

Abstract
Mutations in coiled-coil-helix-coiled-coil-helix-domain containing 10 (CHCHD10), a mitochondrial twin CX9C protein whose function is still unknown, cause myopathy, motor neuron disease, frontotemporal dementia, and Parkinson’s disease. Here, we investigate CHCHD10 topology and its protein interactome, as well as the effects of CHCHD10 depletion or expression of disease-associated mutations in wild-type cells. We find that CHCHD10 associates with membranes in the mitochondrial intermembrane space, where it interacts with a closely related protein, CHCHD2. Furthermore, both CHCHD10 and CHCHD2 interact with p32/GC1QR, a protein with various intra and extra-mitochondrial functions. CHCHD10 and CHCHD2 have short half-lives, suggesting regulatory rather than structural functions. Cell lines with CHCHD10 knockdown do not display bioenergetic defects, but, unexpectedly, accumulate excessive intramitochondrial iron. In mice, CHCHD10 is expressed in many tissues, most abundantly in heart, skeletal muscle, liver, and in specific CNS regions, notably the dopaminergic neurons of the substantia nigra and spinal cord neurons, which is consistent with the pathology associated with CHCHD10 mutations. Homozygote CHCHD10 knockout mice are viable, have no gross phenotypes, no bioenergetic defects or ultrastructural mitochondrial abnormalities in brain, heart or skeletal muscle, indicating that functional redundancy or compensatory mechanisms for CHCHD10 loss occur in vivo. Instead, cells expressing S59L or R15L mutant versions of CHCHD10, but not WT, have impaired mitochondrial energy metabolism. Taken together, the evidence obtained from our in vitro and in vivo studies suggest that CHCHD10 mutants cause disease through a gain of toxic function mechanism, rather than a loss of function.
Introduction
In recent years, several mutations in the gene encoding Coiled-Coil-Helix-Coiled-Coil-Helix Domain Containing 10 (CHCHD10) have been identified in families with amyotrophic lateral sclerosis (ALS) or ALS-frontotemporal lobar dementia (ALS-FTLD) (1–8). Mutations in this gene have also been associated with other diseases (9), including mitochondrial myopathy (10), spinal muscular atrophy (11), Charcot-Marie-Tooth disease (12), late onset Alzheimer’s disease (13) and Parkinson’s disease (14). Despite ample evidence that mutations in CHCHD10 cause neurodegenerative diseases in humans, the function of the protein remains unknown.
CHCHD10 contains a twin CX9C domain, which in mitochondria allows for import and retention of proteins mostly located in the inter membrane space (IMS) or the inner membrane (IM), through the action of the Mia40-Erv1 disulfide relay system (15,16). A genome-wide analysis of eukaryotic twin CX9C proteins suggested that proteins in this family play diverse functions, and are frequently involved in the structural organization of molecular scaffolds in mitochondria (17,18). For example, many twin CX9C proteins with described functions are respiratory chain complex IV (cytochrome c oxidase, COX) assembly factors (19–23). Two other twin CX9C proteins, CHCHD3 (24) and CHCHD6 (25), are components of the mitochondrial contact site and cristae organizing system (MICOS).
Here, we explore the physiological role of CHCHD10 in vitro and in vivo by analysing its localization in mitochondria, its physical interactions with other mitochondrial proteins, the impact of manipulating its expression by gene silencing in cultured cells and gene ablation in mice, and the effects of disease-linked CHCHD10 mutants in cells.
Results
The mitochondrial localization of CHCHD10 requires the N-terminal domain and the twin CX9C domain
We first investigated the mitochondrial localization of CHCHD10. By immunofluorescence in HeLa cells, we detected endogenous CHCHD10 and confirmed the mitochondrial localization by co-immunostaining with the IMS protein cytochrome c (Fig. 1A).
Figure 1.
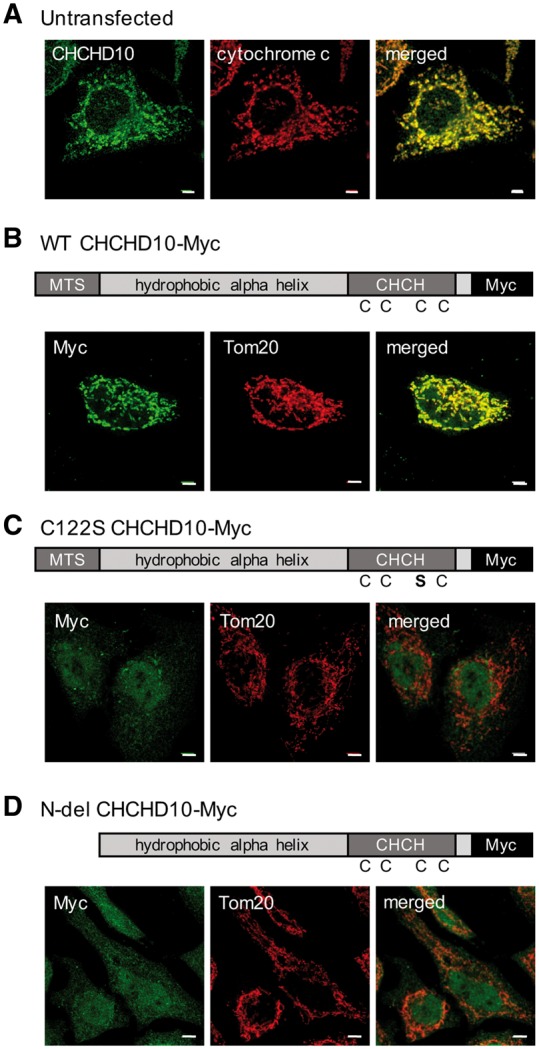
CHCHD10 requires the twin CX9C domain and MTS to localize to mitochondria. (A) Immunocytochemistry of HeLa cells for CHCHD10 (green) and cytochrome c (red). (B) Immunocytochemistry of HeLa cells transfected with WT CHCHD10-Myc and immunostained for Myc (green) and Tom20 (red). (C) C122S CHCHD10-Myc transfected HeLa cells immunostained for Myc (green) and Tom20 (red) (D) N-del CHCHD10-Myc transfected HeLa cells immunostained for Myc (green) and Tom20 (red). Bar = 5 μm.
Since most twin CX9C proteins are retained in the mitochondria by virtue of disulfide formation through the Mia40-Erv1 disulfide relay system (15,16), we investigated whether the twin CX9C domain of CHCHD10 was required for its mitochondrial localization. We generated a wild type (WT) CHCHD10 construct with a C-terminal Myc tag, as well as a C122S mutant CHCHD10 Myc-tagged construct. The WT CHCHD10-Myc localized to mitochondria (Fig. 1B), whereas the substitution to serine of one of the cysteine residues was sufficient to prevent mitochondrial import or retention, and resulted in cytosolic and nuclear localization of the protein (Fig. 1C), indicating that an intact twin CX9C domain is required for mitochondrial localization of CHCHD10. The four cysteines in the twin CX9C motif of members of this family of proteins are usually equally important for protein import (26). Thus, the C122S mutation prevents CHCHD10 mitochondrial import/retention, as would be expected for mutations in any of the three other cysteine residues in the twin CX9C motif. Furthermore, a previous study by Aras and colleagues (27) showed that all four cysteine residues in the twin CX9C motif of CHCHD2, which is highly homologous to that of CHCHD10, impaired mitochondrial localization, with the homolog of C122S having a strong effect.
Since some of the disease-associated mutations in CHCHD10 are found in its putative mitochondrial targeting sequence (MTS) at the N-terminus, we also investigated whether this domain was required for mitochondrial localization by generating a Myc-tagged CHCHD10, in which amino acids 1–16 were deleted. The 1–16 N-terminal deletion resulted in cytosolic and nuclear localization of the protein, indicating that, in addition to the twin CX9C domain, the N-terminus of CHCHD10 is also necessary for mitochondrial localization (Fig. 1D).
CHCHD10, CHCHD2 and p32 co-sediment in sucrose gradient fractionation
To investigate the protein-protein interactions that CHCHD10 normally establishes within mitochondria to perform its function, we surveyed the information available from the literature. It was suggested that CHCHD10, together with CHCHD3 and CHCHD6, interacts with the protein mitofilin, within the mitochondrial contact site and cristae organizing system (MICOS) complex (28). Furthermore, another member of the mitochondrial twin CX9C domain protein family, CHCHD2, was found to interact with CHCHD10 in a mass spectrometry-based interactome study (29). Interestingly, CHCHD10 and CHCHD2 are 58% identical at the protein level, and are thought to be paralogs arisen from a gene duplication event that occurred in mammals (17). Moreover, analysis of CHCHD2 interactome by a mass spectrometry approach in HEK293 cells expressing ectopic FLAG-tagged CHCHD2 and by proximity-dependent in vivo biotinylation (BioID) identified the mitochondrial protein p32 (GC1QR/C1QBP) as an interacting partner (30). However, whether CHCHD2, CHCHD10, and p32 form a protein complex was not explored.
To further investigate the interactome of CHCHD10 and CHCHD2, we performed sucrose gradient sedimentation analyses of HEK293 mitochondrial extracts prepared using a variety of extraction conditions, in the presence of the detergents digitonin or NP-40. When the extracts were prepared in the presence of increasing digitonin concentrations, complexes with different sedimentation properties were detected. A protein: digitonin ratio 1: 1 did not extract mitofilin, but extracted CHCHD10, CHCHD2 and p32 (Fig. 2A). Protein: digitonin ratio of 1: 2 extracted all the proteins in native complexes (Fig. 2B). However, while all mitofilin was found in the higher density fractions, CHCHD10, CHCHD2, and p32 co-sedimented in the same lighter fractions (containing complexes of MW less than 250 kDa). With protein: digitonin ratio of 1: 4 (Fig. 2C), the majority of CHCHD10 and p32 co-sedimented in the lower density fractions (less than 250 kDa), but a subset of these proteins, plus CHCHD2 and mitofilin, co-sedimented in higher density fractions (protein complexes of MW greater than 250 kDa). In NP-40 extracts (protein: NP-40 ratio 1: 2), all CHCHD10 sedimented in low-density fractions (complexes of MW less than 130 kDa) with CHCHD2 and p32, plus a subset of mitofilin (Fig. 2D). In SDS extracts (protein: SDS 1: 4), as expected proteins were completely solubilized and sedimented in the lower density fractions (Fig. 2E). Taken together, the sucrose gradient sedimentation assays demonstrated that the association of the proteins of interest in different fractions was highly dependent on the solubilization method, with mitofilin co-sedimenting with the other proteins only in a minority of the fractions, under specific detergent conditions.
Figure 2.

