

Abstract
In Prader-Willi syndrome (PWS), obesity is caused by the disruption of appetite-controlling pathways in the brain. Two PWS candidate genes encode MAGEL2 and necdin, related melanoma antigen proteins that assemble into ubiquitination complexes. Mice lacking Magel2 are obese and lack leptin sensitivity in hypothalamic pro-opiomelanocortin neurons, suggesting dysregulation of leptin receptor (LepR) activity. Hypothalamus from Magel2-null mice had less LepR and altered levels of ubiquitin pathway proteins that regulate LepR processing (Rnf41, Usp8, and Stam1). MAGEL2 increased the cell surface abundance of LepR and decreased their degradation. LepR interacts with necdin, which interacts with MAGEL2, which complexes with RNF41 and USP8. Mutations in the MAGE homology domain of MAGEL2 suppress RNF41 stabilization and prevent the MAGEL2-mediated increase of cell surface LepR. Thus, MAGEL2 and necdin together control LepR sorting and degradation through a dynamic ubiquitin-dependent pathway. Loss of MAGEL2 and necdin may uncouple LepR from ubiquitination pathways, providing a cellular mechanism for obesity in PWS.
Introduction
Prader-Willi syndrome (PWS) and Schaaf-Yang syndrome are neurodevelopmental disorders with overlapping clinical presentations that include hypotonia, intellectual disability/autism spectrum disorder, maladaptive behavior, and endocrine dysfunction (1). Schaaf-Yang syndrome is caused by inactivating mutations in the melanoma antigen L2 (MAGEL2) gene. MAGEL2 mutations have also been identified in children with congenital contractures (arthrogryposis) or Opitz-C syndrome (2–6). Both MAGEL2 and NDN, a second melanoma antigen (MAGE) gene, are inactivated in PWS along with other contiguous genes on human chromosome 15q11-q13. Energy imbalance and endocrine dysfunction are consistently observed in PWS. Increased appetite and weight gain with excessive accumulation of fat mass are found in both disorders, but are more common and more severe in PWS than in Schaaf-Yang syndrome.
Mice lacking Magel2 are obese with high levels of leptin, an adipose-derived hormone that regulates body weight by inhibiting hunger pathways in the brain (7–11). Consistent with their failure to decrease food intake in response to high endogenous leptin levels, Magel2-null mice fail to reduce their food intake when treated with exogenous leptin (12). Magel2-null mice also have endocrine deficits and reduced muscle mass, activity, strength and endurance (7,9,11–19). Loss of Ndn in mice causes neurodevelopmental defects and increased fat mass, suggesting that the encoded necdin protein may likewise be important for leptin response pathways (20–22). MAGEL2 and NDN are expressed at high levels in brain regions that regulate energy intake and expenditure. These data strongly support the hypothesis that a combined loss of MAGEL2 and NDN contributes to obesity in individuals with PWS through appetitive pathways that involve leptin sensing in the brain (23).
Leptin receptor (LepR) activity in hypothalamic neurons is critical for the regulation of appetite and energy balance. The LepR intracellular domain functions through JAK-STAT and phosphatidylinositol 3 kinase (PI3K) signaling pathways (24–26). PI3K activity and adapter proteins like insulin receptor substrate (IRS) and sarcoma homology 2 B adaptor protein 1 (SH2B1) are required for leptin-mediated activation of anorexigenic pro-opiomelanocortin (POMC) neurons (27,28). Leptin-induced activation of anorexigenic POMC neurons in the arcuate nucleus of the hypothalamus is absent in adult Magel2-null mice, suggesting that MAGEL2 is required for one or more leptin-mediated responses that are critical for POMC neuron-mediated suppression of food intake (12,16). LepR activity is also moderated by cell surface receptor abundance (29). LepR is internalized by endocytosis, then transported via endosomal sorting complexes required for transport (ESCRT)-positive endosomes either to lysosomes or to the cell surface. Reversible ubiquitination of LepR regulates its sorting and degradation. The deubiquitinase (DUB) USP8 regulates this trafficking through interactions with tethering complexes that recognize and target endosomes, including the HRS and STAM proteins in the ESCRT-0 complex (30,31). USP8 interacts with, deubiquitinates, and stabilizes the E3 ligase RNF41, increasing its activity (32,33). In turn, RNF41 ubiquitinates and destabilizes USP8, ultimately regulating LepR sorting through effects on ESCRT-0 stability (25,30,34).
In this study, we found that MAGEL2 and necdin link LepR to the USP8-RNF41 ubiquitination complex through the LepR adapter protein IRS4. Moreover, MAGEL2 alters the stability and intracellular localization of LepR, USP8, and RNF41. The abundance of necdin, LepR, Usp8, Rnf41, and ESCRT-0 complex protein Stam1 is altered in the hypothalamus of Magel2-null mice. We propose that loss of Magel2 disrupts the normal equilibrium of LepR cell surface expression, internalization, and degradation. This mechanism likely accounts for leptin resistance, obesity, and infertility in the Magel2-null mouse model of Schaaf-Yang and Prader-Willi syndromes.
Results
Endogenous levels of leptin receptor, Rnf41, Usp8, Stam1 and necdin are altered in the brain of mice with a loss of function of Magel2
Leptin receptors (LepR) are internalized into sorting endosomes, ultimately leading to recycling to the plasma membrane, retromer-mediated recycling to the trans-Golgi network, or lysosomal degradation. The ubiquitin conjugating enzyme RNF41 and deubiquitinating enzyme USP8 regulate LepR sorting through effects on ESCRT-0 stability (25,30,34). MAGEL2 is required in vivo for the leptin-mediated hypothalamic control of energy balance, and for intracellular retromer-mediated transport. We therefore measured levels of key LepR pathway proteins in brain samples from Magel2-null mice (13). Hypothalamic protein samples from Magel2-null mice had less LepR, more Usp8, and less Rnf41 than wildtype (WT) mice (Fig. 1A, B). More Stam1 was detected in both the cortex and hypothalamus of Magel2-null mice (Fig. 1C). These results suggest that Magel2 is required to maintain normal levels of many proteins involved in LepR regulation, including LepR itself, in brain regions important for energy homeostasis. Heterodimerization of MAGE proteins controls their activity, adding to the complexity of MAGE protein function (35–37). Brain tissues (cortex and hypothalamus) from Magel2-null mice had less of the MAGE protein necdin than WT mice (Fig. 1D).
Figure 1.

LepR-RNF41-Usp8-Escrt-0 complex proteins are altered in abundance in brain tissues from Magel2-null mice. Protein lysates were prepared from adult mice that were either wildtype (WT) or mutant for Magel2 and processed for immunoblotting (IB) to detect endogenous proteins. (A) LepR levels are reduced in Magel2-null hypothalamus but not cortex compared to wildtype. (B) Rnf41 protein levels are reduced and Usp8 protein levels are increased in Magel2-null hypothalamus. (C) Stam1 levels are increased in Magel2-null hypothalamus and cortex. Samples in C) were electrophoresed, immunoblotted and imaged on the same gel/blot with intervening lanes removed for clarity. (D) Necdin levels are reduced in Magel2-null cortex and hypothalamus. Graphs represent mean ± SD of signal intensity relative to β-actin signal intensity, from n = 3–6 mice per genotype with representative samples shown. *P < 0.05 difference in abundance between genotypes by Student t-test.
