

Abstract
Deletion of the 16p11.2 CNV affects 25 core genes and is associated with multiple symptoms affecting brain and body, including seizures, hyperactivity, macrocephaly, and obesity. Available data suggest that most symptoms are controlled by haploinsufficiency of two or more 16p11.2 genes. To identify interacting 16p11.2 genes, we used a pairwise partial loss of function antisense screen for embryonic brain morphology, using the accessible zebrafish model. fam57ba, encoding a ceramide synthase, was identified as interacting with the doc2a gene, encoding a calcium-sensitive exocytosis regulator, a genetic interaction not previously described. Using genetic mutants, we demonstrated that doc2a+/− fam57ba+/− double heterozygotes show hyperactivity and increased seizure susceptibility relative to wild-type or single doc2a−/− or fam57ba−/− mutants. Additionally, doc2a+/− fam57ba+/− double heterozygotes demonstrate the increased body length and head size. Single doc2a+/− and fam57ba+/− heterozygotes do not show a body size increase; however, fam57ba−/− homozygous mutants show a strongly increased head size and body length, suggesting a greater contribution from fam57ba to the haploinsufficient interaction between doc2a and fam57ba. The doc2a+/− fam57ba+/− interaction has not been reported before, nor has any 16p11.2 gene previously been linked to increased body size. These findings demonstrate that one pair of 16p11.2 homologs can regulate both brain and body phenotypes that are reflective of those in people with 16p11.2 deletion. Together, these findings suggest that dysregulation of ceramide pathways and calcium sensitive exocytosis underlies seizures and large body size associated with 16p11.2 homologs in zebrafish. The data inform consideration of mechanisms underlying human 16p11.2 deletion symptoms.
Introduction
Identifying multigenic contribution and relevant genetic interactions contributing to neurodevelopmental disorders is a huge challenge facing the field. In these disorders, physical comorbidities are frequently associated with mental health disorders and compound challenges faced by people with these diagnoses. Multigenic regions associated with neurodevelopmental disorders include copy number variants (CNVs). Loss of one copy of the 16p11.2 is the CNV is highly prevalent, occurring in 1 in 2000 people (1), and is linked to many symptoms affecting brain and body function, including autism spectrum disorders (ASDs), intellectual disability, developmental delay, language impairment, seizures, macrocephaly, obesity, hypotonia, motor impairment, and gastrointestinal problems (2–10).
The 16p11.2 CNV affects 25 core genes, and we considered four models to account for the connection between loss of function in core genes and specific phenotypes or symptoms. First, a single gene from 16p11.2 could be linked to all symptoms (Fig. 1A). This model is not supported by the extensive sequencing analysis that has been done for people with autism and schizophrenia. No statistically significant variants for any of the 25 single genes from this region have been recovered (11–15), making it unlikely that a single gene could account for all the other associated symptoms as well. Second, two or more genes from the 16p11.2 region interact and mediate all symptoms (Fig. 1B). Explorations for smaller internal CNVs associated with autism have not recovered significant hits in the 16p11.2 interval (16–18), but these would not be able to account for multiple critical genes if they are not syntenic. Third, single genes from this region may each mediate a distinct symptom (Fig. 1C). Extensive sequence analysis on large samples from people with autism has not revealed statistically significant variants for any of the 25 single genes from this region (11,12,14,15). However, single genes have been implicated in some of the other symptoms associated with 16p11.2 CNVs. PRRT2 has been associated, as heterozygous or homozygous mutations, with paroxysomal neurological disorders, benign familial infantile seizures, febrile convulsions, and epileptic seizures (19). A single case of epilepsy was found in a person with a KIF22 heterozygous mutation (20). TBX6 is a candidate for vertebral anomalies associated with 16p11.2 CNVs as hypomorphic/null compound mutations (21,22). However, overall, there are not many single gene associations from the 16p11.2 interval, and these cannot account for the full spectrum of symptoms associated with the CNV. Therefore, we consider a fourth model where interaction of two or more genes each mediates one symptom (Fig. 1D). These could include either distinct or overlapping gene groups that interact to affect each symptom. This is a complex model that has not been addressed experimentally. There are additional models that would be composites of the third and fourth scenarios.
Figure 1.
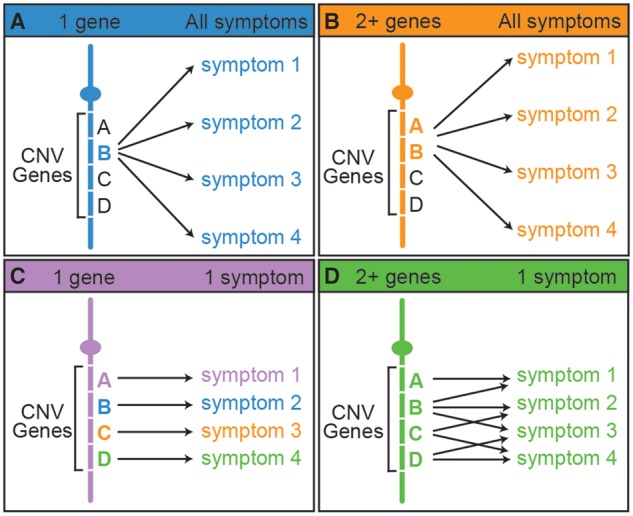
Possible hypotheses for gene-phenotype relationships in multi-symptomatic CNV disorders. (A) A single gene accounts for all of the associated symptoms. (B) The interaction of two or more genes accounts for all of the associated symptoms. (C) Single genes account for a single symptom each. (D) Two or more genes interact to affect one symptom each. These interacting genes can be in different and overlapping sets. There are also additional models that would be composites of the last two hypotheses.
Animal studies have explored the relationship between genes and phenotypes for the 16p11.2 region. Although in these models it is difficult to assess human-specific symptoms, such as language disorders, animal systems can give insight into evolutionarily conserved phenotypes, including seizures and body size control, which are a result of shared physiology, cell biology and molecular biology (23–25). With regard to the ability of single genes to mediate 16p11.2 deletion phenotypes, of eight mouse mutants for single homologs, only one (PPP4C) had a haploinsufficient phenotype: retarded growth (26). Some homozygous mouse mutants for other 16p11.2 homologs showed phenotypes with potential relevance to CNV deletion symptoms, including Doc2a−/− mice, which exhibit defects in synaptic transmission (27), and Coro1a−/− and Mapk3−/− mice, which have some immune deficiencies (28,29). Other mouse studies on 16p11.2 homologs have deleted the entire syntenic interval, and therefore do not address single gene activity (30–32). After complete loss-of-function (LOF) in single genes using antisense techniques, we previously found that 22 of 25 homologs were active during early zebrafish development, but only two (aldoaa and kif22) showed haploinsufficient effects on early brain morphology (33). A small 16p11.2 screen in Drosophila found that a homolog for KIF22 mediated axonal branching patterns (34). A single study has reported that suppressing and overexpressing kctd13 has reciprocal effects on head size in zebrafish, and the suggestion was made that this reflected the opposing effects of 16p11.2 CNV deletion and duplication on people (35). Overall, between human genetics and animal data, there are few single 16p11.2 genes or homologs that regulate distinct symptoms after single copy gene deletion, and only a few of the symptoms associated with 16p11.2 CNVs can be associated with single genes.
Based on these considerations, we explored the fourth possibility outlined above (Fig. 1D), that multiple genes interact to impact each 16p11.2 deletion symptom. We used the zebrafish as a tool to address the most parsimonious hypothesis that two 16p11.2 genes interact to elicit each phenotype. We have previously noted the value of the zebrafish as a tool to study neurodevelopmental disorders (23–25,36). Genetic manipulations are effective, while the wide range of assays and rapid embryonic development in zebrafish means several phenotypes can be studied simultaneously. Although zebrafish cannot demonstrate human-specific phenotypes associated with 16p11.2 CNVs, their genes are orthologous to human disease genes (37). This facilitates the discovery of the biology that triggers pathological mechanisms. Beginning with the simplest interaction hypothesis of just two genes involved in symptomatology, we employed an antisense pair-wise partial LOF screen and double heterozygote lines. Our data identify unpredicted 16p11.2 homolog interactions and new gene functions underlying brain and body phenotypes. These data support the hypothesis that two or more 16p11.2 genes interact to mediate distinct symptoms, and identify lipid metabolism and calcium-sensitive exocytosis as pivotal elements in certain 16p11.2 deletion symptoms.
Results
A subset of interacting 16p11.2 pairs affect early brain development and other phenotypes
We were particularly interested in genetic interactions, which are defined as a combination of two mutations that leads to a phenotype that cannot be predicted from the effects of either mutation alone (38). In other words, a genetic interaction between two gene products results from a non-additive effect of the single gene phenotypes. An additive phenotype resulting from summation of each single gene LOF would not be considered an interaction. To test the hypothesis that two genes from the 16p11.2 CNV interact to impact brain development, we compared the effects of partial LOF in individual zebrafish 16p11.2 homologs to that of two homologs together.
