

Abstract
Duchenne muscular dystrophy (DMD) is a muscle wasting disease in which inflammation influences the severity of pathology. We found that the onset of muscle inflammation in the mdx mouse model of DMD coincides with large increases in expression of pro-inflammatory cytokines [tumor necrosis factor-α (TNFα); interferon gamma (IFNγ)] and dramatic reductions of the pro-myogenic protein Klotho in muscle cells and large increases of Klotho in pro-regenerative, CD206+ macrophages. Furthermore, TNFα and IFNγ treatments reduced Klotho in muscle cells and increased Klotho in macrophages. Because CD206+/Klotho+ macrophages were concentrated at sites of muscle regeneration, we tested whether macrophage-derived Klotho promotes myogenesis. Klotho transgenic macrophages had a pro-proliferative influence on muscle cells that was ablated by neutralizing antibodies to Klotho and conditioned media from Klotho mutant macrophages did not increase muscle cell proliferation in vitro. In addition, transplantation of bone marrow cells from Klotho transgenic mice into mdx recipients increased numbers of myogenic cells and increased the size of muscle fibers. Klotho also acted directly on macrophages, stimulating their secretion of TNFα. Because TNFα is a muscle mitogen, we tested whether the pro-proliferative effects of Klotho on muscle cells were mediated by TNFα and found that increased proliferation caused by Klotho was reduced by anti-TNFα. Collectively, these data show that pro-inflammatory cytokines contribute to silencing of Klotho in dystrophic muscle, but increase Klotho expression by macrophages. Our findings also show that macrophage-derived Klotho can promote muscle regeneration by expanding populations of muscle stem cells and increasing muscle fiber growth in dystrophic muscle.
Introduction
Duchenne muscular dystrophy (DMD) is a progressive, muscle wasting disease that is caused by mutation of the dystrophin gene, which encodes a membrane-associated structural protein at the cytosolic face of muscle membranes (1). Although muscle-membrane weakness caused by the loss of the dystrophin protein is a primary functional defect underlying muscle fiber death in DMD (2), other secondary features of the pathology play major roles in determining the extent of pathology. In particular, the inflammatory response to muscle injury has large influences on the course and severity of pathology. For example, depletion of macrophages from dystrophin-deficient mdx mice, a model for DMD, reduces the number of injured fibers in dystrophic muscle by 70% (3) and anti-inflammatory drugs such as corticosteroids are the most-common, successful palliative treatment for DMD (4).
Other features of the pathology of muscular dystrophy are caused by the secondary loss of other proteins, which occurs as a consequence of dystrophin-deficiency. Most prominently, dystrophin-deficiency leads to a reduction in prevalence of proteins that normally exist in a dystrophin-associated protein complex but become less stable and more rapidly degraded in dystrophin-deficiency. Because the individual loss of any single member of the complex can cause muscle disease, their collective loss can amplify the pathology of dystrophin-deficiency. In addition, other proteins are down-regulated at the transcriptional level as a result of dystrophin-deficiency (5). Transcription of neuronal nitric oxide synthase (nNOS), which normally associates with the dystrophin-associated protein complex, is greatly reduced in dystrophin-deficient muscle (6) and ensuing deficiencies in nitric oxide production can contribute to misregulation of blood flow (7,8), defects in synapse formation, increased fatigability (9,10), increases in muscle inflammation (3) and perturbations in cardiac function (11,12).
More recent investigations have also shown that dystrophin-deficiency causes epigenetic silencing of the gene that encodes Klotho. Klotho is a transmembrane protein from which the extracellular domain can be cleaved and released to function as a circulating hormone. Alternatively, it can be expressed as a truncated form that is secreted or retained in the cytoplasm (13–15). Klotho has been studied primarily in the context of aging biology because its genetic deletion causes premature death and rapid changes in several organs that resemble premature senescence (13). In addition, its expression normally declines in tissues as they age (13). Although Klotho is expressed at highest levels in kidney, skin and brain, it is also expressed at low levels in skeletal muscle (13) and reduced expression in healthy, non-dystrophic muscles causes reductions in muscle mass and strength (13,16).
Silencing of Klotho in dystrophic muscles contributes to several, major components of the dystrophic pathology. For example, expression of a Klotho transgene that is not silenced in dystrophic mdx mice increased their longevity, reduced muscle loss, reduced muscle fibrosis and increased the numbers of muscle-resident stem cells that are required for muscle regeneration (17). However, the absence of dystrophin is not directly responsible for the reduction in Klotho expression, because neonatal mdx mice express normal levels of Klotho. Instead, Klotho expression levels in mdx mice plummet at the acute onset of pathology which occurs at approximately 1-month of age and is characterized by extensive inflammation of the dystrophic muscle (17). This suggests that inflammatory mediators may play a role in Klotho down-regulation in dystrophic muscle.
Previous investigations have shown that inflammation can suppress Klotho expression in other tissues. That relationship has been most thoroughly documented in the kidneys where the inflammation associated with diabetes, colitis or endotoxemia causes large reductions in Klotho expression, contributing to kidney pathology (18–20). Similarly, endotoxemia causes reductions in myocardial Klotho that are associated with cardiac dysfunction (21). Pro-inflammatory cytokines, especially tumor necrosis factor-alpha (TNFα), reduce Klotho expression in vivo, at least in some disease models. For example, the suppression of Klotho signaling in kidneys that is caused by colitis does not occur in mice treated with neutralizing antibodies to TNFα (19). In addition, in vitro studies show that TNFα acts directly on kidney cells to reduce Klotho expression (19,22) and that TNFα-mediated suppression can be further amplified by the presence of other pro-inflammatory cytokines, especially interferon-gamma (IFNγ) (19). Furthermore, genetic deletion of interleukin-10 (IL10), an anti-inflammatory cytokine, reduces Klotho expression in kidneys of colitic mice (19). Because IL10 reduces the expression of TNFα and IFNγ by macrophages (23) and deactivates pro-inflammatory (M1) macrophages (24,25), reduced Klotho expression in IL10 mutant colitic mice is attributable to elevations of pro-inflammatory mediators.
Although inflammation can cause reductions in Klotho expression, Klotho can inhibit pro-inflammatory pathways, in return. Administration of recombinant Klotho to endotoxemic mice reduced activation of the pro-inflammatory NFκB pathway in myocardial cells, leading to lower levels of the pro-inflammatory molecules TNFα, IL1β, IL6 and CCL2 in the myocardium (21). Similarly, Klotho suppresses TNFα induced activation of NFκB in kidney cells in vitro, leading to reductions in expression of pro-inflammatory CCL2 and IL8 (20). Thus, pathological conditions that reduce Klotho expression would diminish a negative regulator of inflammation, which could potentially lead to pathological amplification of chronic inflammatory diseases. In a chronic inflammatory disease such as DMD, loss of muscle Klotho could lead to increased inflammation and inflammatory cell mediated damage to muscle tissue as the disease progresses.
In this investigation, we test the hypothesis that the acute onset of inflammation in mdx muscular dystrophy leads to an abrupt increase in production of pro-inflammatory cytokines that reduce Klotho expression in muscle cells. We also assay whether macrophages that accumulate in inflammatory lesions in dystrophic muscle express Klotho and whether their expression of Klotho is regulated by pro-inflammatory or anti-inflammatory cytokines. Further experiments test whether macrophage-derived Klotho influences myogenesis and assay whether over-expression of Klotho by leukocytes affects myogenesis in mdx muscles. Finally, we test whether the effects of macrophage-derived Klotho on myogenic cells are mediated by TNFα.
Results
Proinflammatory cytokines reduce Klotho expression in muscle
Klotho expression in muscle is reduced at the onset of pathology in mdx dystrophy and during aging. Quantitative PCR (QPCR) of Klotho mRNA in mdx muscle shows a large reduction in Klotho expression at the acute onset of muscle inflammation that then continues to decline as the mice age, until at least 24 months of age (Fig. 1A). Wild-type muscles also experience age-related reductions in Klotho expression between 1-month and 24-months of age, although expression levels in wild-type muscle are always higher than in age-matched mdx muscles during that period (Fig. 1A). Western blots also show that secreted (60 kDa) and transmembrane (120 kDa) Klotho is undetectable in mdx muscles after 1 month of age (Fig 1B). Because soluble Klotho can act on cells through interactions with its receptor that are enhanced by the presence of the receptor co-ligand fibroblast growth factor-23 (FGF23) (26), we also assayed whether expression levels of FGF23 in muscle were affected by the dystrophin mutation at any stage of pathology. However, FGF23 expression does not significantly differ between wild-type and mdx muscles and does not differ between ages within a single genotype (Supplementary Material, Fig. S1). Thus, Klotho signaling is not influenced by modulating FGF23 expression in mdx muscle.
Figure 1.