MAGEL2 regulates the surface abundance of the LepR
Receptor signaling depends on cell surface receptor availability, which in turn depends on rates of receptor internalization and degradation. To determine whether MAGEL2 regulates LepR cell surface expression, we generated a HEK293 cell line carrying a stably-integrated, tetracycline-inducible, MAGEL2 expression construct. We then performed cell surface biotinylation assays to measure the abundance of the LepR at the cell surface. After labeling cell surface proteins with an amine-reactive biotinylation reagent, biotinylated proteins were captured by streptavidin affinity purification and the cytosolic, non-biotinylated proteins were also recovered. The amount of endogenous LepR in the cytosolic fraction compared to the total amount of LepR in the cell lysate is a measure of the proportion of LepR that was exposed to the biotinylation reagent at the cell surface. Tetracycline-treated HEK293 cells expressing MAGEL2 had more endogenous LepR at the cell surface than uninduced HEK293 cells (26 ± 8%, compared to 11 ± 4%, P = 0.04) (Fig. 2A). Biotinylated ‘bound’ LepR was captured in the MAGEL2-expressing cells but was undetectable in uninduced cells. As a control, actin was detected in the total and unbound/cytosolic fractions but not in the bound, biotinylated fraction. As well, MAGEL2-expressing cells had more total LepR than uninduced cells (2.2-fold, P < 0.01).
Figure 2.

MAGEL2 increases the abundance and cell surface localization of the leptin receptor. (A) FLAG-tagged MAGEL2 expression was induced by tetracycline in stably transfected HEK293 cells. The abundance of endogenous LepR was measured relative to β-actin. **Different between uninduced and tetracycline-induced, P < 0.01. The percent cell surface biotinylation of endogenous LepR was derived from the ratio of the ‘total minus unbound’ to ‘total’ LepR. different between uninduced and tetracycline induced, P = 0.04. Immunoblots show results from protein lysates from three wells for each condition [uninduced (U) or treated with tetracycline to induce expression of MAGEL2 (+Tet)]. Graphs represent mean ± SD from three wells of cells. Total and unbound samples were electrophoresed on the same gel and immunoblotted and ‘bound’ samples were electrophoresed on a second gel and immunoblotted. As a control, β-actin was detected in the total and unbound/cytosolic fractions but not in the bound, biotinylated fraction. (B) FLAG-tagged MAGEL2p.R1187C or MAGEL2p.LL1031AA expression was induced by tetracycline in stably transfected HEK293 cells and both the abundance of endogenous LepR and the cell surface biotinylation of endogenous LepR were measured in triplicate samples.
We next generated two mutant MAGEL2 expression constructs carrying mutations in the MAGE homology domain (MHD, pfam01454), a protein-protein interaction domain that contains two tandem winged helix motifs (38). These mutations were modeled after mutations generated for studies of related MAGE proteins (Supplementary Material, Fig. S1). A pathogenic missense mutation in MAGED2 (p.R446C) was identified in a patient with antenatal kidney disease (39). The corresponding arginine in the MAGEL2 protein (R1187) is highly conserved among mammalian MAGEL2 proteins, and the mutation MAGEL2p.R1187C is predicted to be very deleterious by the bioinformatics tool PROVEAN (Protein Variation Effect Analyzer) (40). A second MAGEL2 mutation, p.LL1031AA, replaces a highly conserved dileucine motif with two alanines, analogous to an engineered mutation in the MAGEG1 protein that disrupts its ability to bind to NSE1 (41). In contrast to the effect of wildtype (WT) MAGEL2 on LepR cell surface abundance, there was no change in either proportion of LepR at the cell surface or the total amount of LepR in cells expressing MAGEL2p.R1187C or MAGEL2p.LL1031AA (Fig. 2B). Biotinylated LepR was not detected in the affinity-purified ‘bound’ samples from cells expressing mutant MAGEL2 constructs (not shown). Thus, the integrity of the MHD is required for MAGEL2 to promote cell surface exposure of LepR in cultured cells.
MAGEL2 regulates the subcellular localization and lysosomal degradation of leptin receptor
We next evaluated the intracellular distribution of LepR by transient transfection of C-terminally tagged LepR, in the presence or absence of epitope tagged RNF41 or MAGEL2 into U2OS cells, followed by immunofluorescence confocal microscopy. LepR adopts a dispersed pattern throughout the cell, with minimal co-localization with early endosomes (labeled with anti-EEA1, Fig. 3A) or lysosomes (labeled with anti-LAMP1, Fig. 3B). The majority of cells expressing LepR also expressed the other co-transfected proteins in immunofluorescence microscopy on slides processed in parallel (not shown). Consistent with the cell surface biotinylation assay, expression of MAGEL2 or co-expression of RNF41 and MAGEL2 appeared to increase the localization of LepR at the cell surface. Lysosomal targeting can be detected by the appearance of degradation products of the receptor, the C-terminal stubs (CTS). Further lysosomal degradation of CTS is inhibited by incubation of the cells in chloroquine, so that in the presence of chloroquine, LepR CTS formation reflects lysosomal degradation (30). MAGEL2 reduced the formation of the LepR CTS in the presence of chloroquine (Fig. 3C). RNF41 blocks the formation of the LepR CTS through a USP8-dependent mechanism that involves the destabilization of the ESCRT-0 complex components Hrs, Stam1 and Stam2 (34). MAGEL2 reduced endogenous Stam1 levels, consistent with the ability of MAGEL2 to reduce lysosomal degradation of LepR (Fig. 3D) and with the increase in Stam1 in Magel2-null mouse brain (Fig. 1C).
Figure 3.

Localization and degradation of LepR. (A) Recombinant C-terminally tagged LepR (red) and endogenous early endosomes (anti-EEA1, green) were localized in transiently co-transfected U2OS cells by immunofluorescence confocal microscopy. Localization of LepR was also visualized in cells co-transfected with RNF41 (+R), MAGEL2 (+M) or both RNF41 and MAGEL2 (+R + M). Nuclei were counterstained blue with Hoechst dye. (B) Recombinant LepR (red) and lysosomes (anti-LAMP1, green) were localized by immunofluorescence microscopy. Localization of LepR was also visualized in cells co-transfected with RNF41, MAGEL2, or both RNF41 and MAGEL2. (C) Protein lysates from HEK293T cells transiently co-transfected with LepR and MAGEL2 were immunoblotted to detect full length (FL) recombinant leptin receptor and C-Terminal Stubs (CTS) derived from C-terminally HA-epitope tagged recombinant LepR. Cells were cultured overnight in the absence (-) or presence (+) of 25 μM chloroquine. n.s. indicates a protein that non-specifically cross-reacts with the anti-HA antibody, present in all lanes. (D) FLAG-MAGEL2 was transfected into HEK293T ells and endogenous Stam1 detected with an anti-Stam1 antibody.
In vivo identification of proteins interacting with MAGEL2 and necdin
MAGEL2 interacts with USP7 and TRIM27 to regulate retromer-mediated recycling through WASH ubiquitination (42,43). Necdin interacts with a related E3 ligase, TRIM28 (41). We therefore investigated protein-protein interactions among MAGEL2, necdin, and components of the LepR trafficking system. We initially used heterologous expression of epitope-tagged MAGEL2 or necdin in cultured cells followed by immunoaffinity purification to measure protein-protein interactions. However, these studies were limited by the poor solubility of MAGEL2 under the mild cell lysis conditions used for immunoprecipitation. Consequently, we used two systems that detect protein-protein interactions in intact mammalian cells. In the BioID system, a bait cDNA is fused in frame with biotin ligase (BirA) to produce a fusion protein that biotinylates adjacent proteins, which are isolated by denaturing affinity capture on streptavidin resin, then identified by immunoblotting (44,45). MAPPIT (mammalian protein-protein interaction trap) is a ‘two-hybrid’ technique whereby recombinant cytokine receptors are expressed in cultured cells to identify in vivo interactions among proteins. Interaction of a bait (chimeric with a signaling-deficient receptor) and prey (fused to a functional cytokine receptor domain) restores receptor signaling, causing transcription of a reporter gene (30,46). We refer to the relationship between bait and prey proteins as protein-protein interactions, while recognizing that these approaches do not evaluate whether putative interactions are direct, indirect, transient, or stable.