Unlike our previous study, where we found phenotypes associated with 70–95% single gene knockdown for the majority of 16p11.2 homologs, the partial LOF used here for interaction analysis was ≤50% for each gene to more closely reflect the hemizygous state of people with 16p11.2 deletion CNVs. We defined interacting gene pairs as those that gave no or little phenotype for single partial LOF, but did affect the embryonic brain formation with double partial LOF (Fig. 2A). LOF was induced by injection of antisense morpholino-modified oligonucleotides (MOs) defined in our previous study (33), with the amount of MO titrated so approximately 50% normal RNA remained as determined by qPCR (splice-blocking MOs) or a sub-phenotypic dose (translation blocking MOs) (Supplementary Material, Table S1). We analysed interaction between all 162 possible pairwise combinations of 19 genes, by assaying early brain and ventricle morphology at 24 hours post fertilization (hpf). We chose this assay for two reasons. First, nervous system development is impacted by multiple signaling pathways and is sensitive to perturbations (39–41). Second, radiological examinations of people with 16p11.2 CNVs indicate changes in the volume of certain brain regions and associated brain ventricle size (5,42–44). However, zebrafish analyses are performed at the equivalent of first trimester human embryos, and human screening for 16p11.2 deletions at these stages has not been done.
Figure 2.

A subset of pairwise combinations of 16p11.2 homologs alter brain and ventricle morphology. (A) Definition of an interacting pair. Dorsal views of 24 hpf embryos with red dye injected into the ventricles. Single gene LOFs were induced to about ∼50% normal RNA remaining (or a sub-phenotypic dose) and generally resembled control MO-injected embryos. If a pair of genes interacted, then suppressing them together resulted in abnormal brain and ventricle morphology. (B) Pie chart of all 162 pairwise combinations tested. (C) Map of interacting pairs of genes, with the lines drawn between two genes if they interact, and scaling the font size of the gene name according to the number of interactions it has.
Of the 162 gene pairs tested, 16 (10%) resulted in a ventricle phenotype after double partial LOF (Fig. 2B, Supplementary Material, Fig. S1). A pair of genes was scored as interacting if >50% of the double LOF embryos showed a brain morphology phenotype after normalization. To normalize numbers, background levels of a phenotype after single gene LOF were subtracted from the levels after double gene LOF. Most gene combinations (146, 90%) did not yield any phenotype, suggesting that interactions observed were specific. Additional controls for MO specificity included injection with a p53 MO, since activation of p53 has been associated with off target MO effects, and prevention of a phenotype by RNA injection (45). Furthermore, other 16p11.2 homolog expression levels were not affected by double LOF injections (Supplementary Material, Fig. S2).
We used these data to map a 16p11.2 homolog genetic interaction network with lines drawn between interacting genes and scaling the font size of the gene name relative to the number of interactions (Fig. 2C). Fourteen of nineteen genes screened were part of this interaction map. Six of these genes emerged as highly interactive in impacting early brain morphology: fam57ba and kif22 with four interactions each; and asphd1, hirip3, kctd13, and sez6l2 with three interactions each.
Because people with 16p11.2 CNV deletions exhibit multiple symptoms and because defects in early brain morphology may indicate additional nervous system involvement, we conducted a secondary screen for four additional phenotypes: neuronal specification using a NeuroD:GFP line, cranial motor neuron patterning using an islet1:GFP line, enteric neuron density, and muscle fiber organization. Of the 16 gene pairs identified from our initial screen, 11 LOF pairs impacted at least one secondary screen phenotype (Supplementary Materials, Fig. S3, Tables S2 and S3). Overall, there was enrichment for positive interactions, showing a linkage between brain and body phenotypes. Assaying gene pairs that did not interact with the primary screen gave no interactions for any of the secondary screen phenotypes, indicating the strength of brain and ventricle morphology as a first pass assay.
In sum, these initial assays identified pairs of interacting genes in zebrafish, which were associated with phenotypes that could be reflective of symptoms in people with 16p11.2 deletion CNVs. None of these gene interactions has been previously described and could not have been predicted from database analysis, demonstrating the importance of functional studies for discovery of genetic interactions.
doc2a+/− fam57ba+/− double heterozygotes show hyperactivity and increased seizure propensity
Mutants allow examination of gene function in older fish when MOs would be less effective. We generated TALEN mutant lines for some of the more interactive genes uncovered in our initial antisense screen, including fam57ba and doc2a. Zebrafish have two copies of the fam57b gene, and previous analysis has supported the fam57ba copy as most active (33). Mouse Fam57b encodes a ceramide synthase (46) and Doc2a encodes a calcium sensor that primes vesicles for exocytosis (47,48). While there are strong indications of synaptic defects in the literature for some of the associated 16p11.2 CNV symptoms, particularly epilepsy (49,50) and ASD (51), as well as links to metabolic defects (52), there is little about ceramides in particular (53,54). Therefore, this seemed an intriguing gene combination to analyse further. There are reported roles for calcium and ceramide signaling at the synapse (55–57), and because attention deficit-hyperactivity disorder (ADHD) and seizures are prevalent among people with 16p11.2 deletion CNVs (2,6,8,10,58,59), we analysed mutant zebrafish larvae for their baseline movement level and seizure propensity.
Despite evolutionary distance, zebrafish have been a valuable tool for studying human epilepsies. Several assays have been used to validate the fish as a model for human epilepsies. Electrophysiological recordings of larval brain activity after PTZ treatment show epileptiform discharges of brief interictal bursts and long duration bursts similar to those seen in humans (60). The more easily measured increased swimming speed after PTZ treatment is directly correlated to seizure electrophysiology. Swimming velocity can be readily measured in an automated, high throughput system. Both swimming speed and electrophysiological recordings show attenuated response to human anti-epileptic drugs (60,61). Zebrafish epilepsy models have led to identification of anti-convulsant compounds for drug-resistant Dravet syndrome (62), and of other novel anti-convulsants (63–68). Zebrafish have also been widely used to provide functional confirmation for gene candidates linked to seizures in people (69–74).
Movement of seven days post fertilization (dpf), the standard age at which these assays are conducted (60,75), wild-type and doc2a+/−fam57ba+/− larvae was analysed, comparing average velocity as an output of brain activity, with and without the seizure-inducing GABA-A antagonist pentylenetetrazol (PTZ). Across four independent experiments, doc2a+/−fam57ba+/− larvae exhibited higher baseline movement levels as well as increased velocity with a minimal PTZ treatment relative to wild-type. The increased PTZ sensitivity was significant even with normalization to account for the increased baseline movement present in double heterozygotes (Fig. 3A–D). This normalization was done by subtracting the baseline velocity from the PTZ-induced velocity for each larva.
Figure 3.

doc2a and fam57ba interact to increase hyperactivity and seizure propensity. (A,B) Movement traces of individual 7 dpf larvae in wells of a 96-well plate before and after addition of the seizure-inducing drug PTZ (dose is 0.3 mM for top and bottom panels in B, J, and N and 5 mM for top and bottom panels in F and R). Traces show wild-type larval paths during a 10-min recording. Top panels are wild-type, bottom panels are doc2a+/− fam57ba+/− double heterozygous larvae, which show increased movement compared to wild-type both with and without PTZ. (C) Average velocity of baseline movement (no PTZ) shows the increased movement of doc2a+/− fam57ba+/− larvae (yellow bar) compared to wild-type controls (blue bar), with n’s shown above bars. (D) After PTZ addition, doc2a+/− fam57ba+/− show increased velocity compared to wild-type, even after taking baseline movement differences into account by subtracting the baseline velocity from the velocity with PTZ for each larva. (E,F) Movement traces before and after 5 mM PTZ addition for wild-type and fam57ba−/− larvae. (G) Average velocity of baseline swimming is significantly higher in fam57ba−/− (purple bar) compared to wild-type larvae (blue bar). (H) After 5 mM PTZ addition, fam57ba−/− larvae show significantly increased velocity, even after subtracting the increased baseline velocity for each individual fish. (I–P) Analysis of fam57ba+/− (green bars) and doc2a+/− (orange bars) single heterozygotes indicate an interaction for doc2a+/− fam57ba+/− larvae double heterozygous larvae because no significant differences in baseline activity or PTZ response are observed in single heterozygotes. (Q–R) doc2a−/− larvae do not show increased velocity before or after 5 mM PTZ addition (pink bars). *P < 0.05, ***P < 0.001.
To determine the individual contributions of doc2a and fam57ba to this interaction, we examined a spectrum of doc2a and fam57ba genotypes, including doc2a−/− and fam57ba−/− single homozygous mutants (Table 1). fam57ba−/− mutants displayed higher baseline movement and PTZ sensitivity, but not to the same degree as double heterozygote doc2a+/−fam57ba+/− larvae (Fig. 3E–H). The double heterozygotes showed significantly higher activity than wild-type after a minimal 0.3 mM PTZ dose, which is far below the established dosing range of 2.5 to 25 mM required to induce seizures in wild-type fish (60,76). In contrast, fam57ba−/− mutants needed 5 mM PTZ to show a nominally significant increase in velocity compared to wild-type. Single heterozygous doc2a+/− and fam57ba+/− fish did not show differences in baseline activity or 0.3 mM PTZ sensitivity compared to wild-type, indicating an interaction in the double heterozygote (Fig. 3I–P). doc2a−/− mutants alone did not exhibit changes in baseline movement or seizure susceptibility relative to wild-type at 0.3 mM (data not shown) or 5 mM PTZ (Fig. 3Q–T). Single heterozygotes were also tested at the higher 5 mM PTZ dose, but no significant differences were observed (data not shown). All genotypes tested except for fam57ba+/− exhibited a significant (P < 0.0001) response to PTZ treatment at the tested doses indicated in Figure 3.