CHCHD10 interacts with CHCHD2 and p32. (A-E) Sucrose gradient sedimentation analyses of CHCHD10, CHCHD2, mitofilin and p32 in linear sucrose gradients, extracted from HEK293 mitochondria using the indicated extraction conditions. Hemoglobin (Hb) and lactate dehydrogenase (LDH) were used to calibrate the gradients. M, mitochondria; Ex, total mitochondrial extract. (F) FLAG Co-IP in mitochondria from CHCHD10-FLAG transfected cells. Immunoblot: FLAG and CHCHD2. (G) FLAG Co-IP in mitochondria from CHCHD2-FLAG transfected cells. Immunoblot: FLAG and CHCHD10. (H) FLAG Co-IP in mitochondria from mock (pcdna) or CHCHD10-FLAG transfected cells. Immunoblot: CHCHD10. (I) FLAG Co-IP using CHCHD2-FLAG or CHCHD10-FLAG transfected cells. Immunoblot: FLAG and p32. (J) FLAG Co-IP using CHCHD10-FLAG transfected cells. Immunoblot: FLAG, mitofilin. (K) Mitofilin Co-IP. Immunoblot: mitofilin, CHCHD10, Tim23.
CHCHD10, CHCHD2, and p32 physically interact
Since sucrose gradient fractionations of mitochondrial extracts suggested that CHCHD10 was part of a molecular complex with other proteins, we used co-immunoprecipitation (Co-IP) assays to further explore the binding partners of CHCHD10. We performed Co-IP in mitochondria fractions of HEK293 cells expressing CHCHD10-FLAG and detected CHCHD2 in the FLAG Co-IP eluate, but not in the IgG control Co-IP, demonstrating that the two proteins physically interact (Fig. 2F). CHCHD10-FLAG migrates at a higher molecular weight (approximately 20 kDa) than endogenous CHCHD10 (approximately 15 kDa). We confirmed the interaction between CHCHD2 and CHCHD10 by performing the reverse Co-IP, in which cells were transfected with CHCHD2-FLAG and Co-IP performed with either FLAG antibody or control IgG, and identified CHCHD10 only in the FLAG Co-IP eluate (Fig. 2G).
Next, we investigated whether CHCHD10 forms homo-oligomers. Because of the C-terminal tags in the CHCHD10 construct, we could distinguish the recombinant CHCHD10 from the endogenous, as it migrates higher in an SDS-PAGE gel. When we performed Co-IP of CHCHD10-FLAG using the FLAG antibody, we did not detect endogenous CHCHD10 in the Co-IP eluate (Fig. 2H), indicating that CHCHD10 does not form homo-oligomers.
To study CHCHD2 and CHCHD10 interactions with p32 we transfected HEK293 cells with either CHCHD2-FLAG or CHCHD10-FLAG, and performed Co-IP in mitochondrial fractions, using the FLAG antibody. We were able to confirm the interaction between CHCHD2 and p32, but also to detect p32 in the CHCHD10-FLAG Co-IP eluate (Fig. 2I), but not in the control IgG eluate, indicating that p32 and CHCHD10 physically interact in mitochondria. Taken together, these Co-IP experiments indicated the existence in mitochondria of a protein complex containing CHCHD10, CHCHD2, and p32.
To further investigate the potential interaction between CHCHD10 and mitofilin, we transfected HEK293 cells with CHCHD10-FLAG or empty vector, and prepared enriched mitochondrial fractions for Co-IP with the FLAG antibody. The Western blot confirmed the presence of CHCHD10-FLAG in the eluate (Fig. 2J). Mitofilin was detected in the IP eluate, but also in the control IgG eluate (Fig. 2J), indicating non-specific binding of mitofilin to IgGs. Therefore, we performed reverse Co-IP and pulled down mitofilin. We did not detect CHCHD10 in the mitofilin Co-IP eluate (Fig. 2K), but we were able to detect Tim23, which was previously shown to interact with mitofilin (25). Taken together, the Co-IP experiments did not confirm the interaction of CHCHD10 with mitofilin. Although our data do not exclude a transient interaction between them, they clearly indicate that CHCHD10 is not an integral part of the MICOS complex.
CHCHD2 and CHCHD10 are associated with the mitochondrial inner membrane
To investigate the localization and topology of CHCHD10 and CHCHD2 in mitochondria we utilized multiple complementary approaches. First, we used proteinase K protection assays that showed that both CHCHD2 and CHCHD10 are protease-resistant in mitochondria but not in mitoplasts (i.e. mitochondria devoid of outer membrane, OM) prepared by hypotonic swelling of mitochondria (Fig. 3A). The proteins behaved in this assay as TIMM50, an inner membrane protein previously shown to face the IMS (31,32). This result suggested that globular domains of both CHCHD10 and CHCHD2 face the IMS side of the IM. Differently, p32 had a proteinase K resistance pattern similar to HSP60, a mitochondrial matrix protein, indicating that p32 is localized in the matrix.
Figure 3.

Submitochondrial topology of CHCHD2 and CHCHD10. (A) Proteinase K protection assay in mitochondria (M) and mitoplasts (Mp) prepared by hypotonic swelling of mitochondria. As a control, a mitochondrial sample was sonicated (Son) to give full access to the protease. In this assay, the controls used were TIMM50 (inner transmembrane protein facing the intermembrane space) and HSP60 (matrix protein). (B) CHCHD2 (D2) and CHCHD10 (D10) solubilization by sonication and alkaline carbonate (pH 11.5) extraction. Mitochondria (M) isolated from HEK293 cells were sonicated, and the soluble (S), and membrane-bound fractions were separated by centrifugation. The pellet was subsequently extracted with alkaline sodium carbonate and fractionated into supernatant (CS) and pellet (CP). The different fractions were analysed by immunoblotting using antibodies that recognize CHCHD2, CHCHD10, p32, and the controls COA3 and SCO1 (membrane proteins), SDHA and CMC1 (loosely bound to IM), and LON-P (soluble protein) (C) Isolated mitochondria were fractionated into inner and outer membranes by sonication and centrifugation. The crude membrane fractions were analysed by sucrose gradient sedimentation in a linear 30–60% sucrose gradient. In each case, following ultracentrifugation, 15 gradient fractions were collected, separated by SDS-PAGE and analysed by immunoblotting using antibodies against VDAC (OM marker), COX1 (IM marker), CHCHD2 and CHCHD10. (D) Digitonin solubilization of mitochondria using increasing detergent: protein ratios. Following solubilization, soluble (S) and insoluble material (P) were separated by centrifugation and analysed by immunoblotting using the indicated antibodies.
Second, brief sonication of mitochondria (M) followed by centrifugation yielded a supernatant consisting of soluble proteins (S) and a pellet containing membrane-associated proteins (P) that was subsequently extracted with alkaline carbonate (pH 11.5). Following centrifugation, solubilized extrinsic proteins were collected in the supernatant (CS), whereas intrinsic proteins remained in the pellet (CP). Most CHCHD2 and CHCHD10 were detected in the pellet fraction following carbonate extraction (CP in Fig. 3B), suggesting that they are membrane intrinsic proteins. In this experiment, which was controlled by following the distribution of proteins with known solubility behavior (soluble matrix LON-P, intrinsic trans-IM COA3 and SCO1, and extrinsic IM-bound SDHA and CMC1), p32 was largely recovered in the soluble fraction.
Next, we separated IM and OM by sonication of isolated mitochondria and differential centrifugation, followed by sucrose gradient sedimentation. By immunoblot of these fractions, we determined that CHCHD2 and CHCHD10 are strongly associated with the mitochondrial IM and confirmed that p32 is not (Fig. 3C), although a transient or labile association cannot be discarded. Preferential solubilization of the mitochondrial OM with increasing concentrations of digitonin indicated that CHCHD2, and particularly CHCHD10, are solubilized with low concentrations of the detergent that are insufficient to solubilize VDAC or the single transmembrane domain IM proteins ATAD3A and COA3 (Fig. 3D), suggesting that the membrane association of CHCHD10 and CHCHD2 is more labile than expected for transmembrane proteins. Their behavior in this assay was similar to that of p32 and the extrinsic membrane proteins SDHA and CMC1 (Fig. 3D).
CHCHD10 and CHCHD2 have rapid turnover
To further characterize the properties of these interacting proteins we investigated the turnover of CHCHD10, CHCHD2, and p32 in HEK293 cells. We treated cells with cycloheximide (CHX) to inhibit protein translation for 0.5, 1.5, 3, 6 and 12 h. Immunoblot analysis of whole cell homogenates was used to examine the turnover of these proteins over time. We observed that CHCHD10 and CHCHD2 levels decreased rapidly (Fig. 4A and B). While p32 had a much slower decline, CHCHD10 was degraded with a half-life of approximately 4 h, and the turnover of CHCHD2 was even faster at approximately 1.5 h (Fig. 4B). This result suggested that the degradation of these two proteins can be differentially regulated, and that CHCHD10 and CHCHD2 can exist independently from each other. Addition of the proteasome inhibitor MG132 did not affect the degradation rate of CHCHD10 or CHCHD2 (Fig. 4C), indicating that the proteasome is not involved in the degradation of these proteins.
Figure 4.

CHCHD10 and CHCHD2 turnover rates and silencing. (A) Western blot using antibodies against CHCHD10, CHCHD2, p32 in homogenates from cells treated with cycloheximide (CHX) for the indicated number of hours. (B) Quantification of band intensity following CHX treatment. Data are expressed as percentage of the band intensity at 0 h for each protein. n = 3. (C) Western blot for CHX-treated cells for the indicated number of hours, with or without MG132 treatment. (D–E) Western blot of cell homogenates following 48 h of treatment with scrambled (scram) siRNA or CHCHD10 siRNA. n = 5 *P < 0.05. (F-G) Representative blue native gel and quantification of OXPHOS complexes, in HEK293 cells after 72 h of treatment with scram or siD10 siRNA. The following antibodies were used to detect the OXPHOS complexes: Complex I-39 kDa, Complex V- subunit α, Complex III-core2, Complex IV-2, Complex II, 70 kDa. Data are represented as percentage of scram. n = 3. (H) Intact cell respiration in HEK293 cells treated with scram or CHCHD10 siRNA. n = 6. (I) COX activity in HEK293 cells treated with scram or CHCHD10 siRNA. n = 3. (J) ATP synthesis measured in HEK293 cells treated with scram or CHCHD10 siRNA. n = 3. (K) Iron levels measured by graphite furnace atomic absorption spectrometry in medium (n = 5) and mitochondrial or cytosolic fractions. In mitochondrial and cytosolic fractions (n = 4) iron levels are expressed as fold change relative to scram. (L) Representative electron micrographs of mitochondria from stable CHCHD10 knockdown or scram shRNA control HEK293 cell lines. Bar = 500 nm. (M) Traces of mitochondrial calcium uptake measurements using Fura-FF in HEK293 CHCHD10 knockdown or scram shRNA controls.