We used different MAPPIT bait receptors (BR) in HEK293T cells to examine interactions between necdin or MAGEL2 and proteins important for leptin receptor function. These bait proteins include the leptin receptor itself, its adapter protein IRS4, ubiquitination enzymes USP8 and RNF41, and VPS52, a component of both the Golgi-associated retrograde protein (GARP) and endosome associated recycling protein (EARP) tethering complexes that interacts with RNF41 (30,46) (Fig. 4A). In contrast to MAGEL2, necdin interacts with the LepR intracellular tail, independent of the bait protein attached to it (BR1-empty). Necdin does not interact with bait receptors in which the largest part of the LepR tail is deleted (BR2-empty and BR3-FKBP12) (Fig. 4B). Necdin recruitment to LepR was narrowed down to between amino acids E1041-L1092, which includes Y1077, a tyrosine residue important for LepR signaling through STAT5 (25). This 51 aa region spans the IRS4 recruitment site. The interaction with necdin is independent of phosphorylation at Y1077, because necdin still interacts with LepR carrying the mutation F1077 (Fig. 4C). Interestingly, MAGEL2, and to a lesser extent necdin, interact with IRS4 (Fig. 4C). RNF41 also interacts with IRS4, and IRS4 co-expression increases the interaction between RNF41 and MAGEL2 (Fig. 4D). The RNF41 interaction partners, USP8 and VPS52, strongly interact with MAGEL2. Co-expression of either of these proteins (USP8 Fig. 4E or VPS52, Fig. 4F), increases the interaction between RNF41 and MAGEL2. In summary, necdin interacts with LepR at the IRS4 binding site. IRS4, as well as USP8 and VPS52, interact with both RNF41 and MAGEL2. Thus, MAGEL2 and necdin could act together to facilitate an interaction between the leptin receptor and its adapter proteins, and the ubiquitiylation complex that contains USP8 and RNF41.
Figure 4.

Necdin interacts with LepR and IRS4, while USP8 and VPS52 bridge RNF41 to MAGEL2, by MAPPIT. (A) Schematic representation of the MAPPIT chimeric bait receptor (BR) constructs used. For all MAPPIT experiments, HEK293T cells transfected with the indicated bait, prey and co-expression (50 or 250 ng) constructs were left untreated or stimulated with Epo (BR1 and BR3) or leptin (BR2). Luciferase data of triplicate measurements from a representative experiment are expressed as fold induction (stimulated/non-stimulated) ± SD. FLAG-tagged prey and BR expression was verified by immunoblotting and interaction with a LepR binding prey, respectively. (B) NDN interacts with the LepR tail present in BR1, but absent in BR2 or BR3 constructs. (C) NDN recruitment to LepR was narrowed down to between amino acids E1041-L1092 using BR3 carrying truncated regions of the LepR intracellular tail as bait protein. The NDN prey interacts with a construct carrying wildtype LepR amino acids 986-1137 (LR Y1077), as well as the same construct with a mutation at Y1077 that prevents phosphorylation of this tyrosine residue (LR F1077). MAGEL2, and to a lesser extent NDN, interact with BR3-IRS4. IRS4 (D), USP8 (E) and VPS52 (F) interact with both RNF41 and MAGEL2 and their co-expression increases RNF41 interaction with MAGEL2. Asterisks indicate non-specific bands.
Necdin and MAGEL2 connect the leptin receptor to USP8-RNF41
To further elucidate interactions among the proteins tested by MAPPIT, we performed proximity-dependent biotin identification (BioID) in U2OS cells transiently co-transfected with a FLAG-tagged biotin ligase-fusion protein (BirA-NDN or BirA-MAGEL2), and a second epitope-tagged protein as prey (Fig. 5). After affinity purification, streptavidin-bound biotinylated proteins were recovered and immunoblotted to detect proteins in proximity to the BirA-fusion baits in vivo. BirA-NDN biotinylated both LepR and MAGEL2, but we did not detect an interaction between BirA-NDN and either RNF41 or USP8 by BioID (Fig. 5A). Consistent with previous studies that identified a MAGEL2-TRIM27-USP7 complex, BirA-MAGEL2 biotinylated both TRIM27 and USP7 (Fig. 5B and C). BirA-MAGEL2 also biotinylated USP8 (Fig. 5B and C), consistent with the MAPPIT results (Fig. 4E). The biotinylation of RNF41 by BirA-MAGEL2 could be detected in the presence of co-transfected TRIM27, USP8 or LepR (Fig. 5D), but not in the absence of these other proteins (Fig. 5C). BirA-MAGEL2 also biotinylated NDN (not shown). The combined MAPPIT and BioID results demonstrate a set of interactions that begin at the intracellular domain of the LepR (residues E1041-L1092), the site to which both IRS4 and necdin bind. Necdin interacts with MAGEL2, which interacts with USP8 and RNF41, which interacts with ESCRT-0 and VPS52. These data support a role for necdin and MAGEL2 in the regulation of the leptin receptor, in cooperation with RNF41 and USP8.
Figure 5.

Necdin and MAGEL2 link the leptin receptor to USP8 and RNF41 as detected by proximity-dependent biotinylation (BioID). U2OS cells were transiently co-transfected with cDNA constructs encoding epitope-tagged proteins and incubated with excess biotin. After 24 h, cell lysates were collected, a sample retained as ‘input’ and the remainder processed by streptavidin affinity purification to recover proteins biotinylated by BirA fusion proteins (bound). Input and bound samples were immunoblotted (IB) to detect recombinant proteins. Representative experiments are shown. (A) BirA-FLAG-NDN (N) was co-transfected with V5-tagged RNF41 (R), USP8 (U), LepR (L) or MAGEL2 (M). (B) BirA-FLAG-MAGEL2 (M) was co-transfected with FLAG-tagged USP7 or USP8 (U) and bound proteins detected by immunoblotting. (C) BirA-FLAG-MAGEL2 (M) was co-transfected with V5-tagged TRIM27 (T), RNF41 (R), USP8 (U), or with KANK4 (K), which was used as a non-interacting negative control. (D) BirA-FLAG-MAGEL2 was co-transfected with Etag-tagged RNF41 (R) in the presence of V5-tagged LepR (L), USP8 (U) or TRIM27 (T).
MAGEL2 regulates the stability of USP8 and RNF41
RNF41 ubiquitinates USP8 to reduce USP8 levels (32,34). Conversely, USP8 stabilizes RNF41. USP8 also stabilizes RNF41AE, which can no longer bind USP8, and stabilizes an RNF41 variant that lacks E3 ligase activity (RNF41SQ, RNF41p.[(C34S; H36Q)]) (Fig. 6). Interestingly, co-expression of MAGEL2 reversed the stabilizing effect of USP8 on RNF41 and its mutant forms. MAGEL2 reduced the abundance of USP8 (Fig. 7A) and increased the abundance of RNF41 (Fig. 7B). To determine if MAGEL2 stabilizes RNF41 through a process that involves ubiquitination, we co-transfected constructs encoding V5-MAGEL2, FLAG-RNF41, and HA-tagged ubiquitin. Immunoprecipitation of RNF41 with anti-FLAG antibodies, followed by immunoblotting for HA (ubiquitin) revealed that RNF41 is ubiquitinated (Fig. 7C). MAGEL2 reduced the extent of RNF41 ubiquitination, suggesting that MAGEL2 stabilizes RNF41 by either diminishing its auto-ubiquitination or by increasing de-ubiquitination by endogenous DUBs. Last, co-expression of MAGEL2 significantly extended the half-life of RNF41 from 87 to 179 min in a cycloheximide chase assay (2-way repeated measures ANOVA, F(1, 28)=20.16, n = 3, P = 0.0001, Supplementary Material, Fig. S2). Together, these experiments demonstrate that MAGEL2 increases RNF41 abundance by increasing its stability.
Figure 6.

