation of the construct prior to electroporation. The point mutation on exon 19 coding for the replacement of asparagine at position 965 for a serine (p.Asn965Ser) was generated by extension PCR. The targeting vector was confirmed by restriction analysis after each modification step and by sequencing using primers designed to read from the selection cassette into the genomic sequence. No errors were introduced into the PCR modified region. The targeting vector construct was linearized using Not I prior to transfection into C57BL/6 embryonic stem (ES) cells by electroporation. After selection with G418 antibiotic, surviving clones were expanded for PCR analysis to identify recombinant ES clones. The integrity of the clones was confirmed by Southern blots, and DNA sequencing reconfirmed the point mutation in exon 19 and the absence of spurious mutations. Selected clones were microinjected into C57BL/6 blastocytes. Resulting chimeras were mated to C57BL/6 mice and subsequently mated with C57BL/6 FLP mice to remove the Neo cassette. DNA sequencing was carried out to confirm the presence of the p.Asn965Ser mutation, removal of the Neo cassette and absence of FLP transgene. The homozygous mice for the p.Asn965Ser mutation were obtained by cross-breeding of the heterozygous mice and DNA sequencing was used in confirm the integrity of the mutant ABCA4. PCR primers used for genotyping homozygous and heterozygous p.Asn965Ser mice were as follows:
Forward primer: GAA CTT CGG AAA AGA TGC TGT GGT GG
Reverse primer: GCC CAA GAC TGA GAG ACA TCA AGT GTT C
All procedures were approved by the UBC Animal Care Committee and in accordance with NIH guidelines.
Antibodies
Monoclonal antibodies to ABCA4 (Rim-3F4 and Rim-5B4), CNGA1 (PMc-1D1), peripherin-2 (Per-5H2), GARP (GARP-4B1), rhodopsin (Rho -1D4), and RetGC1 (RetGC-8A5) were produced in our lab and have been previously described (4,16,53–56). Monoclonal antibody to the ER retention sequence KDEL was purchased from Cedarlane Labs (Burlington, ON) and a polyclonal antibody to KDEL was obtained from Abcam (Toronto, ON); polyclonal antibody to M/L cone opsin was from Millipore (Temecula, CA); polyclonal antibodies to glial fibrillary acidic protein (GFAP) and calnexin were from Sigma-Aldrich (Oakville, ON).
Immunofluorescence microscopy
Immunofluorescence microscopy was carried out on retinal cryosections as previously described (56,57). Briefly, retina from 2-month-old homozygous p.Asn965Ser mice and WT litter mates were fixed for 2 h in 2% paraformaldehyde, 0.1 m phosphate buffer, pH 7.3. Cryosections (10–12 μm) were permeabilized and blocked with 0.2% Triton X-100 and 10% normal goat serum and labeled overnight with hybridoma fluid containing primary antibody to photoreceptor proteins at a 1:2 – 1:10 dilution with the exception of Rho-1D4 antibody which was diluted 1:100 in phosphate buffered saline (PBS). The M/L opsin antibody was diluted 1:2000, the KDEL antibody at 1:200, GFAP antibody at 1:500, and calnexin antibody at 1:200. After washing in PBS-0.1% Tween 20, the sections were labeled with the secondary antibody conjugated with Alexa 568 diluted 1:1000. Sections were counterstained with 4’, 6-diamidino-2-phenylindole (DAPI) nuclear stain and visualized on a Zeiss LSM700 confocal microscope using a 40X objective (aperture of 1.3). Images were analyzed using Zeiss Zen software. The fluorescence intensity of the inner and outer segments was determined from the average pixel intensity obtained for 10 squares (6 × 6 µm) over different regions of the retina using NIH Image J software. The background intensity derived from the ONL region and other regions of the retina was typically less than 6% of the intensity in the outer segment layer for each antibody and was subtracted from the measurements in the inner and outer segment. Similar data were obtained for two different sets of p.Asn965Ser and WT mice. Immunofluorescence labeling of paraformaldehyde-fixed COS7 cells was carried out as previously described (27). Composite figures were generated using Photoshop.