Proinflammatory cytokines reduce Klotho expression in muscle in vivo. (A) QPCR analysis of Klotho expression in quadriceps muscles over the course of mdx pathology. Values normalized to 1-month-old wild-type mice. N = 5 per data set. * indicates significant difference from 1-month-old wild-type muscles at P < 0.05. # indicates significant difference from age-matched, wild-type muscles at P < 0.05. n = 5 per data set. (B) Western blot of Klotho in mdx muscle homogenates collected over the course of pathology. Relative Klotho levels are shown for full-length, transmembrane (120 kDa) and cytosolic/secreted (60 kDa) Klotho. “Loading” shows membrane used for western blotting that was stained with Ponceau Red to show relative loading of each lane. (C) QPCR analysis of TNFα expression in quadriceps muscles over the course of mdx pathology. Values normalized to 1-month-old wild-type mice. N = 5 per data set. * indicates significant difference from 1-month-old wild-type muscles at P < 0.05. # indicates significant difference from age matched, wild-type muscles at P < 0.05. (D) QPCR analysis of IFNγ expression in quadriceps muscles over the course of mdx pathology. Values normalized to 1-month-old wild-type mice. N = 5 per data set. * indicates significant difference from 1-month-old wild-type muscles at P < 0.05. # indicates significant difference from age matched, wild-type muscles at P < 0.05. (E) QPCR analysis of Klotho expression in quadriceps muscle of 5-month-old wild-type or TNFα–null mice. Values normalized to wild-type control mice. N = 5 per data set. * indicates significant difference from age-matched, wild-type muscles at P < 0.05. (F) QPCR analysis of Klotho expression in myotubes stimulated with TNFα and IFNγ in vitro. Values normalized to untreated control myotubes. * indicates significant difference from untreated control myotubes at P < 0.05. (G) Western blot of myotube extracts for Klotho. “Load” shows membrane used for western blotting that was stained with Ponceau Red to show relative loading of each lane. “Klotho” shows 60 kDa cytosolic/secreted Klotho in western blot of membrane. (H) Western blot of myotube samples treated with proinflammatory cytokines. Myotubes were treated with IFNγ or TNFα or IFNγ and TNFα together or with vehicle-only controls (Cont.). The image labeled “Loading” shows membrane used for western blotting that was stained with Ponceau Red, to show relative loading of each lane. The anti-Klotho reactive band labeled “60 kDa” shows cytosolic/secreted Klotho in western blot of the membrane.
The abrupt reduction in Klotho mRNA expression in 1-month-old mdx mice corresponds with a similarly abrupt increase in TNFα and IFNγ expression, when muscle inflammation reaches its peak (Fig. 1C and D). TNFα expression then declines progressively in mdx muscles, until at least 24-months of age, although the level of IFNγ expression in mdx muscle remains continuously elevated from 1 to 24-months of age. In contrast, both TNFα and IFNγ undergo a continuous increase in expression in wild-type muscles between 3 and 24 months of age, although expression of each cytokine in wild-type muscles is always less than in mdx muscles.
The finding that the reduction of Klotho expression coincided with increases in expression of proinflammatory cytokines suggested that proinflammatory cytokines could negatively regulate Klotho expression in muscle. We tested this possibility in vivo by assaying the effect of TNFα mutation on Klotho expression in muscle and observed a greater than 2-fold increase in Klotho mRNA in TNFα-null muscles at 6 months of age (Fig. 1E). We also found that direct application of recombinant TNFα and IFNγ to myotubes in vitro reduced Klotho expression by half (Fig. 1F). We further confirmed that TNFα and IFNγ reduced Klotho protein in myotubes in vitro, in which Klotho occurred primarily at 60 kDa (Fig. 1G). Although treatment with IFNγ alone did not affect Klotho expression, treatment with TNFα alone or treatment with TNFα and IFNγ together significantly reduced Klotho (Fig. 1H).
Macrophages in inflammatory lesions in mdx muscles express Klotho
Immunohistochemistry confirmed that Klotho was not present at detectible levels in muscle fibers in 4-week-old mdx mice (Fig. 2A), although previous investigations have shown that Klotho is present in muscle fibers of wild-type muscles at the same age (17). However, mononucleated cells in inflammatory lesions in mdx muscle expressed elevated levels of Klotho (Fig. 2A). We assayed for the identity of the Klotho expressing cells in inflammatory lesions and found that the majority of the Klotho+ cells expressed CD206 (Fig. 2B), a marker for pro-regenerative, M2 macrophages (27).
Figure 2.

Macrophages in dystrophic muscle in vivo and in vitro express Klotho. (A) A cross-section of 4-week-old mdx muscle labeled with antibodies to Klotho shows mononucleated cells in an inflammatory lesion express KL (red) although no Klotho is detectible in neighboring, healthy muscle fibers. (B) A cross-section of 4-week-old mdx muscle labeled with antibodies to Klotho (red) and CD206 (green). No Klotho is detectible in muscle fibers. Most labeled cells in the inflammatory lesion (between brackets) appear yellow or orange, indicating that they express both Klotho and CD206. CD206+ macrophages (green) that are within the muscle in the perimysium (between arrows), but not within the inflammatory lesion do not express Klotho. Nuclei appear blue (DAPI). Bars = 40 µm. (C) Flow cytometry of BMDMs labeled with anti-F4/80 after growth in M-CSF supplemented media show that over 93% of the cells differentiated into mature monocytes/macrophages. (D) Western blot of homogenates of BMDMs with anti-Klotho or with anti- Klotho that had been preabsorbed with full length Klotho shows that the 60 kDa protein in macrophages is Klotho. (E) Western blot of homogenates of BMDMs that were untreated, or treated with Klotho siRNA or scrambled RNA controls show that the 60 kDa protein in macrophage extracts is Klotho. (F–H) BMDMs treated with proinflammatory cytokines TNFα and IFNγ are activated to express elevated levels of TNFα (F), iNOS (G) and a strong trend for increased expression of IFNγ (H). * indicates significant difference from unstimulated control. (I,J) BMDMs treated with anti-inflammatory cytokines IL10 and IL4 are activated to express elevated levels of CD206 (I) and CD163 (J). * indicates significant difference from unstimulated control. (K) Stimulation of BMDMs with either proinflammatory (Th1; TNFα and IFNγ) or anti-inflammatory (Th2; IL10 and IL4) cytokines increases Klotho expression. * indicates significant difference from unstimulated control at P < 0.05.
Macrophage activation with pro-inflammatory or anti-inflammatory cytokines increases Klotho expression
Because pro-inflammatory cytokines reduced Klotho expression in muscle cells, we assayed whether they similarly suppressed Klotho in macrophages. In addition, we tested whether anti-inflammatory cytokines that can promote activation of macrophages to a pro-regenerative, M2 phenotype (IL4 and IL10) influenced Klotho expression in macrophages because of the high levels of expression of Klotho in CD206+ M2 macrophages observed in vivo. We also chose to assay the effects of these anti-inflammatory cytokines on Klotho expression by macrophages because they are elevated in the regenerative stage of mdx dystrophy (27).
Macrophages were generated by isolating bone marrow cells (BMCs) from wild-type mice and then culturing them in the presence of macrophage colony stimulating factor (MCSF). We then confirmed that the cells had differentiated into macrophages using flow cytometry, which showed that 93% of the cultured cells expressed F4/80, a marker for mature monocytes/macrophages (Fig. 2C). Western blots for Klotho in cell extracts showed a prominent 60 kDa protein that was recognized by anti-Klotho (Fig. 2D). We confirmed that the polypeptide was Klotho by also probing western blots of the same samples using anti-Klotho that had been preabsorbed by recombinant Klotho, which depleted reaction of the antibody with the 60 kDa polypeptide (Fig. 2D). Finally, we further confirmed the 60 kDa polypeptide was Klotho by performing a Klotho knockdown in bone-marrow-derived macrophages (BMDMs), which nearly eliminated the 60 kDa immunoreactive band (Fig. 2E), although a control preparation using a scrambled siRNA construct caused no reduction in the 60 kDa polypeptide.
We confirmed induction of the M1 phenotype in macrophages by TNFα and IFNγ by showing that stimulation produced large increases in the expression of transcripts that are elevated in M1 macrophages (TNFα; inducible nitric oxide synthase [iNOS]) and a strong trend for elevated IFNγ expression (Fig. 2F–H). We also confirmed elevated expression of transcripts that are markers of the M2 phenotype (CD206; CD163) in macrophages activated by IL10 and IL4 (Fig. 2I and J). QPCR data show that activation to the M2 phenotype significantly increased Klotho expression (Fig. 2K). However, unexpectedly, activation to the M1 phenotype also increased Klotho expression in macrophages (Fig. 2K), in contrast to the suppression of KL expression in muscle cells experiencing the same treatment protocol (Fig. 1F). Thus, a pro-inflammatory environment in dystrophic muscle can reduce overall Klotho expression in muscle by reductions in muscle-derived Klotho but lead to local increases in Klotho production by macrophages in inflammatory lesions.
Klotho increases the production of TNFα, IL10 and IL4 by macrophages
The finding that the reduction of Klotho expression in mdx muscles coincided with increased expression of pro-inflammatory cytokines (TNFα and IFNγ; Fig. 1C and D) and increased expression of IL10 (27) suggested that Klotho may regulate the expression of cytokines in dystrophic muscle. We found that stimulation of BMDM with Klotho alone produced significant increases in the secretion of TNFα, IL10 and IL4 and a strong trend for elevated IFNγ (Fig. 3A–D). Co-stimulation of BMDM with Klotho combined with FGF23 produced even larger increases in TNFα in the conditioned media, reaching 530.3 pg/ml (S.E.M. = 3.6; n = 6) at the end of the 48-h treatment period. However, co-stimulation with FGF23 combined with Klotho produced only an insignificant trend for increased IL10 (6.0 pg/ml, S.E.M. = 0.6, n = 6) and IL4 (20.6 pg/ml, S.E.M. = 2.9; n = 6) compared with Klotho alone (IL10: 4.5 pg/ml, S.E.M. = 0.7, n = 6; IL4: 12.7 pg/ml, S.E.M. = 5.2, n = 6). Klotho secretion of IFNγ did not differ between BMDMs stimulated with both Klotho and FGF23 (1.1 pg/ml, S.E.M. = 0.7, n = 6) and BMDMs stimulated with Klotho alone (0.9 pg/ml, S.E.M. = 0.5; n = 6).