MAGEL2 reverses the stabilization of RNF41 conferred by USP8. U2OS cells were transiently co-transfected with the indicated cDNAs. RNF41WT is a higher molecular weight because of the Etag epitope tag, whereas the RNF41 mutant constructs RNF41AE (binding to USP8 disabled) and RNF41SQ (E3 ligase activity disabled) are not epitope-tagged. Cell lysates were collected 24 h after transfection and subjected to immunoblotting (IB) to detect RNF41. Graph represents mean ± SD of the RNF41 signal intensity relative to β-actin signal intensity, from three wells of transfected cells for each set of constructs. *P < 0.05 difference in RNF41 abundance with or without USP8 by Student t-test. A representative experiment is shown.
Figure 7.

MAGEL2, RNF41, and USP8 abundance is reciprocally regulated. (A) Protein lysates from transiently co-transfected U2OS cells were immunoblotted (IB) to detect recombinant proteins and the abundance of USP8 quantified. Graph represents mean ± SD of USP8 signal intensity relative to β-actin signal intensity, from three wells of co-transfected cells for each set of constructs. (B) Graph represents mean ± SD of RNF41 signal intensity relative to β-actin signal intensity, from three wells of co-transfected cells for each set of constructs. (C) U2OS cells were transiently co-transfected with FLAG-RNF41, HA-tagged ubiquitin, and V5-MAGEL2. After retaining a sample as input, cell lysates were subjected to immunoprecipitation with mouse anti-FLAG antibodies to recover FLAG-RNF41. Immunoprecipitated (IP) material was immunoblotted to detect HA-ubiquitin (with anti-HA) or RNF41 (with anti-RNF41).
USP8 and RNF41 alter the abundance of MAGEL2
The abundance of MAGEL2 also varied depending on whether either RNF41 or USP8 was heterologously co-expressed (Fig. 8). Co-expression of wildtype RNF41 (RNF41WT) or RNF41AE, but not co-expression of RNF41SQ, lowered MAGEL2 protein levels (Fig. 8A). This suggests that the ability of RNF41 to destabilize MAGEL2 requires ubiquitination activity. Conversely, USP8 increased the abundance of MAGEL2 (Fig. 8B). In summary, MAGEL2, RNF41, and USP8 reciprocally regulate protein stability.
Figure 8.

MAGEL2 alters USP8 and RNF41 abundance. (A) FLAG-MAGEL2 was co-transfected with RNF41WT, RNF41AE, RNF41SQ or empty vector (-) and FLAG-MAGEL2 signal intensity was measured. Anti-RNF41 detects both endogenous and recombinant RNF41. (B) FLAG-MAGEL2 was co-transfected with V5-USP8 or empty vector (-) and FLAG-MAGEL2 signal intensity was measured. (C) Mutant FLAG-MAGEL2 constructs carrying missense mutations in the MHD [MAGEL2p.LL1031AA (LLAA) or MAGEL2p.R1187C (RC)] were co-transfected with Etag-RNF41 or empty vector (-) and MAGEL2 signal intensity was measured. (D) Etag-RNF41 was co-transfected with mutant FLAG-MAGEL2 constructs carrying missense mutations in the MHD or with empty vector (-) and the RNF41 signal intensity was measured. All graphs represent mean±SD relative to β-actin signal intensity, from three wells of transfected cells for each set of constructs. *P < 0.05 difference in abundance with or without co-expressed protein by Student t-test. Representative experiments are shown.
Mutations that disrupt the MAGE homology domain (MHD) of MAGEL2 interfere with the mutual effect of RNF41 and MAGEL2 on each other‘s stability
We next used MAGEL2 expression constructs carrying mutations in the MHD that abrogate the promotion of LepR cell surface expression by MAGEL2 (Fig. 2B, Supplementary Material, Fig. S1). Both mutant FLAG-tagged MAGEL2 constructs produced correctly sized recombinant proteins at similar levels to MAGEL2WT when transiently transfected (Fig. 8C, D). RNF41 lowers levels of MAGEL2WT (Fig. 8A), but increases levels of MAGEL2p.LL1031AA and MAGEL2p.R1187C (Fig. 8C). Likewise, while MAGEL2WT increases levels of RNF41 (Fig. 7B), co-expression of either MAGEL2 mutant construct reduced RNF41 levels (Fig. 8D). These results demonstrate that MAGEL2 MHD defects interfere with the mutual effect of RNF41 and MAGEL2 on each other‘s stability.
MAGEL2 also altered the subcellular localization of USP8-RNF41 (Fig. 9). In transfected U2OS cells, MAGEL2 is mostly dispersed in the cytoplasm, while RNF41 adopts a more punctate pattern throughout the cytoplasm (Fig. 9A). Co-expression of MAGEL2 with RNF41 relocalized pools of both proteins to perinuclear structures (examples in Fig. 9A). USP8 is located diffusely throughout the cytoplasm with some protein located at the plasma membrane (Fig. 9B). Consistent with previous results, co-expression of RNF41 relocalized USP8 from a diffuse cytoplasmic location to large intracellular vesicles (33,34). This relocalization was blocked by co-expression of MAGEL2 (Fig. 9B). In summary, MAGEL2 reverses the stabilizing effect of USP8 on RNF41 and reverses the ability of RNF41 to relocalize USP8 to intracellular vesicles.
Figure 9.

RNF41 and MAGEL2 modulate the subcellular localization of USP8. (A) Recombinant RNF41 (red) and MAGEL2 (green) were detected in either singly transfected (single) or co-transfected (double, two examples) U2OS cells by immunofluorescence microscopy 24 h after transfection. (B) Recombinant USP8 (green) was detected in transfected U2OS cells. Middle: RNF41 (R, red) was co-expressed with USP8 (U, green) in double transfected cells. Bottom: USP8 (U, green) was co-expressed with RNF41 (R, not labeled) and MAGEL2 (M, red) in triple transfected cells. Nuclei are counterstained blue with Hoechst dye. Representative cells are shown. Scale bar, 10 µm.
Discussion
Inactivating mutations in MAGEL2 cause a complex neurodevelopmental disorder called Schaaf-Yang syndrome (2–5), while inactivation of both MAGEL2 and NDN along with other genes causes Prader-Willi syndrome. Concurrent loss of MAGEL2 and necdin proteins in children with PWS could alter the cell surface abundance and activity of the leptin receptor and possibly other receptors important for growth, development and homeostasis, providing a unifying theory for the physiological basis for hyperphagia, obesity, maladaptive behavior and other endophenotypes in this clinically complex disorder. Notably, loss of Magel2 in mice causes a physiological and neuronal phenotype similar to that caused by deficiencies in the LepR-SH2B1-insulin receptor substrate-PI3K axis (12,16). This axis is essential for the leptin-mediated activation of POMC anorexigenic neurons in the arcuate nucleus of the hypothalamus (47). Inactivation of SH2B1 in humans or in mice causes severe early onset hyperphagia and obesity. Loss of IRS2 in LepR-positive neurons causes obesity, and inhibition of PI3K precludes the normal leptin-induced activation of POMC neurons. Our data suggest that MAGEL2 and necdin regulate the activity of LepR through a ubiquitin-dependent mechanism, by modifying LepR cell surface abundance, by binding LepR adapter proteins that couple the receptor to intracellular signaling pathways, and by regulating receptor internalization, intracellular trafficking and degradation.
MAGE proteins assemble into multi-protein complexes with E3 RING ubiquitin ligases to regulate cellular processes (38,48). Heterodimerization of MAGE proteins controls their activity, adding to the complexity of MAGE protein function (35–37). MAGEL2 interacts with the DUB USP7 and the E3 ligase TRIM27 to regulate the ubiquitination of the actin nucleating protein WASH, forming a complex involved in retromer-mediated protein sorting (42,43). Other MAGE proteins interact with USP8-regulated receptors: necdin binds auto-phosphorylated EGFR through its tyrosine kinase domain to inhibit binding of SH2/SH3 adapter protein GRB2 and control signaling in primary cortical progenitor cells (49), while necdin, MAGEH1, MAGEG1, and MAGED1 (Dlxin-1) interact with the p75/TRKA neurotrophin receptors in signaling endosomes and enhance nerve growth factor induced differentiation (37,50–52). Thus, MAGE proteins modulate intracellular signaling and, in some cases, prevent receptor degradation after endocytosis during retrograde signaling.