Table 1.
fam57ba and doc2a genetic mutants affect brain and body size phenotypes. doc2a+/− fam57ba+/− has a stronger effect on increasing hyperactivity and seizures compared to wild-type, while fam57ba−/− has a stronger effect on increasing body size, lipid content, and head size compared to wild-type. Single heterozygotes for each gene as well as doc2a homozygotes do not affect any of these phenotypes, although doc2a−/− larvae have decreased head sizes in one dimension, and increased in other dimensions. This indicates a genuine interaction between doc2a and fam57ba to increase hyperactivity and seizures. Two arrows indicate a stronger effect relative to wild-type compared to a single arrow. A dash indicates no significant differences from wild-type.
| doc+/−fam+/− | fam57ba−/− | fam57ba+/− | doc2a+/− | doc2a−/− | |
|---|---|---|---|---|---|
| Hyperactivity | ↑↑ | ↑ | – | – | – |
| Seizures | ↑↑ | ↑ | – | – | – |
| Body size | ↑ | ↑↑ | – | – | – |
| Lipid content | ↑ | ↑↑ | – | – | – |
| Head Size | ↑ | ↑↑ | – | – | – |
In order to confirm the specificity of the seizure response in our lines, we treated the larvae with human anti-epileptic drugs (AEDs). We also examined for musculature and caudal motoneuron differences. We used valproic acid (VPA), the most widely prescribed AED worldwide, and a broad spectrum drug that is effective against all seizure types (77), and carbamazepine, another first choice AED for partial epilepsies (78). Both drugs have been previously assessed in zebrafish as inhibiting PTZ-induced epileptiform discharges and increased swimming (60,61). We found that VPA treatment before PTZ treatment decreased average swimming velocity to around 28% of velocity after PTZ alone in wild-type, and 22% in doc2a+/−fam57ba+/− and fam57ba−/− larvae. Carbamazepine treatment suppressed movement to about 42% of the PTZ baseline in wild-type, 35% in doc2a+/−fam57ba+/−, and 53% in fam57ba−/− larvae (Supplementary Material, Fig. S4). Immunohistochemical analysis of muscle fibers and primary caudal motoneurons exhibited no obvious differences between wild-type, doc2a+/−fam57ba+/−, and fam57ba−/−, indicating that these would not contribute to movement differences (Supplementary Material, Fig. S5).
These data demonstrate a robust genetic interaction between the fam57ba and doc2a genes resulting in hyperactivity and seizure susceptibility.
doc2a+/− fam57ba+/− heterozygotes and fam57ba−/− mutants show increased body and head size
Since large body size, macrocephaly and obesity are significant symptoms among people with the 16p11.2 deletion, we next examined body size and head size in doc2a+/−fam57ba+/− larvae and other doc2a and fam57ba genotypes. In people with the 16p11.2 deletion, incidence of obesity increases with age, becoming significantly different from non-carriers by 3.5 years. There is also a correlated increase in pre-pubertal height above the population average, but only in obese 16p11.2 CNV deletion carriers. The occurrence of increased head circumference is also positively correlated with age and with obesity (10). With these considerations, head and body size of fish about halfway through the larval period (12 dpf) were assayed to assess a later developmental time point, perhaps more relevant to onset of human phenotypes.
A significant increase in body length was found in doc2a+/−fam57ba+/− larvae at 12 dpf (Fig. 4A–C). To analyse lipid content, larvae were stained with the lipid-binding dye Oil Red O (ORO), and the ORO was then extracted and quantified. While ORO concentrations were significantly elevated in doc2a+/−fam57ba+/− double heterozygotes (Fig. 4D), this difference was not seen after accounting for body length (Fig. 4E). In analysis of additional genotypes, we found that fam57ba−/− larvae displayed a much greater increase in body length versus wild-type (Fig. 4F–H). These fish also showed a significant increase in lipid content, even after normalizing to body length (Fig. 4I–J). Since doc2a+/−, fam57ba+/−, and doc2a−/− larvae did not show changes in body size or lipid content (Fig. 4K–Y), we conclude that with regard to body size, there is an interaction in the double heterozygote for body size but not normalized lipid content.
Figure 4.

Body size and lipid amounts are affected by doc2a and fam57ba. A representative experiment is shown from the 5 conducted. (A,B) 12 dpf larvae stained with Oil Red O (ORO) are shown for wild-type and doc2a+/− fam57ba+/−. (C) A significant increase in body length as measured from the tip of the nose to the end of the tail is found for doc2a+/− fam57ba+/− larvae (n = 19) compared to wild-type (n = 19). (D) ORO was extracted back out of the stained larvae and quantified, showing a significant increase in the concentration of lipids in the double heterozygotes (yellow bar) compared to wild-type (blue bar). (E) After accounting for length, there is no longer a significant increase in ORO concentration in doc2a+/− fam57ba+/− larvae. (F,G) Wild-type and fam57ba−/− larvae stained with ORO. (H) A significant increase in body length was found comparing fam57ba−/− larvae (n = 30) to wild-type (n = 30). (I) ORO quantification showed a significant increase in the concentration of lipids for fam57ba−/− homozygotes (purple bar) compared to wild-type (blue bar). (J) When accounting for length, ORO concentration is still significantly elevated in fam57ba−/− fish. (K–Y) No significant changes in size or ORO concentration were observed for fam57ba+/− (n = 29), doc2a+/− (n = 26), and doc2a−/− (n = 36) larvae compared to their respective wild-type controls. **P < 0.01, ***P < 0.001, ****P < 0.0001. Scale bar = 500 µm.
To assess head size, various measurements of mutants were made relative to wild-type larvae at 12 dpf (Fig. 5). doc2a+/−fam57ba+/− fish showed slight increases in a subset of the dimensions, while fam57ba−/− larvae showed significant changes in all dimensions measured, indicating that losing both copies of fam57ba has the more powerful effect on head size as well as body size. Single heterozygous doc2a+/− and fam57ba+/− larvae did not show changes in head size in any dimension, while doc2a−/− larvae showed a decrease in inter-eye width and an increase in two other dimensions (Fig. 5). If increased head dimensions are normalized by dividing by the body length of doc2a+/−fam57ba+/− and fam57ba−/− larvae, then no significant differences from wild-type are observed (not shown). Therefore, we predict DOC2A and in particular FAM57B contribute to general growth patterns, rather than specifically to head size, although species differences may play into the results.
Figure 5.

doc2a and fam57ba affect head size. A representative experiment from 4 experiments conducted is shown for each genotype. (A) Dorsal schematic of 12 dpf larval head, with panel letters indicating lines for the 4 measurements taken. (B–E) Fold-change relative to respective wild-type controls (blue bars) is shown for doc2a+/− fam57ba+/− (yellow bars, n = 19), fam57ba−/− (purple bars, n = 19), fam57ba+/− (green bars, n = 29), doc2a+/− (orange bars, n = 26), and doc2a−/− (pink bars, n = 36) larvae for inter-eye width, dorsal head length (D), hindbrain width, and forebrain width. (F) Lateral schematic of 12 dpf larval head, with panel letters indicating the four measurements taken. (G–J) Fold-change relative to respective wild-type controls for each genotype as described above for lateral head length (L), head height, eye width and eye height. (K,L) Live dorsal and lateral images of representative larvae for each genotype. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Scale bar = 100 μm.
These data demonstrate that doc2a+/−fam57ba+/− heterozygotes and fam57ba−/− mutants show increased head size, body size, and lipid content – phenotypes that are consistent with obesity.
Discussion
A major challenge in human genetics is the definition of how multigenic copy number variant regions connect to multiple associated symptoms. Deletion of the 16p11.2 CNV is paradigmatic of this challenge, presenting a global burden for four million affected people. We used the tractable zebrafish system and a functional screen to uncover genotype/phenotype connections amongst 16p11.2 homologs, reaching three major conclusions. 1) A subset of 16p11.2 genes interact after partial loss of function and define a functional network, 2) the doc2a and fam57ba genes interact to regulate hyperactivity and seizure propensity, and 3) doc2a and fam57ba genes also interact to control head and body size, with greater contribution from fam57ba. These conclusions add new understanding to the mechanism underlying 16p11.2 deletion phenotypes, and further suggest previously undescribed correlations between brain and body health.
Gene interactions among the 16p11.2 interval do not converge on functional networks predicted by gene-set enrichment analysis (79). Furthermore, no evidence for physical interactions between 16p11.2 genes has been found (80). These findings imply indirect mechanisms of gene interaction, which could only be uncovered by functional assays. Genetic interaction data do not indicate mechanism, but are invaluable in focusing subsequent molecular or cellular assays.
The subset of highly interactive 16p11.2 zebrafish homologs that our pairwise assays reveal have diverse predicted functions, and include asphd1, fam57ba, hirip3, kctd13, kif22 and sez6l2 (Table 2). The initial hypothesis that each pair of genes would be sufficient to regulate a specific 16p11.2 phenotype may be too simple, as suggested by the network of interactions uncovered that affect a single phenotype. Rather, each 16p11.2 deletion symptom may depend on haploinsufficiency of multiple genes (Fig. 1). It is possible that even if multiple 16p11.2 genes regulate each symptom, these converge functionally on a few common biochemical or cell biological targets.
Table 2.
