

Abstract
UBTF (upstream binding transcription factor) exists as two isoforms; UBTF1 regulates rRNA transcription by RNA polymerase 1, whereas UBTF2 regulates mRNA transcription by RNA polymerase 2. Herein, we describe 4 patients with very similar patterns of neuroregression due to recurrent de novo mutations in UBTF (GRCh37/hg19, NC_000017.10: g.42290219C > T, NM_014233.3: c.628G > A) resulting in the same amino acid change in both UBTF1 and UBTF2 (p.Glu210Lys [p.E210K]). Disease onset in our cohort was at 2.5 to 3 years and characterized by slow progression of global motor, cognitive and behavioral dysfunction. Notable early features included hypotonia with a floppy gait, high-pitched dysarthria and hyperactivity. Later features included aphasia, dystonia, and spasticity. Speech and ambulatory ability were lost by the early teens. Magnetic resonance imaging showed progressive generalized cerebral atrophy (supratentorial > infratentorial) with involvement of both gray and white matter. Patient fibroblasts showed normal levels of UBTF transcripts, increased expression of pre-rRNA and 18S rRNA, nucleolar abnormalities, markedly increased numbers of DNA breaks, defective cell-cycle progression, and apoptosis. Expression of mutant human UBTF1 in Drosophila neurons was lethal. Although no loss-of-function variants are reported in the Exome Aggregation Consortium (ExAC) database and Ubtf−/− is early embryonic lethal in mice, Ubtf+/− mice displayed only mild motor and behavioral dysfunction in adulthood. Our data underscore the importance of including UBTF E210K in the differential diagnosis of neuroregression and suggest that mainly gain-of-function mechanisms contribute to the pathogenesis of the UBTF E210K neuroregression syndrome.
Introduction
UBTF (upstream binding transcription factor) is a multi-faceted protein that contains a dimerization domain, six High Mobility Group B (HMGB) Boxes, which are involved in DNA binding, and a transcriptional activation domain. UBTF was initially identified as a transcriptional activator of ribosomal RNA (rRNA) expression (1) and that function has been extensively characterized (2). UBTF exists as two isoforms called UBTF1 (or isoform a) and UBTF2 (or isoform b). UBTF2 lacks exon 8 of UBTF1 which encodes a portion (37 amino acids) of HMGB Box 2 (3). UBTF1 and UBTF2 can form homo- and heterodimers. UBTF1 regulates ribosomal RNA (rRNA) transcription by RNA polymerase 1 (Pol I) employing several mechanisms that involve assembly of transcription complexes (4,5), regulation of chromatin state (6,7), modulating promoter escape (8) and transcriptional elongation (6,9). In contrast, UBTF2 regulates mRNA transcription by RNA polymerase 2 (Pol II) (10,11). UBTF can be regulated by post-translational modification (12,13). In particular, an immediate response of ribosomal transcription to growth factor stimulation is mediated by ERK phosphorylation of UBTF (14,15). The mouse model Ubtf−/− is embryonic lethal (16). Depletion of UBTF leads to abnormalities of cell-cycle progression and response to DNA damage (11,17).
UBTF activates rRNA transcription within the nucleolus (18–20). The nucleolus is also the site of ribosomal particle biogenesis and sequestration of signaling molecules such as p53. Induction of rRNA and other nucleolar activity play a role in normal neural processes such as neurite outgrowth and memory consolidation during spatial training (21). Nucleolar stress, which involves aberrant rRNA expression, has been associated with neurodegenerative disorders including Huntington disease, Parkinson disease, and amyotrophic lateral sclerosis (22–24).
Although characteristically associated with late-onset neurological deterioration, neurodegeneration can begin in childhood and mutations in some genes (e.g. HTT) can cause heredodegeneration throughout the lifespan. Neurodegeneration in childhood is commonly referred to as developmental regression, neurodevelopmental regression or developmental neuroregression since the nervous system is typically the sole site of overt clinical involvement. Developmental neuroregression is characterized by the loss of acquired functions or failure to progress beyond a developmental plateau after a period of seemingly normal development (25). With the increasing use of trio whole-exome sequencing (WES) in children with developmental neuroregression the genetics and pathobiologies of early-onset neurodegeneration can be exposed. Recently, trio WES was used to identify a de novo missense variant in UBTF (Chr17: g.42290219C > T; NM_014233.3: c.628G > A, p.Glu210Lys [p.E210K]) in seven subjects with very similar patterns of neuroregression (26). The UBTF E210K variant was associated with increased expression of rRNA and nucleolar abnormalities in fibroblasts from a single subject (26). Herein, we rigorously detail clinical features of the UBTF E210K neuroregression syndrome in four affected individuals and expand upon the molecular and cellular pathobiology of UBTF E210K-related neuroregression using an integrated collection of cellular, invertebrate, and vertebrate model systems.
Results
Trio WES identifies a recurrent de novo autosomal-dominant UBTF mutation in four probands
Trio WES was supplemented by additional WES of siblings in 3 of 4 pedigrees harboring single individuals with developmental neuroregression (Fig. 1; Supplementary Material, Figs S1 and S2, Tables S1 and S2). De novo, autosomal recessive (compound heterozygous and homozygous), and X-linked inheritance models were considered to generate potential variants (Supplementary Material, Figs S1 and S2, Table S2). A de novo missense variant in UBTF [Chr17: g.42290219C > T; c.628G > A(p.Glu210Lys, p.E210K)] was common to four probands with similar clinical phenotypes and confirmed with Sanger sequencing (Supplementary Material, Fig. S3). Other variants were excluded based upon one or more parameters including population frequency in the Exome Aggregation Consortium (ExAC, exac.broadinstitute.org) database, in silico predictions of pathogenicity and deleteriousness, expression in neural tissues, and known disease associations. Based upon analysis with dbNSFPv3.3 (Database for Nonsynonymous SNP’s Functional Predictions), UBTF E210K was predicted to be highly deleterious to protein function with a MetaLR_rankscore of 0.982, CADD_phred score of 32, and REVEL score of 0.683 (27–29). In addition, UBTF has an ExAC missense Z-score of 5.61 (highly intolerant of variation) and pLI (probability of loss-of-function intolerance) of 1.00.
Figure 1.

UBTF E210K pedigrees. All family members analyzed with WES are labeled on the pedigree diagrams. DNA was not available on the sister of TGen_0328.
UBTF E210K is associated with a novel neuroregression syndrome in humans
In our cohort of four subjects, heterozygous UBTF E210K was characterized by neuroregression with onset at 2 to 3 years (Table 1). There were no dysmorphic features with the exception of pectus carinatum and kyphosis in one subject and no overt evidence of extra-neural disease manifestations. However, affected individuals did tend to lose weight after 2 years. Neuroregression was global with decline in cognition, behavior, and sensorimotor functioning. Patients tended to lose previously acquired developmental milestones after 3 years of age. Early motor dysfunction included hypotonia, gait ataxia, and dysarthria. Speech was high-pitched in at least two of the subjects. Later motor dysfunction was dominated by hypertonia with spasticity and dystonia and postural instability with loss of ambulatory abilities. Cognitive decline first manifested as expression dysphasia progressing to expressive aphasia and, then, eventual inability to follow verbal commands.
Table 1.