We reasoned that MAGEL2 could be required for leptin receptor-mediated activation of POMC neurons through a pathway involving USP8 and RNF41 that regulates trafficking of LepR. Consistent with this, inactivation of Magel2 in mice changes levels of endogenous proteins in the leptin receptor recycling pathway. Magel2-null hypothalamus samples have less LepR, Rnf41 and necdin and more Usp8 and Stam1 than wildtype hypothalamus; Magel2-null cortex has less necdin and more Stam1 than cortex from wildtype mice. The effect of loss of Magel2 on levels of its interacting proteins could be age-dependent, as another model of Magel2-deficiency was found to have increased necdin in the hypothalamus, but at age P0 rather than in the adult (53). Usp8 promotes lysosomal degradation, so increased Usp8 and reduced Rnf41 (which increases Usp8) would enhance lysosomal degradation of cytokine receptors (Fig. 10). This imbalance could reduce the availability of functional leptin receptors by reducing receptor level and the proportion at the cell surface. This would account for leptin-dependent phenotypes in Magel2 mutant mice, in particular the defects in hypothalamic POMC neurons (12,16). This hypothesis provides a possible mechanism to explain leptin resistance, obesity and other leptin-regulated phenotypes in Magel2-deficient mice, such as reduced activity, infertility, defective glucose regulation, and reduced bone mass.
Figure 10.
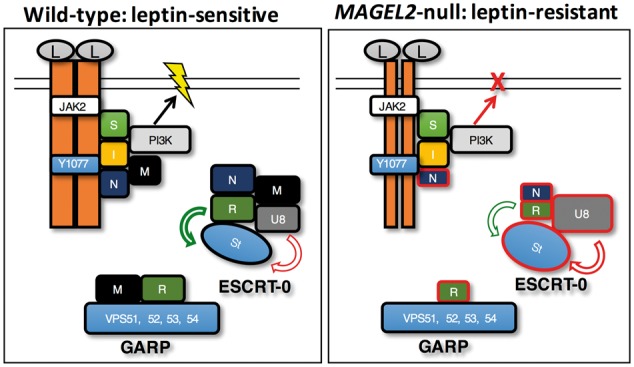
Model for MAGEL2 regulation of leptin receptor processing and effect of loss of MAGEL2. The various complexes in which MAGEL2 participates are shown together with a representation of the leptin receptor and associated proteins (left). Inactivation of MAGEL2 occurs in people with Prader-Willi syndrome, Schaaf-Yang syndrome, or with forms of arthrogryposis or Opitz-C syndrome caused by MAGEL2 mutations, or in Magel2-null gene-targeted mice. Loss of MAGEL2 function cause changes in the abundance and ubiquitination of proteins in the leptin receptor internalization pathway, with affected proteins outlined in red (right). Proteins are labelled as follows: leptin (L), leptin receptors (orange bars, reduced at cell surface in Magel2-null state) with critical residue tyrosine Y1077, SH2B1 (S), IRS4 (I), necdin (N), MAGEL2 (M), phosphatidylinositol 3 kinase (PI3K), TRIM27 (T), USP7 (U7), RNF41 (R), USP8 (U8), Stam1 (St). Yellow lightning symbol indicates neuronal activation.
Interestingly, POMC neurons in Magel2-null mice activate in response to leptin in mice analyzed up to 4 weeks after birth but are unresponsive in adult animals (16). Likewise, after a period of normal weight gain, children with PWS slowly gain excess fat mass, then develop excessive hunger and obesity at a median age of 4.5 years (54). In contrast, children with genetic mutations that ablate leptin receptor signaling entirely typically become obese before a year of age. We hypothesize that in PWS, loss of MAGEL2 and necdin impair leptin receptor processing, but that energy homeostasis can be maintained until a functional threshold for PI3K signaling in POMC neurons is reached. At this point, key hypothalamic neurons may no longer be able to respond adequately to endogenous leptin in children with PWS, causing hyperphagia and severe obesity. Alternatively, MAGEL2 may only be required for leptin receptor recycling in mature neurons, accounting for the progressive nature of obesity and hyperphagia in PWS.
Autophagy defects in POMC hypothalamic neurons and muscle in Magel2–null mice could be linked to changes in Usp8 abundance or activity, as autophagy is deregulated in Usp8-depleted cells (55,56). USP8 also regulates the activity of other receptors important for neuronal function, such as the neurotrophin receptor TrkA (57), vascular endothelial growth factor receptor (58), and epidermal growth factor receptor EGFR (59). Indeed, MAGEL2 and necdin could regulate the activity of receptors other than the leptin receptor. Loss of MAGEL2, with concurrent loss of necdin in PWS, could affect the activity of other receptor tyrosine kinases, such as insulin, neurotrophin, and EGF receptors or of G-protein-coupled receptors such as oxytocin, cannabinoid, dopamine, melanocortin, and ghrelin receptors (37,49,50,52,60–62). In support of this hypothesis, mice lacking Magel2 have impaired function of many of these receptor pathways (7,8,17,18,63–65).
The balance between the activity of USP8 and RNF41 defines the cell surface expression and trafficking of cytokine receptors, including leptin receptors (34). However, the mechanism by which the USP8-RNF41 ubiquitination complex is recruited to the receptor on sorting endosomes remained unclear. Our identification of necdin and MAGEL2 as adapter proteins bridging the intracellular tail of the leptin receptor through the IRS4 adapter protein to the USP8-RNF41-ESCRT-0 complex rationalizes the role of necdin and MAGEL2 in leptin receptor regulation (Fig. 10). MAGEL2 promotes deubiquitination and stabilizes RNF41, and destabilizes USP8 and STAM1. MAGEL2 interferes with the ability of RNF41 to relocalize USP8 to large intracellular vesicles and reduces the stabilization of RNF41 by USP8. MAGEL2 also increases the abundance of LepR and its retention at the cell surface. Like RNF41 (34), MAGEL2 inhibits the formation of the LepR CTS, a marker of lysosomal degradation. Some of these effects require MAGE homology domain integrity, because MAGEL2 variants with MHD mutations no longer act like MAGEL2. Mutants do not stabilize RNF41 nor do they promote cell surface localization of endogenous LepR in cultured cells. The abundance and subcellular localization of many of the proteins in this system appear to be acutely sensitive to the levels of their cognate interacting proteins. It will be important to investigate how these various effects and interactions that we outline change with over-expression or reduced expression of additional members of this complex system.
Intriguingly, faulty trafficking and reduced signaling of the LepR is proposed to be the mechanism for obesity in the genetic neurodevelopmental disorder, Bardet-Biedl syndrome (BBS). In BBS, disease arises secondary to loss of components of the BBSome, a protein trafficking complex with which MAGEL2 and necdin proteins interact (66,67). Thus, a variety of genetic mutations can impact POMC neurons causing pediatric obesity: loss of function of leptin, leptin receptor, or leptin receptor adaptor proteins (28), deficiency of POMC or the POMC processing enzyme proprotein convertase 1/3 (68), and a more recently recognized mechanism impacting leptin receptor trafficking (BBS proteins in Bardet-Biedl syndrome and MAGEL2/necdin in Prader-Willi syndrome). It remains to be seen whether mutations in pediatric obesity syndrome genes encoding intracellular trafficking or ubiquitination proteins, such as CUL4B (69), PHIP (70), and VPS13B/COH1 (71), likewise impair leptin receptor trafficking in POMC neurons. Interestingly, intellectual disability and hypotonia are found in the syndromes associated with this last category of genes, suggesting roles in the trafficking of proteins in other tissues beyond the brain, thus giving rise to pleiotropic phenotypes.