CHCHD10 silencing does not affect mitochondrial OXPHOS in HEK293 cells, but it alters iron homeostasis
We then transfected HEK293 cells with siRNA to silence CHCHD10 or a scrambled siRNA control. The siRNA treatment resulted in robust CHCHD10 silencing (Fig. 4D), with approximately 80% decrease in protein expression (Fig. 4E), while expression levels of CHCHD2 and p32 were unchanged. Since OXPHOS is a critical mitochondrial function and some twin CX9C proteins are involved in OXPHOS complex assembly (21,22), we investigated OXPHOS complex assembly and function in HEK293 cells upon CHCHD10 silencing. No differences were found in the levels of assembled complexes, as assessed by blue native PAGE (Fig. 4F and G). Despite apparently normal assembly, we detected a moderate decrease in the activity of complex IV (cytochrome c oxidase, COX) (Fig. 4H). However, the COX deficiency caused by CHCHD10 silencing did not affect intact cell oxygen consumption rate (OCR, Fig. 4I) or mitochondrial ATP synthesis (Fig. 4J).
Silencing of CHCHD10 has been associated with phenotypes related to iron homeostasis (33,34). Therefore, we investigated whether iron levels were affected by CHCHD10 silencing using atomic absorption spectrometry (Fig. 4K). When cells were treated with CHCHD10 siRNA, iron levels in the culture medium decreased. Iron levels in mitochondrial fractions from CHCHD10 silenced cells were increased relative to scrambled control, but iron levels in the cytosol fraction were not different than controls, indicating that CHCHD10 is involved in the regulation of mitochondrial iron.
To address the effects of chronic CHCHD10 silencing, we generated stable HEK293 cell lines, in which CHCHD10 was knocked down using lentiviral shRNA transduction and stable selection. All stable clones generated showed approximately 80% CHCDH10 silencing (Supplementary Material, Fig. S1A and B). Stable CHCHD10 silencing did not affect CHCHD2 and p32 expression levels (Supplementary Material, Fig. S1A and B), or the interaction of CHCHD2 with p32 (Supplementary Material, Fig. S1C), suggesting that this interaction can occur independently of CHCHD10.
Because of the proposed role for CHCHD10 in maintaining mitochondrial cristae structure (28), we assessed mitochondrial ultrastructure in these cell lines using electron microscopy, but did not detect differences between CHCHD10 silenced and scrambled control cells (Fig. 4L).
Since p32 has been shown to play a role in mitochondrial calcium handling (35) and proposed as a mitochondrial permeability transition pore candidate (36), we also assessed mitochondrial calcium handling exposing mitochondrial preparations from stable CHCHD10 silenced and scrambled control cells to sequential calcium additions in the presence of the ratiometric calcium probe Fura-FF. No differences in the rate of mitochondrial calcium uptake or mitochondrial calcium capacity were observed (Fig. 4M).
To further analyse the global OXPHOS function of cells with stable CHCHD10 knockdown, we tested their ability to grow in medium containing galactose as the main carbon source and normalized it to the growth in glucose. After 3 days of growth in galactose (GAL) medium or in glucose (GLU) medium, there was no difference in the cell number ratio GAL: GLU between stable CHCHD10 silenced and scrambled control cells (Supplementary Material, Fig. S1D). Taken together with the results in CHCHD10 transiently silenced cells, this finding indicated that reduction in CHCHD10 expression levels did not affect OXPHOS capacity in HEK293 cells.
We also tested the sensitivity to the pro-apoptotic agents staurosporine and actinomycin D, and the oxidizing agents paraquat and H2O2. There were no differences in cell viability between stable CHCHD10 silenced and scrambled control cells after treatment with any of these agents (Supplementary Material, Fig. S1E), suggesting that reduction in CHCHD10 expression levels did not affect apoptosis or antioxidant defenses in HEK293 cells.
CHCHD10 silencing in human fibroblasts causes mild mitochondrial abnormalities
Since it was reported that fibroblasts from patients harboring mutant CHCHD10 have mitochondrial dysfunction (28), to test the effects of loss of CHCHD10 in this cell type, we transiently silenced CHCHD10 in human skin fibroblasts for five days. As expected, CHCHD10 was effectively silenced, and CHCHD2 and p32 were normally expressed (Fig. 5A). Silenced fibroblasts were cultured in growth medium containing glucose (GLU), or in galactose-containing medium (GAL), in order to force the cells to produce ATP through OXPHOS. The number of cells after 5 days of growth in GLU or GAL medium did not differ between CHCHD10-silenced and scrambled control fibroblasts (Fig. 5B). The baseline OCR of fibroblasts cultured in GAL medium was comparable in CHCHD10 silenced and scrambled control cells (Fig. 5C), but the uncoupled respiration (i.e. OCR after FCCP) was significantly decreased (Fig. 5D), suggesting that CHCHD10 silencing impairs the maximal respiration rate induced by the uncoupler.
Figure 5.

CHCHD10 silencing in human skin fibroblasts. (A) Western blot of human skin fibroblasts treated with scram or CHCHD10 siRNA and cultured in GLU or GAL-containing medium for 5 days. (B) Cell number after 5 days in GLU or GAL medium. n = 3. (C) Intact cell oxygen consumption rate (OCR). n = 6. (D) Uncoupled OCR following FCCP (1 μM) addition. n = 6; *P < 0.05. (E) Representative images of scram and siD10 human skin fibroblasts immunostained for CHCHD10 (green) and cytochrome c (red). Bar = 50 μm (F–H). Aspect ratio (AR), form factor (FF), number of mitochondria per cell (Nc), expressed as percentage of scram. n = 30 cells for scrambled an n = 28 cells for CHCHD10 siRNA; *P < 0.05.
We then investigated if the mitochondrial morphology of fibroblasts cultured in GLU medium is affected by CHCHD10 silencing. Figure 5E shows representative pictures of cells immunostained for endogenous CHCHD10 and cytochrome c. As expected, after 5 days of silencing, the expression of CHCHD10 was virtually absent. Mitochondrial aspect ratio (Fig. 5F) and form factor (Fig. 5G) were unchanged in control and siD10 cells, whereas the number of mitochondria per cell was slightly but significantly increased in siD10 cells compared with scrambled controls (Fig. 5H), suggesting that silencing of CHCHD10 may induce an adaptive mitochondrial biogenesis program.
Tissue expression of CHCHD10 is highly variable, and in the CNS CHCHD10 is strongly expressed in dopaminergic neurons of the substantia nigra and in spinal cord neurons
To evaluate the distribution of CHCHD10 in the context of an organism, we generated crude mitochondrial fractions from tissues of WT adult C57Bl6J mice, and analysed CHCHD10 and CHCHD2 content by Western blot (Fig. 6A). CHCHD10 and CHCHD2 were expressed in all tissues examined, but the levels were highly variable with highest content of both proteins in heart, liver, pancreas and skeletal muscle. The patterns of tissue expression of CHCHD10 and CHCHD2 were clearly very similar, consistent with the finding that they form a multimeric protein complex.
Figure 6.

CHCHD10 expression in mouse tissues. (A) Representative Western blot using antibodies against CHCHD10, CHCHD2, and Tim23 (as mitochondrial loading control), in crude mitochondrial extracts prepared from WT mouse tissues. 50 μg of protein was loaded per sample. SC = spinal cord. (B) Input, flow through, and eluate fractions from CHCHD10, CHCHD2, and IgG control Co-IP in heart mitochondria. Immunoblot: CHCHD10, CHCHD2, p32, mitofilin. (C) CHCHD10 immunoreactivity in brain section containing substantia nigra (SN) region from WT mouse. PAG = periaqueductal gray, IPN = interpeduncular nucleus. Left panel: bar = 50 μm. Right panel: bar = 10 μm. (D–E) Immunohistochemistry of brain sections stained with CHCHD10 and TH antibodies. Bar = 100 µm in D, bar = 10 µm in E. (F–G). Immunohistochemistry of spinal cord sections stained with CHCHD10 and ChAT antibodies. Ba r = 100 µm in F, bar = 30 µm in G.
We sought to confirm the physical interaction between CHCHD2 and CHCHD10 in mouse heart. Mitochondria were subjected to Co-IP with antibodies against CHCHD10, CHCHD2 or control IgG. As in the cell culture Co-IP experiment using tagged constructs, also in mouse heart we demonstrated interactions between endogenous CHCHD10 and endogenous CHCHD2 and p32, but not with mitofilin (Fig. 6B), suggesting that these interactions occur in vivo. The amount of CHCHD2 and CHCHD10 in the flow through fraction was at the lower limit of detection, indicating that the majority of each protein Co-IPed with its partner. In contrast, there was a large portion of p32 in the flow through fraction, which corresponded approximately to 95% of the total protein, suggesting that only a minority of p32 interacts with CHCHD10 or CHCHD2.
CHCHD10 and CHCHD2 expression in mitochondria obtained from whole forebrain and spinal cord was relatively low compared with other tissues (Fig. 6A). This was unexpected because these tissues are targets of pathology in patients carrying CHCHD10 (1–7,14) and CHCHD2 (14,37,38) mutations. Therefore, to better define the expression of these proteins in the central nervous system, we performed immunohistochemistry with CHCHD10 antibodies, followed by peroxidase staining. In brain, we found highly enriched CHCHD10 expression in the substantia nigra (Fig. 6C). Immunofluorescence of CHCHD10 in the substantia nigra imaged at high magnification showed a colocalization of CHCHD10 and the tyrosine hydroxylase (TH) marker of dopaminergic neurons (Fig. 6D and E). In spinal cord, CHCHD10 was expressed in neurons, notably co-localizing with the motor neuron marker choline acetyltransferase (ChAT) in the anterior horn (Fig. 6F and G). These results indicated that CHCHD10 is highly expressed in cells affected by the disease mutations.
CHCHD10KO mice are born at Mendelian ratio and are viable
We generated a CHCHD10 knockout animal (CHCHD10KO) to investigate the effects of the lack of CHCHD10 in vivo. Using CRISPR/Cas9, we introduced a single adenine nucleotide in exon 2 of chchd10, which resulted in a frameshift mutation and premature stop codon (Fig. 7A). Breeding of heterozygote animals resulted in homozygote KO animals born at Mendelian ratios. We confirmed the absence of CHCHD10 expression by immunoblot of homogenates from brain, skeletal muscle and heart of CHCHD10 homozygote KO mice (Fig. 7B). Heterozygote and homozygote males and females were viable and did not exhibit any gross phenotypic abnormalities from birth through early adulthood (up to 200 days of age). Furthermore, the level of CHCHD2 in these tissues was not affected by the lack of CHCHD10 expression.
Figure 7.