Clinical features of UBTF p.E210K probands
| TGen_0328 | UDP_2009 | UDP_7621 | UDP_5762 | |
|---|---|---|---|---|
| Sex | Male | Male | Female | Female |
| Ethnicity | Caucasian of European descent | Ashkenazi Jewish | Caucasian of European descent | Caucasian of European descent |
| Prenatal abnormalities | None | None | None | None |
| Paternal age at conception | 29 | NA | 32 | NA |
| Maternal age at conception | 30 | 33 | 31 | 39 |
| Birth parameters | Weight - 3.52 kg (47%) | Weight- 3.63 kg (56%) | NA | Weight - 3.29 kg (40%); |
| Length - 50.8 cm (69%); OFC 33.5 cm (19%) | ||||
| Age at onset of neuroregression | 2.5 years | 2–2.5 years | 3 years | 3 years |
| Developmental history | Achieved standard developmental milestones until 2.5 years | Achieved standard developmental milestones until 2 years | Achieved standard developmental milestones until 3 years | Achieved standard developmental milestones until 2.5–3 years |
| Seizures | None | None | None | None |
| Growth parameters | 9.3 years: weight 18.7 kg (<1%), height 115.8 cm (< 1%), OFC 51.5 cm (21%) | 2 years: weight 12 kg (29%), height 91 cm (86%) | NA | 12.3 years: weight 33.7 kg (9%), height 152.1cm (53%) |
| 10.8 years: weight 27.4 kg (8%), height 131cm (6%) OFC 52.7 cm (41%) | 3.1 years: weight 13.9 kg (36%), height 98 cm (69%), OFC 48 cm (16%) | |||
| 11.7 years: height 136 cm (8%), weight 28.5 kg (4%) | 5 years: weight 16.2 kg (19%), height 102.1 cm (11%), OFC 49 cm (17%) | |||
| 12.2 years: weight 30.8 kg (5%), height 140 cm (11%), OFC 54 cm (56%) | 6.2 years: weight 17 kg (5%), height 109 cm (9%), OFC 49 cm (3%) | |||
| Behavior | Mildly autistic, mildly self-injurious, not fully toilet trained. Inattentive. | Mouthing of hands | Some compulsive and repetitive behaviors. Distractible but did not respond to methylphenidate | Exhibited parallel but not group play |
| Hyperactivity | Moderate | None | None | Moderate |
| Impulsivity | Mild | None | None | Mild |
| Sensation | Reduced sensation to noxious stimuli | Reduced responses to noxious stimuli | NA | Normal |
| Motor power | Mildly reduced (9 years) and progressive | Normal | Normal | Normal |
| Postural instability | Yes. Onset 4 years and progressive. | Normal | Yes | Yes |
| Tone | Hypotonia (6 years) | Axial hypotonia with pectus carinatum & kyphosis, hypertonic Extremities | Spasticity (3 years) eventually leading to severe contractures and scoliosis at 33 years | Axial hypotonia, extremities hypertonic |
| Spastic tone (12 years) | ||||
| Reflexes | 1+/4+ (6 years); 2+/4+, ankle clonus (12 years) | Hyperreflexia | Hyperactive muscle stretch reflexes | Muscle stretch reflexes 1+/4+ throughout, bilateral |
| Babinski responses | ||||
| Spasticity | Mild (12 years) | Mild 6 years | Yes | Yes |
| Dystonia | Mild (12 years) | None | Yes | Yes |
| Chorea | Mild (12 years) | None | None | None |
| Gait ataxia | Moderate (12 years) | Mild 6 years | Yes | Yes |
| Appendicular ataxia | Mild (12 years) | Mild 6 years | N/A | Mild 12 years |
| Speech | Moderate dysarthria, high pitched | Dysarthria | Normal until 3 years. Subsequently high-pitched, evolving to dysarthria and anarthria. | Delayed |
| Language | Expression dysphasia > receptive dysphasia | Expression dysphasia > receptive dysphasia | Dysphasia progressing to aphasia | Expression dysphasia > receptive dysphasia. Aphasic by 8 years. |
| Swallowing | Moderate dysphagia | Normal | Dysphagia. PEG tube feedings. | Dysphagia. PEG tube feedings. |
| Fundal examination | Normal | Normal | Normal | Normal |
| MRI | 10 years, 7 months: diffuse cerebral atrophy both gray and white matter (supratentorial > infratentorial) with periventricular gliosis, ex vacuo dilatation of the ventricles, thinning of the corpus callosum | 6.2 years: thinning of the corpus callosum, mild cerebellar vermian atrophy, mild-to moderate cerebral hemispheric atrophy with ventriculomegaly | 33 years: extreme atrophy of the cerebral hemispheres and midline cerebellum with less involvement of cerebellar hemispheres | 12 years, 4 months: thinning of the corpus callosum, cerebral hemispheric atrophy, mild cerebellar atrophy (mainly vermis), and ventriculomegaly |
Abbreviations: OFC, occipital front circumference; PEG, percutaneous endoscopic gastrostomy; NA, not available.
TGen_0328 (Table 1) rolled over at 5 months, sat alone at 6 months, crawled at 7.5 months, and walked at 1 year. He was able to say “mama” and “dada” at 10 months and single words such as “ball” and “dog” at 12 months. He used simple sentences by 3 years and conversed with two or more exchanges on the same topic by 3.5 years. He was never able to ride a tricycle or bicycle. He was only partially toilet trained at 4.6 years, had to wear pull-ups, and was incontinent on many nights. He did not toilet spontaneously and did not seem bothered when he wet or soiled himself.
TGen_0328 showed some autistic behavior patterns at 4.6 years (Supplementary Material, Table S3). He was attending a pre-kindergarten program at the time of this assessment. He was affectionate with his family but not with peers. He would sometimes smack himself or bang his head when frustrated. He exhibited motor apraxia and postural instability. He was treated with clonidine, amphetamine/dextroamphetamine, and lisdexamfetamine for impulsivity and attention deficit with minimal benefit. A normal awake electroencephalogram (EEG) was recorded at 8 years. He could laugh at 8 years. Mild limitation of upgaze, mild dysmetria, and mild action tremor were noted at 9 years. Action-induced chorea was present when attempting to run. By age 11, he was chewing almost everything put in his mouth and having difficulty swallowing. At age 11, he could no longer speak clearly or walk for more than a few steps without falling. His parents had to limit the amount of food that he puts in his mouth at one time. He has appeared relatively insensitive to noxious stimuli and has had cool, mottled extremities. At 11.6 years, he could follow simple commands, reach and hold, and transfer objects. At that time, speech was severely dysarthric and only 10% of words were understandable. At 11.6 years, he was able to recognize letters and read a few words and could play some simple games on a computer touch screen.
UDP_2009 (Table 1) showed initial evidence of neurological regression at 2.5 years. He achieved normal developmental milestones in the first year of life. At 3 years, his developmental milestones included rolling at 4 months, sitting at 4 months, babbling at 6 months, crawling at 8 months, pulling to stand at 8 months, eating finger foods by 10 months, and walking at 14 months. However, he was not able to run well or go up and down stairs until 2.5 years. He was speaking single words at 18 months and combining words at age 2.5 years. At 2.5 years, his vocabulary included approximately 20 words, and his speech was described as somewhat dysarthric. His receptive language seemed better than his expressive language. He exhibited social behavior and had no stereotypies. Speech therapy was initiated at approximately 2.5 years.
At 3.3 years, there was mild overall developmental delay with abilities approximating that of a 26-month-old child. It was noted that he could state his age, use two word phrases, able to follow directions, and mouthed many objects with drooling. He exhibited no atypical behaviors, and he did evidence some symbolic play, as well as imitate language. On the Bayley Scales of Infant Development, his score was less than 50 revealing that his overall development was that of a 26 months old at a chronological age of 40 months. His highest passes were at the 32- to 34-month level comparing sizes (big/little) and counting to seven. He could sort pegs by color, placing him at the 42-month level. He could scribble but not imitate a horizontal or vertical line. He could not string small beads or place a string of beads in a small tube. He could name animals in a book. He understood the preposition “on.” Progressive dysphasia progressed to aphasia by 4 years. Spasticity and ataxia progressed more slowly.
UDP_7621 (Table 1) was the full-term product of an uncomplicated birth and delivery. Her early development was overtly normal until approximately 3 years when she developed slowly progressive loss of gross and fine motor skills with spasticity and postural instability.
Communication skills also declined gradually after 3 years. A high-pitched voice gradually declined in intelligibility and progressed to severe dysarthria and dysphagia culminating in the need for percutaneous endoscopic gastrostomy (PEG) tube placement.
She exhibited compulsive behavior and was easily distractible but did not benefit from methylphenidate. She became unable to spell her name by age 6 and was never able to ride a bicycle. At the time of evaluation (33 years of age), she was institutionalized and required total care. She was mute and did not follow commands but could track moving objects. Generalized dystonia, spasticity with contractures, and scoliosis were noted. There had been no history of seizures. Electromyoneurography was normal. Her parents and younger sister (30 years old) of Northern-European descent were healthy. Specialty labs including coenzyme Q10, a white blood cell lysosomal panel (13 enzymes) and chronic granulomatous disease screen were normal. Skin biopsy evaluated with electron microscopy was negative for neuronal ceroid-lipofuscinosis, and showed normal mitochondrial morphology and no evidence of abnormal lysosomal storage.
UDP_5762 (Table 1) was evaluated at 12 years of age. She had an unremarkable birth history and age-appropriate early development. She was sitting, standing and walking at appropriate ages, but at 3 years she was noted to have expressive language delay and mild dysarthria upon starting her preschool classes. At 4 years, an educational assessment revealed difficulty maintaining attention, poor auditory processing, and speech delay. Hypotonia was apparent at 5 years. She continued to have worsening expressive speech and difficulty with her gross and fine motor skills. At age 6, her parents felt that the patient had reached her developmental peak. She could zip up clothing and put on shoes with Velcro, but could not tie her laces. She could recognize and identify a few letters of the alphabet. She could count to 20 and was able to perform simple math. She was toilet trained, with occasional accidents overnight. She recognized some colors, could sight read a few words, and sound out several letters. By age 7, she has lost several developmental milestones and was no longer toilet trained. The patient continued to attend a specialized school until the age of 8. Overall, the subsequent neurological course was characterized by neuroregression with progressive global cognitive impairment, gait disturbance, and severe dysphagia with eventual gastrostomy tube dependence, troublesome sialorrhea and poor management of secretions. Electroencephalograpy (EEG) showed some mild slowing and disorganization of the background rhythms but no focal discharges.
Magnetic resonance imaging shows progressive neurodegeneration in subjects harboring the UBTF E210K mutation
In aggregate, analysis of magnetic resonance imaging (MRI) images (Fig. 2, Supplementary Material, Fig. S4) showed progressive brain atrophy (supratentorial > infratentorial with gray matter > white matter). Gyral patterns were normal and there were no heterotopias, abnormal sulcation, or dysplastic cortical regions (Supplementary Material, Figs S4A and B). Thinning of the corpus callosum was seen in all subjects and progressed in severity with increasing age (Fig. 2A, E, I, M, Q). MRI evidence for periventricular gliosis was apparent on FLAIR sequences (Fig. 2L). Cerebellar atrophy mainly affected the vermis with much lesser involvement of the cerebellar hemispheres (Fig. 2T). Marked ex vacuo ventriculomegaly was apparent in older subjects (Supplementary Material, Fig. S4C).
Figure 2.