Diverse predicted functions for most interactive 16p11.2 genes. Little is known about ASPHD1 function, although it contains an aspartate beta-hydroxylase domain, and aspartate beta-hydroxylase is important for regulating calcium homeostasis (112). FAM57B is a ceramide synthase that regulates adipogenesis in mouse tissue culture cells (46). HIRIP3 may be part of a complex involved in chromatin and histone metabolism (113). KCTD13 encodes a ubiquitin ligase adaptor (114) that is reported to have reciprocal effects on zebrafish head size with gain- and loss-of-function (35). KIF22 is a kinesin-like protein that links chromosomes to the mitotic spindle (115,116), and is necessary for proper embryonic chromosome segregation (118) and synaptic development (34). SEZ6L2 is a seizure-related protein that modulates neurite outgrowth (117).
The interaction of doc2a and fam57ba in genetic mutants suggests a mechanism by which brain activity is regulated. Although both genes are expressed predominantly in the head of larval zebrafish (Supplementary Material, Fig. S6) and are apparently co-expressed in the adult mouse brain (81) and the prenatal human brain (82), it is unclear whether they function in the same cells. The Doc2a gene encodes a calcium sensor that interacts with the docking machinery to prime vesicles for exocytosis (47,48,83), including at the synapse (57). Mutations in other synaptic vesicle docking components are associated with ADHD and epilepsy (72,84,85).
Fam57b has a Tram-Lag1-Cln8 (TLC) domain associated with ceramide synthases (86), and is capable of ceramide synthesis in vitro (46). Based on this ceramide synthase activity, we predict that there is a change in the ceramide cohort in fam57ba mutant fish, and possibly in people with 16p11.2 CNVs. Serum levels of particular sphingomyelins, a ceramide derivative, have been shown to be decreased in people with ADHD (87). Mice treated with fumonisin B1, a general ceramide synthase inhibitor, are sensitized to PTZ-induced seizures (88), and mutations in two ceramide synthase genes in humans are associated with progressive myoclonus epilepsy (54,89).
Dysregulated ceramides could interact with DOC2A to impact synaptic signaling in several ways (Fig. 6). Ceramides are critical structural components of cell membranes and dysregulation could affect vesicle fusion and endocytic recycling. In cell culture, ceramide application was found to enhance exocytosis (90). Various ceramide derivatives interact with components of the SNARE vesicular docking complex (91,92) and recruit Munc13 (93) – the binding partner of DOC2A (47,48) – and enhance intracellular calcium flow (94), which could help activate DOC2A. These findings point to future analyses addressing the molecular pathways by which these genes control synaptic activity and their cellular targets.
Figure 6.
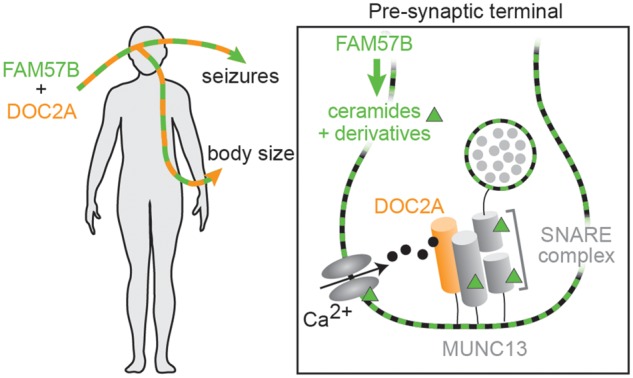
Hypothesis: FAM57B and DOC2A are critical regulators of brain and body health. Loss of a single copy of both FAM57B and DOC2A genes may lead to increased body size and seizures through the brain, where both genes are predominantly expressed. Inset of pre-synaptic terminal: Ceramides potentially interact with DOC2A to impact the vesicular release. Ceramides and ceramide derivatives represented by green squares (in cell and vesicular membranes) and triangles. Docking and release of neurotransmitters (gray circles) from synaptic vesicles may be influenced by ceramides and DOC2A in several ways. The ceramide derivative sphingosine promotes vesicular fusion via activation of the SNARE component SYNAPTOBREVIN (92). Sphingosine also influences another SNARE component, SYNTAXIN, to enhance its interaction with MUNC18, which promotes an active conformation of SYNTAXIN (91). Sphingosine-1-phosphate may contribute to neurotransmitter release via recruitment of MUNC13 (93), the vesicle docking component that is activated by DOC2A binding after calcium influx (47,48). Ceramide-1-phosphate has been shown to enhance intracellular calcium concentrations by acting on voltage-gated calcium channels (94).
No 16p11.2 gene has previously been linked to the increased body size observed in people with deletion of one 16p11.2 copy. The longer body length we observe in doc2a+/−fam57ba+/− double heterozygotes is consistent with increased height in people with 16p11.2 deletion CNVs. While single heterozygote and double heterozygote genetic data support the necessity of doc2a in the haploinsufficient interaction, the strong phenotype observed in fam57ba−/− single mutants suggests that this is the pivotal gene driving body size phenotypes. In our studies, fam57ba showed a clear function in regulating lipid content, suggesting that this gene may promote normal amounts of fat. While obesity is generally defined as having too much body fat, a body mass index (BMI) of ≥30 is used as the clinical indicator of obesity. Because there is no universally accepted value of an obese zebrafish BMI value in larvae and because weighing larvae at this stage is not consistently accurate, we analysed the total lipid content in relation to body length, and by these criteria, fam5ba−/− fish are obese. Interaction of FAM57B with another 16p11.2 gene(s) such as DOC2A may modulate body size. Consistent with this suggestion, manipulating other synaptic vesicle docking components like DOC2A has been associated with metabolic changes contributing to obesity, presumably from disruption of hormonal and/or neurotransmitter release dynamics (95,96). While fam57ba−/− mutants also had significantly increased lipid content, even after normalizing to body size, doc2a+/−fam57ba+/− double heterozygotes did not, implying that in the human hemizygous condition, FAM57B could be interacting with another as yet to be identified 16p11.2 gene(s) to regulate lipid content.
Macrocephaly is generally defined as an absolute measure and does not take body size into account. Disproportionate macrocephaly refers to an increase in head size relative to the body length (97). In people with 16p11.2 CNV deletion, disproportionate macrocephaly is observed, as head circumference is elevated even after taking body mass index (BMI) into account (10). In zebrafish, doc2a+/−fam57ba+/− and fam57ba−/− larvae only show indications of absolute macrocephaly, indicating somatic overgrowth. Together, the increased body length, lipid content, and head size indicate that doc2a and fam57ba impact growth patterns and overall size, consistent with phenotypes seen in people with 16p11.2 deletion (10).
Since both doc2a and fam57ba are predominantly expressed in the head (Supplementary Material, Fig. S6), we hypothesize that their effects on body size may be exerted through changes in the brain. For example, signaling deficits, arising from potential ceramide and synaptic vesicle docking mechanisms discussed above, in neural circuits regulating appetite could contribute to differences in body size. This is in contrast to the suggestion that mouse Fam57b acts directly in cultured adipocytes, where overexpression and exogenous ceramide application inhibited lipid accumulation, and knockdown resulted in increased lipids (46). However, previous work in zebrafish has shown that adipocytes are only starting to appear at 12 dpf, and do not fully differentiate in larvae until 17 dpf (98), five days after our size measurements were done. This does not preclude a possible contribution from premature adipocyte differentiation in fam57ba−/− mutants, but we did not observe significant differences in white adipose tissue depots at 12 dpf (data not shown).
The effects of mutating fam57ba and doc2a in zebrafish are more similar to increased body and head size in people with 16p11.2 deletions than phenotypes seen in mouse models. Instead, mouse 16p11.2 deletion models exhibit reduced body and head size as well as decreased adiposity and do not recapitulate this human phenotype (30–32). Mouse models do recapitulate other aspects of human symptoms, including hyperactivity, impaired learning, and social deficits (30–32,99), illustrating the utility of using multiple animal models to address human gene function.
Our results indicate for the first time that FAM57B functions in both brain and body health, through interaction with DOC2A and perhaps together with other, as yet unidentified genes (summarized in Fig. 6). Together, these data suggest that in people with 16p11.2 deletions, ceramide dysregulation and altered exocyotosis may be mechanisms underlying seizures and obesity. The identification of a potential lipid mechanism underlying pathology suggests a role for metabolic regulation of the 16p11.2-related phenotypes addressed. Because we focused on this single interaction for further analysis, there is nothing to rule out that other 16p11.2 genes also contribute to seizure propensity and body size, as well as other phenotypes. Future directions include molecular analysis of fam57ba and doc2a interaction, assessment of whether function of these genes is dysregulated in human 16p11.2 deletion cells, and exploring interactions of additional 16p11.2 homologs with doc2a and/or fam57ba related phenotypes.
Materials and Methods
Fish lines and maintenance
Adult zebrafish of the wild-type AB strain were maintained at 28˚C on 12h/12h light/dark cycle. Embryos were obtained from natural spawnings and staged as previously described by Kimmel et al. (1995). The transgenic lines used, islet1:GFP and NeuroD:GFP, have been previously characterized (100,101). Due to the polygenic nature of sex determination and timing of gonadal development in zebrafish (102,103), we are unable to determine the sexes of the embryos and larvae for our assays. However, because our assays utilized large numbers of embryos and larvae, both sexes should be adequately represented.