Brain, skeletal muscle and heart mitochondrial bioenergetics and ultrastructure in CHCHD10KO mice. (A) Schematic representation of the strategy utilized to knock down CHCHD10 expression in the CHCHD10KO mouse. (B) Representative Western blot of brain, skeletal muscle (gastrocnemius) and heart homogenates from WT and CHCHD10KO (KO) mice at 100 days of age. 100 μg protein was loaded per lane and the membrane was probed with antibodies against CHCHD10, CHCHD2 and Tim23 (mitochondrial loading control). (C) Oxygen consumption rates in WT and CHCHD10KO brain mitochondria before (State 2) and after (State 3) ADP stimulation. n = 3. (D) ATP synthesis rate in WT and CHCDH10KO brain mitochondria. n = 3. (E) Representative electron micrographs of WT and CHCHD10KO substantia nigra. Peroxidase labeling indicates TH-immunoreactive dendrites. Bar = 500 nm. (F) Oxygen consumption rate of WT and CHCHD10KO in skeletal muscle mitochondria. n = 7; *P < 0.05, (G) ATP synthesis rate in WT and CHCDH10KO skeletal muscle mitochondria. n = 7. (H) Representative electron micrograph of WT and CHCHD10KO skeletal muscle (soleus). (I) Oxygen consumption rate in WT and CHCHD10KO heart mitochondria. n = 4. (J) ATP synthesis rate in WT and CHCDH10KO heart mitochondria. n = 4. (K) Representative electron micrograph of WT and CHCHD10KO heart right ventricle wall. Arrowheads indicate electron-dense structures present in CHCHD10KO but not in WT. m = mitochondria. Bar = 500 nm.
We studied mitochondrial bioenergetics in brain, skeletal muscle, and heart of CHCHD10KO at 100 days of age. Mitochondria isolated from forebrain did not display differences in oxygen consumption (Fig. 7C) or ATP synthesis (Fig. 7D), in CHCHD10KO compared with WT. Since it was previously suggested that CHCHD10 plays a role in mitochondrial cristae maintenance using cultured cells from patients affected by CHCHD10 mutations (28), we investigated mitochondrial ultrastructure by electron microscopy in CHCHD10KO tissues. In the substantia nigra of WT and CHCHD10KO mice, dopaminergic neuronal processes were identified by TH immunoreactivity. We did not detect mitochondrial ultrastructural abnormalities in dopaminergic processes (Fig. 7E) or in the other cell types in this brain region.
Mitochondria isolated from skeletal muscle (gastrocnemius) had decreased ADP-stimulated respiration (Fig. 7F), but no ATP synthesis differences (Fig. 7G) relative to WT or abnormal mitochondrial ultrastructure (Fig. 7H). There were no differences in respiration (Fig. 7I) or ATP synthesis (Fig. 7J) between CHCHD10KO and WT heart mitochondria. While we did not observe abnormal cristae or other mitochondrial structural abnormalities in heart, we detected numerous electron-dense round structures (Fig. 7K) localized between the sarcomeres. The nature of these structures remains to be fully elucidated, but the morphology and localization suggest that they could be lysosomes in close proximity to mitochondria.
Taken together, these data suggest that CHCHD10KO has tissue-specific effects, resulting in a mild decline in respiration in skeletal muscle, but not in other tissues examined. This decline in muscle, however, does not result in ATP synthesis defects. Importantly, the lack of changes in mitochondrial ultrastructure in any of the tissues studied does not support the hypothesis that CHCHD10 is required for mitochondrial cristae maintenance.
Disease-associated CHCHD10 mutants mislocalize and induce mild bioenergetics defects in cultured cells
Since downregulation of CHCHD10 did not result in significant mitochondrial functional defects, to assess whether disease-associated mutants of CHCHD10 could exert toxic effects, we introduced the R15L and S59L mutations in the CHCHD10-FLAG plasmid by in situ mutagenesis. Mutant CHCHD10 was expressed in HEK293 cells and expression of the recombinant proteins detected by Western blot in mitochondria (Fig. 8A). Next, we generated stable lines expressing WT or mutant CHCHD10, and demonstrated that R15L CHCHD10 and S59L CHCHD10 can bind to CHCHD2 (Fig. 8B). Furthermore, we determined that the mutant proteins did not differ from the WT protein in their turnover rate (Fig. 8C).
Figure 8.

Mutant CHCHD10 overexpression in HEK293 cells. (A) Western blot of crude mitochondrial fractions from control HEK293 cells and cells transfected with WT CHCHD10-FLAG, R15L CHCHD10-FLAG or S59L CHCHD10-FLAG. (B) Co-IP of WT, R15L or S59L CHCHD10-FLAG with FLAG antibody or IgG control. Immunoblot: FLAG, CHCHD2. (C) Quantification of Western blot band intensity following CHX treatment. Data are expressed as percentage of the FLAG intensity at 0 h. n = 4. (D) Representative images of HeLa cells transfected with WT CHCHD10-Myc, R15L CHCHD10-Myc or S59L CHCHD10-Myc and immunostained for Myc (green) or Tom20 (red). Bar = 5 μm. (E) CHCHD10 and Tom20 percentage of colocalization n = 15 cells for WT CHCHD10-Myc, n = 22 cells for R15L CHCHD10-Myc, n = 25 cells for S59L CHCHD10-Myc. (F–G) Oxygen consumption rate (OCR) in cells transfected with empty vector (pcdna) or CHCHD10-FLAG, before or after FCCP treatment (1 μM). n = 3. (H) ATP synthesis in cells transfected with empty vector (pcdna) or CHCHD10-FLAG. n = 3 to 5.
We then investigated the effect of CHCHD10 mutations on mitochondrial localization of the protein by generating and expressing Myc-tagged constructs in HeLa cells. Like WT CHCHD10-Myc, R15L CHCHD10-Myc immunostaining mostly colocalized with the mitochondrial protein Tom20 (Fig. 8D). Interestingly, S59L CHCHD10-Myc immunostaining showed a more punctate distribution, with most puncta colocalizing with mitochondria, but some found in the cytosol. We quantified the colocalization of CHCHD10 with Tom20, and found that in R15L and S59L CHCHD10-expressing cells, the colocalization of CHCHD10 with Tom20 was decreased (Fig. 8E).
Expression of R15L and S59L mutant CHCHD10 did not affect baseline or FCCP-uncoupled OCR in intact cells (Fig. 8F and G). However, ATP synthesis measured in permeabilized cells was decreased in the mutant cells (Fig. 8H), indicating an impairment of the OXPHOS system in these cells. In this experimental paradigm, the recombinant proteins were overexpressed (5–10 fold relative to endogenous CHCHD10). However, only mutant proteins resulted in mislocalization and ATP synthesis defects. Taken together, these data indicate that overexpression of disease-associated CHCHD10 but not WT protein causes OXPHOS defects, and that two different mutants affect the localization and distribution of the protein.
Discussion
Mitochondrial topology and protein–protein interactions of CHCHD10
This study demonstrated a relationship between two mitochondrial twin CX9C proteins, CHCHD10 and CHCHD2. The homology between CHCHD10 and CHCHD2 led us to investigate their relationship. We found that these proteins bind to each other, and to the mitochondrial protein p32. In heart tissue, the majority of CHCHD10 and CHCHD2 interact with each other. However, only a small percentage of p32 is pulled down with either CHCHD10 or CHCHD2. This is not surprising because, p32 is a very abundant matrix protein with no membrane spanning domains, and it is likely that only a portion of the protein interact with inner membrane bound CHCHD2/CHCHD10 complex.
Since we found in our studies that CHCHD10 and CHCHD2 are tethered to the IM, facing the IMS and p32 localizes to the mitochondrial matrix (39), it is possible that they may interact while p32 is en route to the matrix, or that they may interact through the IM or other IM proteins since p32 can associate with membranes (40). The first possibility may suggest that CHCHD10 and CHCHD2 act as molecular chaperones in p32 import. However, since silencing of CHCHD10 does not result in p32 deficiency, this interpretation does not appear likely. The second possibility is particularly intriguing in light of the results on the topology of CHCHD10 and CHCHD2 relative to the IM. These results suggest that CHCHD2 and CHCHD10 might not possess a complete transmembrane domain, since they are solubilized from the IM at lower digitonin concentrations than other known IM transmembrane proteins (Fig. 3D). At the same time, however, both CHCHD2 and CHCHD10 are bound to the membrane in a manner that largely resists sonication and alkaline carbonate extraction (Fig. 3B). Because digitonin is a steroidal saponin that selectively disrupts the lipid membranes enriched in sterols, it could be hypothesized that CHCHD2 and CHCHD10 are located in a cholesterol-rich compartment in the mitochondrial IM. Most mitochondrial cholesterol is concentrated in specialized structures spanning both the IM and the OM, forming the endoplasmic reticulum (ER)-mitochondrial junctions (41), which are relevant to calcium homeostasis, lipid transport, mtDNA maintenance and mitochondrial dynamics. However, ATD3A, a single-transmembrane IM protein located in the ER-mito junctions was as poorly extracted as COA3, a single-transmembrane IM protein that acts as a COX assembly factor. Therefore, the hydrophobic patches observed in the hydrophobicity plots for CHCHD2 and CHCHD10 (Fig. 9A) and indicated in the models of the protein structures (Fig. 9B) may not be transmembrane helices but protein–protein interacting domains. Based on these considerations, we can propose two models for the potential interactions and localization for CHCHD2, CHCHD10 and p32 (Fig. 9C). In model 1, CHCHD10 and CHCHD2 have membrane-spanning helices and interact with p32 in the matrix at their N-termini. In model 2, CHCHD10 and CHCHD2 have membrane bound regions, but both ends face the IMS, and the interaction with p32 occurs through additional, still unknown, IM proteins.
Figure 9.

Proposed models of protein-protein interactions in the mitochondrial IM. (A) Kyte and Doolitle hydrophobicity plot of D2 and D10 obtained with the ProtScale server (58). (B) Structure of D2 and D10 modeled with RaptorX structure prediction server (59). The putative transmembrane helices were predicted using the TMPred server (60). The cysteine residues forming the twin CX9C motifs in CHCHD2 and CHCHD10 are indicated. (C) Hypothetical models of the interaction of CHCHD2 and CHCHD10 with the IM. The schematic incorporates a simulation of the interaction between D2 (green) and D10 (blue) obtained with GRAMMX (Vakser.compbio.ku.edu) and the structure of p32 (PDB # 1P32) from Jiang et al. (40).
Although we did not find evidence that modulating the expression of CHCHD10 or CHCHD2 affects the levels of p32 in cells, we cannot yet exclude that it may affect its localization and function. This possibility is complicated by the multiplicity of proposed functional roles of p32, which range from complement response in immunity to apoptosis. Interestingly, mutations in p32 have been recently associated with multisystem mitochondrial diseases with variable presentation, ranging from infantile lactic acidosis to childhood (cardio)myopathy and late-onset progressive external ophthalmoplegia (42).