Retinal extracts and photoreceptor outer segments for Western blotting
Retinas were dissected from 10 eyes of 2-month old p.Asn965Ser mice and 10 eyes of age-matched WT litter mates and immersed in 1 ml of Tris buffer saline [TBS: 20 mm Tris, pH 7.4, 0.1 m NaCl and ProteaseArrest (Biosciences)] for 20 min on ice. A retinal membrane extract was prepared by passing the suspension through 22G and 28G needles. The extract (400 μl) was layered on top of 1.6 ml of 60% (w/w) sucrose/TBS and centrifuged at 25 000 rpm for 30 min in a TLS-55 rotor using a Beckman TLX Optima centrifuge. The retinal membrane fraction which banded on top of the 60% sucrose was removed, diluted with three volumes of TBS, and pelleted by centrifugation at 30 000 rpm for 10 min in a TLA 110 rotor. The membranes were resuspended with 100 μl of TBS. Photoreceptor outer segments were isolated from retinal membrane extracts using Optiprep (Sigma-Aldrich) according to the manufacturer’s protocol. The protein concentrations were determined by BCA assay (Thermo Fisher Scientific, Waltham, MA).
SDS gel electrophoresis and western blotting was performed as previously described (55). Briefly, the retinal membrane and photoreceptor outer segment preparations (20–50 µg protein) were solubilized in an equal volume of 4% SDS containing 4% 2-mercaptoethanol, resolved on a 8% SDS polyacrylamide gel, and transferred to PVDF Immobilon-FL membranes (Millipore, Billerica, MA). The blots were blocked in 1% milk/PBS, rinsed in PBS, and subsequently labeled for 1 h at 25 °C with hybridoma culture fluid containing the primary antibodies to ABCA4 (Rim 3F4) and the CNGA1 (PMc1D1) diluted 1:20 with PBS. The blots were then rinsed in PBS and subsequently labeled for 1 h at 25 °C with goat anti-mouse IgG conjugated to IRdye 680. After extensive washing in PBS-0.1% Tween 20, the blots were imaged and quantified on an Odyssey Licor infrared imaging system.
ATPase assays
ABCA4 was purified from CHAPS-solubilized retinal membranes obtained from 28 eyes of 2-month old p.Asn965Ser and 14 eyes of age-matched WT mice using a Rim-3F4 immunoaffinity column as previously published (20). Briefly, the retinal membranes were solubilized in column buffer [10 mm HEPES, 1 mm dithiotreitol, 5 mm MgCl2, 0.2 mg/ml brain polar lipids (BPL)] and 0.002% cholesteryl hemisuccinate from Avanti Polar Lipids (Alabaster, AB) in the presence of 18 mm CHAPS detergent and applied to an immunoaffinity matrix in which the purified Rim-3F4 antibody was directly coupled to CNBr-activated Sepharose 2B. The reaction mixture was gently mixed for 1 h at 4 °C, washed extensively with column buffer containing 10 mm CHAPS, and eluted in the same buffer containing 0.2 mg/ml 3F4 peptide. The sample (1 µg) was added to BPL at a ratio 1:200 and dialyzed against reconstitution buffer (20 mm HEPES, pH 8.0, 0.15 m NaCl, and 1 mm dithiothreitol) for 16 h with at least 1 change (1 l each) to remove the detergent and reconstitute the protein into liposomes. The protein concentration was determined on Coomassie blue stained SDS gels using bovine serum albumin as a standard.