Figure 3.

Klotho increases the expression of pro-inflammatory and anti-inflammatory cytokines. (A–D) Conditioned media from BMDMs stimulated in vitro with Klotho were assayed by ELISA for levels of TNFα (A), IFNγ (B), IL10 (C) and IL4 (D). * indicates significant difference from cytokine concentration in control cultures at P < 0.05. (E–G) Expression levels of TNFα (E), IFNγ (F) and IL10 (G) mRNA in 6-month-old quadriceps muscles from wild-type, mdx and KL+/mdx mice. N = 8 per control data set. N = 6 per Klotho treated data set. * indicates significant difference from expression in wild-type control muscles at P < 0.05. # indicates significant difference from expression in mdx muscles at P < 0.05.
We then assayed whether increases in Klotho in vivo could similarly affect cytokine expression by assaying whether expression of a Klotho transgene in mdx mice influenced cytokine mRNA levels. We observed elevations in TNFα, IL10 and IFNγ expression in 6-month-old quadriceps muscles of mdx mice, compared with wild-type controls, and found that expression of a Klotho transgene further elevated TNFα expression, although the transgene had no effect on the expression of either IL10 or IFNγ in mdx mice (Fig. 3E–G). Thus, in vivo and in vitro data show that increases in Klotho are sufficient to increase TNFα in muscle.
Klotho and CD206+ M2 macrophages are co-regulated in vivo
The observation that CD206+ macrophages are the primary sites of detectible expression of Klotho in mdx muscles and the finding that stimulation of BMDMs with IL10 and IL4 promoted the expression of both Klotho and CD206 suggested that the two transcripts may also be co-regulated in vivo. We tested that possibility first by assaying the effect of Klotho transgene expression on CD206 expression in mdx muscles and found a significant increase in CD206 mRNA in the transgenic muscles (Fig. 4A). We also found that Klotho transgene expression increased the concentration of CD206+ macrophages in mdx muscle while reducing numbers of CD68+ macrophages (Fig. 4B), indicating that Klotho promotes a shift of macrophages toward the pro-regenerative M2 phenotype in vivo. In addition, the expression of both CD206 and Klotho were positively influenced by IL10 expression in mdx muscles; null mutation of IL10 in mdx mice caused similar reductions in the expression of both transcripts (Fig. 4C and D).
Figure 4.

Klotho and CD206 expressing macrophages are co-regulated in vivo. (A) QPCR assay of CD206 expression levels in 6-month-old quadriceps muscle from mdx and KL+/mdx mice. N = 5 per data set. * indicates significant difference from expression in control muscles at P < 0.05. (B) Numbers of CD68+ macrophages and CD206+ macrophages in quadriceps muscles from 6-month-old mdx and KL+/mdx mice. N = 5 per data set. * indicates significant difference from expression in mdx muscles at P < 0.05. (C) Numbers of CD206+ macrophages in quadriceps muscles from 6-month-old mdx and IL10 null mutant mdx mice. N = 5 per data set. * indicates significant difference from expression in mdx muscles at P < 0.05. (D) QPCR assay of Klotho expression levels in 6-month-old quadriceps muscle from mdx and IL10 null mutant mdx mice. N = 5 per data set. * indicates significant difference from expression in mdx muscles at P < 0.05.
Macrophage-derived Klotho increases muscle cell proliferation and M2 macrophage numbers
M2 macrophages can increase muscle growth and regeneration in vivo (28) and increase muscle cell proliferation in vitro (29–31). Because expression of a Klotho transgene in mdx mice caused increases in the numbers of Pax7-expressing muscle stem cells, called satellite cells (17), we tested whether macrophage-derived Klotho could increase satellite cell numbers in vivo and in vitro. We tested this possibility first in vivo by transplanting BMCs from wild-type mice or Klotho transgenic mice into mdx recipients. QPCR data showed that at 6-months post-transplantation, muscles from mdx mice that received bone marrow transplantation (BMT) of cells from Klotho transgenic mice expressed 32-times more Klotho than mdx recipients that received wild-type BMT (Fig. 5A). In addition, mdx mice that received BMCs from Klotho transgenic donors showed a 91% increase in the number of Pax7+ cells/muscle fiber and a 61% increase in Pax7+ cells per unit volume of muscle, compared with mdx mice receiving wild-type BMCs (Fig. 5B and C). Those increases in satellite cell numbers were associated with increased muscle fiber size (Fig. 5D), where the cross-sectional area of mdx recipients of Klotho transgenic BMCs were 41% larger than recipients of wild-type BMCs (Fig. 5E). Despite the increase in fiber size, Klotho transgenic BMT did not affect the muscle mass to body mass ratio, compared with wild-type BMC recipients (Supplementary Material, Fig. S2A). Unlike the previously reported findings which showed that systemic expression of a Klotho transgene increased numbers of muscle fibers in mdx mice (17), transplantation of Klotho transgenic BMCs did not affect numbers of muscle fibers, compared with wild-type recipients (Supplementary Material, Fig. S2B). However, transplantation of Klotho transgenic BMCs into mdx recipients increased the numbers of CD206+ M2 macrophages in muscle, compared with mdx mice that received BMT of wild-type cells (Fig. 5F), which is similar to the increase achieved by systemic expression of a Klotho transgene in mdx mice. Collectively, these observations show that macrophage-derived Klotho can regulate satellite cell numbers and muscle fiber growth in vivo and increase numbers of macrophages that can promote muscle regeneration.
Figure 5.

Transplantation of BMCs from Klotho transgenic mice increases Klotho delivery to mdx muscles and increases satellite cell numbers, muscle fiber size and CD206+ M2 macrophages. (A) QPCR analysis of Klotho expression in tibialis anterior muscles of mdx mice that received BMT from wild-type mice or from Klotho transgenic mice show BMT transplantation from transgenic mice causes a 32-fold greater level of Klotho expression in the muscle. For all panels in Figure 5, white bars are data from mdx mice receiving wild-type BMT. Black bars are data from mdx mice receiving Klotho transgenic BMT. (B,C) BMT of Klotho transgenic BMCs caused significantly higher numbers of satellite cells per muscle fiber (B) and per unit volume of muscle (C), compared with mdx mice receiving wild-type BMT. (D,E) BMT of Klotho transgenic BMCs caused significantly larger muscle fiber cross-sectional areas, compared with mdx mice receiving wild-type BMT. Data are shown as frequency distribution of fiber cross-sectional areas for tibialis anterior muscles from mdx mice receiving wild-type BMT (white bars) or Klotho transgenic BMT (black bars) (D) and shown as mean value of cross-sectional areas of the same groups (E). (F) Transplantation of Klotho transgenic BMCs caused significantly more CD206+ M2 macrophages to occur in mdx muscles at 6 months BMT, compared with mice receiving wild-type BMT. N = 5 per data set. * indicates significant difference from expression in mdx muscles at P < 0.05.
We next assayed whether macrophage-derived Klotho acted directly on myogenic cells to affect their proliferation. Conditioned media were collected from cultures of wild-type control BMDMs, Klotho transgenic BMDMs and Klotho hypomorphic mutant BMDMs and transferred to myoblast cultures for 48 h. Conditioned media from Klotho transgenic cultures caused large increases in myogenic cell numbers that were prevented by anti-Klotho, but unaffected by treatment with isotype control antibodies (Fig. 6A). In contrast, conditioned media from Klotho mutant BMDM cultures produced no increase in myogenic cell numbers compared with cultures that did not receive conditioned media (Fig. 6A). In addition, cell numbers in myogenic cell cultures that received media from Klotho mutant BMDM cultures treated with anti-Klotho or isotype control antibodies did not differ from Klotho mutant media that received no antibody treatment (Fig. 6A). These data show that much of the pro-proliferative effect of macrophages on myogenic cells is attributable to Klotho.
Figure 6.

Klotho increases satellite cell proliferation through a TNFα-mediated mechanism. (A) Myoblast cell numbers per well after growth in culture media only (Control; white bar) or conditioned media from wild-type BMDMs, Klotho transgenic BMDMs (KL Tg) or Klotho hypomorphic mutants (KL mutants). For each sample group grown with conditioned media, 5 wells contained conditioned media only (grey bars), 5 wells contained conditioned media to which isotype control IgG had been added (black bars) and 5 wells contained conditioned media with anti-Klotho (striped bars). * indicates significant difference from control cultures at P < 0.05. # indicates significant difference from isotype control cultures from same conditioned media culture group at P < 0.05. (B) Myoblast cell numbers per well after growth in culture media only or culture media containing recombinant Klotho (KL treated). For each of those treatment groups, 5 wells contained no antibody (white bars), 5 wells contained isotype control antibody (grey bars) and 5 wells contained anti-TNFα (black bars). * indicates significant difference from control cultures at P < 0.05. # indicates significant difference from isotype control cultures at P < 0.05. (C) Protein concentration in isovolumetric homogenates of myotubes grown and stimulated under same conditions as myoblasts in Figure 6C. * indicates significant difference from control cultures at P < 0.05.