Our Co-IP and co-sedimentation studies of CHCHD10 binding partners revealed interactions with CHCHD2 and p32 in stronger ionic detergent conditions, and a more labile interaction with mitofilin when milder detergent was used, suggesting the possibility that CHCHD10 and CHCHD2 may be involved in multiple protein complexes. Unbiased co-IP and proteomics studies will help to identify potential additional players.
Putative functions of CHCHD10
Relative to other mitochondrial membrane proteins, CHCHD10 and CHCHD2 are rapidly turned over, with a half-life of a few hours. This observation does not support a structural role for either protein. Instead, it suggests that they could serve as chaperones, either for protein import, similar to other CXnC Tim proteins (43,44) or for metal transport, such as the CXnC proteins involved in copper transport and respiratory chain complex assembly (21,22). It is worth noting that CHCHD10 does not have specific metal binding motifs, beyond the structural cysteines. However, it is possible that it interacts with iron transporters to modulate their action, similarly to what has been shown for COX19 acting on COX11 for copper trafficking (45). In addition or alternatively, CHCHD10 and CHCHD2 could be involved in signaling processes, as suggested by the nuclear translocation of CHCHD2 under hypoxic conditions to regulate COX isoforms (27).
Mitochondrial iron accumulation occurs in several neurodegenerative diseases, including Parkinson’s disease, and Alzheimer’s disease (46), and our findings that CHCHD10 silencing modulates mitochondrial iron homeostasis suggest that CHCHD10 could be involved in regulating iron transport in mitochondria. Future structural and functional studies of the interactions of CHCHD10 with putative mitochondrial iron chaperones and transporters are warranted to better delineate the role that CHCHD10 plays in mitochondrial iron homeostasis.
In vivo tissue distribution of CHCHD10
The expression of CHCHD10 in mouse tissues appears to be similar to what has been previously shown in human tissue, with higher expression in heart and liver (1). However, since both CHCHD10 and CHCHD2 mutations are associated with neurodegeneration, we were interested in better exploring CHCHD10 expression in the central nervous system. We demonstrated that CHCHD10 in the brain is localized in specific cell types, especially the TH-expressing dopaminergic neurons of the substantia nigra, which is the vulnerable region in Parkinson’s disease. Furthermore, spinal cord motor neurons, the cells that are most affected in ALS, also showed abundant CHCHD10 expression. Therefore, the cell-type specificity of protein expression in the nervous system could explain the disease phenotypes associated with CHCHD10 mutations.
CHCHD10 loss of function versus gain of toxic function in vitro and in vivo
Here, we investigated cellular models of CHCHD10 downregulation and a mouse model of CHCHD10KO. While there was no mitochondrial bioenergetic dysfunction in immortalized cells, such as HEK293, in which the protein was silenced, CHCHD10 silencing in primary human fibroblasts caused mild mitochondrial abnormalities, suggesting that there could be a cell type specificity involving compensatory processes for the loss of CHCHD10.
CHCHD10KO mice do not display symptoms reminiscent of human neurodegenerative diseases, at least until 200 days of age, the latest time at which we have observed their gross phenotype during breeding and handling for experiments. Furthermore, in brain, muscle, and heart of the CHCHD10KO mouse we did not detect the mitochondrial abnormalities that were described in mutant CHCHD10 patient fibroblasts (28). These mice will have to be monitored for longer time, as age-related phenotypes may arise in the future. The data presented here suggest that CHCHD10 does not play a major role in maintenance of mitochondrial structure in vivo.
It was proposed that CHCHD10 has a protective role in maintaining mitochondrial function and synaptic integrity, and the disease-associated mutations display a loss of function phenotype in both mammalian systems and C. elegans, and accumulation of TDP-43 in the cytoplasm (47). However, the mutations may not cause a loss of function, especially because most of the familial forms of ALS, FTD, and myopathy reported were autosomal dominant with variable penetrance and phenotypes. Therefore, it is likely that the mutants act by disrupting mitochondrial functions and structure in a manner independent from the normal function of CHCHD10, as has been shown for the well-characterized forms of ALS caused by autosomal dominant SOD1 mutations (48).
In summary, this study provides novel insight into CHCHD10 interactions with the mitochondrial proteins CHCHD2 and p32. It also generated and characterized novel genetic models of CHCHD10 modulation in vitro and in vivo. In the future, these findings will be extended to investigating other potential mitochondrial partners of CHCHD10 to better elucidate its function. Moreover, in vivo models of pathogenic CHCHD10 mutations will have to be studied to better understand the mechanisms underlying CHCHD10-associated ALS and related diseases.
Materials and Methods
Cell culture
HEK293 and HeLa cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies) supplemented with 5% fetal bovine serum (Atlanta Biologicals) and 1% antibiotic-antimycotic (Life Technologies). Human skin fibroblasts from a healthy individual were cultured for 5 days in glucose medium (GLU) or in galactose medium (GAL), containing 10% dialyzed FBS, 10 mM galactose and 1% sodium pyruvate. Cells were treated with cycloheximide (50 μg/ml) and MG132 (10 μm) to assess protein stability.
Immunocytochemistry
Coverslips were washed with PBS, fixed in 4% paraformaldehyde for 20 min and washed with 20 mM glycine in PBS (PBS-glycine). Next, cells were permeabilized with 0.1% Triton X-100, washed with PBS-glycine, and blocked in 0.5% BSA in PBS-glycine. Coverslips were incubated in primary antibody for 45 min at 37 °C, washed in PBS glycine, and incubated in secondary antibody (Cy2 or Cy3, Jackson ImmunoResearch) for 45 min at 37 °C. Primary antibodies were used for CHCHD10 (Sigma), CHCHD2 (Proteintech), Myc (Abcam), cytochrome c (BioLegend), and Tom20 (SantaCruz FL-145). Coverslips were washed with PBS-glycine and mounted using Fluoromount-G (SouthernBiotech). To verify the specificity of the CHCHD10 and CHCHD2 antibodies, immunocytochemistry was performed in HeLa cells transfected with siRNA against CHCHD10 and CHCHD2 or scrambled siRNA (Supplementary Material, Fig. S2A and B).
For the quantification of mitochondrial morphology, human skin fibroblasts were stained with cytochrome c antibody. Images from ten random fields per coverslip were acquired using a Leica TCS SP5 confocal microscope with an oil-immersion 63X [1.4 (NA)] lens. Mitochondrial morphology was measured using ImagePro software (49,50). For CHCHD10 and Tom20 colocalization, ten random fields were imaged and the colocalization of the two fluorochromes was assessed using the colocalization function in MetaMorph.
Co-immunoprecipitation, western blot and blue native page
To prepare total lysates, cells were harvested in RIPA buffer [50 mM Tris-HCl (pH 7.4)], 50 mM NaCl, 1% Triton-X, 5 mM EDTA, 10 mM Na4P2O7, 50 mM NaF) for 30 min on ice and then centrifuged 13 000g for 10 min at 4 °C to obtain protein supernatants. To prepare the enriched mitochondrial fractions, cells were harvested and homogenized in mannitol-sucrose buffer (225 mM mannitol, 75 mM sucrose, 5 mM HEPES, 1 mM EGTA, 1 mg/ml fatty-acid free BSA, pH 7.4), and centrifuged at 4 °C at 800g for 5 min. Supernatants were centrifuged at 10 000g for 10 min, and pellets washed in BSA-free mannitol-sucrose buffer and then resuspended in mannitol-sucrose buffer. Protein concentrations were quantified using a BCA assay kit (Pierce).
Co-Immunoprecipitation (Co-IP) experiments were performed on HEK293 cell mitochondrial fractions using a co-Immunoprecipitation kit (Pierce), which contains an IP buffer with 1% NP-40. Samples were separated by SDS-PAGE on AnyKD Tris-acrylamide/bis-acrylamide gels (BioRad). Membranes were probed overnight with the following primary antibodies: CHCHD10 (Sigma, HPA003440), CHCHD2 (Proteintech, 19424–1-AP), OXPHOS subunit cocktail (MitoSciences), p32 (Abcam ab24733, Cell Signaling 6502), mitofilin (ThermoFisher, PA3–870), TIMM50 (Abcam ab109527), VDAC1 (Abcam ab14734), Hsp60 (LSBio LS-B2040/15499), ATAD3 (Abcam ab188386), Tim23 (BD Transduction Laboratories 611222), β-actin (Sigma A5316), CMC1 (Sigma HPA043333), COA3 (Sigma HPA031966), SDHA (Abcam Ab14715), LON-P (Abcam ab103809), and SCO1 [previously reported in (51)]. Membranes were incubated with secondary antibodies [anti-mouse or anti-rabbit IgG (H + L)], 1: 10 000, LiCOR), and signal was detected using the Odyssey CLX Imager (LiCOR). Blue native gels were performed as previously described (52).
Submitochondrial localization and topology of CHCHD2 and CHCHD10
The submitochondrial localization and topology of CHCHD2 and CHCHD10 was analysed basically as described previously (53,54) and briefly described below.
Proteinase K protection assay
To prepare mitoplasts by hypotonic swelling of mitochondria, 250 µg were washed once and then resuspended in 200 µl of 20 mM Hepes pH 7.5. In a parallel control sample, mitochondria were disrupted by brief sonication. The samples were incubated 50 min at 4 °C with proteinase K at final 1 µg/ml, and the reaction was stopped with 5 µl of PMSF 0.2 M. Mitoplasts were centrifuged at 15 000g for 15 min at 4 °C and the resulting pellet was resuspended in 80 µl of Laemmli buffer 2X. Samples of sonicated mitochondria were also supplemented with Laemmli buffer. All samples were loaded on an SDS-PAGE and analysed by immunoblotting.
Mitochondrial protein solubility assay
Two hundred µg of mitochondria in 200 µl of STE buffer (0.6 M sorbitol, 20 mM Tris pH 7.5, 1 mM EDTA, 1 mM PMSF) were sonicated for 3 s at intensity 2 using a Virtis Virsonic 100 ultrasonic cell disrupter and then centrifuged at 35 000g for 15 min at 4 °C. The supernatant containing the soluble proteins (S) was removed. The pellet was then resuspended in 100 µl of 200 mM Na2CO3 (pH 11.5), to extract extrinsic proteins, incubated 30 min on ice and centrifuged at 35 000g for 15 min at 4 °C. The pellet was extracted a second time in the same conditions. The 200 µl of supernatant after alkaline carbonate extraction represent the extrinsic proteins loosely associated with membranes (CS). The pellet, containing intrinsic transmembrane proteins, was resuspended in 200 µl STE (CP). Equivalent samples from the different fractions were analysed by immunoblotting.