Antisense morpholino-modified oligonucleotide design and use
We chose to focus on only those 16p11.2 zebrafish homologs that gave a brain phenotype in our single gene study. This means we did not test prrt2 or tbx24 or the b copies for aldoa, fam57b, and ppp4c. The antisense morpholino-modified oligonucleotides (MOs) utilized were the same as our previous study (sequences and specificity assays in (33)). Concentrations for splice-blocking MOs that resulted in approximately 50% normal RNA remaining at 24 hpf are in Supplementary Material, Table S1. These doses were determined by qPCR using the same primers as previously described (33), except for hirip3 primers, which were F: 5’-GTTGTGGCTCAACTGCAGAA and R: 5’-GCACTGAGCGACTCTCTTCC. For the start site MOs, doses that did not result in a phenotype when injected alone were used (Supplementary Material, Table S1). Because aldoaa and kif22 LOF give a phenotype at 50% LOF, non-phenotypic doses at ∼25% LOF were used in interaction assays. To control for MO load in testing interactions, single MO injections were balanced with the same amount of control MO as would be used for the test partner MO. For the control condition, the same amount of control MO was used as the combined amounts in the double LOF condition.
Morphological assays
Brain ventricle morphology was evaluated at 24 hpf. Texas Red dextran (Sigma) was injected into ventricle space as done previously (104). Dye injection does not affect morphology (105). Neuronal specification and cranial motor neuron patterning were analysed at 2 dpf using transgenic lines. Embryos were scored in 2–4 independent experiments, with the combined number of embryos scored across experiments shown in the figures.
Microscopy
Bright-field and fluorescent images were captured on a Zeiss Stereo Discovery V8 microscope with an AxioCam MRc digital camera using ZEN software (Zeiss). Confocal imaging was performed on an inverted Zeiss LSM710 laser-scanning microscope with a 25X (0.8 NA) LD LCI Plan Apochromat oil-immersion objective.
Immunohistochemistry
Embryos and larvae were fixed in 4% PFA. Anti-HuC/D monoclonal antibody (Invitrogen) was used at 1:500, znp1 (anti-SYT2, Abcam) monoclonal antibody was used at 1:200, and Alexa Fluor 488 secondary antibody (Jackson) was used at 1:400. Alexa Fluor 635 phalloidin (Invitrogen) was used at 1:40. Cell counting was done in Imaris (Bitplane) with manual threshold adjustment.
TALEN generation and identification of TALEN mutants
Target sites for TALENs were identified using Mojo Hand (106) and TALE-NT 2.0 (107). All sites chosen had a restriction enzyme site at the cut site to be used for genotyping (Supplementary Material, Table S4). TALENs were cloned and injected into one-cell zebrafish embryos as previously described (108,109). F0 fish with high germline transmission rates were identified by loss of restriction site, sequenced, and outcrossed. The resulting heterozygous F1 fish were in-crossed if they carried the same mutation to generate wild-type, heterozygous, and mutant siblings. These F2 fish were bred to generate embryos and larvae used in the experiments described below.
Seizure assays
Wild-type and doc2a+/−fam57ba+/− double heterozygous cousin embryos were obtained from separate crosses of sibling wild-type and single homozygous mutant fish, respectively. Single heterozygous fish were generated by crossing homozygous to wild-type fish. At 7 dpf, larvae were pipetted into 96-well plates in 200 µl E3 media, allowed to acclimate to the plate for 15 min, and then moved to the Noldus Daniovision for another 10 min habituation period. Baseline activity was then recorded for 10 min. Following this, 100 µl E3 was removed from each well, and replaced with 100 µl of varying concentrations of PTZ to test a range of doses. Plates were immediately placed back on the Daniovision system for a 10-min habituation period, followed by another 10- min recording. Average velocity was calculated using the Ethovision XT 11 software from Noldus. The numbers shown in the figures are combined from 2 to 5 independent experiments. Even though there may not necessarily be a correlation between baseline movement and velocity after PTZ addition, we still accounted for this possibility by normalization via subtracting the baseline velocity from the PTZ-induced velocity for each larva.
Anti-epileptic drug treatments
Human anti-epileptic drugs were added to larvae for 15 min before PTZ addition and seizures were assayed as above. Valproic acid (Sigma) was used at 3 mM and carbamazepine (Sigma) was used at 100 µM.
Oil red O staining and quantification
To control for variable growth rates, bowl larval density was checked daily to ensure equal numbers of wild-type and test genotype larvae within experiments. Larvae were fixed at 12 dpf in 4% PFA and washed in PBS. Pigment was bleached with 1% KOH/3% H2O2 solution. The 0.5% stock Oil Red O (ORO, Sigma) solution was made in isopropanol. Immediately before use, a working 0.3% solution was made with dH2O, allowed to stand for 10 min after mixing, and then filtered. Larvae were incubated in the fresh working solution for 30 min to 2 h, rinsed quickly twice in 60% isopropanol, and rinsed again in PBS (110,111).
To quantify ORO amount, ORO was extracted back out of 15–25 stained larvae per genotype by soaking 2–4 fish in 300 µl isopropanol for at least 60 min with occasional agitation until larvae appeared white in appearance. The supernatant from these 6–8 pools per genotype was removed, split in triplicate, and absorbance was measured at 492 nm (averaged from 10 readings per well). Average concentration was calculated per larva by comparison to a standard curve analysed on the same plate.
Statistical analysis
Exact binomial tests were used for statistical analysis comparing qualitative phenotypes. Unpaired nonparametric t-tests were used for statistical analysis comparing quantitative phenotypes. Unless noted otherwise, error bars on bar graphs reflect standard error, while error bars on scatter plots show standard deviation.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We thank Olivier Paugois and Hugo Perdomo for excellent fish care. Thanks to Wendy Salmon (Whitehead Institute, Keck Imaging Facility) for help with confocal imaging, and George Bell (Whitehead Institute, Bioinformatics and Research Computing) for useful advice on statistical analysis. Thanks to Brad Carter, George Bell, Limor Freifeld, Ambroise Vuaridel, Amin Allalou, Dimira Tambunan, Ann Poduri, Brooke April, and Jonathan Dale for assistance with setting up and analysing seizure experiments. The Fink lab kindly shared use of their plate reader. We are grateful to Sive lab members, Ed Scolnick, and Jon Madison for discussion and Ryann Fame for feedback on the manuscript.
Conflict of Interest statement. None declared.
Funding
This work was supported by kind contributions from Jim and Pat Poitras, Len and Ellen Polaner, and the Markell-Balkin-Weinberg postdoctoral fellowship to JMM. Funding to pay the Open Access publication charges for this article was provided by the Whitehead Institute for Biomedical Research.
References
- 1. Walsh K.M., Bracken M.B. (2011) Copy number variation in the dosage-sensitive 16p11.2 interval accounts for only a small proportion of autism incidence: a systematic review and meta-analysis. Genet. Med., 13, 377–384. [DOI] [PubMed] [Google Scholar]
- 2. Hanson E., Bernier R., Porche K., Jackson F.I., Goin-Kochel R.P., Snyder L.G., Snow A.V., Wallace A.S., Campe K.L., Zhang Y.. et al. (2015) The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol. Psychiatry, 77, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hippolyte L., Maillard A.M., Rodriguez-Herreros B., Pain A., Martin-Brevet S., Ferrari C., Conus P., Mace A., Hadjikhani N., Metspalu A.. et al. (2015) The number of genomic copies at the 16p11.2 locus modulates language, verbal memory, and inhibition. Biol. Psychiatry, 80,129–39. [DOI] [PubMed] [Google Scholar]
- 4. Jacquemont S., Reymond A., Zufferey F., Harewood L., Walters R.G., Kutalik Z., Martinet D., Shen Y., Valsesia A., Beckmann N.D.. et al. (2011) Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature, 478, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Qureshi A.Y., Mueller S., Snyder A.Z., Mukherjee P., Berman J.I., Roberts T.P., Nagarajan S.S., Spiro J.E., Chung W.K., Sherr E.H.. et al. (2014) Opposing brain differences in 16p11.2 deletion and duplication carriers. J. Neurosci., 34, 11199–11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reinthaler E.M., Lal D., Lebon S., Hildebrand M.S., Dahl H.H., Regan B.M., Feucht M., Steinbock H., Neophytou B., Ronen G.M.. et al. (2014) 16p11.2 600 kb Duplications confer risk for typical and atypical Rolandic epilepsy. Hum. Mol. Genet., 23, 6069–6080. [DOI] [PubMed] [Google Scholar]