The ATPase activity of the reconstituted protein was determined using the ADP-Glo Kinase Assay Kit (Promega) according to the manufacturer’s protocol. Briefly, 18 µl of ABCA4 reconstituted into liposomes and immersed in reconstitution buffer was incubated with 100 μM ATP with or without all-trans retinal at a final concentration of 20 or 40 µm. After 30 min at 37 °C, the sample was added to an equal volume of ADP-Glo reagent and incubated for 60–90 min at RT. The ADP-Glo kinase detection reagent was then added and the sample was incubated for another 30 min prior to luminescence detection in a microtiter plate reader. Each sample was measured in triplicate. At least three independent samples were analyzed. ABCA4 containing a 1D4 tag was expressed and purified from HEK293 cells as previously described (27) for ATPase activity measurements as described above. ATPase measurements of WT and p.Asn965Ser variants were carried out using the same protein concentration.
A2E quantification and SLO autofluorescence
A2E was generated and purified by the method of Parish et al. (58) for use as a standard. A2E was quantified from eyecups of age-matched WT, heterozygous, and homozygous ABCA4 p.Asn965Ser mice by the method of Kim et al. (59). Briefly, frozen eyecups in PBS were homogenized and added to an equal volume of chloroform/methanol (2:1). After three extractions, the sample was passed through a Teflon filter and dried under nitrogen. The sample was dissolved in methanol-0.1% trifluoroacetic acid (TFA) and 5–20 µl was applied to a C18 column (4 × 150 mm) connected to an Agilent HP 1100 HPLC. A gradient of acetonitrile in water containing 0.1% TFA was used as follows: 90–100% (0–10 min), 100% acetonitrile (10–20 min) with a flow rate of 0.8 ml/min. A2E monitored at 430 nm and confirmed by UV-Vis spectroscopy.
Autofluorescence was measured in 6-month-old mice using a scanning laser ophthalmoscope (SLO) that was designed and built for in vivo retinal/RPE imaging with an excitation wavelength of 532 nm and a broadband emission filter. Prior to SLO imaging the mice were anesthetized with a subcutaneous injection of ketamine (100 mg/kg of body weight) and dexmedetomidine (0.1 mg/kg of body weight). The eyes were dilated with a drop of 1% Tropicamide and a zero-diopter contact lens was placed on the cornea to prevent dehydration. The SLO images covered a 50 degree field-of-view, which was centered at the optic nerve head and manually focused on the RPE layer for each eye. After imaging, the mice were recovered with an injection of atipamezole (1.8 mg/kg of body weight). For six mice, the fluorescence intensity was determined by calculating the mean pixel value from each of the SLO images.
Molecular modeling and statistics
ABCA1 (PDB 5XJY) was used as a template for modeling of ABCA4 using Swiss-Model modelling program (60). Graphics and comparative structures between ABCA1 and ABCA4 in NBD1 were obtained using Chimera software (61). Graph-Pad was used for statistical analysis.
Supplementary Material
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health (EY002422 to R.S.M.); Canadian Institutes of Health Research (PJT-148649 to R.S.M. and PJT-378123 to M.V.S. and R.S.M.).