Much of the mitogenic activity of Klotho on myogenic cells occurs through TNFα-mediated signaling
TNFα is a strong mitogen for myogenic cells (32) which suggests the possibility that the increase in myogenic cell numbers caused by Klotho may be mediated by Klotho induction of TNFα. Because previous findings also showed that Klotho has anabolic effects on myotubes (17), we assayed whether the effects of Klotho on myotube protein content are also mediated by TNFα. We found that the increase in myoblast numbers in vitro that resulted from Klotho stimulation was significantly reduced by the presence of anti-TNFα in the culture media, but not influenced by addition of isotype control antibody (Fig. 6B). In addition, myoblast numbers in cultures that were not treated with Klotho were unaffected by addition of anti-TNFα, indicating that the anti-proliferative effect of anti-TNFα negatively affected proliferation promoted by Klotho but did not inhibit proliferation that occurred independent of Klotho stimulation (Fig. 6B). We also found that direct application of Klotho to myotube cultures increased protein content of the myotubes, as demonstrated previously (17), although addition of anti-TNFα did not diminish the anabolic effects of Klotho on myotubes (Fig. 6C). Thus, the mitogenic effect of Klotho on muscle cells is largely mediated by TNFα, although Klotho induction of protein accumulation is not.
Klotho is most highly expressed in CD206+ macrophages at sites where muscle undergoes early stages of regeneration
Because Klotho can promote myogenesis in vitro, we tested whether the distribution of Klotho-expressing M2 macrophages in dystrophic muscle related to the stage of mdx pathology. At 1-month of age, nearly all Klotho-expressing cells in mdx muscle were CD206+ macrophages (Fig. 7A). At 3-months of age, clusters of CD206+ macrophages within the mdx muscle parenchyma expressed Klotho, although CD206+ macrophages residing in the epimysium, the layer of connective tissue at the muscle surface, did not express detectible Klotho (Fig. 7B). We tested whether the clusters of KL+/CD206+ macrophages were associated with sites of muscle regeneration by co-labeling with antibodies to Klotho and to developmental myosin heavy chain (dMyHC), and found that the highest concentrations of Klotho+ cells were located at sites where muscle regeneration was at early stages of myogenesis, occupied by small, dMyHC+ myotubes (Fig. 7C). In contrast, relatively little Klotho was expressed by cells that were located on the surface of large, regenerative, central-nucleated muscle fibers at late stages of regeneration (Fig. 7D). Finally, we assayed for the distribution of Klotho+/CD206+ cells in 2-year-old mdx mice that are at advanced stages of pathology when muscle regeneration is deficient and found little Klotho expression in CD206+ macrophages (Fig. 7E and F), suggesting an age-related decline in Klotho expression by myeloid cells, as occurs in other tissues (13).
Figure 7.
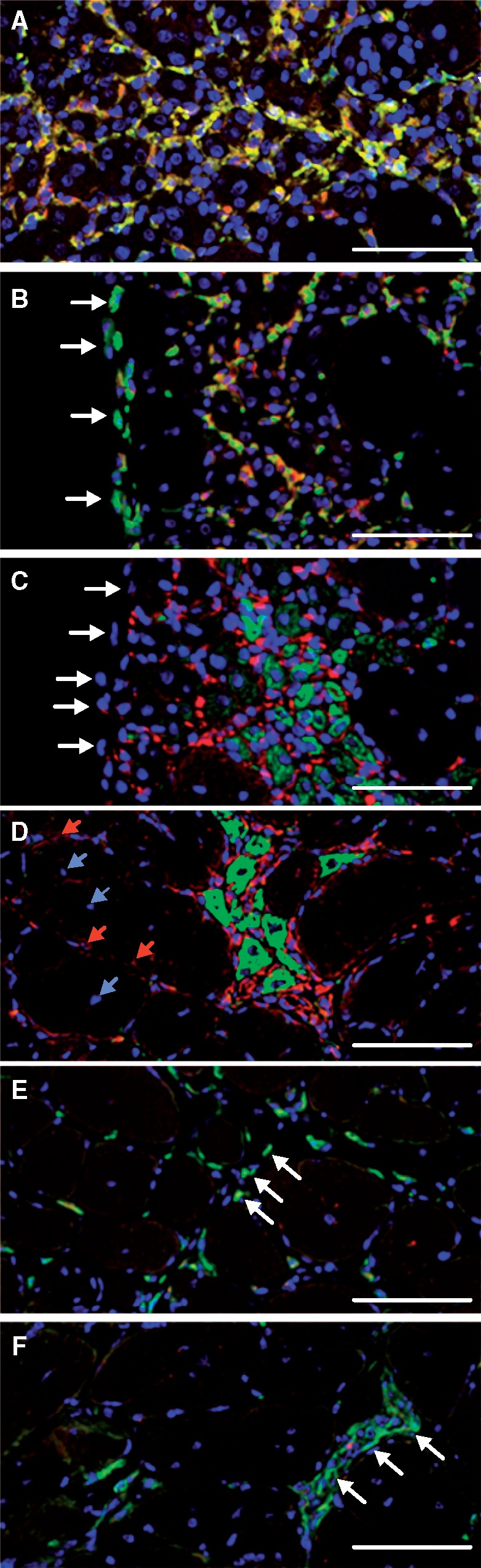
Klotho is expressed by M2 macrophages at sites of muscle regeneration in adult mdx muscles but not senescent mdx muscle. (A) Section of 4-week-old mdx quadriceps muscle labeled with antibodies to CD206 (green) and Klotho (red). Nuclei are labeled with DAPI (blue). Double-positive KL+/CD206+ macrophages are yellow or orange. (B) Section of 3-month-old mdx quadriceps muscle with antibodies to CD206 (green) and Klotho (red). Macrophages in the epimysium at the surface of the muscle (arrows) are KL-/CD206+. Most macrophages in inflammatory site in muscle (center of figure) are KL+/CD206+ (yellow or orange). (C) Section from muscle shown in Figure 6B that was taken adjacent to the section in Figure 6B. Section was stained with antibodies to Klotho (red) or developmental myosin heavy chain (green) to indicate sites of muscle regeneration. Cells in the epimysium at the surface of the muscle (arrows) do not express Klotho. (D) Section of a 3-month-old quadriceps muscle stained with antibodies to Klotho (red) or developmental myosin heavy chain (green) showed the high concentration of M2 macrophages expressing elevated levels of Klotho at sites of muscle regeneration. Central nuclei of large, regenerative muscle fibers are indicated with blue arrows. Macrophages at surface of regenerative fibers express low levels of Klotho (red arrows) (E) Section of 24-month-old mdx quadriceps stained with antibodies to CD206 (green) and Klotho (red) showing that CD206+ macrophages distributed throughout the thickened endomysial tissue (arrows) do not express Klotho. (F) Section of 24-month-old mdx quadriceps stained with antibodies to CD206 (green) and Klotho (red) showing an inflammatory lesion (arrows) occupied by elevated numbers of KL-/CD206+ macrophages. Bars = 50 µm.
Discussion
The results of the present investigation show that macrophage-derived Klotho can have significant influences on both myogenesis and inflammation in dystrophic muscle. These findings were particularly unexpected by us, because previous work has shown that the expression of Klotho by muscle cells is silenced in dystrophic muscle (17), unlike the elevated expression of Klotho in CD206+ M2 macrophages in the same tissue that is reported here. Several observations show that macrophage-derived Klotho can enhance the regenerative capacity of dystrophic muscle. First, expression of Klotho by leukocytes that are derived from BMCs increases the number of satellite cells in dystrophic muscle in vivo. In addition, Klotho+/CD206+ macrophages are concentrated at sites of regeneration in dystrophic muscle and macrophage-derived Klotho can act directly on muscle cells to increase their proliferation and growth. Finally, elevated expression of Klotho in dystrophic muscle biases macrophage populations toward an M2 macrophage phenotype that can promote muscle growth and regeneration (33). These observations may be particularly significant in DMD because loss of the renewal capacity of satellite cells in dystrophic muscle may contribute to impaired regeneration of dystrophic muscle (17,34). Collectively, these findings support a model in which M2-biased macrophages specifically target Klotho to sites of regeneration in dystrophic muscle, which increases the regenerative capacity of muscle (Fig. 8).
Figure 8.

Schematic representation of the regulatory interactions between inflammation and dystrophic muscle cells that are influenced by Klotho. Direct action of pro-inflammatory cytokines on muscle cells reduces Klotho expression (present investigation) which contributes to loss of satellite cells in aging, dystrophic muscle.
Although the mechanism through which Klotho increases the numbers of satellite cells in dystrophic muscles is unknown, previous work has shown that the effect is attributable to increased proliferation and is not a consequence of reductions in myoblast necrosis or apoptosis (17). As shown by the current findings, at least part of the pro-proliferative effect is mediated by TNFα. Several previous investigations have shown that TNFα can increase muscle growth and regeneration by direct actions on myogenic cells. For example, TNFα promotes myoblast proliferation (32) and inhibits differentiation (35–37) through a process that depends in part on activation of NFκB and the subsequent increase in the expression and stability of cyclin D1 (35,38,39). Our finding that the direct application of Klotho to myogenic cells in vitro increases cell numbers but that the increase is diminished by TNFα neutralizing antibodies shows that myogenic cells themselves are a source of the TNFα that then acts through an autocrine/paracrine pathway.