Materials and Methods
Plasmids
RNF41 constructs [RNF41WT (wildtype, pMET7-Etag-hRNF41), RNF41SQ (carrying C34S and H36Q mutations that inactivate the RING E3 ligase domain), RNF41AE (carrying Q266A and R269E mutations that abolish binding to USP8)], a leptin receptor construct (LepR-HA), and a USP8 construct (FLAG-USP8) were previously described (30,34). HA-Ub was expressed from pRK5-HA-Ubiquitin-WT, a gift from Ted Dawson (Addgene plasmid # 17608) (72). For the remaining constructs, cDNAs in entry vectors from the ORFeome consortium were obtained from the DNASU plasmid repository (73) and transferred into mammalian expression vectors by Gateway cloning. pcDNA3.1/nV5-DEST or pcDNA3.2/V5-DEST were used to generate V5-RNF41, V5-LepR, V5-USP8, V5-TRIM27, V5-MAGEL2, and V5-KANK4. pDEST-pcDNA5-FLAG Destination vectors were generously provided by Dr. A-C Gingras (74) and used to generate FLAG-MAGEL2 and FLAG-USP7. A modified pCI-based vector was used to generate HA-MAGEL2. BirA-MAGEL2-FLAG and BirA-NDN-FLAG were created in pDEST-pcDNA5-BirA-FLAG, generously provided by Dr. A-C. Gingras (74). pENTR clones carrying MAGEL2p.LL1031AA and MAGEL2p.R1187C were created in the wildtype pENTR-MAGEL2 cDNA (DNASU Clone: HsCD00295122) by site-directed mutagenesis, confirmed by sequencing and then transferred to pDEST-pcDNA5-FLAG. Appropriate empty vector plasmids were used to equalize the amount of DNA transfected for co-transfections. Details of plasmid construction are available on request.
MAPPIT method and plasmids
The MAPPIT bait receptor plasmids BR1-empty (pSEL1-empty), BR3-FKBP12, BR3-LR Y1077, BR3-LR Y1077F, BR3-IRS4, BR1-RNF41, BR1-USP8, and the MAPPIT empty, IRS4 and USP8 prey constructs as well as FLAG-IRS4, FLAG-USP8 and FLAG-VPS52 co-expression constructs were previously described (25,46). BR2-empty was generated by BamHI/NotI digest of the pCLG-MyD88 vector (75), which was blunt ended using Pfu polymerase and self-ligated. The mouse LepR motifs 986-1092 and 986-1040 were amplified by PCR with forward primer TGCGAGCTCAGCAACTCTGGTCAGCAATG and reverse primers ATCGCGGCCGCCTACAAAAGCACACCACTCTCTCTTC and ATCGCGGCCGCCTAGTCTGATAAAAGGAAAAATGTCTGG respectively. The LepR motif 1041-1092 was amplified by PCR with forward primer TGCGAGCTCACAGCAACCCATCATGATTTCACC and the same reverse primer as for LepR motif 986-1092. Forward primers contained a SacI site, while reverse primers contained a NotI site and a stop codon, allowing in-frame coupling to the BR3 chimeric bait receptor. BR1-VPS52 and VPS52, NDN, MAGEL2 prey constructs were generated via an LR reaction (Invitrogen) to transfer the VPS52 gene from a Gateway entry clone of the human ORFeome v5.1 collection or the NDN and MAGEL2 cDNAs to a previously described pSEL1 bait (76) or pMG1 prey destination vector (77). For MAPPIT experiments, HEK293T cells were seeded at 1 × 104 cells/well in 96 well plates and 24 h later transfected using the calcium phosphate method with 250 ng bait receptor plasmid, 250 ng prey plasmid and 50 ng of the STAT3-dependent pXP2d2-rPAPI luciferase reporter plasmid. 24 h after transfection, cells were left untreated or stimulated with human Epo (5 ng/ml) or mouse leptin (100 ng/ml). Luciferase activity from triplicate samples was measured by chemiluminescence in an EnVision luminometer (PerkinElmer) and expressed as fold induction (stimulated/non-stimulated relative light units). All luciferase data shown are based on at least three independent experiments (n = 3).
Cell lines and transfections
Human osteosarcoma cells (U2OS cells) and human embryonic kidney cells (HEK293T cells) were cultured in Dulbecco‘s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 1% l-glutamine and 1% penicillin/streptomycin at 37 °C with 5% CO2. Tissue culture reagents were from Thermo-Fisher Scientific. U2OS cells were seeded at 4 × 105 cells/well in 6 well plates, 24 h before transfection of recombinant constructs encoding epitope-tagged proteins using Effectene (Qiagen cat#301425). HEK293T cells were seeded at 2 × 105 cells/well in a 6 well plate, 24 h before transfection using FuGENE6 (Promega E2691). Lysates were collected 24 h after transfection. To measure protein half-life, cells were treated with cycloheximide (final concentration 10 μg/ml) and lysates recovered at specific time intervals after treatment. Protein levels were quantified by ImageJ, normalized to levels of β-actin, and plotted versus the collection times. Decay curves were fit to the data points and the protein half-life was determined using the equation of the line.
Immunoblotting and immunofluorescence
Preparation of cell protein lysates and immunoblotting was performed essentially as described (66). Primary antibodies were: rabbit anti-HA (Santa Cruz sc-805, 1:500), rabbit anti-FLAG (Sigma F7425 1:1000), rabbit anti-RNF41 (Bethyl A300-048 A, 1:5000), rabbit anti-USP8 (Bethyl A302-929 A, 1:2000), rabbit anti-V5 (Millipore AB3792, 1:3000), mouse anti-V5 (Sigma MA5-15253, 1:3000), rabbit anti-STAM1 (Santa Cruz H-175, 1:2000), mouse anti-necdin (Sigma AB9372, 1:2000), rabbit anti-LepR (Abcam ab104403, 1:500), mouse anti-β-actin (Sigma A2228, 1:50 000). Secondary antibodies were: HRP-linked donkey anti-rabbit (Amersham 1:5000) and HRP-linked sheep anti-mouse (Amersham 1:5000). Signals on immunoblots were visualized on a Kodak Image station, quantified using ImageJ and reported as a ratio of protein signal pixel intensity to signal intensity of β-actin. A representative set of results from thrice replicated experiments is presented. Processing of U2OS cells plated on coverslips for immunofluorescence microscopy was performed essentially as described (66). Primary antibodies were as above: rabbit anti-HA (1:500), rabbit anti-FLAG (1:500), rabbit anti-RNF41 (1:5000), rabbit anti-USP8 (1:500) mouse anti-V5 (1:500), and mouse anti-LAMP1 (Abcam ab25630, 1:500), mouse anti-EEA1 (Abcam ab70521, 1:500), mouse anti-RNF41 (Santa Cruz A-6, 1: 500) and appropriate Alexa Fluor 594- or 488-linked secondary antibodies from Life Technologies. Additional slides were processed in parallel to confirm that the large majority of cells expressing one recombinant protein also expressed the other recombinant protein(s) in co-transfected cells.