Supplementary Material
References
- 1. Stargardt K. (1909) Uber familiare, progressive degeenration under makulagegend des augen. Albrecht. Von Graefes. Arch. Ophthalmol., 71, 534–550. [Google Scholar]
- 2. Allikmets R., Singh N., Sun H., Shroyer N.F., Hutchinson A., Chidambaram A., Gerrard B., Baird L., Stauffer D., Peiffer A.. et al. (1997) A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet., 15, 236–246. [DOI] [PubMed] [Google Scholar]
- 3. Nasonkin I., Illing M., Koehler M.R., Schmid M., Molday R.S., Weber B.H. (1998) Mapping of the rod photoreceptor ABC transporter (ABCR) to 1p21-p22.1 and identification of novel mutations in Stargardt's disease. Hum. Genet., 102, 21–26. [DOI] [PubMed] [Google Scholar]
- 4. Illing M., Molday L.L., Molday R.S. (1997) The 220-kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily. J. Biol. Chem., 272, 10303–10310. [DOI] [PubMed] [Google Scholar]
- 5. Kang Derwent J.J., Derlacki D.J., Hetling J.R., Fishman G.A., Birch D.G., Grover S., Stone E.M., Pepperberg D.R. (2004) Dark adaptation of rod photoreceptors in normal subjects, and in patients with Stargardt disease and an ABCA4 mutation. Invest. Ophthalmol. Vis. Sci., 45, 2447–2456. [DOI] [PubMed] [Google Scholar]
- 6. Weleber R.G. (1994) Stargardt's macular dystrophy. Arch. Ophthalmol., 112, 752–754. [DOI] [PubMed] [Google Scholar]
- 7. Rotenstreich Y., Fishman G.A., Anderson R.J. (2003) Visual acuity loss and clinical observations in a large series of patients with Stargardt disease. Ophthalmology, 110, 1151–1158. [DOI] [PubMed] [Google Scholar]
- 8. Mantyjarvi M., Tuppurainen K. (1992) Color vision in Stargardt's disease. Int. Ophthalmol., 16, 423–428. [DOI] [PubMed] [Google Scholar]
- 9. Fishman G.A., Farber M., Patel B.S., Derlacki D.J. (1987) Visual acuity loss in patients with Stargardt's macular dystrophy. Ophthalmology, 94, 809–814. [DOI] [PubMed] [Google Scholar]
- 10. Klevering B.J., Blankenagel A., Maugeri A., Cremers F.P., Hoyng C.B., Rohrschneider K. (2002) Phenotypic spectrum of autosomal recessive cone-rod dystrophies caused by mutations in the ABCA4 (ABCR) gene. Invest. Ophthalmol. Vis. Sci., 43, 1980–1985. [PubMed] [Google Scholar]
- 11. Cremers F.P., van de Pol D.J., van Driel M., den Hollander A.I., van Haren F.J., Knoers N.V., Tijmes N., Bergen A.A., Rohrschneider K., Blankenagel A.. et al. (1998) Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR. Hum. Mol. Genet., 7, 355–362. [DOI] [PubMed] [Google Scholar]
- 12. Franceschetti A., F J. (1965) Fundus flavimaculatus. Arch. Ophthalmol., 25, 505–530. [PubMed] [Google Scholar]
- 13. Fishman G.A., Stone E.M., Eliason D.A., Taylor C.M., Lindeman M., Derlacki D.J. (2003) ABCA4 gene sequence variations in patients with autosomal recessive cone-rod dystrophy. Arch. Ophthalmol., 121, 851–855. [DOI] [PubMed] [Google Scholar]
- 14. Lee W., Schuerch K., Zernant J., Collison F.T., Bearelly S., Fishman G.A., Tsang S.H., Sparrow J.R., Allikmets R. (2017) Genotypic spectrum and phenotype correlations of ABCA4-associated disease in patients of south Asian descent. Eur. J. Hum. Genet., 25, 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schulz H.L., Grassmann F., Kellner U., Spital G., Ruther K., Jagle H., Hufendiek K., Rating P., Huchzermeyer C., Baier M.J.. et al. (2017) Mutation Spectrum of the ABCA4 Gene in 335 Stargardt Disease Patients From a Multicenter German Cohort-Impact of Selected Deep Intronic Variants and Common SNPs. Invest. Ophthalmol. Vis. Sci., 58, 394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Molday L.L., Rabin A.R., Molday R.S. (2000) ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat. Genet., 25, 257–258. [DOI] [PubMed] [Google Scholar]