The observation that CD206+ macrophages in dystrophic muscles express high levels of Klotho while Klotho is silenced in dystrophic muscle fibers at the onset of pathology shows that Klotho silencing in muscular dystrophy is cell type selective. Previous work has already demonstrated that Klotho silencing in mdx dystrophy was tissue selective, in which Klotho silencing in mdx muscle tissue coincided with increased methylation at the promoter region of the gene and a significant increase in a repressive histone mark, dimethylated lysine 9 on histone 3 (H3K9) (17). Klotho silencing in mdx muscle is also associated with increases in methylation of the Klotho gene promoter region at sites enriched in cytosines linked to guanine nucleotides by a single phosphate (CpG sites) (17). Klotho gene methylation at CpG sites in kidneys, cancer cells and neurons has also been linked to Klotho silencing (40–45). However, dystrophin-deficiency did not cause Klotho silencing in other tissues in mdx mice, including kidney, spleen and liver, showing that Klotho silencing was caused by local conditions in the pathological muscle (17). We expect that local elevations in oxidative stress underlie Klotho silencing, at least in part, because increased dimethylation of H3K9 on the Klotho gene in muscle cells was associated with increased oxidative stress (17) and oxidative stress occurs at high levels in inflammatory lesions in dystrophic muscle (46,47). In addition, others have shown that oxidative stress is sufficient to silence Klotho in other tissues and pathologies (45). However, we show in the present investigation that inflammatory lesions in dystrophic muscle are the sites of highest expression of Klotho by macrophages. That observation could suggest the possibility that Klotho was expressed before the macrophages migrated into inflammatory lesions and were exposed to elevated oxidative stress. However, the epimysium is a major source for myeloid cells that migrate to sites of muscle injury after their activation (48) and we found that epimysial CD206+ macrophages did not express Klotho, although CD206+ macrophages that expressed high levels of Klotho were located in inflammatory lesions within 100 µm from the epimysium. That finding indicates that Klotho is not elevated in CD206+ macrophages before their migration to inflammatory sites.
An alternative mechanism through which macrophages may be spared Klotho silencing by oxidative stress would be through their high levels of expression of anti-oxidants that protect them from free radical-mediated effects. This self-protection of macrophages from damage by oxidative stress is an adaptive trait that enables macrophages to survive the cytotoxic levels of free radicals they generate which are sufficient to kill infectious organisms. For example, macrophages and other leukocytes in inflammatory lesions can produce hypochlorous acid, which is a potentially cytotoxic free radical that can induce tremendous oxidative stress in tissues (49,50). Macrophages also produce nitric oxide and superoxide at concentrations that are sufficient to lyse muscle cells in co-cultures without causing damage to macrophages in the cultures (51). Macrophages are better protected from detrimental effects of hypochlorous acid and other free radicals by their relatively high levels of expression of anti-oxidants, especially glutathione (52). Furthermore, Klotho induces expression of manganese superoxide dismutase which can also increase resistance to oxidative stress by removal of reactive oxygen species, at least in HeLa cells (53). Thus, muscle fibers may experience more oxidative stress than experienced by macrophages in the same lesion, and their subsequent loss of Klotho expression would further increase their susceptibility to oxidative stress. The mechanism through which macrophages in dystrophic muscle escape Klotho silencing is under continuing study.
Our findings that the pro-inflammatory cytokines TNFα and IFNγ increase Klotho expression in macrophages show that regulatory relationships between pro-inflammatory cytokines and Klotho in inflammatory cells differs from other cells, at least in vitro. For example, we found that identical treatment conditions using TNFα and IFNγ reduced Klotho expression in myotubes by more than 50% but increased Klotho expression in macrophages by more than 50%. Similarly, treatments of cultured kidney tubular cells with TNFα at a range of concentrations that bracket the concentration used in the present investigation, produced reductions of Klotho expression by 50% or more (22). Likewise, TNFα treatment of a distal convoluted tubule cell line at the same concentration as used in the present investigation caused reductions in Klotho expression by nearly 50%, that was further reduced by co-stimulation with IFNγ (19). However, an earlier investigation found no reduction of Klotho expression in renal proximal tubular epithelial cells caused by TNFα, but the concentration of TNFα was lower in that investigation (18). Although the mechanism through which pro-inflammatory cytokines reduce Klotho expression in kidney and muscle cells is not known definitively, several observations suggest that the effect is attributable to TNFα induction of free radical production. In particular, TNFα and IFNγ induction of iNOS expression leads to large increases in nitric oxide (NO) production and NO can be further metabolized to other free radicals that can increase oxidative stress in tissues. Treatment of kidney cells in vitro with TNFα and IFNγ produced elevations in iNOS expression that accompany reductions in Klotho expression (19). In addition, treatment of kidney cells with an NO donor caused reductions in Klotho expression that were similar to reductions caused by TNFα and IFNγ treatments (19). Furthermore, the TNFα/IFNγ induced down-regulation of Klotho occurs at the transcriptional level, again indicating Klotho gene silencing by oxidative stress (19).
Although our findings indicate that M2-biased macrophages escape silencing of the Klotho gene in dystrophic muscle and that macrophage-derived Klotho increases satellite cell populations and promotes muscle growth, other investigations have shown that M2 macrophages can also worsen the pathology of muscular dystrophy by increasing muscle fibrosis. M2 macrophages express arginase which metabolizes arginine to produce pro-fibrotic molecules such as polyamines and ornithine, which is further metabolized to produce proline (54–57), creating a pro-fibrotic environment (58). During progressive stages of mdx dystrophy when muscle fibrosis becomes more prominent, the levels of arginase expression in intramuscular M2 macrophages increase continuously (59). Furthermore, ablation of arginase-2 expression significantly reduces fibrosis in mdx muscles at advanced stages of the pathology, showing that arginine metabolism contributes significantly to mdx pathology (59). Other observations implicate CD206+ M2 macrophages with increased muscle fibrosis in DMD, where they are enriched at sites of increased endomysial fibrosis (46). Thus, M2 macrophages can play beneficial roles in mdx dystrophy through Klotho-mediated promotion of muscle growth and regeneration, or detrimental roles through arginase-mediated muscle fibrosis. Unfortunately, the age-related loss of Klotho expression in mdx macrophages (current investigation) and age-related increase in arginase expression (59) appear to produce an M2 macrophage population that may be primarily pathogenic at late stages of the disease. However, experimental interventions that increase expression of Klotho counter the elevated expression of pro-fibrotic transcripts and prevent the accumulation of connective tissue in dystrophic muscle at progressive stages of mdx dystrophy (17). Thus, Klotho delivery to dystrophic muscle can reduce the pathology of muscular dystrophy at early and progressive stages of the pathology by reducing fibrosis, as well as improving growth and maintaining satellite cell populations.
Materials and Methods
Mice
All animals were handled according to guidelines provided by the Chancellor’s Animal Research Committee at the University of California, Los Angeles. C57BL/10ScSn-Dmdmdx/J mice (mdx mice), B10.129P2(B6)-Il10tm1Cgn/J mice (IL10 mutant mice), B6.129S-Tnftm1Gkl/J (TNFα mutant mice) and C57BL/6 (wild-type mice) were purchased from The Jackson Laboratory (Bar Harbor, ME) and bred in specific pathogen-free vivaria at the University of California, Los Angeles. The production of Klotho hypomorphic mutants and the generation of transgenic mice that overexpress Klotho (EFmKL46) have been described previously (13). The Klotho transgene is under control of the elongation factor 1α promoter that causes systemic expression of the transcript. Mice carrying the Klotho transgene were crossed into the C57BL/6 background for a minimum of six generations and then crossed into the mdx background using a previously described breeding strategy (3,60). Expression of the Klotho transgene was confirmed by qPCR and Western blot. Null mutation of the dystrophin gene was confirmed using mdx-amplification-resistant mutation system PCR (61). Primers are listed in Supplementary Material, Table S1.
IL10 mutant mice were back-crossed onto a C57/BL6 background and then crossed with mdx mice to generate mice that lacked both IL-10 and dystrophin using a breeding strategy previously described (3,27). Null mutation of IL-10 was confirmed by PCR using an upstream primer that was common for both wild-type and mutant DNA, a wild-type downstream primer and a downstream primer for the neomycin cassette.
RNA isolation and quantitative PCR
RNA was isolated from muscle homogenates (17) and then electrophoresed on agarose gels and its quality assessed by 28S and 18S ribosomal RNA integrity. RNA samples (2 μg) were reverse transcribed with Super Script Reverse Transcriptase II using oligo dTs to prime extension (Invitrogen) to produce cDNA. Expression of selected transcripts was assayed using SYBR green qPCR Supermix according to the manufacturer’s protocol (BioRad) and an iCycler thermocycler system equipped with iQ5 optical system software (BioRad). We used established guidelines for experimental design, data normalization and data analysis for quantitative PCR (QPCR) to maximize the rigor of quantifying the relative levels of mRNA (62,63). We empirically identified reference genes that did not vary between experimental groups using geNorm 3.5 software (27). The normalization factor for each sample was calculated by geometric averaging of the Ct values of reference genes using the geNorm software. Expression for each gene in control samples was set to 1 and the other values were to scaled the control. Primers used for QPCR are listed in Supplementary Material, Table S1.
Western blots
Expression levels of Klotho were assayed by western blot analysis, using 30 μg total protein per lane. Equal loading of samples was confirmed by staining nitrocellulose blots with 0.1% Ponceau S solution (Sigma). Nitrocellulose membranes were blocked with 50 mM sodium phosphate buffer pH 7.4 containing 200 mM sodium chloride (phosphate-buffered saline; PBS) and 0.1% Tween-20 and 3% non-fat dried milk for 24 h at 4 °C. The membranes were probed with rabbit anti-mouse Klotho (Abcam #75023) for 2 h at room temperature. Membranes were washed with PBS containing 0.05% Tween-20 and probed with horseradish peroxidase (HRP) conjugated-donkey anti-rabbit IgG (Amersham) for 1 h. Membranes were washed and Klotho was visualized with chemiluminescent substrate (FemtoGlow; Michigan Diagnostics) and a fluorochem imaging system (Syngene PXi). Specificity of the anti-Klotho was tested by comparing blots treated with anti-Klotho or with anti-Klotho that had been pre-incubated with recombinant Klotho (R&D Systems) to deplete Klotho specific immunoglobins from the solution.