BioID
U2OS cells were seeded at 3 × 105 cells/well in 6 well plates, with 2 wells seeded per BioID experiment. After transfection, biotin was added to a final concentration of 50 µM. 24 h after transfection, media was removed from the plates and the cells were washed twice with 3 ml of PBS per well to remove excess biotin. Lysis buffer [400 μl, 50 mM Tris HCl (pH 7.5), 500 mM NaCl, 0.2% SDS, 2% Triton-X, 1 mM DTT with Complete Mini Protease Inhibitor (Roche 11836153001)] was added to the plates, which were then incubated at room temperature for 10 min on a rocker. Cell lysates were transferred to 1.5 ml eppendorf tubes, sonicated on ice (3 times for 5 s with 5 s pauses) and centrifuged at 16 500×g for 10 min at 4 °C. Supernatants were filtered using Amicon Ultra Centrifugal units (Millipore # UFC500324) and 30 μl was retained (input). The remaining filtrate was added to pre-equilibrated streptavidin sepharose beads (GE product code: 17-5113-01) and incubated at 4 °C overnight on a rotator. Samples were centrifuged at 800×g at RT for 2 min and the supernatants discarded. The beads were then incubated at room temperature for 8 min in three sequential wash buffers: wash buffer 1 (2% SDS), wash buffer 2 (0.1% deoxycholic acid, 1% Triton X-100, 1 mM EDTA, 500 mM NaCl, 50 mM HEPES, pH 7.5), wash buffer 3 (0.5% deoxycholic acid, 0.5% NP-40, 1 mM EDTA, 250 mM LiCl, 10 mM Tris Cl, pH 7.4). The tubes were centrifuged at 800×g at RT for 2 min, then beads were resuspended in 30 μl 50 mM Tris Cl and mixed with 30 μl 2× modified sample buffer (20% glycerol, 4% SDS, 0.13 M Tris, pH 7.8) with beta-mercaptoethanol, and 5 μl of 1 mM biotin. Supernatants were then used for immunoblot analysis (bound).
Ubiquitination assay
HEK293T cells were seeded at 8 × 105 cells/well in 100 mm dishes. 24 h after transfection, cells were incubated overnight with 25 µM chloroquine and 5 µM MG 132 in serum-free OPTIMEM media (Life Technologies). The media was removed and the cells were washed with PBS. 250 µl of modified lysis buffer (2% SDS, 150 mM NaCl, 10 mM Tris-HCl, pH 8.0, 1 mM sodium orthovanadate, 1 mM sodium fluoride, 20 mM β glycerophosphate phosphatase, 10 mM N-ethylmaleimide and Complete Mini Protease Inhibitor) was added to each well, followed by a 30 min incubation on a rocker at room temperature to lyse the cells. Lysates were collected into 1.5 ml eppendorf tubes and sonicated on ice (3 times for 5 s with 5 s pauses), then boiled for 10 min. After removing 40 μl (input), the remaining lysate was incubated with 2250 µl dilution buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100) for 1 h in a rotator at 4 °C, centrifuged at 20 000×g for 30 min and pre-cleared by incubating with 20 μl Sepharose 4B beads (Sigma) for 1 h at 4 °C on a rocker. After centrifugation, the supernatant was incubated with 30 μl of anti-FLAG M2 Affinity Gel (Sigma) at 4 °C on a rocker overnight. The beads were recovered by centrifugation, washed twice with wash buffer (10 mM Tris-HCl, pH 8.0, 1 M NaCl, 1 mM EDTA, 1% NP-40), then used for immunoblot analysis.
Cell surface biotinylation
Stable cell lines expressing BirA-MAGEL2WT-FLAG, BirA-MAGEL2p.R1187C-FLAG or BirA-MAGEL2p.LL1081AA-FLAG were created in HEK293 T-Rex Flp-In cells (Invitrogen) by selection in 200 μg/ml hygromycin B. In these cells, single copy integration is mediated by the Flp recombinase at a unique recognition target site. To induce expression of MAGEL2, cells were incubated in 1 μg/ml tetracycline for 24 h with control (uninduced) cells not treated with tetracycline. Cells were then incubated with 1 mg/ml Sulfo-NHS- SS-Biotin* (Pierce) in borate buffer (10 mM boric acid, 145 mM NaCl, 7.2 mM KCl, 1.8 mM CaCl2, pH 9) for 15 min at 4 °C. Biotinylation solution was renewed for another 15 min incubation. Excess biotin was quenched with 100 mM glycine in borate buffer, then the cells were washed twice with 1 ml of cold PBS. The cells were lysed in 300 μl TNT lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.2% SDS, and Complete Mini Protease Inhibitor) for 20 min at 4 °C. Ten percent of the lysate was retained as total lysate and the remaining 90% was incubated with 100 μl Streptavidin Sepharose affinity chromatography beads (GE product code: 17-5113-01) for 1 h at 4 °C on a rotator. After centrifugation, the supernatant (cytosolic fraction) was removed and mixed 1: 1 with 2× modified sample buffer. The beads were washed three times with 1 ml of TNT lysis buffer. Cell surface proteins were eluted with 50 μl 2× modified sample buffer that contained 15% β-mercaptoethanol. After immunoblotting, the LepR signal intensity from the cytosolic fraction was compared to the signal intensity from the total lysate to infer the percentage of LepR at cell surface (biotinylated).
Mouse brain tissues
All animal studies were conducted in accordance with the Canadian Council on Animal Care Guidelines and Policies with approval from the Animal Care and Use Committee: Health Sciences for the University of Alberta. Adult Magel2+m/-p (Magel2-null) and control littermate Magel2+m/+p mice [Jackson Laboratories C57BL/6-Magel2tm1Stw/J, stock 009062 (13)] were genotyped by PCR of ear notch biopsies at weaning (9). Mice were euthanized and brains removed then the cortex and hypothalamus were dissected and snap frozen in liquid nitrogen. The tissues were ground using pre-chilled disposable plastic pestles and resuspended in 500 µl of 2× modified sample buffer with Complete Mini Protease Inhibitor. The tissues were sonicated on ice (3× for 5 s each with 5 s pauses), incubated at 65 °C for 5 min, and centrifuged 10 min at 20 000×g at room temperature, and supernatants retained for immunoblot analysis.
Statistics
Continuous data are presented as mean ± SD of 3-4 replicates per experiment. Differences between means were evaluated using a Student t-test and considered significant if P <0.05. The half-life of RNF41 with and without co-expression of MAGEL2 was compared by 2-way repeated measures ANOVA with time and expression as variables.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We thank Jocelyn Bischof, Herman Cortes and Kelly Fagan for technical assistance and advice and Dr. Emmanuelle Cordat for assistance with the cell surface biotinylation assay.
Conflict of Interest statement. None declared.
Funding
This work was supported by the Canadian Institutes of Health Research (grant number MOP 130367 to RW); and by a European Research Council Advanced Grant (grant CYRE, N°340941 to JT). TMW was funded in part by a Graduate Studentship from the Faculty of Medicine and Dentistry, University of Alberta. MRS was funded by a NSERC Canada Graduate Scholarships-Master's Program. KVC had generous support from the Stollery Children's Hospital Foundation through a Women and Children‘s Health Research Institute Graduate Studentship.