- 17. Papermaster D.S., Reilly P., Schneider B.G. (1982) Cone lamellae and red and green rod outer segment disks contain a large intrinsic membrane protein on their margins: an ultrastructural immunocytochemical study of frog retinas. Vision Res., 22, 1417–1428. [DOI] [PubMed] [Google Scholar]
- 18. Sun H., Nathans J. (1997) Stargardt's ABCR is localized to the disc membrane of retinal rod outer segments. Nat. Genet., 17, 15–16. [DOI] [PubMed] [Google Scholar]
- 19. Beharry S., Zhong M., Molday R.S. (2004) N-retinylidene-phosphatidylethanolamine is the preferred retinoid substrate for the photoreceptor-specific ABC transporter ABCA4 (ABCR). J. Biol. Chem., 279, 53972–53979. [DOI] [PubMed] [Google Scholar]
- 20. Quazi F., Lenevich S., Molday R.S. (2012) ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat. Commun., 3, 925. doi: 10.1038/ncomms1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Quazi F., Molday R.S. (2014) ATP-binding cassette transporter ABCA4 and chemical isomerization protect photoreceptor cells from the toxic accumulation of excess 11-cis-retinal. Proc. Natl Acad. Sci. U S A, 111, 5024–5029.http://dx.doi.org/10.1073/pnas.1400780111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sparrow J.R., Boulton M. (2005) RPE lipofuscin and its role in retinal pathobiology. Exp. Eye Res., 80, 595–606. [DOI] [PubMed] [Google Scholar]
- 23. Weng J., Mata N.L., Azarian S.M., Tzekov R.T., Birch D.G., Travis G.H. (1999) Insights into the function of rim protein in photoreceptors and etiology of Stargardt's Disease from the phenotype in abcr knockout mice. Cell, 98, 13–23. [DOI] [PubMed] [Google Scholar]
- 24. Sun H., Molday R.S., Nathans J. (1999) Retinal stimulates ATP hydrolysis by purified and reconstituted ABCR, the photoreceptor-specific ATP-binding cassette transporter responsible for Stargardt disease. J. Biol. Chem., 274, 8269–8281. [DOI] [PubMed] [Google Scholar]
- 25. Molday R.S., Zhong M., Quazi F. (2009) The role of the photoreceptor ABC transporter ABCA4 in lipid transport and Stargardt macular degeneration. Biochim. Biophys. Acta, 1791, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sun H., Smallwood P.M., Nathans J. (2000) Biochemical defects in ABCR protein variants associated with human retinopathies. Nat. Genet., 26, 242–246. [DOI] [PubMed] [Google Scholar]
- 27. Zhong M., Molday L.L., Molday R.S. (2009) Role of the C terminus of the photoreceptor ABCA4 transporter in protein folding, function, and retinal degenerative diseases. J. Biol. Chem., 284, 3640–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang N., Tsybovsky Y., Kolesnikov A.V., Rozanowska M., Swider M., Schwartz S.B., Stone E.M., Palczewska G., Maeda A., Kefalov V.J.. et al. (2015) Protein misfolding and the pathogenesis of ABCA4-associated retinal degenerations. Hum. Mol. Genet., 24, 3220–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wiszniewski W., Zaremba C.M., Yatsenko A.N., Jamrich M., Wensel T.G., Lewis R.A., Lupski J.R. (2005) ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies. Hum. Mol. Genet., 14, 2769–2778. [DOI] [PubMed] [Google Scholar]
- 30. Quazi F., Molday R.S. (2013) Differential phospholipid substrates and directional transport by ATP-binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants. J. Biol. Chem., 288, 34414–34426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cideciyan A.V., Swider M., Aleman T.S., Tsybovsky Y., Schwartz S.B., Windsor E.A., Roman A.J., Sumaroka A., Steinberg J.D., Jacobson S.G.. et al. (2009) ABCA4 disease progression and a proposed strategy for gene therapy. Hum. Mol. Genet., 18, 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bungert S., Molday L.L., Molday R.S. (2001) Membrane topology of the ATP binding cassette transporter ABCR and its relationship to ABC1 and related ABCA transporters: identification of N-linked glycosylation sites. J. Biol. Chem., 276, 23539–23546. [DOI] [PubMed] [Google Scholar]