Representative MRI slices from four UBTF E210K probands. (A) Mid-sagittal T1-weighted image, (B) axial T1-weighted image, (C) axial T2-weighted image, (D) coronal fluid attenuation inversion recovery [FLAIR] image, (E) mid-sagittal T1-weighted image, (F) axial T1-weighted image, (G) axial T2-weighted image, (H) coronal T2-FLAIR image, (I) mid-sagittal T1-weighted image, (J) axial T2-FLAIR image, (K) axial T2-weighted image, (L) coronal T2-FLAIR image, (M) mid-sagittal T1-weighted image, (N) axial magnetization prepared rapid acquisition gradient echo (MPRAGE)-weighted image, (O) axial T2-weighted image, (P) coronal short-T1 inversion recovery [STIR] image, (Q) mid-sagittal T1-weighted image, (R) axial T1-weighted image, (S) axial T2-weighted image, and (T) axial T1-weighted image.
MRIs of UDP_2009 at 3 years (Supplementary Material, Fig. S4A and B) and 5 years (Fig. 2) showed enlarged ventricles, generalized cerebral atrophy with involvement of gray and white matter, thinning of the corpus callosum, and mild cerebellar vermian atrophy. A third brain MRI at 8.5 years of age showed further progression of supratentorial volume loss marked by increase in the size of the lateral and third ventricles and subarachnoid spaces. Flattening along the lateral margins of the anterior lateral ventricles was consistent with volume loss of the caudate nuclei. There was more convexity along the borders of the third ventricle correlating with deep white matter volume loss. White matter signal was isointense to cortical gray matter signal, indicating axonal loss and demyelination representing Wallerian degeneration. There was an atrophic vermis and thin corpus callosum.
Comparison of MRIs on TGen_0328 (Fig. 2E–L) at 6 years, 5 months, and 10 years, 7 months, showed interval progression of sulcal prominence. At 10 years, 7 months, the periventricular and deep white matter showed hyperintensity. Magnetic resonance spectroscopy (short echo time [TE]) at 10 years, 7 months, obtained at the level of the right posterior white matter, showed a relative decrease of N-acetyl aspartate (NAA) in relation to choline and creatine, mild reversal of the choline-creatine ratio, and elevation of myoinositol. Spectra at the level of the posterior gray matter were unremarkable. Spectra at the level of the basal ganglia and thalamus showed mildly decreased NAA.
MRI examination of UDP_5672 showed marked atrophy of the cerebral hemispheres with relatively mild involvement of the cerebellum. Magnetic resonance spectroscopy showed increased choline-creatine ratio and reduced NAA in the basal ganglia. Despite advanced volume loss in the cerebral hemispheres (Fig. 2N, O, R, S), there was relative preservation of the lateral cerebellar hemispheres, even in advanced disease (Fig. 2T).
UBTF E210K patient fibroblasts exhibit increased expression of rRNA and nucleolar abnormalities
There was no effect of the UBTF mutation on expression of total UBTF or UBTF1 transcripts (Fig. 3A, Supplementary Material, Table S4). However, our patients harboring the UBTF E210K mutation showed marked increases in expression of pre-rRNA (> 3X) and 18S rRNA (> 2X) compatible with a molecular gain-of-function mechanism (Fig. 3A, Supplementary Material, Table S4). To survey the effects of UBTF2 E210K on Pol II gene expression, relative quantitative RT-PCR (QRT-PCR) was used to examine the expression of putative UBTF2 targets previously identified via UBTF siRNA knock-down in 3T3 cells (11). In 3T3 cells, knock-down of UBTF was associated with up-regulation of PPARGC1A and HYAL1, and down-regulation of SLC4A4, HIST1H4B, and FANCB. Relative to controls, PPARGC1A was up-regulated and HIST1H4B and SLC4A4 were down-regulated in UBTF E210K human fibroblasts (Fig. 3B, Supplementary Material, Table S5). In contrast, there was no significant effect of UBTF E210K on expression of HYAL1 or FANCB. In this context, it should be noted that conditional inactivation of Ubtf in floxed MEFs had no significant effect on any of these genes, or indeed any other protein coding gene (16).
Figure 3.
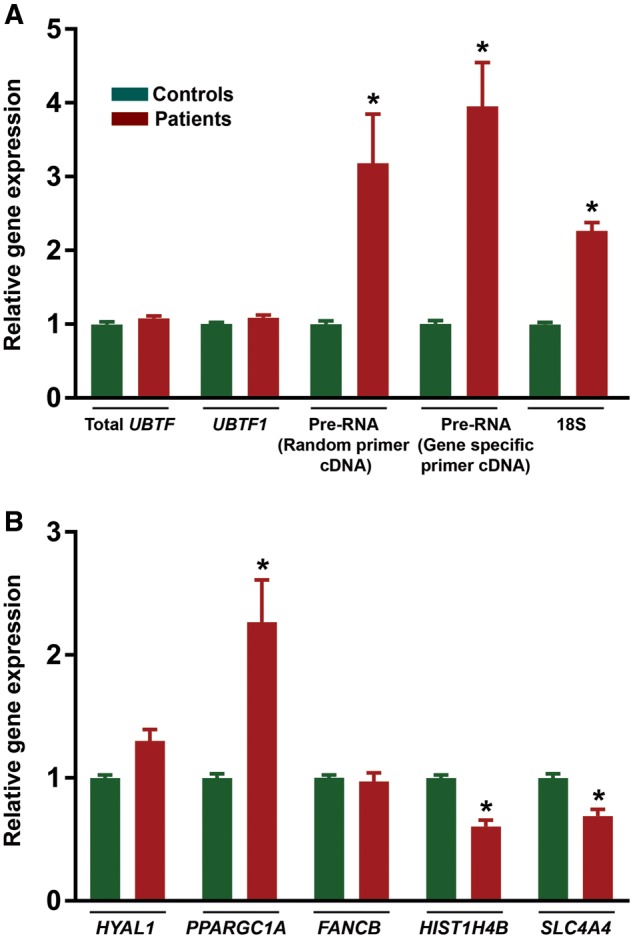
Gene expression in human control and UBTF E210K fibroblasts. (A) Expression of UBTF and rRNA. (B) Expression of genes regulated by UBTF2.
UBTF E210K is associated with DNA double-strand breaks, defective cell-cycle progression, and apoptosis
Comet assays showed clear evidence of increased DNA breaks in UBTF E210K fibroblasts and Ubtf+/− MEFs (Fig. 4). Using an established marker of DNA double-strand breaks (DSBs), 53BP1, we found that the E210K mutation was associated with markedly increased numbers of 53BP1-positive foci in fibroblasts (Fig. 5). UBTF E210K fibroblasts also showed failure of progression to the G2 phase of the cell cycle and a tendency towards apoptotic cell death in comparison to normal wild-type (WT) control fibroblasts (Fig. 6). The UBTF E210K mutation did not alter the localization of UBTF (Fig. 7A). UBTF co-localized with the nucleolar marker fibrillarin in control and UBTF E210K fibroblasts (Fig. 7A). There was a trend for UBTF E210K fibroblasts to harbor fewer nucleoli per nucleus than normal control fibroblasts (P = 0.084, Fig. 7B), but there was no significant effect of the UBTF E210K mutation on nucleolar areas (P = 0.25, Fig. 7C). Though less extreme, this resembles the reduction in numbers of nucleoli, but not total nucleolar volume observed when Ubtf was fully inactivated in MEFs (16).
Figure 4.
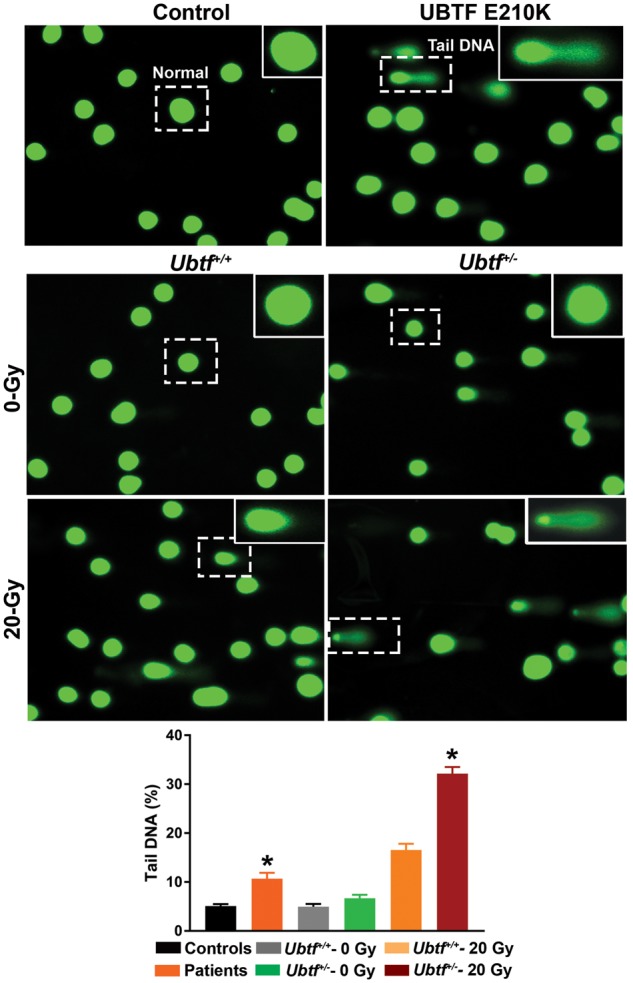
Alkaline comet assay of human fibroblasts in normal controls and UBTF E210K subjects (n = 4/group), and Ubtf+/+ and Ubtf+/− MEFs before and after γ-irradiation (IR) (n = 4/group). *P < 0.01, for differences between genotypes.
Figure 5.

UBTF E210K is associated with DNA double-strand breaks (DSBs) in fibroblasts. (A) Fibroblasts from a normal control and E210K UBTF patient were stained with 53BP1, a marker of DSBs. Nuclei were visualized with DAPI. Scale bar, 50 µm. (B) Quantification of 53BP1+foci was used for analysis of DSBs. Values are expressed as means ± SEM. *P = 0.0012 (n = 4/genotype).