Purification of mitochondrial outer and inner membranes by sonication and sucrose gradient sedimentation
Mitochondria were swollen, shrunken and sonicated as described (55,56) except that I mM PMSF and a complete EDTA-free protease inhibitor cocktail were included throughout. Basically, three mg of mitochondria isolated from HEK293 cells were suspended in 400 μl of l0 mM Tris-phosphate buffer, pH 7.5 to induce swelling. Following 5 min of incubation on ice, the suspension was diluted with 1V of 1.8 M sucrose containing 2 mM ATP and 2 mM Mg2SO4 to induce shrinking. Incubate 5 min. Following 5 min of incubation on ice, the suspension was sonicated, 3×10 s at intensity 2 using a Virtis Virsonic 100 ultrasonic cell disrupter. The mixture was then centrifuged at 27 000g. for 15 min and the resulting pellet was resuspended in buffer B (5 mM HEPES-KOH, 10 mM KCl, 1 mM MgCl2, 2 mM DTT, pH 7.4). The resuspended pellet and the supernatant fraction from the same centrifugation were separately centrifuged at 140 000g for 1 h. The pellet obtained from the resuspended pellet is the crude inner membrane; the pellet obtained from the supernatant is the crude outer membrane. The crude membrane fractions were each resuspended in 400 μl of buffer B and layered over linear sucrose gradients in buffer B (0.85–1.6 M) and centrifuged for 14–16 h at 95 000g. Fractions were collected from the bottom by puncturing the gradient tubes and analysed by immunoblotting.
Digitonin solubilization of mitochondrial outer membrane
The mitochondrial outer membrane was solubilized basically as described (57). Briefly, aliquots of 500 μg of mitochondria isolated from HEK293 cells were pelleted and re-suspended in 500 μl of mitochondrial isolation (MTiso) buffer containing 3 mM HEPES-KOH (pH 7.4), 210 mM mannitol, 70 mM sucrose, 0.2 mM EGTA and complete Mini EDTA-free protease inhibitor cocktail, supplemented with increasing concentrations of digitonin to reach final protein: digitonin ratios of 4: 1, 2.7: 1, 2.1: 1, 1.3: 1 and 1: 1. The suspension was mixed intensely and kept on ice for 15 min with intermittent 30 s vortexing every 3 min. Subsequently an equal volume of MTiso-buffer was added to stop digitonin extraction and the suspension was centrifuged at 10 000g (10 min, 4 °C) to isolate the pellet containing mostly mitoplast (IM plus matrix) and the supernatant containing solubilized OM and inter-membrane space (IMS) proteins.
Sucrose gradient sedimentation analysis of CHCHD2 and CHCHD10 native molecular weights
Mitochondrial extracts were prepared in various conditions. Purified mitochondria (400 μg) were solubilized in 0.08 ml of extraction buffer (20 mM Tris-HCl, pH 7.4, 100 mM KCl, 1 mM MgSO4 and 0.5 mM PMSF) containing digitonin (0.25 to 1%, at several protein: detergent ratios indicated in the figures), on ice for 15 min. In a different set of experiments, mitochondria (500 μg) were solubilized in 0.392 ml of extraction buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, protease inhibitor cocktail and 0.5 mM PMSF) containing either 1% NP-40 or 2% SDS.
Clarified extracts were obtained by centrifugation at 21, 000g for 15 min at 4 °C, mixed with standard proteins (hemoglobin and lactate dehydrogenase) and applied to a linear 7–20%, 5–15% or 5–10% sucrose gradients prepared in extraction buffer containing 0.1% of the corresponding detergent. After centrifugation for 12 h at 28 000g (7–20% gradient) or 14 h at 30 000g (5–10% and 5–15% gradients) in a Beckman 55Ti rotor, the gradients were fractionated in 12 or 13 equal fractions. Each fraction was assayed for hemoglobin by absorption at 409 nm and for lactate dehydrogenase activity by measuring NADH-dependent conversion of pyruvate to lactate. Subsequently, the proteins contained in each fraction were concentrated by trichloroacetic acid (TCA) precipitation and analysed by immunoblotting.
In silico analyses
Hydropathy analyses (Kyte and Doolitle) of D2 and D10 was performed with the ProtScale server (58). The structures of D2 and D10 were modeled with RaptorX structure prediction server (59). Potential transmembrane helices in D2 and D10 were predicted using the TMPred server (60).
Modulation of protein expression in cultured cells
CHCHD10 silencing was achieved Silencer Select siRNAs (Life Technologies): s53405 (in the 3’ untranslated region of CHCHD10), and the Silencer Select scrambled negative control (#4390846). Transient transfection was performed using Lipofectamine RNAiMAX (ThermoFisher) according to manufacturer’s instructions. HEK293 cell lines with CHCHD10 stable knockdown were created by generating lentiviral particles with the Mission shRNA CHCHD10 silencing construct (Sigma, SHCLNG-NM_213720) and selecting for transduced clones using puromycin (1 μg/ml) in the culture medium. Expression plasmids with c-terminal FLAG tags, CHCHD10-FLAG (Vigene, Cat. CH805072) and CHCHD2-FLAG (Sino Biological Inc., Cat. HG14156-CF), were commercially available. A Myc-tagged WT CHCHD10 expression construct as well as the C122S CHCHCD10-Myc, R15L CHCHD10-Myc, R15L CHCHD10-FLAG, S59L CHCHD10-Myc and S59L CHCHD10-FLAG constructs were generated by plasmid mutagenesis using the Agilent QuickChange II Site-Directed Mutagenesis kit, or by PCR with appropriate primer sets. The N-del CHCHD10-Myc plasmid was generated by DNA synthesis (GenScript). All plasmids were introduced by transfection with Lipofectamine 2000 (ThermoFisher Scientific) according to manufacturer’s instructions.
Mitochondrial functional analyses
Measurement of oxygen consumption
HEK293 cells were trypsinized, washed and resuspended in 100 μl of MS-EGTA buffer. Part of this suspension (3 μl) was used for protein concentration determination and the rest was added to the chamber. O2 consumption was measured at 37 °C using polarographic oxygen sensors in a two-chamber Oxygraph (OROBOROS Instruments, Innsbruck, Austria). After the recording of the endogenous respiration in the presence of pyruvate (5 mM), uncoupled O2 consumption was determined by adding carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, 1 µM). At the end of the experiment, potassium cyanide (KCN, 1 mM) was added to completely inhibit the respiration. Oxygraphic measurements of isolated mitochondria from brain, muscle and heart were performed using specific buffers and substrates: pyruvate (5 mM) and malate (2 mM) in MS-EGTA buffer with 4 mM H2KPO4, 1 mM MgCl2 for brain; pyruvate (5 mM), malate (2 mM) and glutamate (5 mM) in KCl buffer (125 mM KCl, 20 mM HEPES, 4 mM H2KPO4, 1 mM MgCl2, pH 7.2) for muscle; malate (2 mM) and glutamate (5 mM) in MS-EGTA buffer with 4 mM H2KPO4, 1 mM MgCl2 for heart. Mitochondrial proteins (125 µg) were loaded in the chamber in the presence of substrates to determine non-stimulated, State 2 respiration rate. After addition of 0.5 mM ADP, stimulated State 3 respiration rate was assessed. The ATPase inhibitor carboxiactrattilosite (CATR, 1 µM) was then added to inhibit mitochondrial respiration. To determine the uncoupled respiration, 2, 4-Dinitrophenol (DNP, 40 µM) was added at the end of the experiment. Data analysis was performed using the Oroboros O2k DatLab software.
Measurement of ATP synthesis
ATP synthesis in HEK293 digitonin permeabilized cells (1.5 million), or mitochondria freshly isolated from tissues (20–60 μg) was measured using a rapid kinetic luminescence assay, as described in detail previously (61). This assay monitors continuous ATP synthesis using a luciferase-luciferase system. In cells, measurements were performed in buffer containing 5 mM pyruvate and malate and expressed as nmol ATP/min/million cells. In tissue, the data were expressed as nmol ATP/min/mg protein and the following substrates were used: pyruvate (5 mM) and malate (2mM) in brain, pyruvate (5 mM), malate (2 mM) and glutamate (5 mM) in muscle, and malate (2 mM) and glutamate (5 mM) in heart.
Measurement of COX activity
Cytochrome c oxidase (COX) enzymatic activity was measured spectrophotometrically (at 550 nm) using a previously described assay in isolated mitochondria from tissue or from cells permeabilized with n-dodecyl-maltoside (62). Sample was suspended in 10mM KH2PO4 (pH 7.4), and reduced cytochrome c (0.025 mM) was added to the cuvette. The rate of decrease of absorbance was calculated as nmol cytochrome c/min/million cells or mg protein using the extinction coefficient E = 0.0195 x µM−1 × cm−1.
Calcium capacity measurement
Calcium capacity was measured fluorimetrically using the ratiometric probe Fura-FF. In these studies, calcium capacity in cells was measured in buffer containing 125 mM KCl, 20 mM HEPES, 1 mM MgCl2 (pH 7.2), 200 μg/ml fatty-acid free BSA, 4 mM KH2PO4, 0.2 mM ATP, 1 μM rotenone and 5 mM succinate. In brain, mitochondrial calcium capacity was measured using the same buffer and substrates described for brain mitochondrial oxygen consumption experiments above (with 25 μM EGTA, 200 μg/ml fatty-acid free BSA and 0.2 mM ATP). A single 25 μM calcium addition followed by consecutive 10 μM additions were added to 0.15 mg mitochondria in the presence of Fura-FF (0.2 μM, Life Technologies). In all experiments, a final addition of EGTA (1 mM) was added.
Graphite furnace atomic absorption spectrometry
Crude mitochondrial and cytosolic fractions were digested in 50% nitric acid supplemented with 1% digitonin for 2 h at 60 °C. Iron was measured in 20 μl of digested fractions, as well as in the extracellular media (after centrifugation at 1000 g for 5 min) using a graphite furnace atomic absorption spectrophotometer (Perkin Elmer PinAAcle 900z). Values were compared with a standard curve of known iron concentrations (1000 PPM in 2% nitric acid). Final concentrations in the digested fractions were normalized to the amount of protein in each fraction.
Animal studies
CHCHD10 knockout animals in the C57Bl/6J background were generated using CRISPR/Cas9 technology. A mutation (a single nucleotide insertion of an A residue within exon 2, P16fs46ter) in the chchd10 gene and the cas9 nuclease were introduced into the cytoplasm C57BL/6J-derived fertilized eggs, and correctly targeted embryos were transferred to pseudopregnant females. Correctly targeted pups were bred to C57BL/6J (Stock No. 000664) to develop the colony. Heterozygote males and females were bred to obtain CHCHD10KO animals and littermate controls, which were born according to Mendelian ratio.
All experiments were approved by the Institutional Animal Care and Use Committee of the Weill Cornell Medicine and carried out in compliance with the National Institute of Health guidelines for the care and use of laboratory animals.