- 33. Rosenberg T., Klie F., Garred P., Schwartz M. (2007) N965S is a common ABCA4 variant in Stargardt-related retinopathies in the Danish population. Mol. Vis., 13, 1962–1969. [PubMed] [Google Scholar]
- 34. Jiang F., Pan Z., Xu K., Tian L., Xie Y., Zhang X., Chen J., Dong B., Li Y. (2016) Screening of ABCA4 Gene in a Chinese Cohort With Stargardt Disease or Cone-Rod Dystrophy With a Report on 85 Novel Mutations. Invest. Ophthalmol .Vis. Sci., 57, 145–152. [DOI] [PubMed] [Google Scholar]
- 35. Ahn J., Wong J.T., Molday R.S. (2000) The effect of lipid environment and retinoids on the ATPase activity of ABCR, the photoreceptor ABC transporter responsible for Stargardt macular dystrophy. J. Biol. Chem., 275, 20399–20405. [DOI] [PubMed] [Google Scholar]
- 36. Sparrow J.R., Blonska A., Flynn E., Duncker T., Greenberg J.P., Secondi R., Ueda K., Delori F.C. (2013) Quantitative fundus autofluorescence in mice: correlation with HPLC quantitation of RPE lipofuscin and measurement of retina outer nuclear layer thickness. Invest. Ophthalmol. Vis. Sci., 54, 2812–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Charbel Issa P., Barnard A.R., Singh M.S., Carter E., Jiang Z., Radu R.A., Schraermeyer U., MacLaren R.E. (2013) Fundus autofluorescence in the Abca4(-/-) mouse model of Stargardt disease–correlation with accumulation of A2E, retinal function, and histology. Invest. Ophthalmol. Vis. Sci., 54, 5602–5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boyer N.P., Higbee D., Currin M.B., Blakeley L.R., Chen C., Ablonczy Z., Crouch R.K., Koutalos Y. (2012) Lipofuscin and N-retinylidene-N-retinylethanolamine (A2E) accumulate in retinal pigment epithelium in absence of light exposure: their origin is 11-cis-retinal. J. Biol. Chem., 287, 22276–22286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Radu R.A., Mata N.L., Bagla A., Travis G.H. (2004) Light exposure stimulates formation of A2E oxiranes in a mouse model of Stargardt's macular degeneration. Proc. Natl Acad. Sci. U S A, 101, 5928–5933.http://dx.doi.org/10.1073/pnas.0308302101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Radu R.A., Hu J., Yuan Q., Welch D.L., Makshanoff J., Lloyd M., McMullen S., Travis G.H., Bok D. (2011) Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J. Biol. Chem., 286, 18593–18601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ahn J., Beharry S., Molday L.L., Molday R.S. (2003) Functional interaction between the two halves of the photoreceptor-specific ATP binding cassette protein ABCR (ABCA4). Evidence for a non-exchangeable ADP in the first nucleotide binding domain. J. Biol. Chem., 278, 39600–39608. [DOI] [PubMed] [Google Scholar]
- 42. Walker J.E., Saraste M., Runswick M.J., Gay N.J. (1982) Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. embo J., 1, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dean M., Hamon Y., Chimini G. (2001) The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res., 42, 1007–1017. [PubMed] [Google Scholar]
- 44. Utech M., Hobbel G., Rust S., Reinecke H., Assmann G., Walter M. (2001) Accumulation of RhoA, RhoB, RhoG, and Rac1 in fibroblasts from Tangier disease subjects suggests a regulatory role of Rho family proteins in cholesterol efflux. Biochem. Biophys. Res. Commun., 280, 229–236. [DOI] [PubMed] [Google Scholar]