Figure 6.
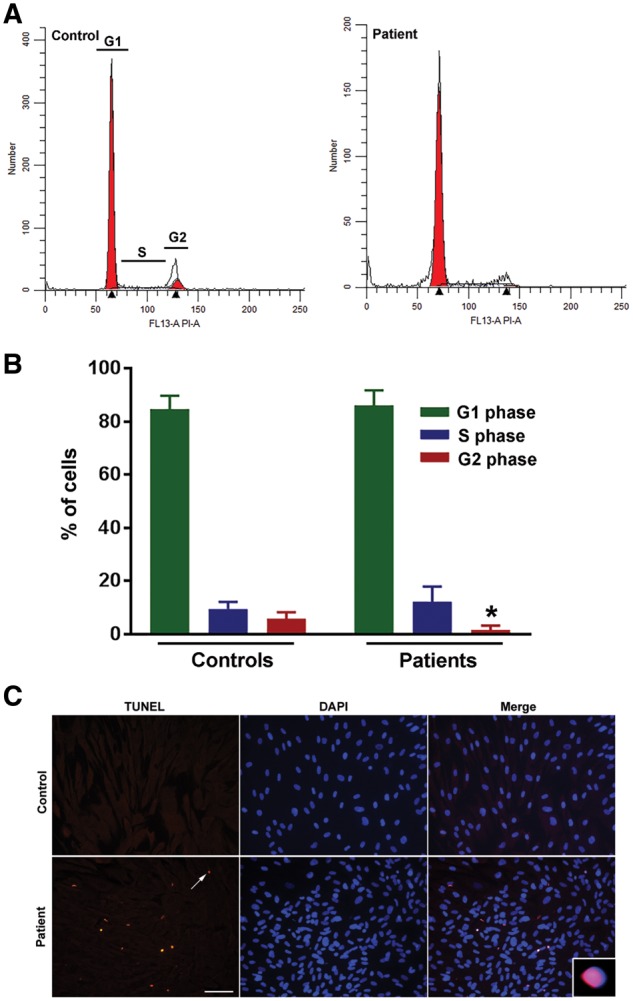
UBTF E210K is associated with cell-cycle defects and apoptosis in human fibroblasts. (A) Representative fibroblast cell-cycle histograms from normal control and UBTF E210K patient. (B) Percentage of cells present in G1, S and G2 phases. Values are expressed as means ± SEM. *P < 0.05 (n = 4–7/genotype). (C) Representative photomicrographs show TUNEL labeling of fibroblasts from a normal control and UBTF E210K patient. Scale bar, 100 µm.
Figure 7.

Effect of UBTF E210K on nucleolar localization of UBTF, nucleolar numbers/nucleus, and nucleolar size. (A) UBTF co-localizes with the nucleolar protein fibrillarin in control and UBTF E210K fibroblasts. Scale bar, 50 µm. (B) Numbers of nucleoli per cell. (C) Nucleolar area. (n = 4 sets of fibroblasts/genotype).
Expression of UBTF E210K in Drosophila neurons is lethal
Wildtype (WT) UAS-UBTF1/CyO or UAS-UBTF1-E210K/CyO flies were crossed to the pan-neuronal driver c155-GAL4 to express WT or UBTF1-E210K in all neurons throughout the fly. Offspring from each cross were counted and, because flies were heterozygous for the balancer chromosome CyO, we expected 50% of the offspring to receive the UAS-UBTF1 construct and 50% to receive the balancer chromosome with curly (Cy) wings. With WT UBTF1 expression, we observed the expected ratio. However, with UBTF1-E210K, we observed no c155 > UBTF1-E210K offspring (P < 0.0001; Supplementary Material, Table S6), indicating that neuronal expression of UBTF-E210K is lethal in Drosophila.
Next, we restricted UBTF1 expression to the eye using Eye3.5-GAL4. Upon overexpression of WT UBTF1 we observed a dramatic small-eye phenotype (Fig. 8A) indicating loss of photoreceptor development in the eye. Additionally, we observed a semi-lethal phenotype in Eye3.5 > UBTF1 flies (Supplementary Material, Table S6; P = 0.0052). Overexpression of UBTF1-E210K resulted in complete lethality (Supplementary Material, Table S6; P < 0.0001), occurring predominantly in the pupal stages. Upon inspection of Eye3.5 > UBTF1-E210K pupae, the heads of these flies were undeveloped and missing inside the pupal case (Fig. 8B). While expression of wildtype UBTF with Eye3.5-GAL4 resulted in eye defects and semi-lethality, these phenotypes were accentuated with UBTF1-E210K expression.
Figure 8.

Expression of human UBTF1 E210K in Drosophila. (A) Expression of WT or UBTF1 E210K results in anatomical abnormalities with Eye3.5-GAL4. Control eye3.5-GAL4 alone flies display normal eye development (left), however expression of UBTF1 with Eye3.5-GAL4 results in a dramatic small-eye phenotype with a complete loss of eye development (right). (B) Expression of UBTF1-E210K resulted in lethality at the pupal stage or earlier. Control Eye3.5-GAL4 alone (left) or Eye3.5 > UBTF1 (center) produced well-recognizable structures including a fully-formed head. However, Eye3.5 > UBTF1-E210K pupae displayed a complete failure of head development within the pupal case.
Adult Ubtf+/− mice exhibit mild motor and behavioral abnormalities
In mouse brain, total Ubtf and Ubtf1 showed higher expression in the cerebellum than in cerebral cortex (Supplementary Material, Table S7). Expression in liver was approximately 60% of that found in the brain. Although, the expression of total Ubtf was reduced by approximately half in cerebral cortex, cerebellum and liver, Ubtf1 levels were not significantly reduced in Ubtf+/− mice (Supplementary Material, Table S7). There were no differences between Ubtf+/− mice and WT littermates in expression of putative UBTF2 targets (Supplementary Material, Table S5).
No evidence of involuntary movements or seizures was noted while mice were routinely observed in their home cages, during open field behavior, and while handling from the early postnatal period through 3 months of age. Ubtf+/− mice showed no overt differences from their WT littermates on righting during the preweanling period (Supplementary Material, Fig. S5). There was a large overall effect of sex on weight (P < 0.0001) but no effect of genotype on weight (Supplementary Material, Table S8). There was no overall effect of genotype on grip strength or normalized rope climbing times (Supplementary Material, Table S8). Overall, Ubtf+/− mice were more aggressive than WT littermates in the dominance tube. However, this effect was only modest in male Ubtf+/− mice (P = 0.080) relative to female Ubtf+/− mice (P < 0.0001).
Both male (P = 0.049) and female (P = 0.058) Ubtf+/− mice tended to fall off the rotarod at shorter latencies than their WT littermates (Fig. 9). Overall, Ubtf+/− mice took more time to traverse raised beams, particularly the 9 mm square and round beams, in comparison to WT littermates. However, no differences in the numbers of slips were detected with either 9 mm or 12 mm beams.
Figure 9.
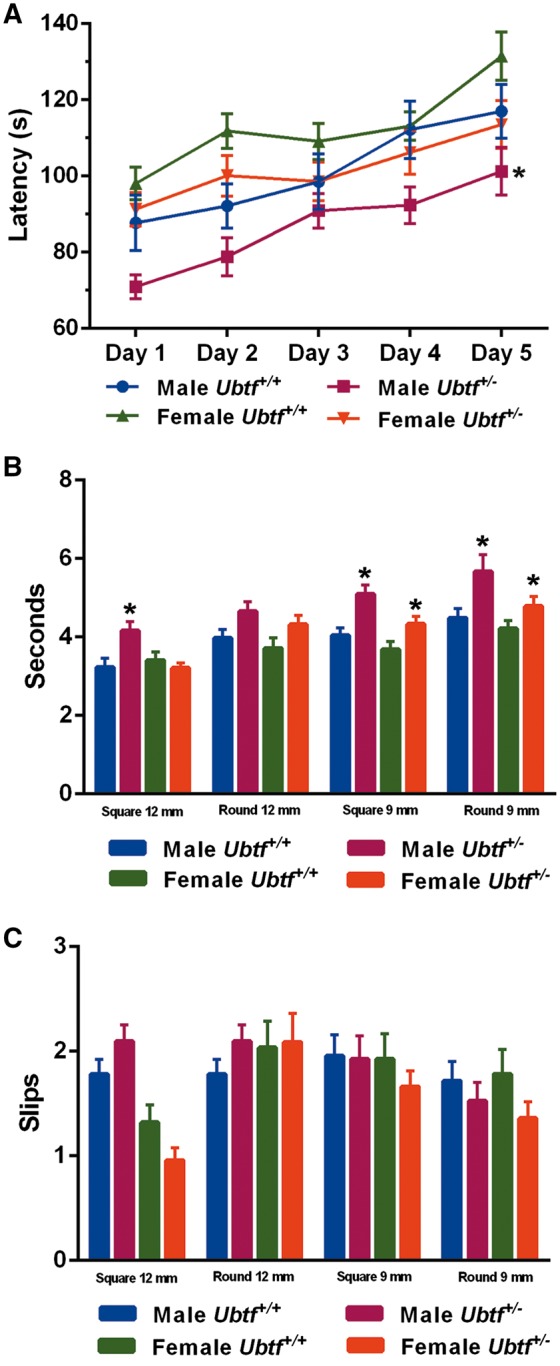
Ubtf +/− mice and sex-matched WT littermates were analyzed with rotarod (A) and the raised beam task (B and C). (A) There was an effect of genotype on the latency to fall from an accelerating rotating drum. (B and C) The raised beam tasks assessed the ability of mice to traverse narrow beams to reach a dark box. There was an effect of genotype on traversal times but no effect on the numbers of slips. *P < 0.05.