Mitochondrial isolation from tissue
Isolation and purification of mouse forebrain mitochondria was performed using a Percoll gradient as previously described (63,64). The muscle (gastrocnemius) was mashed with scissors in a petri dish on ice. The tissue was homogenized with a glass-glass homogenizer on ice (30 times loose pestle, 30 times tight pestle) in MS-EGTA BSA buffer (210 mM mannitol, 70 mM sucrose, 1 mM EGTA, 5 mM HEPES, 0.5% fatty acid free BSA). The homogenate was filtered with gauze in order to remove fat and fibrous tissue and then was centrifuged at 600g for 10 min at 4 °C. The supernatant was collected and the pellet was homogenized a second time. The combined supernatants were finally centrifuged at 12 000g for 10 min at 4 °C. Heart mitochondria were prepared by homogenizing tissue with a Teflon-glass drill homogenizer on ice for 1 min in MS-EGTA BSA (0.1%) buffer and spinning at 1000g for 10 min at 4 °C. The supernatant was centrifuged at 8000g for 10 min at 4 °C, and pellet resuspended in MS-EGTA and centrifuged again. The final mitochondrial pellets were resuspended in MS-EGTA without BSA and protein concentration was determined using a BCA Assay Kit (Pierce).
Immunohistochemistry and electron microscopy
Immunohistochemistry in brain tissue sections was performed using a CHCHD10 primary antibody (Sigma) and 3, 3’-diaminobenzidine peroxidase labeling for detection, as described in (65). Immunofluorescence staining was performed in mouse brain and spinal cord tissue sections using primary antibodies against CHCHD10 (Sigma), Tyrosine Hydroxylase (Millipore) and Choline Acetyltransferase (Novus), and fluorescent secondary antibodies (Jackson ImmunoResearch). Briefly, sections were washed in 0.1 M phosphate buffer, blocked in 1% BSA, washed, incubated in primary antibody (in 0.5% BSA with 0.25% Triton-X 100) at 4 °C for two nights, washed, incubated in secondary antibody (in 0.5% BSA with 0.25% Triton-X 100) for 2 h at room temperature, washed, and mounted using Fluoromount G.
Preparation of cell cultures for electron microscopy was as described (66). Perfusion of animals and fixation of tissue for electron microscopy was performed as described (65). An anti-Tyrosine Hydroxylase antibody (Millipore) was used for 3, 3’-diaminobenzidine peroxidase labeling of brain sections prior to embedding for electron microscopy.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
We are grateful to Dr. Werner Koopman for providing the mitochondrial image-analysis software. We thank the Neuroanatomy EM Core in the BMRI.
Conflict of Interest statement. None declared.
Funding
Muscular Dystrophy Association (MDA382033 to GM and MDA381828 to AB), NIH/NHLBI (K99-HL125899 to SMC), American Lung Association Biomedical Research (RG-348928 to SMC) and grant DA08259 to TAM.
References
- 1. Bannwarth S., Ait-El-Mkadem S., Chaussenot A., Genin E.C., Lacas-Gervais S., Fragaki K., Berg-Alonso L., Kageyama Y., Serre V., Moore D.G. (2014) A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain, 137, 2329–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johnson J.O., Glynn S.M., Gibbs J.R., Nalls M.A., Sabatelli M., Restagno G., Drory V.E., Chio A., Rogaeva E., Traynor B.J. (2014) Mutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosis. Brain, 137, e311.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Perrone F., Nguyen H.P., Van Mossevelde S., Moisse M., Sieben A., Santens P., De Bleecker J., Vandenbulcke M., Engelborghs S., Baets J.. et al. (2017) Investigating the role of ALS genes CHCHD10 and TUBA4A in Belgian FTD-ALS spectrum patients. Neurobiol. Aging, 51, 177 e179–177 e116. [DOI] [PubMed] [Google Scholar]
- 4. Dols-Icardo O., Nebot I., Gorostidi A., Ortega-Cubero S., Hernandez I., Rojas-Garcia R., Garcia-Redondo A., Povedano M., Llado A., Alvarez V.. et al. (2015) Analysis of the CHCHD10 gene in patients with frontotemporal dementia and amyotrophic lateral sclerosis from Spain. Brain, 138, e400.. [DOI] [PubMed] [Google Scholar]
- 5. Zhang M., Xi Z., Zinman L., Bruni A.C., Maletta R.G., Curcio S.A., Rainero I., Rubino E., Pinessi L., Nacmias B.. et al. (2015) Mutation analysis of CHCHD10 in different neurodegenerative diseases. Brain, 138, e380.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Muller K., Andersen P.M., Hubers A., Marroquin N., Volk A.E., Danzer K.M., Meitinger T., Ludolph A.C., Strom T.M., Weishaupt J.H. (2014) Two novel mutations in conserved codons indicate that CHCHD10 is a gene associated with motor neuron disease. Brain, 137, e309.. [DOI] [PubMed] [Google Scholar]
- 7. Ronchi D., Riboldi G., Del Bo R., Ticozzi N., Scarlato M., Galimberti D., Corti S., Silani V., Bresolin N., Comi G.P. (2015) CHCHD10 mutations in Italian patients with sporadic amyotrophic lateral sclerosis. Brain, 138, e372.. [DOI] [PubMed] [Google Scholar]
- 8. Jiao B., Xiao T., Hou L., Gu X., Zhou Y., Zhou L., Tang B., Xu J., Shen L. (2016) High prevalence of CHCHD10 mutation in patients with frontotemporal dementia from China. Brain, 139, e21.. [DOI] [PubMed] [Google Scholar]
- 9. Pasanen P., Myllykangas L., Poyhonen M., Kiuru-Enari S., Tienari P.J., Laaksovirta H., Toppila J., Ylikallio E., Tyynismaa H., Auranen M. (2016) Intrafamilial clinical variability in individuals carrying the CHCHD10 mutation Gly66Val. Acta Neurol. Scand., 133, 361–366. [DOI] [PubMed] [Google Scholar]
- 10. Ajroud-Driss S., Fecto F., Ajroud K., Lalani I., Calvo S.E., Mootha V.K., Deng H.X., Siddique N., Tahmoush A.J., Heiman-Patterson T.D.. et al. (2015) Mutation in the novel nuclear-encoded mitochondrial protein CHCHD10 in a family with autosomal dominant mitochondrial myopathy. Neurogenetics, 16, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Penttila S., Jokela M., Bouquin H., Saukkonen A.M., Toivanen J., Udd B. (2015) Late onset spinal motor neuronopathy is caused by mutation in CHCHD10. Ann. Neurol., 77, 163–172. [DOI] [PubMed] [Google Scholar]
- 12. Auranen M., Ylikallio E., Shcherbii M., Paetau A., Kiuru-Enari S., Toppila J.P., Tyynismaa H. (2015) CHCHD10 variant p.(Gly66Val) causes axonal Charcot-Marie-Tooth disease. Neurol. Genet., 1, e1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xiao T., Jiao B., Zhang W., Pan C., Wei J., Liu X., Zhou Y., Zhou L., Tang B., Shen L. (2016) Identification of CHCHD10 mutation in Chinese patients with Alzheimer disease. Mol. Neurobiol., 54,5243–5247. [DOI] [PubMed] [Google Scholar]
- 14. Rubino E., Brusa L., Zhang M., Boschi S., Govone F., Vacca A., Gai A., Pinessi L., Lopiano L., Rogaeva E.. et al. (2017) Genetic analysis of CHCHD2 and CHCHD10 in Italian patients with Parkinson's disease. Neurobiol. Aging, 53, 193.e7–193.e8. [DOI] [PubMed] [Google Scholar]
- 15. Koehler C.M., Tienson H.L. (2009) Redox regulation of protein folding in the mitochondrial intermembrane space. Biochim. Biophys. Acta, 1793, 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hell K. (2008) The Erv1-Mia40 disulfide relay system in the intermembrane space of mitochondria. Biochim. Biophys. Acta, 1783, 601–609. [DOI] [PubMed] [Google Scholar]
- 17. Cavallaro G. (2010) Genome-wide analysis of eukaryotic twin CX9C proteins. Mol. Biosyst., 6, 2459–2470. [DOI] [PubMed] [Google Scholar]
- 18. Longen S., Bien M., Bihlmaier K., Kloeppel C., Kauff F., Hammermeister M., Westermann B., Herrmann J.M., Riemer J. (2009) Systematic analysis of the twin cx(9)c protein family. J. Mol. Biol., 393, 356–368. [DOI] [PubMed] [Google Scholar]
- 19. Arnesano F., Balatri E., Banci L., Bertini I., Winge D.R. (2005) Folding studies of Cox17 reveal an important interplay of cysteine oxidation and copper binding. Structure, 13, 713–722. [DOI] [PubMed] [Google Scholar]
- 20. Banci L., Bertini I., Ciofi-Baffoni S., Janicka A., Martinelli M., Kozlowski H., Palumaa P. (2008) A structural-dynamical characterization of human Cox17. J. Biol. Chem., 283, 7912–7920. [DOI] [PubMed] [Google Scholar]
- 21. Horn D., Al-Ali H., Barrientos A. (2008) Cmc1p is a conserved mitochondrial twin CX9C protein involved in cytochrome c oxidase biogenesis. Mol. Cell Biol., 28, 4354–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horn D., Zhou W., Trevisson E., Al-Ali H., Harris T.K., Salviati L., Barrientos A. (2010) The conserved mitochondrial twin Cx9C protein Cmc2 Is a Cmc1 homologue essential for cytochrome c oxidase biogenesis. J. Biol. Chem., 285, 15088–15099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rigby K., Zhang L., Cobine P.A., George G.N., Winge D.R. (2007) Characterization of the cytochrome c oxidase assembly factor Cox19 of Saccharomyces cerevisiae. J. Biol. Chem., 282, 10233–10242. [DOI] [PubMed] [Google Scholar]
- 24. Li H., Ruan Y., Zhang K., Jian F., Hu C., Miao L., Gong L., Sun L., Zhang X., Chen S.. et al. (2016) Mic60/Mitofilin determines MICOS assembly essential for mitochondrial dynamics and mtDNA nucleoid organization. Cell Death Differ., 23, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ding C., Wu Z., Huang L., Wang Y., Xue J., Chen S., Deng Z., Wang L., Song Z., Chen S. (2015) Mitofilin and CHCHD6 physically interact with Sam50 to sustain cristae structure. Sci. Rep., 5, 16064.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bourens M., Dabir D.V., Tienson H.L., Sorokina I., Koehler C.M., Barrientos A. (2012) Role of twin Cys-Xaa9-Cys motif cysteines in mitochondrial import of the cytochrome C oxidase biogenesis factor Cmc1. J. Biol. Chem., 287, 31258–31269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aras S., Bai M., Lee I., Springett R., Huttemann M., Grossman L.I. (2015) MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion, 20, 43–51. [DOI] [PubMed] [Google Scholar]