- 45. Tsybovsky Y., Orban T., Molday R.S., Taylor D., Palczewski K. (2013) Molecular organization and ATP-induced conformational changes of ABCA4, the photoreceptor-specific ABC transporter. Structure, 21, 854–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Qian H., Zhao X., Cao P., Lei J., Yan N., Gong X. (2017) Structure of the Human Lipid Exporter ABCA1. Cell, 169, 1228–1239 e1210. [DOI] [PubMed] [Google Scholar]
- 47. Rando R.R., Bangerter F.W. (1982) The rapid intermembraneous transfer of retinoids. Biochem. Biophys. Res. Commun., 104, 430–436. [DOI] [PubMed] [Google Scholar]
- 48. Han Z., Conley S.M., Naash M.I. (2014) Gene therapy for Stargardt disease associated with ABCA4 gene. Adv. Exp. Med. Biol., 801, 719–724. [DOI] [PubMed] [Google Scholar]
- 49. Trapani I., Colella P., Sommella A., Iodice C., Cesi G., de Simone S., Marrocco E., Rossi S., Giunti M., Palfi A.. et al. (2014) Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol. Med., 6, 194–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Auricchio A., Trapani I., Allikmets R. (2015) Gene Therapy of ABCA4-Associated Diseases. Cold Spring Harb. Perspect. Med., 5, a017301.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Maeda A., Golczak M., Chen Y., Okano K., Kohno H., Shiose S., Ishikawa K., Harte W., Palczewska G., Maeda T.. et al. (2011) Primary amines protect against retinal degeneration in mouse models of retinopathies. Nat. Chem. Biol., 8, 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Charbel Issa P., Barnard A.R., Herrmann P., Washington I., MacLaren R.E. (2015) Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin A dimerization. Proc. Natl Acad. Sci. U S A, 112, 8415–8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hsu Y.T., Molday R.S. (1994) Interaction of calmodulin with the cyclic GMP-gated channel of rod photoreceptor cells. Modulation of activity, affinity purification, and localization. J. Biol. Chem., 269, 29765–29770. [PubMed] [Google Scholar]
- 54. Connell G., Bascom R., Molday L., Reid D., McInnes R.R., Molday R.S. (1991) Photoreceptor peripherin is the normal product of the gene responsible for retinal degeneration in the rds mouse. Proc. Natl Acad. Sci. U S A, 88, 723–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Poetsch A., Molday L.L., Molday R.S. (2001) The cGMP-gated channel and related glutamic acid-rich proteins interact with peripherin-2 at the rim region of rod photoreceptor disc membranes. J. Biol. Chem., 276, 48009–48016. [DOI] [PubMed] [Google Scholar]
- 56. Molday L.L., Djajadi H., Yan P., Szczygiel L., Boye S.L., Chiodo V.A., Gregory-Evans K., Sarunic M.V., Hauswirth W.W., Molday R.S. (2013) RD3 gene delivery restores guanylate cyclase localization and rescues photoreceptors in the Rd3 mouse model of Leber congenital amaurosis 12. Hum. Mol. Genet., 22, 3894–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cheng C.L., Djajadi H., Molday R.S. (2013) Cell-specific markers for the identification of retinal cells by immunofluorescence microscopy. Methods Mol. Biol., 935, 185–199. [DOI] [PubMed] [Google Scholar]
- 58. Parish C.A., Hashimoto M., Nakanishi K., Dillon J., Sparrow J. (1998) Isolation and one-step preparation of A2E and iso-A2E, fluorophores from human retinal pigment epithelium. Proc. Natl Acad. Sci. U S A, 95, 14609–14613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim S.R., Fishkin N., Kong J., Nakanishi K., Allikmets R., Sparrow J.R. (2004) Rpe65 Leu450Met variant is associated with reduced levels of the retinal pigment epithelium lipofuscin fluorophores A2E and iso-A2E. Proc. Natl Acad. Sci. U S A, 101, 11668–11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Gallo Cassarino T., Bertoni M., Bordoli L.. et al. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res., 42, W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem., 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.