Quantitative measures of gait exposed modest yet notable differences between Ubtf+/− mice and their WT littermates (Supplementary Material, Table S8). There was an overall effect of genotype on hindlimb stride length and hindlimb paw areas (P < 0.05, for both). Ubtf+/− mice had longer hindlimb stride lengths and smaller hindlimb paw areas in comparison with their WT littermates.
Discussion
The differential diagnosis of neuroregression in early childhood is broad and includes Rett syndrome, storage disorders such as Krabbe disease and GM1-gangliosidosis type II, the neuronal ceroid lipofuscinoses, multiple carboxylase deficiency, mitochondrial disorders such as Leigh syndrome, and numerous metabolic, infectious, nutritional and toxic etiologies. The search term “developmental regression” generates over 90 hits on Online Inheritance in Man (OMIM). Most genetic etiologies are autosomal recessive with X-linked and mtDNA inheritance patterns being much less common. Historically, a clear etiology has eluded many parents with a child experiencing developmental regression. However, with the advent of trio WES and shared phenotype-genotype databases, the etiologies for developmental delay and/or regression are being exposed in an increasing percentage of cases given that novel de novo mutations are impossible to uncover using classic genetic approaches such as linkage analysis, positional cloning and co-segregation analyses. Recent examples of successful trio WES for the identification of genetic etiologies in children with developmental delay and/or regression include PURA (30), KAT6A (31), ANK3 (32), TCF20 (33), and UBTF E210K (26).
The key features of UBTF E210K-related neuroregression that may facilitate diagnosis include age of onset at 2.5 to 3 years, gait ataxia and hypotonia, mixed cognitive and behavioral abnormalities, absence of extra-neural manifestations, normal head circumference until onset of regression, and normal funduscopic examination (Table 1). Seizures were described in 3/7 of the previously reported UBTF E210K cases but were not present in our cohort of four patients. In similar fashion, acquired microcephaly was reported in 6/7 of the previously reported cases but should not be considered universal since one of our patients (TGen_0328) maintained normal head circumference until 12 years of age. Relative declines in height and weight after 2.5 years of age were documented in three of our patients and may be a common feature of the UBTF E210K neuroregression syndrome. Gait ataxia in our patients correlated with mild-to-moderate midline cerebellar atrophy with relative preservation of the cerebellar hemispheres. Expressive dysphasia was documented to precede receptive dysphasia in three of our affected subjects. High-pitched dysarthria was reported in two of our patients and may be another useful diagnostic clue for clinicians. Progression of speech and language difficulties appeared to correlate with dysphagia and older affected individuals can be expected to develop global aphasia and ultimately require PEG tube feedings.
MRI findings in our cohort of four subjects were very similar to those previously reported (26). First, supratentorial atrophy exceeds infratentorial atrophy. Second, cerebellar atrophy mainly affects the vermis. Third, atrophy is consistently progressive after 3 years of age and affects both gray and white matter. Fourth, thinning of the corpus callosum can be readily detected soon after disease onset. Fifth, high-signal abnormalities can be seen in periventricular white matter with FLAIR and T2-weighted sequences. When combined with clinical features, these MRI findings should narrow the differential diagnosis in cases of neuroregression and prompt targeted Sanger sequencing of UBTF.
Findings that UBTF E210K exhibits enhanced binding to the rDNA promoter and drives significantly increased expression of 18S rRNA, strongly suggested that E210K operates via a UBTF1 gain-of-function mechanism (26). In large part, our own data support this model and expand upon previous work. First of all, we found markedly increased expression of pre-rRNA and 18S rRNA in human UBTF E210K fibroblasts. Second, lethal phenotypes were identified with neuronal expression of human UBTF1 in Drosophila. By utilizing the PhiC31 integrase system in Drosophila to avoid expression differences based on transgene landing site in the genome (34), we could investigate the pathogenicity of the UBTF1-E210K mutation. Our experiments overexpressing WT UBTF1 or UBTF1-E210K in Drosophila neurons using c155-GAL4 indicates that UBTF1-E210K is pathogenic, since expression of UBTF1-E210K caused lethality while expression of WT UBTF1 did not. We observed small-eye and semi-lethal phenotypes upon expression of WT UBTF1 with a second GAL4 line eye3.5-GAL4, and expression of UBTF1-E210K exacerbated these phenotypes and caused complete lethality and lack of head development. In combination with our molecular and cellular studies in fibroblasts, these findings in Drosophila support the notion that increased levels of rRNA via expression of either UBTF1 E210K or over-expression of WT UBTF1 is toxic to cells.
Our gene expression studies in patient fibroblasts and mouse behavioral experiments suggest possible contributions of UBTF2 gain-of-function and total UBTF loss-of-function, respectively, to the pathogenesis of the UBTF E210K regression syndrome. Although homozygous loss of Ubtf is embryonic lethal, heterozygous germline loss of Ubtf was associated with only modest neurological abnormalities in young adult mice and increased sensitivity to γ-irradiation in MEFs. If accumulation of non-rDNA DNA damage contributes to the pathobiology of UBTF deficiency and/or dysfunction, it is possible that neurological and cellular phenotypes will be more robust in aged Ubtf+/− mice or other models. However, in isolation, it is doubtful that loss of one of UBTF1’s functions significantly contributes to the pathobiology of the UBTF E210K neuroregression. On the other hand, up-regulation of PPARGC1A, down-regulation of HIST1H4B and SLC4A4, and dysregulation of other UBTF2 targets may play a role in neuronal dysfunction and disease manifestations. For instance, recessive loss-of-function mutations of SLC4A4 cause cognitive impairment (35).
Examination of the ExAC and DECIPHER v9.18 (decipher.sanger.ac.uk) databases indicates that UBTF loss-of-function is poorly tolerated. DECIPHER reports 5 duplications/gains and 4 deletions/losses that encompass the entirety of UBTF on Chr 17. Of these 9 structural variants, 8 are either de novo or of unknown inheritance and these 8 are associated with overt neurological phenotypes. Consistent with our findings in Drosophila, mice and MEFs, both over or under expression of UBTF may be deleterious in humans.
In healthy cells, nucleolar size tends to correlate with Pol I transcriptional activity and cell growth (36). However, nucleolar size and increased rDNA transcription may be decoupled by molecular pathways regulated by Pol II transcription (37,38). Nucleolar number is determined, in part, by cell line, cell-cycle stage and various genetic factors (39). In an adjustment of previous work generated with fibroblasts from a single patient and single control, our blinded quantitative analyses of four patients and four controls exposed only modest differences in nucleoli number and area which could be ascribed, in part, to sample size and minor methodologies dissimilarities between studies. Alternatively, UBTF2 E210K dysregulation of genes directly or indirectly involved in the regulation of nucleolar size and number could have dampened the morphological effects of increased Pol I transcriptional activity (37–39). In addition, aberrant regulation of Pol I by mutant UBTF1 may produce downstream events quite disparate from those driven by normal signaling cascades associated with cellular growth.
Overexpression of rRNA and nucleolar dysfunction may contribute to damage of both nucleolar rDNA and non-nucleolar nuclear DNA. Conversely, depletion of Ubtf1/Ubtf2 has been shown to cause DNA damage and genomic instability independent of Pol I and increased expression of rRNA (11,16). The slow temporal accumulation of damaged DNA may be responsible for the fairly precise onset of neurological dysfunction at 2.5 to 3 years of age in humans harboring the UBTF E210K mutation. As suggested by our cell-cycle analyses and TUNEL labeling of fibroblasts, abnormalities of ribosomal biogenesis can lead to activation of p53-dependent cell-cycle arrest and apoptosis (40).
The UBTF E210K neuroregression syndrome is best characterized as a neurodegenerative disorder with an early and relatively precise age of onset compared with more classic adult-onset neurodegenerative disorders like Huntington disease, Alzheimer disease, and Parkinson disease. Ribosomal biogenesis and nucleolar stress may be centrally implicated in the pathogenesis of these and other disorders of the nervous system (41–43). Expression levels of rRNA must be tightly linked to cellular state and either aberrant down- or up-regulation of rRNA expression may be deleterious to cells. For example, mutant Htt transcripts have been shown to interact with nucleolin leading to down-regulation of pre-rRNA and activation of the p53 nucleolar stress pathway (41). Moreover, nucleolar proteins participate in repair of damaged DNA outside the nucleolus (44,45). Therefore, persistent DNA breaks identified with comet assays and 53BP1 immunohistochemistry could be due to either increased expression of rRNA and nucleolar stress or dysregulation of UBTF2 target genes.
In comparison with many other neurodevelopmental and neuroregression disorders, phenotypic features of the UBTF E210K neuroregression syndrome show relatively high consistency among affected individuals; this should facilitate more rapid identification of additional cases. Cellular, invertebrate and vertebrate models of this disorder may be useful as novel and powerful tools to study the relationships among rDNA, rRNA, the nucleolar stress response, and DNA DSB break repair in neurodegeneration, and therapeutic/phenotypic rescue, as well.
Materials and Methods
Research subjects
The individuals or guardians of the individuals participating in this study gave written, informed consent as part of clinical protocols approved by the Institutional Review Boards of the University of Tennessee Health Science Center, National Institutes of Health, National Human Genome Research Institute, Children’s Hospital of Pittsburgh, and Translational Genomics Research Institute (TGen). DNA was harvested from peripheral whole blood. Fibroblast cultures were established from forearm skin punch biopsies.