Production of Pax7 antibody and immunohistochemistry
Pax7 hybridoma cells were purchased from Developmental Studies Hybridoma Bank (Iowa City Iowa). Cells were cultured in complete medium consisting of DMEM with 1% penicillin- streptomycin (Gibco) and 20% heat-inactivated fetal bovine serum (FBS). Conditioned medium was collected from the cultures and used for purification of antibodies to Pax7, as described previously (64).
Muscles were dissected from euthanized mice and rapidly frozen in liquid nitrogen-cooled isopentane. Cross-sections 10-µm thick were taken from the mid-belly of muscles. Sections were then fixed in 2% paraformaldedye for 10 min and then immersed in antigen retrieval buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) at 95–100° C for 40 min. Endogenous peroxidase activity in the sectioned tissue was quenched by immersion in 0.3% H2O2. Sections were then treated with blocking buffer from a mouse-on-mouse immunohistochemistry kit (M.O.M kit; Vector) for 1 h and immunolabeled with mouse anti-Pax7 antibody overnight at 4° C. Specificity of the antibody for Pax7 was confirmed, as described previously (17). Sections were washed with PBS and then incubated with biotin-conjugated anti-mouse IgG for 30 min. Sections were subsequently washed with PBS and then incubated for 30 min with ABC reagents from the M.O.M kit. Staining was visualized with the peroxidase substrate 3-amino-9-ethylcarbazole (AEC kit; Vector), yielding a red reaction product. The number of satellite cells/sectioned muscle fiber was determined by counting the number of Pax7+ cells in mid-belly cross-sections of muscles and the total number of fibers per cross-section.
For identification of Klotho+ or CD206+ cells for bright-field microscopy, acetone-fixed frozen sections of quadriceps were blocked in 3% bovine serum albumin (BSA) and 2% gelatin in 50 mM Tris buffer (pH 7.2) for 1 h and then immunolabeled with rat anti-CD206 (1/50; Serotec) for 3 h at room temperature or with rabbit anti-Klotho (1/50) overnight at 4°C. Sections were washed with PBS and then probed with biotin-conjugated secondary antibodies (1/200; Vector Laboratories) for 30 min. Sections were subsequently washed with PBS and then incubated for 30 min with avidin D-conjugated horseradish peroxidase (1/1000; Vector Laboratories). Staining was visualized with the AEC kit.
Macrophages that were double positive for Klotho and CD206 were identified in muscle sections by indirect immunofluorescence. Sections were incubated overnight at 4° C with anti-Klotho followed by labeling with Texas Red-conjugated anti-rat IgG secondary antibody (Vector). Sections were then washed, blocked, and incubated with rat anti-CD206 for 3 h at room temperature. Sections were subsequently washed and incubated with fluorescein-conjugated anti-rat IgG secondary antibody (Vector). Sections were washed with PBS and treated with RNase A (Sigma, 0.2 ug/ml) and DAPI diluted 1: 100 000 in PBS for 5 min. Following washes with PBS, sections were mounted with Gel Mount (Biomedia) and glass coverslips.
Stereology
The number of cells/volume of muscle was determined by measuring the total volume of each section using a stereological, point-counting technique to determine section area and then multiplying that value by the section thickness (10 μm) (3). The numbers of immunolabeled cells in each section were counted and expressed as the number of cells/unit volume of each section.
Fiber size and number
Cross-sections of tibialis anterior muscles were sectioned at their midbelly and used for fiber cross-sectional area measurements and fiber number counts. Sections were stained with a hematoxylin solution, and all fibers were counted in each cross-section were counted and the cross-sectional area of 500 fibers was measured using a digital imaging system (Bioquant).
TNFα and IFNγ stimulation of myoblasts and myotubes
Murine myoblasts (C2C12; ATCC) were seeded in six-well plates in complete DMEM with 100 U/ml penicillin, 100 µg/ml streptomycin and 10% FBS. When cells reached confluence, cultures were transferred to differentiation medium (DMEM with 100 U/ml penicillin, 100 µg/ml streptomycin) overnight to induce differentiation and fusion to form myotubes. Cultures were then returned to complete medium to allow growth and differentiation for 72 h, before cytokine treatments. Myotube cultures were then treated for two, 24 h periods with 10 ng/ml of IFNγ or 20 ng/ml of TNFα or both IFNγ and TNFα or complete medium only, as control preparation. Myotubes were then collected in reducing sample buffer (80 mM Tris-HCl pH 6.8, 0.1 M dithiothreitol, 70 mM dodecyl sulfate, 1.0 mM glycerol) with protease inhibitor cocktail (1: 100, Sigma). Protein concentration was quantified as previously described (65). Relative quantities of Klotho in treatment groups were then assessed by western blotting as described above. Each group included six replicates.
Preparation of bone marrow-derived macrophages
Bone marrow cells (BMCs) were aseptically flushed from wild-type femurs and tibiae with Dulbecco’s phosphate buffered saline (DPBS; Sigma, St. Louis, MO) and treated with ACK lysing buffer (Gibco, Waltham, MA) to clear red blood cells. Following a DPBS wash and filtration through a 70-µm filter, BMCs were seeded at 5 × 106 per 6-cm dish in RPMI-1640 (Sigma) with 20% heat-inactivated fetal bovine serum (FBS; Omega Scientific, Tarzana, CA), penicillin (100 U/ml; Gibco), streptomycin (100 µg/ml; Gibco) and 10 ng/ml MCSF (R&D, Minneapolis, MN) at 37˚C with 5% C02 for 6 days. For some experiments, adherent cells were released by brief trypsinization (Gibco), washed with DPBS and filtered through a 70-µm mesh filter before blocking with anti-CD16/32 and staining with anti-F4/80-APC (eBioscience, San Diego, CA). Flow cytometry was performed on a FACS Calibur (Becton-Dickinson, San Jose, CA) to confirm F4/80+ bone marrow-derived macrophages (BMDMs). For other experiments, adherent cells were stimulated for 24 h with activation media consisting of Dulbecco’s Modified Eagle Medium (DMEM) with 0.25% heat-inactivated FBS, penicillin–streptomycin, 10 ng/ml MCSF and either Th1 cytokines (10 ng/ml IFNγ and 10 ng/ml TNFα) or Th2 cytokines (10 ng/ml IL-10 + 25 ng/ml IL-4) (BD Pharmingen).
In vitro Klotho siRNA knockdown
BMCs were aseptically prepared and cultured as described above. After 6 days in culture, the BMDMs were switched to DMEM only (Sigma) for 24 h prior to transfection. BMDM were transfected using lipofectamine 2000 (Invitrogen) alone (untreated) or with 100 nM Klotho Stealth RNAi siRNA oligos (Stealth RNAi siRNA MSS205816, Invitrogen) or 100 nM scrambled oligos (Stealth RNAi siRNA 12935–300, Invitrogen) for 6 h in Opti-MEM medium (Gibco). After transfection, cultures were changed to DMEM only for 24 h to allow for gene knockdown. BMDM were collected in reducing sample buffer with protease inhibitors and prepared for Western blot analysis of Klotho expression.
ELISA analysis of BMDM-conditioned media
Cultures of BMDM from wild-type mice were established as described above. On the sixth day of culture, the BMDM were switched to DMEM containing penicillin/streptomycin, 10 µm/ml heparin (Sigma), 0.25% heat-inactivated FBS and M-CSF (control media) or media containing 1 µg/ml recombinant mouse Klotho (R&D) alone, or in combination with 0.5 µg/ml FGF23 (R&D) (stimulation media). After 24 h of stimulation, the BMDM received fresh control media or stimulation media and were cultured for an additional 24 h. At the end of 48-h stimulation, conditioned media were collected, aliquoted, briefly centrifuged to remove particulates and then frozen at −20˚C. Separate aliquots of BMDM-conditioned media from each sample were analysed for expression of pro-inflammatory (TNFα, IFNγ) and anti-inflammatory (IL10, IL4) cytokines by ELISA, according to manufacturer’s instructions (Quantikine ELISA, R&D). Each group contained at least six replicates.
Bone marrow transplantation
Beginning one week prior to bone marrow transplantation (BMT) mouse drinking water was supplemented with trimethoprim/sulfamethoxazole (80 μg/ml trimethoprim and 400 μg/ml sulfamethoxazole) and continued for 3-weeks. Two-month-old female mdx mice underwent myeloablative preconditioning via three consecutive intraperitoneal injections of 1, 4-butanediol dimethanesulfonate (Sigma-Aldrich) (20 mg/kg body weight) 72, 48 and 24 h prior to BMT. On the day of transplantation, male donor mice were sacrificed and their femur and tibia bones were sterilely dissected and flushed of BMCs. BMCs were isolated and recipient mice received 107 donor BMCs by tail-vein injection. At 6-month post-BMT, tissues and BMCs were collected from recipient mice. BMCs were used for chimerism analysis by fluorescent in situ hybridization of the Y-chromosome (Kreatech FISH Probes) (bone marrow leukocytes donor-derived: 83.61%; S.E.M. = 1.24; n = 24).
In vitro assays for muscle cell proliferation
For all proliferation assays, myoblasts were seeded in six-well plates at 60 000 cells per well in complete DMEM (with 100 U/ml penicillin, 100 µg/ml streptomycin and 10% FBS) for 24 h at 37˚C in 5% CO2 before treatments. Myoblasts were approximately 30% confluent at the onset of treatments and received fresh, treatment media at 24 and 48-h post-plating. The myoblasts were approximately 90% confluent and not yet fused when the cells were collected at 72-h post-plating. Myoblasts were trypsinized with 1 ml 0.05% trypsin-EDTA, washed with DPBS, stained with trypan blue and counted using a hemocytometer to determine how the various treatments affected proliferation. Each group contained at least five replicates.