- 7. Roll P., Sanlaville D., Cillario J., Labalme A., Bruneau N., Massacrier A., Delepine M., Dessen P., Lazar V., Robaglia-Schlupp A.. et al. (2010) Infantile convulsions with paroxysmal dyskinesia (ICCA syndrome) and copy number variation at human chromosome 16p11. PloS One, 5, e13750.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shinawi M., Liu P., Kang S.H., Shen J., Belmont J.W., Scott D.A., Probst F.J., Craigen W.J., Graham B.H., Pursley A.. et al. (2010) Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J. Med. Genet., 47, 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Walters R.G., Jacquemont S., Valsesia A., de Smith A.J., Martinet D., Andersson J., Falchi M., Chen F., Andrieux J., Lobbens S.. et al. (2010) A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature, 463, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zufferey F., Sherr E.H., Beckmann N.D., Hanson E., Maillard A.M., Hippolyte L., Mace A., Ferrari C., Kutalik Z., Andrieux J.. et al. (2012) A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J. Med. Genet., 49, 660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Neale B.M., Kou Y., Liu L., Ma'ayan A., Samocha K.E., Sabo A., Lin C.F., Stevens C., Wang L.S., Makarov V.. et al. (2012) Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature, 485, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. O'Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D.. et al. (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature, 485, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ripke S., O'Dushlaine C., Chambert K., Moran J.L., Kahler A.K., Akterin S., Bergen S.E., Collins A.L., Crowley J.J., Fromer M.. et al. (2013) Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet., 45, 1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sanders S.J., He X., Willsey A.J., Ercan-Sencicek A.G., Samocha K.E., Cicek A.E., Murtha M.T., Bal V.H., Bishop S.L., Dong S.. et al. (2015) Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron, 87, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L.. et al. (2012) De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature, 485, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Celestino-Soper P.B., Shaw C.A., Sanders S.J., Li J., Murtha M.T., Ercan-Sencicek A.G., Davis L., Thomson S., Gambin T., Chinault A.C.. et al. (2011) Use of array CGH to detect exonic copy number variants throughout the genome in autism families detects a novel deletion in TMLHE. Hum. Mol. Genet., 20, 4360–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Girirajan S., Dennis M.Y., Baker C., Malig M., Coe B.P., Campbell C.D., Mark K., Vu T.H., Alkan C., Cheng Z.. et al. (2013) Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am. J. Hum. Genet., 92, 221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Poultney C.S., Goldberg A.P., Drapeau E., Kou Y., Harony-Nicolas H., Kajiwara Y., De Rubeis S., Durand S., Stevens C., Rehnstrom K.. et al. (2013) Identification of small exonic CNV from whole-exome sequence data and application to autism spectrum disorder. Am. J. Hum. Genet., 93, 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ebrahimi-Fakhari D., Saffari A., Westenberger A., Klein C. (2015) The evolving spectrum of PRRT2-associated paroxysmal diseases. Brain, 138, 3476–3495. [DOI] [PubMed] [Google Scholar]
- 20. Min B.J., Kim N., Chung T., Kim O.H., Nishimura G., Chung C.Y., Song H.R., Kim H.W., Lee H.R., Kim J.. et al. (2011) Whole-exome sequencing identifies mutations of KIF22 in spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type. Am. J. Hum. Genet., 89, 760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Al-Kateb H., Khanna G., Filges I., Hauser N., Grange D.K., Shen J., Smyser C.D., Kulkarni S., Shinawi M. (2014) Scoliosis and vertebral anomalies: additional abnormal phenotypes associated with chromosome 16p11.2 rearrangement. Am. J. Med. Genet. Part A, 164A, 1118–1126. [DOI] [PubMed] [Google Scholar]
- 22. Wu N., Ming X., Xiao J., Wu Z., Chen X., Shinawi M., Shen Y., Yu G., Liu J., Xie H.. et al. (2015) TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N. Engl. J. Med., 372, 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCammon J.M., Sive H. (2015) Challenges in understanding psychiatric disorders and developing therapeutics: a role for zebrafish. Dis. Model. Mech., 8, 647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCammon J.M., Sive H. (2015) Addressing the genetics of human mental health disorders in model organisms. Annu .Rev. Genomics Hum. Genet., 16, 173–197. [DOI] [PubMed] [Google Scholar]
- 25. Sive H. (2011) ′Model′ or ′tool′? New definitions for translational research. Dis. Model. Mech., 4, 137–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shui J.W., Hu M.C., Tan T.H. (2007) Conditional knockout mice reveal an essential role of protein phosphatase 4 in thymocyte development and pre-T-cell receptor signaling. Mol. Cell. Biol., 27, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sakaguchi G., Manabe T., Kobayashi K., Orita S., Sasaki T., Naito A., Maeda M., Igarashi H., Katsuura G., Nishioka H.. et al. (1999) Doc2alpha is an activity-dependent modulator of excitatory synaptic transmission. Eur. J. Neurosci., 11, 4262–4268. [DOI] [PubMed] [Google Scholar]
- 28. Foger N., Jenckel A., Orinska Z., Lee K.H., Chan A.C., Bulfone-Paus S. (2011) Differential regulation of mast cell degranulation versus cytokine secretion by the actin regulatory proteins Coronin1a and Coronin1b. J. Exp. Med., 208, 1777–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pages G., Guerin S., Grall D., Bonino F., Smith A., Anjuere F., Auberger P., Pouyssegur J. (1999) Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science, 286, 1374–1377. [DOI] [PubMed] [Google Scholar]
- 30. Arbogast T., Ouagazzal A.M., Chevalier C., Kopanitsa M., Afinowi N., Migliavacca E., Cowling B.S., Birling M.C., Champy M.F., Reymond A.. et al. (2016) Reciprocal effects on neurocognitive and metabolic phenotypes in mouse models of 16p11.2 deletion and duplication syndromes. PLoS Genet., 12, e1005709.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Horev G., Ellegood J., Lerch J.P., Son Y.E., Muthuswamy L., Vogel H., Krieger A.M., Buja A., Henkelman R.M., Wigler M.. et al. (2011) Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc. Natl Acad. Sci. U S A, 108, 17076–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Portmann T., Yang M., Mao R., Panagiotakos G., Ellegood J., Dolen G., Bader P.L., Grueter B.A., Goold C., Fisher E.. et al. (2014) Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell Rep., 7, 1077–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blaker-Lee A., Gupta S., McCammon J.M., De Rienzo G., Sive H. (2012) Zebrafish homologs of genes within 16p11.2, a genomic region associated with brain disorders, are active during brain development, and include two deletion dosage sensor genes. Dis. Model. Mech., 5, 834–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Park S.M., Littleton J.T., Park H.R., Lee J.H. (2016) Drosophila homolog of human KIF22 at the autism-linked 16p11.2 loci influences synaptic connectivity at larval neuromuscular junctions. Exp. Neurobiol., 25, 33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Golzio C., Willer J., Talkowski M.E., Oh E.C., Taniguchi Y., Jacquemont S., Reymond A., Sun M., Sawa A., Gusella J.F.. et al. (2012) KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature, 485, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tropepe V., Sive H.L. (2003) Can zebrafish be used as a model to study the neurodevelopmental causes of autism?. Genes Brain Behav., 2, 268–281. [DOI] [PubMed] [Google Scholar]
- 37. Howe K., Clark M.D., Torroja C.F., Torrance J., Berthelot C., Muffato M., Collins J.E., Humphray S., McLaren K., Matthews L.. et al. (2013) The zebrafish reference genome sequence and its relationship to the human genome. Nature, 496, 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perez-Perez J.M., Candela H., Micol J.L. (2009) Understanding synergy in genetic interactions. Trends Genet., 25, 368–376. [DOI] [PubMed] [Google Scholar]
- 39. Cavodeassi F., Houart C. (2012) Brain regionalization: of signaling centers and boundaries. Dev. Neurobiol., 72, 218–233. [DOI] [PubMed] [Google Scholar]
- 40. Dworkin S., Jane S.M. (2013) Novel mechanisms that pattern and shape the midbrain-hindbrain boundary. Cell. Mol. Life Sci., 70, 3365–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lowery L.A., De Rienzo G., Gutzman J.H., Sive H. (2009) Characterization and classification of zebrafish brain morphology mutants. Anat. Rec. (Hoboken), 292, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Maillard A.M., Ruef A., Pizzagalli F., Migliavacca E., Hippolyte L., Adaszewski S., Dukart J., Ferrari C., Conus P., Mannik K.. et al. (2015) The 16p11.2 locus modulates brain structures common to autism, schizophrenia and obesity. Mol. Psychiatry, 20, 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Owen J.P., Chang Y.S., Pojman N.J., Bukshpun P., Wakahiro M.L., Marco E.J., Berman J.I., Spiro J.E., Chung W.K., Buckner R.L.. et al. (2014) Aberrant white matter microstructure in children with 16p11.2 deletions. J. Neurosci., 34, 6214–6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Steinman K.J., Spence S.J., Ramocki M.B., Proud M.B., Kessler S.K., Marco E.J., Green Snyder L., D'Angelo D., Chen Q., Chung W.K.. et al. (2016) 16p11.2 deletion and duplication: Characterizing neurologic phenotypes in a large clinically ascertained cohort. Am. J. Med. Genet. Part A, [DOI] [PubMed] [Google Scholar]