Whole exome sequencing
National Institutes of Health–Undiagnosed Disease Program
Genomic DNA was extracted from peripheral whole blood using the Gentra Puregene Blood Kit (Qiagen) and an AutoGen FlexStar with standard procedures. The DNA of all family members was subjected to an integrated set of genomic analyses including high-density single-nucleotide polymorphism (SNP) arrays and WES as previously described (46–49). To test for copy number variants and to form segregation BED files for exome analysis, Omni Express 12 (hg19) SNP arrays were run on genomic DNA from all family members as described (46). WES was performed on probands and available family members using the Illumina HiSeq2000 platform and the TrueSeqV1 capture kit (UDP_2009; Illumina), SeqCap EZ Exome + UTR Lib (UDP_5762; Roche); TruSeq Human Exome 62 Mb (UDP_7621, Illumina). Sample library preparation, sequencing, and analysis were performed using the standard National Institutes of Health (NIH) Intramural Sequencing Center (NISC) pipeline and Axeq Technologies. WES quality was verified by using Picard (http://broadinstitute.github.io/picard/). WES quality for UDP_2009, UDP_7621, and UDP_5762 was assessed by conjointly using Sambamba 0.6.6 (50) and shell scripts. Sequence data were aligned to human reference genome (hg19) using Novoalign (Novocraft Technologies). The quality of alignment and genotype call of variants were checked using the Integrative Genome Viewer (https://www.broadinstitute.org/igv/home). Variants listed in the Variant Call Files (VCFs) were filtered based on rarity (MAF <0.02, 95% confidence interval and gnomAD homozygote count ≤ 25; Undiagnosed Disease Program (UDP) founders cohort population frequency data with variant allele count < 12), Mendelian segregation, and predicted deleteriousness, and were prioritized based on coding effect (non-synonymous, frameshift, stop gain, stop loss, start loss, in frame), proximity to splice sites (within 20 base pairs of a canonical splice site into the intron, or 5 base pairs into the exon), CADD v1.3 Phred scores (28) and Exomiser (51). Prioritized variants with potential coding effects had CADD Phred scores ≥ 5. Splice site variants did not undergo the CADD Phred score filter of ≥ 5 because non-coding variants usually have low CADD Phred scores (< 5). Top-scoring variants were confirmed by Sanger sequencing on the Cartagenia platform.
TGen. Genomic DNA from the family was extracted from peripheral blood and processed in a CLIA lab using the DNeasy extraction kit (Qiagen). Genomic libraries were prepared for sequencing with the Kapa Biosystems Hyper DNA Prep Kit for Illumina Platforms. Exome capture was performed with the Agilent SureSelectXT Target Enrichment Platform using Clinical Research Exome baits. Sequencing was performed by 100bp paired-end sequencing on a HiSeq2000 instrument (Illumina). Mean target exome coverage was 100–110X. Filtered reads were aligned to the human genome (hg19/GRC37) using the Burrows-Wheeler transform (BWA v.0.5.10). Reads were sorted and PCR duplicates were removed using Picard v1.79. Base quality recalibration and indel realignment were performed using the Genome Analysis Toolkit (GATK v3.1–1). Data were filtered against db SNP137 1000 Genomes, an in-house exome database of over 2600 exomes, and then annotated with SnpEff 3.0a against Ensembl GRCh37.66 to identify novel damaging mutations. Variants were jointly called with HaplotypeCaller, annotated with SnpEff and selected (SnpSift) for protein-coding events. Prediction scores were loaded from dbNSFP (Database for Nonsynonymous SNP’s Functional Predictions) and used for filtering. Relatedness and gender for families was checked using Plink v1.07. The variant was confirmed as present in the proband and absent in the parents at a CLIA lab using DNA extracted from buccal cells. Using genomic DNA from the submitted specimen, the relevant portion of the requested gene was PCR amplified and capillary sequencing was performed. Bi-directional sequence was assembled, aligned to reference gene sequences based on human genome build GRCh37/UCSC hg19 and analyzed for known familial sequence variants.
Cell culture
Mouse embryonic fibroblasts (MEFs) were isolated from Ubtf+/+ and Ubtf+/− embryos at 12 to 14 days gestation. MEFs and fibroblasts from four sex-matched normal controls and four UBTF E210K subjects were grown and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 1% L-glutamine and 1% penicillin-streptomycin. Cells were plated on gelatin-coated T25 tissue-culture flasks in an incubator at 37°C with 5% CO2. After their fourth passage, MEFs were exposed to 20 Gy γ-irradiation (γ-IR) using a 137Cs source.
Immunofluorescence
For assessment of DNA damage, cells were plated on poly-l-lysine coated coverslips and fixed in 4% paraformaldehyde solution for 10 min. After fixation, cells were washed 2X with 1X phosphate-buffered saline (PBS) and permeabilized with 0.3% TritonTM X-100 for 15 min followed by blocking with 1% bovine serum albumin in PBS. Cells were incubated with anti-53BP1 (ab21083; Abcam), anti-UBTF [Moss Lab, anti-UBTF#7, a.a.2–404, (52) or anti-fibrillarin (MMS-581S; BioLegend) antibodies overnight, rinsed, and incubated with Alexa Fluor 488 goat anti-rabbit (A-11008, Thermo Fisher Scientific) or Alexa Fluor 568 anti-mouse (A-11004, Thermo Fisher Scientific) secondary antibodies for 1 h and again washed 3X with PBS. Coverslips were then immersed in mounting medium containing DAPI (H-1200, Vector) and placed on slides. Images were captured with a 710 Zeiss confocal microscope. For quantitative analyses, 53BP1-foci were visually counted under an epifluorescence microscope using a 63X objective. The number of nucleoli per nucleus and nucleolar areas was quantified blindly using NIH ImageJ software and high-resolution digital images. Fibroblasts (12–29/subject) from four controls and four UBTF E210K patients were used for quantitative analysis of nucleoli.
Cell-cycle analysis
For cell-cycle analysis, cells were fixed with ice-chilled 70% ethanol for 1 h at -20°C. Fixed cells were then washed with 3X PBS followed by incubation with propidium iodide (PI) staining buffer (PBS with RNaseA 100 μg/ml and PI 50 μg/ml) for 30 min. After the incubation period, cells were analyzed by flow cytometry (YETI, Propel Lab).
Apoptosis
A TUNEL Apoptosis Kit (Roche) was used to detect apoptosis according to manufacturer’s instructions. Briefly, cells were washed 3X with PBS, fixed with 4% paraformaldehyde for 10 min at room temperature, washed 2X, and then permeabilized with 0.3% TritonTM X-100 (Sigma-Aldrich). After washing (2X), cells were incubated with the TUNEL reaction mixture for 1 h at 37°C and then washed 3X with PBS. Coverslips were then immersed in mounting medium containing DAPI (H-1200, Vector) and placed on slides for subsequent imaging.
Reverse transcription polymerase chain reaction and quantitative real-time polymerase chain reaction
Relative levels of total Ubtf and Ubtf1 mRNA were determined in the cerebral cortex, cerebellum and liver of 3-mo-old WT and Ubtf+/− mice (2 males and 2 females of each genotype). Relative levels of human total UBTF and UBTF1 mRNA, pre-rRNA 45S and 18S were compared between patients and normal controls with RNA extracted from fibroblasts (2 males and 2 females in each group). TaqMan probe based QRT-PCR was performed with Roche’s LightCycler® 480 System using primers to target UBTF1 or Ubtf1 and regions common to both major isoforms (total UBTF or Ubtf) in humans and mice, respectively (Supplementary Material, Table S9). Human pre-rRNA expression levels in patients and normal controls were analyzed with SYBR Green-based QRT-PCR using cDNA derived from reverse transcription using either random primers or gene-specific primers. Expression of 18S RNA was analyzed with cDNA derived from reverse transcription using random primers and the RETROscript™ Reverse Transcription Kit from ThermoFisher Scientific (Supplementary Material, Table S9). Mouse and human β-actin were used as endogenous controls. The expression levels of selected genes shown to be dysregulated by knock-down of UBTF (11) were also examined with TaqMan probe based QRT-PCR in mouse tissues and human fibroblasts using mouse or human β-actin as endogenous controls (Supplementary Material, Table S9). More detailed QRT-PCR protocols are presented in previous work (53,54).
Comet assay
The alkaline comet assays were performed on human fibroblasts (4 probands and 4 sex-matched normal controls) and cultured MEFs (4 in each group of WT and Ubtf+/−) according to standard protocol described by Trevigen's CometAssay kit. MEFs were treated with or without γ-irradiation (20 Gy), and then subjected to comet assays at 20 hrs after IR. Cells were trypsinized and washed in ice-cold Ca2+/Mg2+-free 1X PBS. Cells (1 × 105) were mixed with 500 μl of pre-warmed low melting agarose (1: 10, v/v) and 50 μl of the mixture were plated onto a slide (CometSlide™, Trevigen). After the agarose solidified and attached to the slides, the slides were immersed in pre-chilled lysis solution for 1 h at 4°C, then in alkaline unwinding solution (200 mM NaOH and 1 mM EDTA) for 20 min at room temperature. Electrophoresis was performed in pre-chilled alkaline electrophoresis solution (200 mM NaOH and 1 mM EDTA) at 4°C for 30 min at 21 V. The slides were then washed twice with distilled water for 5 min each time, followed by a five min-incubation with 70% ethanol. The slides were air-dried at 37°C for 15 min in the dark and the agarose gels were stained with SYBRTM Gold for 30 min at room temperature. After SYBR Gold was removed and slides were air-dried, images were acquired with fluorescence microscopy. Images were randomly chosen by an investigator blinded to genotype and processed with an automated tool for comet assay image analysis (OpenComet v1.3.1). OpenComet was deployed as an ImageJ (www.rsbweb.nih.gov/ij/) plugin (55). Three samples were processed from each patient, human control, and mouse, and at least 100 cells were analyzed per sample. Comet analysis is based on the fact that the intensity of emitted light is linearly related to the quantity of DNA in particular regions of a cell. OpenComet identifies comets and delineates the boundaries of their heads and tails. Tail DNA content as a percentage of comet DNA content is deemed tail DNA %.