For myoblast treatments with BMDM-conditioned media, BMDM cultures were established from wild-type, KL Tg or KL mutant mice as detailed above. On day 6 of culture, BMDM were switched to DMEM with penicillin/streptomycin, 0.25% heat-inactivated FBS and MCSF. After 24 h of culture, the BMDM-conditioned medium was collected, briefly centrifuged to remove particulates and applied to previously plated myoblast cultures. Control wells received non-conditioned, complete DMEM that was incubated at 37˚C for 24 h in plates without BMDM. Five wells from each group received 2 µg/ml anti-Klotho neutralizing antibody (rat anti-human, cross-reactive with mouse, clone #775340; R&D) or 2 µg/ml rat IgG2B isotype control antibody (clone #141945, R&D) in the conditioned media. Myoblasts were counted after 48 h of stimulation as described above.
The effect of TNFα depletion on Klotho-stimulated myoblast proliferation was also assayed. Myoblasts were stimulated with 10 µg/ml heparin and 0.5 µg/ml recombinant human FGF23 (control; R&D) or with heparin, FGF23 and 1 µg/ml recombinant mouse Klotho (R&D) in complete DME. Five wells of the control and Klotho-treated groups received 6 µg/ml rat anti-mouse TNFα-neutralizing antibody (clone MP6-XT3; Southern Biotech) and five wells of the Klotho-treated group received 6 µg/ml rat IgG1K isotype control (clone KLH/G1–2-2, Southern Biotech). Proliferation was quantified at 72 h post-plating after 48 h of stimulation, as described above.
In vitro assays of myotube growth
Myoblasts were seeded in six-well plates at 120 000 cells per well and grown in complete DMEM with alternate-day media changes. When cells reached confluence, cultures were serum-starved for 24 h to induce differentiation. Myotubes were stimulated with heparin and FGF23 or heparin, FGF23 and Klotho at 24 and 48 h following differentiation, as described previously. To test whether TNFα affects Klotho-stimulated myotube growth, a TNFα-neutralizing antibody or isotype control antibody were added to cultures as detailed above. Myotubes were collected 72 h following serum starvation in reducing sample buffer with protease inhibitors (Sigma). Protein concentration was quantified as previously described (65). Each group included five replicates.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We thank David Rangel and Katherine Wen for expert assistance in preparing tissue for histology and immunohistochemistry.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health (AR062579 to J.G.T, AR066036 to J.G.T, AG041147 to J.G.T, AR066817 to J.G.T and AR065845 to S.S.W).
References
- 1. Hoffman E.P., Brown R.H. Jr., Kunkel L.M. (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell, 51, 919–928. [DOI] [PubMed] [Google Scholar]
- 2. Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. (1993) Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA, 90, 3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wehling M., Spencer M.J., Tidball J.G. (2001) A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell. Biol, 155, 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gloss D., Moxley R.T. 3rd, Ashwal S., Oskoui M. (2016) Practice guideline update summary: Corticosteroid treatment of Duchenne muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology, 86, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ervasti J.M., Ohlendieck K., Kahl S.D., Gaver M.G., Campbell K.P. (1990) Deficiency of a glycoprotein component of the dystrophin complex in dystrophic mice. Nature, 345, 315–319. [DOI] [PubMed] [Google Scholar]
- 6. Chang W.J., Iannaccone S.T., Lau K.S., Masters B.S., McCabe T.J., McMillan K., Padre R.C., Spencer M., Tidball J.G., Stull J.T. (1996) Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc. Natl. Aca.d Sci. USA, 93, 9142–9147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thomas G.D., Sander M., Lau K.S., Huang P.L., Stull J.T., Victor R.G. (1998) Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc. Natl. Acad. Sci. USA, 95, 15090–15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sander M., Chavoshan B., Harris S.A., Iannaccone S.T., Stull J.T., Thomas G.D., Victor R.G. (2000) Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA, 97, 13818–13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kobayashi Y.M., Rader E.P., Crawford R.W., Iyengar N.K., Thedens D.R., Faulkner J.A., Parikh S.V., Weiss R.M., Chamberlain J.S., Moore S.A., Campbell K.P. (2008) Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature, 456, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wehling-Henricks M., Oltmann M., Rinaldi C., Myung K.H., Tidball J.G. (2009) Loss of positive allosteric interactions between neuronal nitric oxide synthase and phosphofructokinase contributes to defects in glycolysis and increased fatigability in muscular dystrophy. Hum. Mol. Genet., 18, 3439–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bia B.L., Cassidy P.J., Young M.E., Rafael J.A., Leighton B., Davies K.E., Radda G.K., Clarke K.J. (1999) Decreased myocardial nNOS, increased iNOS and abnormal ECGs in mouse models of Duchenne muscular dystrophy. ) Mol. Cell. Cardiol., 31, 1857–1862. [DOI] [PubMed] [Google Scholar]
- 12. Wehling-Henricks M., Jordan M.C., Roos K.P., Deng B., Tidball J.G. (2005) Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum. Mol. Genet., 14, 1921–1933. [DOI] [PubMed] [Google Scholar]
- 13. Kuro-o M., Matsumura Y., Aizawa H., Kawaguchi H., Suga T., Utsugi T., Ohyama Y., Kurabayashi M., Kaname T., Kume E.. et al. (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature, 390, 45–51. [DOI] [PubMed] [Google Scholar]
- 14. Matsumura Y., Aizawa H., Shiraki-Iida T., Nagai R., Kuro-o M., Nabeshima Y. (1998) Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem. Biophys. Res. Commun., 242, 626–630. [DOI] [PubMed] [Google Scholar]
- 15. Li S.A., Watanabe M., Yamada H., Nagai A., Kinuta M., Takei K. (2004) Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell Struct. Funct., 29, 91–99. [DOI] [PubMed] [Google Scholar]
- 16. Phelps M., Pettan-Brewer C., Ladiges W., Yablonka-Reuveni Z. (2013) Decline in muscle strength and running endurance in klotho deficient C57BL/6 mice. Biogerontology, 14, 729–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wehling-Henricks M., Li Z., Lindsey C., Wang Y., Welc S.S., Ramos J.N., Khanlou N., Kuro-O M., Tidball J.G. (2016) Klotho gene silencing promotes pathology in the mdx mouse model of Duchenne muscular dystrophy. Hum. Mol. Genet., 25, 2465–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ohyama Y., Kurabayashi M., Masuda H., Nakamura T., Aihara Y., Kaname T., Suga T., Arai M., Aizawa H., Matsumura Y.. et al. (1998) Molecular cloning of rat klotho cDNA: markedly decreased expression of klotho by acute inflammatory stress. Biochem. Biophys. Res. Comm., 251, 920–925. [DOI] [PubMed] [Google Scholar]
- 19. Thurston R.D., Larmonier C.B., Majewski P.M., Ramalingam R., Midura-Kiela M., Laubitz D., Vandewalle A., Besselsen D.G., Mühlbauer M., Jobin C., Kiela P.R., Ghishan F.K. (2010) Tumor necrosis factor and interferon-gamma down-regulate Klotho in mice with colitis. Gastroenterology, 138, 1384–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao Y., Banerjee S., Dey N., LeJeune W.S., Sarkar P.S., Brobey R., Rosenblatt K.P., Tilton R.G., Choudhary S. (2011) Klotho depletion contributes to increased inflammation in kidney of the db/db mouse model of diabetes via RelA (serine)536 phosphorylation. Diabetes, 60, 1907–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hui H., Zhai Y., Ao L., Cleveland J.C. Jr, Liu H., Fullerton D.A., Meng X. (2017) Klotho suppresses the inflammatory responses and ameliorates cardiac dysfunction in aging endotoxemic mice. Oncotarget, 8, 15663–15676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moreno J.A., Izquierdo M.C., Sanchez-Niño M.D., Suárez-Alvarez B., Lopez-Larrea C., Jakubowski A., Blanco J., Ramirez R., Selgas R., Ruiz-Ortega M.. et al. (2011) The inflammatory cytokines TWEAK and TNFα reduce renal klotho expression through NFκB. J. Am. Soc. Nephrol., 22, 1315–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fiorentino D.F., Zlotnik A., Mosmann T.R., Howard M., O'Garra A. (1991) IL-10 inhibits cytokine production by activated macrophages. J. Immunol., 147, 3815–3822. [PubMed] [Google Scholar]
- 24. Mosser D.M. (2003) The many faces of macrophage activation. J. Leukoc. Biol., 73, 209–212. [DOI] [PubMed] [Google Scholar]
- 25. Murray P.J. (2006) Understanding and exploiting the endogenous interleukin-10/STAT3 mediated anti-inflammatory response. Curr. Opin. Pharmacol., 6, 379–386. [DOI] [PubMed] [Google Scholar]
- 26. Urakawa I., Yamazaki Y., Shimada T., Iijima K., Hasegawa H., Okawa K., Fujita T., Fukumoto S., Yamashita T. (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature, 444, 770–774.17086194 [Google Scholar]