- 28. Genin E.C., Plutino M., Bannwarth S., Villa E., Cisneros-Barroso E., Roy M., Ortega-Vila B., Fragaki K., Lespinasse F., Pinero-Martos E.. et al. (2016) CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med., 8, 58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Floyd B.J., Wilkerson E.M., Veling M.T., Minogue C.E., Xia C., Beebe E.T., Wrobel R.L., Cho H., Kremer L.S., Alston C.L.. et al. (2016) Mitochondrial protein interaction mapping identifies regulators of respiratory chain function. Mol. Cell, 63, 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wei Y., Vellanki R.N., Coyaud E., Ignatchenko V., Li L., Krieger J.R., Taylor P., Tong J., Pham N.A., Liu G.. et al. (2015) CHCHD2 is coamplified with EGFR in NSCLC and regulates mitochondrial function and cell migration. Mol. Cancer Res., 13, 1119–1129. [DOI] [PubMed] [Google Scholar]
- 31. Geissler A., Chacinska A., Truscott K.N., Wiedemann N., Brandner K., Sickmann A., Meyer H.E., Meisinger C., Pfanner N., Rehling P. (2002) The mitochondrial presequence translocase: an essential role of Tim50 in directing preproteins to the import channel. Cell, 111, 507–518. [DOI] [PubMed] [Google Scholar]
- 32. Yamamoto H., Esaki M., Kanamori T., Tamura Y., Nishikawa S., Endo T. (2002) Tim50 is a subunit of the TIM23 complex that links protein translocation across the outer and inner mitochondrial membranes. Cell, 111, 519–528. [DOI] [PubMed] [Google Scholar]
- 33. Mleczko-Sanecka K., Roche F., da Silva A.R., Call D., D'Alessio F., Ragab A., Lapinski P.E., Ummanni R., Korf U., Oakes C.. et al. (2014) Unbiased RNAi screen for hepcidin regulators links hepcidin suppression to proliferative Ras/RAF and nutrient-dependent mTOR signaling. Blood, 123, 1574–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Collinet C., Stoter M., Bradshaw C.R., Samusik N., Rink J.C., Kenski D., Habermann B., Buchholz F., Henschel R., Mueller M.S.. et al. (2010) Systems survey of endocytosis by multiparametric image analysis. Nature, 464, 243–249.http://dx.doi.org/10.1038/nature08779 [DOI] [PubMed] [Google Scholar]
- 35. Chowdhury A.R., Ghosh I., Datta K. (2008) Excessive reactive oxygen species induces apoptosis in fibroblasts: role of mitochondrially accumulated hyaluronic acid binding protein 1 (HABP1/p32/gC1qR). Exp. Cell Res., 314, 651–667. [DOI] [PubMed] [Google Scholar]
- 36. Starkov A.A. (2010) The molecular identity of the mitochondrial Ca2+ sequestration system. febs J., 277, 3652–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ogaki K., Koga S., Heckman M.G., Fiesel F.C., Ando M., Labbe C., Lorenzo-Betancor O., Moussaud-Lamodiere E.L., Soto-Ortolaza A.I., Walton R.L.. et al. (2015) Mitochondrial targeting sequence variants of the CHCHD2 gene are a risk for Lewy body disorders. Neurology, 85, 2016–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shi C.H., Mao C.Y., Zhang S.Y., Yang J., Song B., Wu P., Zuo C.T., Liu Y.T., Ji Y., Yang Z.H.. et al. (2016) CHCHD2 gene mutations in familial and sporadic Parkinson's disease. Neurobiol. Aging, 38, 217 e219213.. [DOI] [PubMed] [Google Scholar]
- 39. Muta T., Kang D., Kitajima S., Fujiwara T., Hamasaki N. (1997) p32 protein, a splicing factor 2-associated protein, is localized in mitochondrial matrix and is functionally important in maintaining oxidative phosphorylation. J. Biol. Chem., 272, 24363–24370. [DOI] [PubMed] [Google Scholar]
- 40. Jiang J., Zhang Y., Krainer A.R., Xu R.M. (1999) Crystal structure of human p32, a doughnut-shaped acidic mitochondrial matrix protein. Proc. Natl Acad. Sci. U S A, 96, 3572–3577.http://dx.doi.org/10.1073/pnas.96.7.3572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gerhold J.M., Cansiz-Arda S., Lohmus M., Engberg O., Reyes A., van Rennes H., Sanz A., Holt I.J., Cooper H.M., Spelbrink J.N. (2015) Human mitochondrial DNA-protein complexes attach to a cholesterol-rich membrane structure. Sci. Rep., 5, 15292.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Feichtinger R.G., Olahova M., Kishita Y., Garone C., Kremer L.S., Yagi M., Uchiumi T., Jourdain A.A., Thompson K., D'Souza A.R.. et al. (2017) Biallelic C1QBP mutations cause severe neonatal-, childhood-, or later-onset cardiomyopathy associated with combined respiratory-chain deficiencies. Am. J. Hum. Genet., 101, 525–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Curran S.P., Leuenberger D., Schmidt E., Koehler C.M. (2002) The role of the Tim8p-Tim13p complex in a conserved import pathway for mitochondrial polytopic inner membrane proteins. J. Cell Biol., 158, 1017–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mesecke N., Terziyska N., Kozany C., Baumann F., Neupert W., Hell K., Herrmann J.M. (2005) A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell, 121, 1059–1069. [DOI] [PubMed] [Google Scholar]
- 45. Bode M., Woellhaf M.W., Bohnert M., Laan M. v d., Sommer F., Jung M., Zimmermann R., Schroda M., Herrmann J.M. (2015) Redox-regulated dynamic interplay between Cox19 and the copper-binding protein Cox11 in the intermembrane space of mitochondria facilitates biogenesis of cytochrome c oxidase. Mol Biol Cell, 26, 2385–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Horowitz M.P., Greenamyre J.T. (2010) Mitochondrial iron metabolism and its role in neurodegeneration. J. Alzheimers Dis., 20 Suppl 2, S551–5S568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Woo J.-A.A., Liu T., Trotter C., Fang C.C., De Narvaez E., LePochat P., Maslar D., Bukhari A., Zhao X., Deonarine A., Westerheide S.D., Kang D.E. (2017) Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat. Commun., 8, 15558.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Igoudjil A., Magrane J., Fischer L.R., Kim H.J., Hervias I., Dumont M., Cortez C., Glass J.D., Starkov A.A., Manfredi G. (2011) In vivo pathogenic role of mutant SOD1 localized in the mitochondrial intermembrane space. J. Neurosci., 31, 15826–15837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Koopman W.J., Visch H.J., Smeitink J.A., Willems P.H. (2006) Simultaneous quantitative measurement and automated analysis of mitochondrial morphology, mass, potential, and motility in living human skin fibroblasts. Cytometry A, 69A, 1–12. [DOI] [PubMed] [Google Scholar]
- 50. Koopman W.J., Visch H.J., Verkaart S., van den Heuvel L.W., Smeitink J.A., Willems P.H. (2005) Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am. J. Physiol. Cell Physiol., 289, C881–C890. [DOI] [PubMed] [Google Scholar]
- 51. Bourens M., Boulet A., Leary S.C., Barrientos A. (2014) Human COX20 cooperates with SCO1 and SCO2 to mature COX2 and promote the assembly of cytochrome c oxidase. Hum. Mol. Genet., 23, 2901–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Valsecchi F., Monge C., Forkink M., de Groof A.J.C., Benard G., Rossignol R., Swarts H.G., van Emst-de Vries S.E., Rodenburg R.J., Calvaruso M.A.. et al. (2012) Metabolic consequences of NDUFS4 gene deletion in immortalized mouse embryonic fibroblasts. Biochim. Biophys. Acta, 1817, 1925–1936. [DOI] [PubMed] [Google Scholar]
- 53. Bourens M., Barrientos A. (2017) A CMC1-knockout reveals translation-independent control of human mitochondrial Complex IV biogenesis. EMBO Rep., 18,477–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bourens M., Barrientos A. (2017) Human mitochondrial cytochrome c oxidase assembly factor COX18 acts transiently as a membrane insertase within the subunit 2 maturation module. J. Biol. Chem., 22, [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sottocasa G.L., Kuylenstierna B., Ernster L., Bergstrand A. (1967) An electron-transport system associated with the outer membrane of liver mitochondria. A biochemical and morphological study. J. Cell Biol., 32, 415–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Riezman H., Hay R., Gasser S., Daum G., Schneider G., Witte C., Schatz G. (1983) The outer membrane of yeast mitochondria: isolation of outside-out sealed vesicles. embo J., 2, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pallotti F., Lenaz G. (2007) Isolation and subfractionation of mitochondria from animal cells and tissue culture lines. Methods Cell Biol., 80, 3–44. [DOI] [PubMed] [Google Scholar]
- 58. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M.R., Appel R.D., Bairoch A. (2005) Walker J.M. (ed.), In The Proteomics Protocols Handbook. Humana Press, New York, in press., pp. 571–607. [Google Scholar]
- 59. Kallberg M., Wang H., Wang S., Peng J., Wang Z., Lu H., Xu J. (2012) Template-based protein structure modeling using the RaptorX web server. Nat. Protoc., 7, 1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hofmann K., Stoffel W. (1993) TMBASE - A database of membrane spanning protein segments. Biol. Chem. Hoppe-Seyler, 374, 166. [Google Scholar]
- 61. Vives-Bauza C., Yang L., Manfredi G. (2007) Assay of mitochondrial ATP synthesis in animal cells and tissues. Methods Cell Biol., 80, 155–171. [DOI] [PubMed] [Google Scholar]
- 62. Birch-Machin M.A., Turnbull D.M. (2001) Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol., 65, 97–117. [DOI] [PubMed] [Google Scholar]
- 63. Damiano M., Starkov A.A., Petri S., Kipiani K., Kiaei M., Mattiazzi M., Flint Beal M., Manfredi G. (2006) Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J. Neurochem., 96, 1349–1361. [DOI] [PubMed] [Google Scholar]
- 64. Kim H.J., Magrane J., Starkov A.A., Manfredi G. (2012) The mitochondrial calcium regulator cyclophilin D is an essential component of oestrogen-mediated neuroprotection in amyotrophic lateral sclerosis. Brain, 135, 2865–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Milner T.A., Waters E.M., Robinson D.C., Pierce J.P. (2011) Degenerating processes identified by electron microscopic immunocytochemical methods. Methods Mol. Biol., 793, 23–59. [DOI] [PubMed] [Google Scholar]
- 66. Cohen-Gould L. (2013), In Microscopy Today. Microscopy Society of America, in press., pp. 10-12.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.