UBTF transgenic flies
Human UBTF1 EZShuttle™ Gateway® PLUS ORF Clone was ordered from GeneCopoeia (Vector #GC-F0235). Site-directed mutagenesis was performed to create mutant UBTF1 (c.628G > A, p.E210K). Both wildtype and mutant UBTF1 cDNAs were shuttled into the pUASg.attb gateway vector obtained from FlyORF by performing an LR clonase reaction according to the manufacturer’s instructions (ThermoFisher Cat# 11791–020). Clones were verified by Sanger sequencing and complete pUASg.attb vectors were injected into the PhiC31 integrase line y[1] M{vas-int.Dm}ZH-2A w[*]; M{3xP3-RFP.attP’}ZH-51C (BDSC Stock # 24482) to generate UAS lines (BestGene). The ZH-51C line harbors an attP site allowing for integration of the pUASg.attB vectors into a known genomic location allowing for similar expression levels of wild-type and mutant UBTF1 constructs. The attP insertion site is located on Chr 2 and lines were balanced by the Chr 2 balancer CyO.
Fly stocks obtained from the Bloomington Drosophila Stock Center (BDSC, NIH P40OD018537) were used in this study. Fly crosses were performed at 25°C on a 12 h light/dark cycle and raised on standard corn meal media (BDSC). The Eye3.5-GAL4 (BDSC stock # 8221) stock was obtained from BDSC and c155-GAL4 was a gift from Dr. Hugo Bellen.
Ubtf null mice
All mouse experiments were performed in accordance with the National Institutes of Health’s Guidelines for the Care and Use of Laboratory Animals and approved by our Institutional Animal Care and Use Committee. Derivation and genotyping of Ubtf+/− mice has been detailed in previous work (16). Adult (3-month-old) Ubtf+/− mice (n = 20 males and 22 females) and sex-matched WT littermates (n = 23 males and 21 females) were subjected to a battery of motor and behavioral examinations including open-field activity, rotarod, vertical rope climbing, raised-beam task, grip strength, gait analysis (DigiGaitTM), dominance tube, and cross-maze test. Righting reflex assays were performed prior to weaning in independent groups of Ubtf+/+ (n = 6 males and 5 females) and Ubtf+/− (n = 4 males and 5 females) mice. Mice were weighed weekly and examined for evidence of dystonia or other involuntary movements during open-field activity and routine handling. Sensorimotor and behavioral methods used in this study have been previously described in detail (53,54).
Statistical analyses
ANOVA with post-hoc tests was used to determine the effects of genotype and gender on parametric behavioral measures. The Mann-Whitney test was used to determine the effects of genotype within gender for a non-parametric behavioral measure (slips on the raised beam task). Fisher’s exact test was used for lethality analyses in Drosophila and to determine the effects of genotype on the results of dominance tube testing in mice. Two-tailed t-tests were used to establish the effects of genotype on cellular measurements. An alpha (α) of 0.05 was chosen for statistical significance.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
The C4RCD Research Group at the Center for Rare Childhood Disorders, TGen, thanks go to Newell Belnap, David Craig, Matt De Both, Matthew Huentelman, Vinodh Narayanan, Sampath Rangasamy, Ryan Richholt, Megan Russell, Isabelle Schrauwen, Ashley Siniard and Szabolcs Szelinger for assistance with data analysis and interpretation; Ana Claasen for assistance with wet-lab sample processing; and Keri Ramsey for enrollment and administrative support. At the University of Tennessee Health Science Center, Phillip Hargrove helped with behavioral analyses of mice.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health [#R21 GM118962 to R.T.H. and M.S.L., #R01 NS082296 and #R56 NS094965 to M.S.L., and #R21 HD091541 to L.T.R.], Dorothy/Daniel Gerwin Parkinson’s Research Fund to M.S.L., the Department of Defense [#W81XWH-17–1-0062 to M.S.L.], Intramural Research Program of the National Human Genome Research Institute, and Common Fund of the NIH Office of the Director. TGen's Center for Rare Childhood Disorders receives funding through private donations made to the TGen Foundation. Funding to pay the Open Access publication charges for this article was provided by National Institutes of Health grant #R21 GM118962 to R.T.H. and M.S.L.
References
- 1. Jantzen H.M., Admon A., Bell S.P., Tjian R. (1990) Nucleolar transcription factor hUBF contains a DNA-binding motif with homology to HMG proteins. Nature, 344, 830–836. [DOI] [PubMed] [Google Scholar]
- 2. Moss T., Langlois F., Gagnon-Kugler T., Stefanovsky V. (2007) A housekeeper with power of attorney: the rRNA genes in ribosome biogenesis. Cell. Mol. Life Sci., 64, 29–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O'Mahony D.J., Rothblum L.I. (1991) Identification of two forms of the RNA polymerase I transcription factor UBF. Proc. Natl. Acad. Sci. U. S. A, 88, 3180–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beckmann H., Chen J.L., O'Brien T., Tjian R. (1995) Coactivator and promoter-selective properties of RNA polymerase I TAFs. Science, 270, 1506–1509. [DOI] [PubMed] [Google Scholar]
- 5. Jantzen H.M., Chow A.M., King D.S., Tjian R. (1992) Multiple domains of the RNA polymerase I activator hUBF interact with the TATA-binding protein complex hSL1 to mediate transcription. Genes Dev., 6, 1950–1963. [DOI] [PubMed] [Google Scholar]
- 6. Stefanovsky V., Langlois F., Gagnon-Kugler T., Rothblum L.I., Moss T. (2006) Growth factor signaling regulates elongation of RNA polymerase I transcription in mammals via UBF phosphorylation and r-chromatin remodeling. Mol. Cell, 21, 629–639. [DOI] [PubMed] [Google Scholar]
- 7. Herdman C., Mars J.C., Stefanovsky V.Y., Tremblay M.G., Sabourin-Felix M., Lindsay H., Robinson M.D., Moss T. (2017) A unique enhancer boundary complex on the mouse ribosomal RNA genes persists after loss of Rrn3 or UBF and the inactivation of RNA polymerase I transcription. PLoS Genet., 13, e1006899.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Panov K.I., Friedrich J.K., Russell J., Zomerdijk J.C. (2006) UBF activates RNA polymerase I transcription by stimulating promoter escape. embo J., 25, 3310–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stefanovsky V.Y., Langlois F., Bazett-Jones D., Pelletier G., Moss T. (2006) ERK modulates DNA bending and enhancesome structure by phosphorylating HMG1-boxes 1 and 2 of the RNA polymerase I transcription factor UBF. Biochemistry, 45, 3626–3634. [DOI] [PubMed] [Google Scholar]
- 10. Grueneberg D.A., Pablo L., Hu K.Q., August P., Weng Z., Papkoff J. (2003) A functional screen in human cells identifies UBF2 as an RNA polymerase II transcription factor that enhances the beta-catenin signaling pathway. Mol. Cell. Biol., 23, 3936–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sanij E., Diesch J., Lesmana A., Poortinga G., Hein N., Lidgerwood G., Cameron D.P., Ellul J., Goodall G.J., Wong L.H.. et al. (2015) A novel role for the Pol I transcription factor UBTF in maintaining genome stability through the regulation of highly transcribed Pol II genes. Genome Res., 25, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pelletier G., Stefanovsky V.Y., Faubladier M., Hirschler-Laszkiewicz I., Savard J., Rothblum L.I., Cote J., Moss T. (2000) Competitive recruitment of CBP and Rb-HDAC regulates UBF acetylation and ribosomal transcription. Mol. Cell, 6, 1059–1066. [DOI] [PubMed] [Google Scholar]
- 13. Voit R., Grummt I. (2001) Phosphorylation of UBF at serine 388 is required for interaction with RNA polymerase I and activation of rDNA transcription. Proc. Natl. Acad. Sci. U. S. A, 98, 13631–13636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stefanovsky V.Y., Pelletier G., Hannan R., Gagnon-Kugler T., Rothblum L.I., Moss T. (2001) An immediate response of ribosomal transcription to growth factor stimulation in mammals is mediated by ERK phosphorylation of UBF. Mol. Cell, 8, 1063–1073. [DOI] [PubMed] [Google Scholar]
- 15. Stefanovsky V.Y., Moss T. (2008) The splice variants of UBF differentially regulate RNA polymerase I transcription elongation in response to ERK phosphorylation. Nucleic Acids Res., 36, 5093–5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hamdane N., Stefanovsky V.Y., Tremblay M.G., Németh A., Paquet E., Lessard F., Sanij E., Hannan R., Moss T., Pikaard C.S. (2014) Conditional inactivation of Upstream Binding Factor reveals its epigenetic functions and the existence of a somatic nucleolar precursor body. PLoS Genet., 10, e1004505.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hamdane N., Herdman C., Mars J.C., Stefanovsky V., Tremblay M.G., Moss T. (2015) Depletion of the cisplatin targeted HMGB-box factor UBF selectively induces p53-independent apoptotic death in transformed cells. Oncotarget, 6, 27519–27536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McStay B., Grummt I. (2008) The epigenetics of rRNA genes: from molecular to chromosome biology. Annu. Rev. Cell. Dev. Biol., 24, 131–157. [DOI] [PubMed] [Google Scholar]