- 45. Robu M.E., Larson J.D., Nasevicius A., Beiraghi S., Brenner C., Farber S.A., Ekker S.C. (2007) p53 activation by knockdown technologies. PLoS Genet., 3, e78.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yamashita-Sugahara Y., Tokuzawa Y., Nakachi Y., Kanesaki-Yatsuka Y., Matsumoto M., Mizuno Y., Okazaki Y. (2013) Fam57b (family with sequence similarity 57, member B), a novel peroxisome proliferator-activated receptor gamma target gene that regulates adipogenesis through ceramide synthesis. J. Biol. Chem., 288, 4522–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mochida S., Orita S., Sakaguchi G., Sasaki T., Takai Y. (1998) Role of the Doc2 alpha-Munc13-1 interaction in the neurotransmitter release process. Proc. Natl Acad. Sci. U S A, 95, 11418–11422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Orita S., Naito A., Sakaguchi G., Maeda M., Igarashi H., Sasaki T., Takai Y. (1997) Physical and functional interactions of Doc2 and Munc13 in Ca2+-dependent exocytotic machinery. J. Biol. Chem., 272, 16081–16084. [DOI] [PubMed] [Google Scholar]
- 49. Casillas-Espinosa P.M., Powell K.L., O'Brien T.J. (2012) Regulators of synaptic transmission: roles in the pathogenesis and treatment of epilepsy. Epilepsia, 53 Suppl 9, 41–58. [DOI] [PubMed] [Google Scholar]
- 50. Fukata Y., Fukata M. (2017) Epilepsy and synaptic proteins. Curr. Opin. Neurobiol., 45, 1–8. [DOI] [PubMed] [Google Scholar]
- 51. Ebrahimi-Fakhari D., Sahin M. (2015) Autism and the synapse: emerging mechanisms and mechanism-based therapies. Curr. Opin. Neurol., 28, 91–102. [DOI] [PubMed] [Google Scholar]
- 52. Frye R.E. (2015) Metabolic and mitochondrial disorders associated with epilepsy in children with autism spectrum disorder. Epilepsy & Behavior, 47, 147–157. [DOI] [PubMed] [Google Scholar]
- 53. Brown C.M., Austin D.W. (2011) Autistic disorder and phospholipids: A review. Prostaglandins Leukot. Essent. Fatty Acids, 84, 25–30. [DOI] [PubMed] [Google Scholar]
- 54. Vanni N., Fruscione F., Ferlazzo E., Striano P., Robbiano A., Traverso M., Sander T., Falace A., Gazzerro E., Bramanti P.. et al. (2014) Impairment of ceramide synthesis causes a novel progressive myoclonus epilepsy. Ann. Neurol., 76, 206–212. [DOI] [PubMed] [Google Scholar]
- 55. Inokuchi J., Mizutani A., Jimbo M., Usuki S., Yamagishi K., Mochizuki H., Muramoto K., Kobayashi K., Kuroda Y., Iwasaki K.. et al. (1998) A synthetic ceramide analog (L-PDMP) up-regulates neuronal function. Ann. N. Y. Acad. Sci., 845, 219–224. [DOI] [PubMed] [Google Scholar]
- 56. Rohrbough J., Rushton E., Palanker L., Woodruff E., Matthies H.J., Acharya U., Acharya J.K., Broadie K. (2004) Ceramidase regulates synaptic vesicle exocytosis and trafficking. J. Neurosci., 24, 7789–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yao J., Gaffaney J.D., Kwon S.E., Chapman E.R. (2011) Doc2 is a Ca2+ sensor required for asynchronous neurotransmitter release. Cell, 147, 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. D'Angelo D., Lebon S., Chen Q., Martin-Brevet S., Snyder L.G., Hippolyte L., Hanson E., Maillard A.M., Faucett W.A., Mace A.. et al. (2016) Defining the effect of the 16p11.2 duplication on cognition, behavior, and medical comorbidities. JAMA Psychiatry, 73, 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lionel A.C., Crosbie J., Barbosa N., Goodale T., Thiruvahindrapuram B., Rickaby J., Gazzellone M., Carson A.R., Howe J.L., Wang Z.. et al. (2011) Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci. Transl. Med., 3, 95ra75.. [DOI] [PubMed] [Google Scholar]
- 60. Baraban S.C., Taylor M.R., Castro P.A., Baier H. (2005) Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c-fos expression. Neuroscience, 131, 759–768. [DOI] [PubMed] [Google Scholar]
- 61. Berghmans S., Hunt J., Roach A., Goldsmith P. (2007) Zebrafish offer the potential for a primary screen to identify a wide variety of potential anticonvulsants. Epilepsy Res., 75, 18–28. [DOI] [PubMed] [Google Scholar]
- 62. Baraban S.C., Dinday M.T., Hortopan G.A. (2013) Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat. Commun., 4, 2410.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Banote R.K., Koutarapu S., Chennubhotla K.S., Chatti K., Kulkarni P. (2013) Oral gabapentin suppresses pentylenetetrazole-induced seizure-like behavior and cephalic field potential in adult zebrafish. Epilepsy & Behavior, 27, 212–219. [DOI] [PubMed] [Google Scholar]
- 64. Baxendale S., Holdsworth C.J., Meza Santoscoy P.L., Harrison M.R., Fox J., Parkin C.A., Ingham P.W., Cunliffe V.T. (2012) Identification of compounds with anti-convulsant properties in a zebrafish model of epileptic seizures. Dis. Model. Mech., 5, 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Buenafe O.E., Orellana-Paucar A., Maes J., Huang H., Ying X., De Borggraeve W., Crawford A.D., Luyten W., Esguerra C.V., de Witte P. (2013) Tanshinone IIA exhibits anticonvulsant activity in zebrafish and mouse seizure models. ACS Chem. Neurosci., 4, 1479–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Menezes F.P., Rico E.P., Da Silva R.S. (2014) Tolerance to seizure induced by kainic acid is produced in a specific period of zebrafish development. Prog. Neuropsychopharmacol. Biol. Psychiatry, 55, 109–112. [DOI] [PubMed] [Google Scholar]
- 67. Orellana-Paucar A.M., Serruys A.S., Afrikanova T., Maes J., De Borggraeve W., Alen J., Leon-Tamariz F., Wilches-Arizabala I.M., Crawford A.D., de Witte P.A.. et al. (2012) Anticonvulsant activity of bisabolene sesquiterpenoids of Curcuma longa in zebrafish and mouse seizure models. Epilepsy & Behavior, 24, 14–22. [DOI] [PubMed] [Google Scholar]
- 68. Siebel A.M., Menezes F.P., da Costa Schaefer I., Petersen B.D., Bonan C.D. (2015) Rapamycin suppresses PTZ-induced seizures at different developmental stages of zebrafish. Pharmacol. Biochem. Behav., 139 Pt B, 163–168. [DOI] [PubMed] [Google Scholar]
- 69. Engel A.G., Selcen D., Shen X.M., Milone M., Harper C.M. (2016) Loss of MUNC13-1 function causes microcephaly, cortical hyperexcitability, and fatal myasthenia. Neurol. Genet., 2, e105.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Johnson M.R., Behmoaras J., Bottolo L., Krishnan M.L., Pernhorst K., Santoscoy P.L., Rossetti T., Speed D., Srivastava P.K., Chadeau-Hyam M.. et al. (2015) Systems genetics identifies Sestrin 3 as a regulator of a proconvulsant gene network in human epileptic hippocampus. Nat. Commun., 6, 6031.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Moortgat S., Desir J., Benoit V., Boulanger S., Pendeville H., Nassogne M.C., Lederer D., Maystadt I. (2016) Two novel EIF2S3 mutations associated with syndromic intellectual disability with severe microcephaly, growth retardation, and epilepsy. Am. J. Med. Genet. Part A, 170, 2927–2933. [DOI] [PubMed] [Google Scholar]
- 72. Schubert J., Siekierska A., Langlois M., May P., Huneau C., Becker F., Muhle H., Suls A., Lemke J.R., de Kovel C.G.. et al. (2014) Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat. Genet., 46, 1327–1332. [DOI] [PubMed] [Google Scholar]
- 73. Sicca F., Ambrosini E., Marchese M., Sforna L., Servettini I., Valvo G., Brignone M.S., Lanciotti A., Moro F., Grottesi A.. et al. (2016) Gain-of-function defects of astrocytic Kir4.1 channels in children with autism spectrum disorders and epilepsy. Sci. Rep., 6, 34325.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Suls A., Jaehn J.A., Kecskes A., Weber Y., Weckhuysen S., Craiu D.C., Siekierska A., Djemie T., Afrikanova T., Gormley P.. et al. (2013) De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am. J. Hum. Genet., 93, 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cunliffe V.T., Baines R.A., Giachello C.N., Lin W.H., Morgan A., Reuber M., Russell C., Walker M.C., Williams R.S. (2015) Epilepsy research methods update: Understanding the causes of epileptic seizures and identifying new treatments using non-mammalian model organisms. Seizure, 24, 44–51. [DOI] [PubMed] [Google Scholar]
- 76. Tiedeken J.A., Ramsdell J.S. (2007) Embryonic exposure to domoic Acid increases the susceptibility of zebrafish larvae to the chemical convulsant pentylenetetrazole. Environ. Health Perspect., 115, 1547–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Perucca E. (2002) Pharmacological and therapeutic properties of valproate: a summary after 35 years of clinical experience. CNS Drugs, 16, 695–714. [DOI] [PubMed] [Google Scholar]
- 78. Marson A.G., Williamson P.R., Hutton J.L., Clough H.E., Chadwick D.W. (2000) Carbamazepine versus valproate monotherapy for epilepsy. Cochrane Database Syst. Rev., CD001030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pinto D., Delaby E., Merico D., Barbosa M., Merikangas A., Klei L., Thiruvahindrapuram B., Xu X., Ziman R., Wang Z.. et al. (2014) Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet., 94, 677–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lin G.N., Corominas R., Lemmens I., Yang X., Tavernier J., Hill D.E., Vidal M., Sebat J., Iakoucheva L.M. (2015) Spatiotemporal 16p11.2 protein network implicates cortical late mid-fetal brain development and KCTD13-Cul3-RhoA pathway in psychiatric diseases. Neuron, 85, 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lein E.S., Hawrylycz M.J., Ao N., Ayres M., Bensinger A., Bernard A., Boe A.F., Boguski M.S., Brockway K.S., Byrnes E.J.. et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature, 445, 168–176. [DOI] [PubMed] [Google Scholar]