- 27. Villalta S.A., Rinaldi C., Deng B., Liu G., Fedor B., Tidball J.G. (2011) Interleukin-10 reduces the pathology of mdx muscular dystrophy by deactivating M1 macrophages and modulating macrophage phenotype. Hum. Mol. Genet., 20, 790–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tidball J.G., Wehling-Henricks M. (2007) Macrophages promote muscle membrane repair and muscle fibre growth and regeneration during modified muscle loading in mice in vivo. J. Physiol., 578, 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cantini M., Massimino M.L., Bruson A., Catani C., Dalla Libera L., Carraro U. (1994) Macrophages regulate proliferation and differentiation of satellite cells. Biochem. Biophys. Res. Commun., 202, 1688–1696. [DOI] [PubMed] [Google Scholar]
- 30. Massimino M.L., Rapizzi E., Cantini M., Libera L.D., Mazzoleni F., Arslan P., Carraro U. (1997) ED2+ macrophages increase selectively myoblast proliferation in muscle cultures. Biochem. Biophys. Res. Commun., 235, 754–759. [DOI] [PubMed] [Google Scholar]
- 31. Malerba A., Pasut A., Frigo M., De Coppi P., Baroni M.D., Vitiello L. (2010) Macrophage-secreted factors enhance the in vitro expansion of DMD muscle precursor cells while preserving their myogenic potential. Neurol. Res., 3232, 55–62. [DOI] [PubMed] [Google Scholar]
- 32. Li Y.P. (2003) TNF-alpha is a mitogen in skeletal muscle. Am. J. Physiol. Cell Physiol., 285, C370–C376. [DOI] [PubMed] [Google Scholar]
- 33. Deng B., Wehling-Henricks M., Villalta S.A., Wang Y., Tidball J.G. (2012) IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J. Immunol., 189, 3669–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heslop L., Morgan J.E., Partridge T.A. (2000) Evidence for a myogenic stem cell that is exhausted in dystrophic muscle. J. Cell Sci., 113, 2299–2308. [DOI] [PubMed] [Google Scholar]
- 35. Guttridge D.C., Albanese C., Reuther J.Y., Pestell R.G., Baldwin A.S. Jr. (1999) NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol., 19, 5785–5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Langden R.C., Schols A.M., Kelders M.C., Wouters E.F., Janssen-Heininger Y.M. (2001) Inflammatory cytokines inhibit myogenic differentiation through activation of nuclear factor-kappaB. FASEB, 15, 1169–1180. [DOI] [PubMed] [Google Scholar]
- 37. Langden R.C., Van Der Velden J.L., Schols A.M., Kelders M.C., Wouters E.F., Janssen-Heininger Y.M. (2004) Tumor necrosis factor-alpha inhibits myogenic differentiation through MyoD protein destabilization. faseb J., 18, 227–237. [DOI] [PubMed] [Google Scholar]
- 38. Hinz M., Krappmann D., Eichten A., Heder A., Scheidereit C., Strauss M. (1999) NF-kappaB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol. Cell. Biol., 19, 2690–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bakkar N., Wang J., Ladner K.J., Wang H., Dahlman J.M., Carathers M., Acharyya S., Rudnicki M.A., Hollenbach A.D., Guttridge D.C. (2008) IKK/NF-kappaB regulates skeletal myogenesis via a signaling switch to inhibit differentiation and promote mitochondrial biogenesis. J. Cell Biol., 180, 787–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sun C.Y., Chang S.C., Wu M.S. (2012) Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int., 81, 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen J., Zhang X., Zhang H., Lin J., Zhang C., Wu Q., Ding X., Dussaule J.-C. (2013) Elevated Klotho promoter methylation is associated with severity of chronic disease. PLoS One, 8, e79856. 10.1371/journal.pone.0079856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee J., Jeong D.-J., Kim J., Lee S., Park J.-H., Chang B., Jung S.-I., Yi L., Han Y., Yang Y.. et al. (2010) The anti-aging gene KLOTHO is a novel target for epigenetic silencing in human cervical carcinoma. Mol. Cancer, 9, 109. 10.1186/1476-4598-9-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pan J., Zhong J., Gan L.H., Chen S.J., Jin H.C., Wang X., Wang L.J. (2011) Klotho, an anti-senescence related gene, is frequently inactivated through promoter hypermethylation in colorectal cancer. Tumour Biol., 32, 729–735. [DOI] [PubMed] [Google Scholar]
- 44. Rubinek T., Shulman M., Israeli S., Bose S., Avraham A., Zundelevich A., Evron E., Gal-Yam E.N., Kaufman B., Wolf I. (2012) Epigenetic silencing of the tumor suppressor klotho in human breast cancer. Breast Cancer Res. Treat., 133, 649–657. [DOI] [PubMed] [Google Scholar]
- 45. Xin Y.J., Yuan B., Yu B., Wang Y.Q., Wu J.J., Zhou W.H., Qiu Z. (2015) Tet1-mediated DNA demethylation regulates neuronal cell death induced by oxidative stress. Sci. Rep., 5, 7645.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Desguerre I., Mayer M., Leturcq F., Barbet J.P., Gherardi R.K., Christov C. (2009) Endomysial fibrosis in Duchenne muscular dystrophy: a marker of poor outcome associated with macrophage alternative activation. J. Neuropathol. Exp. Neurol., 68, 762–773. [DOI] [PubMed] [Google Scholar]
- 47. Kozakowska M., Pietraszek-Gremplewicz K., Jozkowicz A., Dulak J. (2015) The role of oxidative stress in skeletal muscle injury and regeneration: focus on antioxidant enzymes. J. Muscle Res. Cell Motil., 36, 377–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brigitte M., Schilte C., Plonquet A., Baba-Amer Y., Henri A., Charlier C., Tajbakhsh S., Albert M., Gherardi R.K., Chrétien F. (2010) Muscle resident macrophages control the immune cell reaction in a mouse model of notexin-induced myoinjury. Arthritis Rheum., 62, 268–279. [DOI] [PubMed] [Google Scholar]
- 49. Albrich J.M., McCarthy C.A., Hurst J.K. (1981) Biological reactivity of hypochlorous acid: implications for microbicidal mechanisms of leukocyte myeloperoxidase. Proc. Natl. Acad. Sci. USA, 78, 210–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McKenna S.M., Davies K.J. (1988) The inhibition of bacterial growth by hypochlorous acid. Possible role in the bactericidal activity of phagocytes. Biochem. J., 254, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nguyen H.X., Tidball J.G. (2003) Interactions between neutrophils and macrophages promote macrophage killing of rat muscle cells in vitro. J. Physiol., 547, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang Y.T., Whiteman M., Gieseg S.P. (2012) Intracellular glutathione protects human monocyte-derived macrophages from hypochlorite damage. Life Sci., 90, 682–688. [DOI] [PubMed] [Google Scholar]
- 53. Yamamoto M., Clark J.D., Pastor J.V., Gurnani P., Nandi A., Kurosu H., Miyoshi M., Ogawa Y., Castrillon D.H., Rosenblatt K.P.. et al. (2005) Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem., 280, 38029–38034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shearer J.D., Richards J.R., Mills C.D., Caldwell M.D. (1997) Differential regulation of macrophage arginine metabolism: a proposed role in wound healing. Am. J. Physiol., 272, E181–E190. [DOI] [PubMed] [Google Scholar]
- 55. Witte M.B., Vogt N., Stuelten C., Gotoh T., Mori M., Becker H.D. (2003) Arginase acts as an alternative pathway of L-arginine metabolism in experimental colon anastomosis. J. Gastrointest. Surg., 7, 378–385. [DOI] [PubMed] [Google Scholar]
- 56. Witte M.B., Barbul A. (2003) Arginine physiology and its implication for wound healing. Wound Repair Regen., 11, 419–423. [DOI] [PubMed] [Google Scholar]
- 57. Curran J.N., Winter D.C., Bouchier-Hayes D. (2006) Biological fate and clinical implications of arginine metabolism in tissue healing. Wound Repair Regen., 14, 376–386. [DOI] [PubMed] [Google Scholar]
- 58. Satriano J. (2004) Arginine pathways and the inflammatory response: interregulation of nitric oxide and polyamines: Review article. Amino Acids, 23, 321–329. [DOI] [PubMed] [Google Scholar]
- 59. Wehling-Henricks M., Jordan M.C., Gotoh T., Grody W.W., Roos K.P., Tidball J.G. (2010) Arginine metabolism by macrophages promotes cardiac and muscle fibrosis in mdx muscular dystrophy. PLoS One, 21, 10.1371/journal.pone.0010763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Spencer M., Walsh C.M., Dorshkind K.A., Rodriguez E.M., Tidball J.G. (1997) Myonuclear apoptosis in dystrophic mdx muscle occurs by perforin-mediated cytotoxicity. J. Clin. Invest., 99, 2745–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Amalfitano A., Chamberlain J.S. (1996) The mdx-amplification-resistant mutation system assay, a simple and rapid polymerase chain reaction-based detection of the mdx allele. Muscle Nerve, 19, 1549–1553. [DOI] [PubMed] [Google Scholar]
- 62. Nolan T., Hands R.E., Bustin S.A. (2006) Quantification of mRNA using real-time RT-PCR. Nat. Protoc., 1, 1559–1582. [DOI] [PubMed] [Google Scholar]
- 63. Bustin S.A., Benes V., Garson J.A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M.W., Shipley G.L.. et al. (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem., 55, 611–622. [DOI] [PubMed] [Google Scholar]
- 64. Wang Y., Wehling-Henricks M., Samengo G., Tidball J.G. (2015) Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell, 14, 678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Minamide L.S., Bamburg J.R. (1990) A filter paper dye-binding assay for quantitative determination of protein without interference from reducing agents or detergents. Anal. Biochem., 190, 66–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.