- 19. Sanij E., Hannan R.D. (2009) The role of UBF in regulating the structure and dynamics of transcriptionally active rDNA chromatin. Epigenetics, 4, 374–382. [DOI] [PubMed] [Google Scholar]
- 20. Pederson T. (2011) The nucleolus. Cold Spring Harb. Perspect. Biol., 3, a000638.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Capitano F., Gargiuli C., Angerilli A., Maccaroni K., Pelliccia F., Mele A., Camilloni G. (2016) RNA polymerase I transcription is modulated by spatial learning in different brain regions. J. Neurochem., 136, 706–716. [DOI] [PubMed] [Google Scholar]
- 22. Lee J., Hwang Y.J., Boo J.H., Han D., Kwon O.K., Todorova K., Kowall N.W., Kim Y., Ryu H. (2011) Dysregulation of upstream binding factor-1 acetylation at K352 is linked to impaired ribosomal DNA transcription in Huntington's disease. Cell Death Differ., 18, 1726–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Garcia-Esparcia P., Hernandez-Ortega K., Koneti A., Gil L., Delgado-Morales R., Castano E., Carmona M., Ferrer I. (2015) Altered machinery of protein synthesis is region- and stage-dependent and is associated with alpha-synuclein oligomers in Parkinson's disease. Acta. Neuropathol. Commun., 3, 76.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tao Z., Wang H., Xia Q., Li K., Li K., Jiang X., Xu G., Wang G., Ying Z. (2015) Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet., 24, 2426–2441. [DOI] [PubMed] [Google Scholar]
- 25. Kamate M. (2014) Clinical aspects of neuroregression: our experience on Batten disease. Mol. Cytogenet., 7, I37.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Edvardson S., Nicolae C.M., Agrawal P.B., Mignot C., Payne K., Prasad A.N., Prasad C., Sadler L., Nava C., Mullen T.E.. et al. (2017) Heterozygous De Novo UBTF Gain-of-Function Variant Is Associated with Neurodegeneration in Childhood. Am. J. Hum. Genet., 101, 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu X., Wu C., Li C., Boerwinkle E. (2016) dbNSFP v3.0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat., 37, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kircher M., Witten D.M., Jain P., O'Roak B.J., Cooper G.M., Shendure J. (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet., 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ioannidis N.M., Rothstein J.H., Pejaver V., Middha S., McDonnell S.K., Baheti S., Musolf A., Li Q., Holzinger E., Karyadi D.. et al. (2016) REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet., 99, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hunt D., Leventer R.J., Simons C., Taft R., Swoboda K.J., Gawne-Cain M., study D.D.D., Magee A.C., Turnpenny P.D., Baralle D. (2014) Whole exome sequencing in family trios reveals de novo mutations in PURA as a cause of severe neurodevelopmental delay and learning disability. J. Med. Genet., 51, 806–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Millan F., Cho M.T., Retterer K., Monaghan K.G., Bai R., Vitazka P., Everman D.B., Smith B., Angle B., Roberts V.. et al. (2016) Whole exome sequencing reveals de novo pathogenic variants in KAT6A as a cause of a neurodevelopmental disorder. Am. J. Med. Genet. A, 170, 1791–1798. [DOI] [PubMed] [Google Scholar]
- 32. Kloth K., Denecke J., Hempel M., Johannsen J., Strom T.M., Kubisch C., Lessel D. (2017) First de novo ANK3 nonsense mutation in a boy with intellectual disability, speech impairment and autistic features. Eur. J. Med. Genet., 60, 494–498. [DOI] [PubMed] [Google Scholar]
- 33. Schafgen J., Cremer K., Becker J., Wieland T., Zink A.M., Kim S., Windheuser I.C., Kreiss M., Aretz S., Strom T.M.. et al. (2016) De novo nonsense and frameshift variants of TCF20 in individuals with intellectual disability and postnatal overgrowth. Eur. J. Hum. Genet., 24, 1739–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bischof J., Maeda R.K., Hediger M., Karch F., Basler K. (2007) An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc. Natl. Acad. Sci. U. S. A, 104, 3312–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Igarashi T., Inatomi J., Sekine T., Seki G., Shimadzu M., Tozawa F., Takeshima Y., Takumi T., Takahashi T., Yoshikawa N.. et al. (2001) Novel nonsense mutation in the Na+/HCO3- cotransporter gene (SLC4A4) in a patient with permanent isolated proximal renal tubular acidosis and bilateral glaucoma. J. Am. Soc. Nephrol., 12, 713–718. [DOI] [PubMed] [Google Scholar]
- 36. Farley K.I., Surovtseva Y., Merkel J., Baserga S.J. (2015) Determinants of mammalian nucleolar architecture. Chromosoma, 124, 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Neumuller R.A., Gross T., Samsonova A.A., Vinayagam A., Buckner M., Founk K., Hu Y., Sharifpoor S., Rosebrock A.P., Andrews B.. et al. (2013) Conserved regulators of nucleolar size revealed by global phenotypic analyses. Sci. Signal., 6, ra70.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ma T.H., Lee L.W., Lee C.C., Yi Y.H., Chan S.P., Tan B.C., Lo S.J. (2016) Genetic control of nucleolar size: An evolutionary perspective. Nucleus, 7, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hernandez-Verdun D. (2011) Assembly and disassembly of the nucleolus during the cell cycle. Nucleus, 2, 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rubbi C.P., Milner J. (2003) Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. embo J., 22, 6068–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tsoi H., Chan H.Y. (2013) Expression of expanded CAG transcripts triggers nucleolar stress in Huntington's disease. Cerebellum, 12, 310–312. [DOI] [PubMed] [Google Scholar]
- 42. Parlato R., Liss B. (2014) How Parkinson's disease meets nucleolar stress. Biochim. Biophys Acta, 1842, 791–797. [DOI] [PubMed] [Google Scholar]
- 43. Hallgren J., Pietrzak M., Rempala G., Nelson P.T., Hetman M. (2014) Neurodegeneration-associated instability of ribosomal DNA. Biochim. Biophys. Acta, 1842, 860–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. De A., Donahue S.L., Tabah A., Castro N.E., Mraz N., Cruise J.L., Campbell C. (2006) A novel interaction [corrected] of nucleolin with Rad51. Biochem. Biophys. Res. Commun., 344, 206–213. [DOI] [PubMed] [Google Scholar]
- 45. Koike A., Nishikawa H., Wu W., Okada Y., Venkitaraman A.R., Ohta T. (2010) Recruitment of phosphorylated NPM1 to sites of DNA damage through RNF8-dependent ubiquitin conjugates. Cancer Res., 70, 6746–6756. [DOI] [PubMed] [Google Scholar]
- 46. Gahl W.A., Markello T.C., Toro C., Fajardo K.F., Sincan M., Gill F., Carlson-Donohoe H., Gropman A., Pierson T.M., Golas G.. et al. (2012) The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet. Med., 14, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gnirke A., Melnikov A., Maguire J., Rogov P., LeProust E.M., Brockman W., Fennell T., Giannoukos G., Fisher S., Russ C.. et al. (2009) Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol., 27, 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Teer J.K., Bonnycastle L.L., Chines P.S., Hansen N.F., Aoyama N., Swift A.J., Abaan H.O., Albert T.J., Margulies E.H., Green E.D.. et al. (2010) Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res., 20, 1420–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bentley D.R., Balasubramanian S., Swerdlow H.P., Smith G.P., Milton J., Brown C.G., Hall K.P., Evers D.J., Barnes C.L., Bignell H.R.. et al. (2008) Accurate whole human genome sequencing using reversible terminator chemistry. Nature, 456, 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tarasov A., Vilella A.J., Cuppen E., Nijman I.J., Prins P. (2015) Sambamba: fast processing of NGS alignment formats. Bioinformatics, 31, 2032–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smedley D., Jacobsen J.O., Jager M., Kohler S., Holtgrewe M., Schubach M., Siragusa E., Zemojtel T., Buske O.J., Washington N.L.. et al. (2015) Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat. Protoc., 10, 2004–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lessard F., Morin F., Ivanchuk S., Langlois F., Stefanovsky V., Rutka J., Moss T. (2010) The ARF tumor suppressor controls ribosome biogenesis by regulating the RNA polymerase I transcription factor TTF-I. Mol. Cell, 38, 539–550. [DOI] [PubMed] [Google Scholar]
- 53. Xiao J., Vemula S.R., Xue Y., Khan M.M., Kuruvilla K.P., Marquez-Lona E.M., Cobb M.R., LeDoux M.S. (2016) Motor phenotypes and molecular networks associated with germline deficiency of Ciz1. Exp. Neurol., 283, 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xiao J., Vemula S.R., Xue Y., Khan M.M., Carlisle F.A., Waite A.J., Blake D.J., Dragatsis I., Zhao Y., LeDoux M.S. (2017) Role of major and brain-specific Sgce isoforms in the pathogenesis of myoclonus-dystonia syndrome. Neurobiol. Dis., 98, 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gyori B.M., Venkatachalam G., Thiagarajan P.S., Hsu D., Clement M.V. (2014) OpenComet: an automated tool for comet assay image analysis. Redox Biol., 2, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.