- 82. Miller J.A., Ding S.L., Sunkin S.M., Smith K.A., Ng L., Szafer A., Ebbert A., Riley Z.L., Royall J.J., Aiona K.. et al. (2014) Transcriptional landscape of the prenatal human brain. Nature, 508, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Duncan R.R., Betz A., Shipston M.J., Brose N., Chow R.H. (1999) Transient, phorbol ester-induced DOC2-Munc13 interactions in vivo. J. Biol. Chem., 274, 27347–27350. [DOI] [PubMed] [Google Scholar]
- 84. Grone B.P., Marchese M., Hamling K.R., Kumar M.G., Krasniak C.S., Sicca F., Santorelli F.M., Patel M., Baraban S.C. (2016) Epilepsy, behavioral abnormalities, and physiological comorbidities in syntaxin-binding protein 1 (STXBP1) mutant zebrafish. PloS One, 11, e0151148.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sanchez-Mora C., Cormand B., Ramos-Quiroga J.A., Hervas A., Bosch R., Palomar G., Nogueira M., Gomez-Barros N., Richarte V., Corrales M.. et al. (2013) Evaluation of common variants in 16 genes involved in the regulation of neurotransmitter release in ADHD. Eur. Neuropsychopharmacol., 23, 426–435. [DOI] [PubMed] [Google Scholar]
- 86. Winter E., Ponting C.P. (2002) TRAM, LAG1 and CLN8: members of a novel family of lipid-sensing domains?. Trends Biochem. Sci., 27, 381–383. [DOI] [PubMed] [Google Scholar]
- 87. Henriquez-Henriquez M.P., Solari S., Quiroga T., Kim B.I., Deckelbaum R.J., Worgall T.S. (2015) Low serum sphingolipids in children with attention deficit-hyperactivity disorder. Front. Neurosci., 9, 300.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Poersch A.B., Trombetta F., Souto N.S., de Oliveira Lima C., Braga A.C., Dobrachinski F., Ribeiro L.R., Soares F.A., Fighera M.R., Royes L.F.. et al. (2015) Fumonisin B1 facilitates seizures induced by pentylenetetrazol in mice. Neurotoxicol. Teratol., 51, 61–67. [DOI] [PubMed] [Google Scholar]
- 89. Mosbech M.B., Olsen A.S., Neess D., Ben-David O., Klitten L.L., Larsen J., Sabers A., Vissing J., Nielsen J.E., Hasholt L.. et al. (2014) Reduced ceramide synthase 2 activity causes progressive myoclonic epilepsy. Ann. Clin. Transl Neurol., 1, 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tang N., Ong W.Y., Zhang E.M., Chen P., Yeo J.F. (2007) Differential effects of ceramide species on exocytosis in rat PC12 cells. Exp. Brain Res., 183, 241–247. [DOI] [PubMed] [Google Scholar]
- 91. Camoletto P.G., Vara H., Morando L., Connell E., Marletto F.P., Giustetto M., Sassoe-Pognetto M., Van Veldhoven P.P., Ledesma M.D. (2009) Synaptic vesicle docking: sphingosine regulates syntaxin1 interaction with Munc18. PloS One, 4, e5310.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Darios F., Wasser C., Shakirzyanova A., Giniatullin A., Goodman K., Munoz-Bravo J.L., Raingo J., Jorgacevski J., Kreft M., Zorec R.. et al. (2009) Sphingosine facilitates SNARE complex assembly and activates synaptic vesicle exocytosis. Neuron, 62, 683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chan J.P., Sieburth D. (2012) Localized sphingolipid signaling at presynaptic terminals is regulated by calcium influx and promotes recruitment of priming factors. J. Neurosci., 32, 17909–17920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tornquist K., Blom T., Shariatmadari R., Pasternack M. (2004) Ceramide 1-phosphate enhances calcium entry through voltage-operated calcium channels by a protein kinase C-dependent mechanism in GH4C1 rat pituitary cells. Biochem. J., 380, 661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Valladolid-Acebes I., Daraio T., Brismar K., Hokfelt T., Bark C. (2016) Minor differences in the molecular machinery mediating regulated membrane fusion has major impact on metabolic health. Adipocyte, 5, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhang G., Bai H., Zhang H., Dean C., Wu Q., Li J., Guariglia S., Meng Q., Cai D. (2011) Neuropeptide exocytosis involving synaptotagmin-4 and oxytocin in hypothalamic programming of body weight and energy balance. Neuron, 69, 523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Klein S., Sharifi-Hannauer P., Martinez-Agosto J.A. (2013) Macrocephaly as a clinical indicator of genetic subtypes in autism. Autism Res., 6, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Imrie D., Sadler K.C. (2010) White adipose tissue development in zebrafish is regulated by both developmental time and fish size. Dev. Dyn., 239, 3013–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yang M., Lewis F.C., Sarvi M.S., Foley G.M., Crawley J.N. (2015) 16p11.2 Deletion mice display cognitive deficits in touchscreen learning and novelty recognition tasks. Learn Mem., 22, 622–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Higashijima S., Hotta Y., Okamoto H. (2000) Visualization of cranial motor neurons in live transgenic zebrafish expressing green fluorescent protein under the control of the islet-1 promoter/enhancer. J. Neurosci., 20, 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Obholzer N., Wolfson S., Trapani J.G., Mo W., Nechiporuk A., Busch-Nentwich E., Seiler C., Sidi S., Sollner C., Duncan R.N.. et al. (2008) Vesicular glutamate transporter 3 is required for synaptic transmission in zebrafish hair cells. J. Neurosci., 28, 2110–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Liew W.C., Bartfai R., Lim Z., Sreenivasan R., Siegfried K.R., Orban L. (2012) Polygenic sex determination system in zebrafish. PloS One, 7, e34397.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Liew W.C., Orban L. (2014) Zebrafish sex: a complicated affair. Brief. Funct. Genomics, 13, 172–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Gutzman J.H., Sive H. (2009) Zebrafish brain ventricle injection. J. Vis. Exp., 26, 1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lowery L.A., Sive H. (2005) Initial formation of zebrafish brain ventricles occurs independently of circulation and requires the nagie oko and snakehead/atp1a1a.1 gene products. Development, 132, 2057–2067. [DOI] [PubMed] [Google Scholar]
- 106. Neff K.L., Argue D.P., Ma A.C., Lee H.B., Clark K.J., Ekker S.C. (2013) Mojo Hand, a TALEN design tool for genome editing applications. BMC Bioinformatics, 14, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Doyle E.L., Booher N.J., Standage D.S., Voytas D.F., Brendel V.P., Vandyk J.K., Bogdanove A.J. (2012) TAL effector-nucleotide targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res., 40, W117–W122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bedell V.M., Wang Y., Campbell J.M., Poshusta T.L., Starker C.G., Krug R.G. 2nd, Tan W., Penheiter S.G., Ma A.C., Leung A.Y.. et al. (2012) In vivo genome editing using a high-efficiency TALEN system. Nature, 491, 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Cermak T., Doyle E.L., Christian M., Wang L., Zhang Y., Schmidt C., Baller J.A., Somia N.V., Bogdanove A.J., Voytas D.F. (2011) Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res., 39, e82.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Koshimizu E., Imamura S., Qi J., Toure J., Valdez D.M. Jr., Carr C.E., Hanai J., Kishi S. (2011) Embryonic senescence and laminopathies in a progeroid zebrafish model. PloS One, 6, e17688.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Schlombs K., Wagner T., Scheel J. (2003) Site-1 protease is required for cartilage development in zebrafish. Proc. Natl Acad. Sci. U S A, 100, 14024–14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Treves S., Franzini-Armstrong C., Moccagatta L., Arnoult C., Grasso C., Schrum A., Ducreux S., Zhu M.X., Mikoshiba K., Girard T.. et al. (2004) Junctate is a key element in calcium entry induced by activation of InsP3 receptors and/or calcium store depletion. J. Cell Biol., 166, 537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lorain S., Quivy J.P., Monier-Gavelle F., Scamps C., Lecluse Y., Almouzni G., Lipinski M. (1998) Core histones and HIRIP3, a novel histone-binding protein, directly interact with WD repeat protein HIRA. Mol. Cell. Biol., 18, 5546–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Chen Y., Yang Z., Meng M., Zhao Y., Dong N., Yan H., Liu L., Ding M., Peng H.B., Shao F. (2009) Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol. Cell, 35, 841–855. [DOI] [PubMed] [Google Scholar]
- 115. Antonio C., Ferby I., Wilhelm H., Jones M., Karsenti E., Nebreda A.R., Vernos I. (2000) Xkid, a chromokinesin required for chromosome alignment on the metaphase plate. Cell, 102, 425–435. [DOI] [PubMed] [Google Scholar]
- 116. Funabiki H., Murray A.W. (2000) The Xenopus chromokinesin Xkid is essential for metaphase chromosome alignment and must be degraded to allow anaphase chromosome movement. Cell, 102, 411–424. [DOI] [PubMed] [Google Scholar]
- 117. Boonen M., Staudt C., Gilis F., Oorschot V., Klumperman J., Jadot M. (2016) Cathepsin D and its newly identified transport receptor SEZ6L2 can modulate neurite outgrowth. J. Cell Sci., 129, 557–568. [DOI] [PubMed] [Google Scholar]
- 118. Ohsugi M., Adachi K., Horai R., Kakuta S., Sudo K., Kotaki H., Tokai-Nishizumi N., Sagara H., Iwakura Y., Yamamoto T. (2008) Kid-mediated chromosome compaction ensures proper nuclear envelope formation. Cell, 132, 771–782. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.