References
- 1. Irizarry K.A., Miller M., Freemark M., Haqq A.M. (2016) Prader Willi syndrome: genetics, metabolomics, hormonal function, and new approaches to therapy. Adv. Pediatr., 63, 47–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Soden S.E., Saunders C.J., Willig L.K., Farrow E.G., Smith L.D., Petrikin J.E., LePichon J.B., Miller N.A., Thiffault I., Dinwiddie D.L.. et al. (2014) Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci. Transl. Med., 6, 265ra168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mejlachowicz D., Nolent F., Maluenda J., Ranjatoelina-Randrianaivo H., Giuliano F., Gut I., Sternberg D., Laquerriere A., Melki J. (2015) Truncating mutations of MAGEL2, a gene within the Prader-Willi locus, are responsible for severe arthrogryposis. Am. J. Hum. Genet., 97, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schaaf C.P., Gonzalez-Garay M.L., Xia F., Potocki L., Gripp K.W., Zhang B., Peters B.A., McElwain M.A., Drmanac R., Beaudet A.L.. et al. (2013) Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat. Genet., 45, 1405–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fountain M.D., Aten E., Cho M.T., Juusola J., Walkiewicz M.A., Ray J.W., Xia F., Yang Y., Graham B.H., Bacino C.A.. et al. (2017) The phenotypic spectrum of Schaaf-Yang syndrome: 18 new affected individuals from 14 families. Genet. Med., 19, 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Urreizti R., Cueto-Gonzalez A.M., Franco-Valls H., Mort-Farre S., Roca-Ayats N., Ponomarenko J., Cozzuto L., Company C., Bosio M., Ossowski S.. et al. (2017) A de novo nonsense mutation in MAGEL2 in a patient initially diagnosed as Opitz-C: similarities between Schaaf-Yang and Opitz-C syndromes. Sci. Rep., 7, 44138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Igarashi M., Narayanaswami V., Kimonis V., Galassetti P.M., Oveisi F., Jung K.M., Piomelli D. (2017) Dysfunctional oleoylethanolamide signaling in a mouse model of Prader-Willi syndrome. Pharmacol Res., 117, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Knani I., Earley B.J., Udi S., Nemirovski A., Hadar R., Gammal A., Cinar R., Hirsch H.J., Pollak Y., Gross I.. et al. (2016) Targeting the endocannabinoid/CB1 receptor system for treating obesity in Prader-Willi syndrome. Mol. Metab., 5, 1187–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bischof J.M., Stewart C.L., Wevrick R. (2007) Inactivation of the mouse Magel2 gene results in growth abnormalities similar to Prader-Willi syndrome. Hum. Mol. Genet., 16, 2713–2719. [DOI] [PubMed] [Google Scholar]
- 10. Bischof J.M., Van Der Ploeg L.H., Colmers W.F., Wevrick R. (2016) Magel2-null mice are hyper-responsive to setmelanotide, a melanocortin 4 receptor agonist. Br. J. Pharmacol., 173, 2614–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arble D.M., Pressler J.W., Sorrell J., Wevrick R., Sandoval D.A. (2016) Sleeve gastrectomy leads to weight loss in the Magel2 knockout mouse. Surg. Obes. Relat. Dis., 12, 1795–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mercer R.E., Michaelson S.D., Chee M.J.S., Atallah T.A., Wevrick R., Colmers W.F., Yeo G.S.H. (2013) Magel2 is required for leptin-mediated depolarization of POMC neurons in the hypothalamic arcuate nucleus in mice. PLoS Genet., 9, e1003207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kozlov S.V., Bogenpohl J.W., Howell M.P., Wevrick R., Panda S., Hogenesch J.B., Muglia L.J., Van Gelder R.N., Herzog E.D., Stewart C.L. (2007) The imprinted gene Magel2 regulates normal circadian output. Nat. Genet., 39, 1266–1272. [DOI] [PubMed] [Google Scholar]
- 14. Mercer R.E., Kwolek E.M., Bischof J.M., van Eede M., Henkelman R.M., Wevrick R. (2009) Regionally reduced brain volume, altered serotonin neurochemistry, and abnormal behavior in mice null for the circadian rhythm output gene Magel2. Am. J. Med. Genet. B Neuropsychiatric Genet. Am. J. Med. Genet., 150B, 1085–1099. [DOI] [PubMed] [Google Scholar]
- 15. Mercer R.E., Wevrick R. (2009) Loss of magel2, a candidate gene for features of Prader-Willi syndrome, impairs reproductive function in mice. PloS One, 4, e4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pravdivyi I., Ballanyi K., Colmers W.F., Wevrick R. (2015) Progressive postnatal decline in leptin sensitivity of arcuate hypothalamic neurons in the Magel2-null mouse model of Prader-Willi syndrome. Hum. Mol. Genet., 24, 4276–4283. [DOI] [PubMed] [Google Scholar]
- 17. Tennese A.A., Wevrick R. (2011) Impaired hypothalamic regulation of endocrine function and delayed counterregulatory response to hypoglycemia in Magel2-null mice. Endocrinology, 152, 967–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maillard J., Park S., Croizier S., Vanacker C., Cook J.H., Prevot V., Tauber M., Bouret S.G. (2016) Loss of Magel2 impairs the development of hypothalamic Anorexigenic circuits. Hum. Mol. Genet., 25, 3208–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fountain M.D., Tao H., Chen C.A., Yin J., Schaaf C.P. (2017) Magel2 knockout mice manifest altered social phenotypes and a deficit in preference for social novelty. Genes Brain Behav., 16, 592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pagliardini S., Ren J., Wevrick R., Greer J.J. (2005) Developmental abnormalities of neuronal structure and function in prenatal mice lacking the prader-willi syndrome gene necdin. Am. J. Pathol., 167, 175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bush J.R., Wevrick R. (2012) Loss of the Prader-Willi obesity syndrome protein necdin promotes adipogenesis. Gene, 497, 45–51. [DOI] [PubMed] [Google Scholar]
- 22. Fujiwara K., Hasegawa K., Ohkumo T., Miyoshi H., Tseng Y.H., Yoshikawa K. (2012) Necdin controls proliferation of white adipocyte progenitor cells. PloS One, 7, e30948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Colmers W.F., Wevrick R. (2013) Leptin signaling defects in a mouse model of Prader-Willi syndrome: An orphan genetic obesity syndrome no more? Rare Dis., 1, e24421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Allison M.B., Myers M.G. Jr. (2014) 20 years of leptin: connecting leptin signaling to biological function. J. Endocrinol., 223, T25–T35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wauman J., De Smet A.S., Catteeuw D., Belsham D., Tavernier J. (2008) Insulin receptor substrate 4 couples the leptin receptor to multiple signaling pathways. Mol. Endocrinol., 22, 965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ren D., Li M., Duan C., Rui L. (2005) Identification of SH2-B as a key regulator of leptin sensitivity, energy balance, and body weight in mice. Cell Metab., 2, 95–104. [DOI] [PubMed] [Google Scholar]
- 27. Hill J.W., Williams K.W., Ye C., Luo J., Balthasar N., Coppari R., Cowley M.A., Cantley L.C., Lowell B.B., Elmquist J.K. (2008) Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J. Clin. Invest., 118, 1796–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Doche M.E., Bochukova E.G., Su H.-W., Pearce L.R., Keogh J.M., Henning E., Cline J.M., Saeed S., Dale A., Cheetham T.. et al. (2012) Human SH2B1 mutations are associated with maladaptive behaviors and obesity. J. Clin. Invest., 122, 4732–4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Belouzard S., Rouille Y. (2006) Ubiquitylation of leptin receptor OB-Ra regulates its clathrin-mediated endocytosis. EMBO J., 25, 932–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wauman J., De Ceuninck L., Vanderroost N., Lievens S., Tavernier J. (2011) RNF41 (Nrdp1) controls type 1 cytokine receptor degradation and ectodomain shedding. J. Cell Sci., 124, 921–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Clague M.J., Urbe S. (2006) Endocytosis: the DUB version. Trends Cell Biol., 16, 551–559. [DOI] [PubMed] [Google Scholar]
- 32. Wu X., Yen L., Irwin L., Sweeney C., Carraway K.L. 3rd (2004) Stabilization of the E3 ubiquitin ligase Nrdp1 by the deubiquitinating enzyme USP8. Mol. Cell. Biol., 24, 7748–7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cao Z., Wu X., Yen L., Sweeney C., Carraway K.L. 3rd (2007) Neuregulin-induced ErbB3 downregulation is mediated by a protein stability cascade involving the E3 ubiquitin ligase Nrdp1. Mol. Cell. Biol., 27, 2180–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. De Ceuninck L., Wauman J., Masschaele D., Peelman F., Tavernier J. (2013) Reciprocal cross-regulation between RNF41 and USP8 controls cytokine receptor sorting and processing. J. Cell Sci., 126, 3770–3781. [DOI] [PubMed] [Google Scholar]
- 35. Kuwajima T., Taniura H., Nishimura I., Yoshikawa K. (2004) Necdin interacts with the Msx2 homeodomain protein via MAGE-D1 to promote myogenic differentiation of C2C12 cells. J. Biol. Chem., 279, 40484–40493. [DOI] [PubMed] [Google Scholar]
- 36. Lavi-Itzkovitz A., Tcherpakov M., Levy Z., Itzkovitz S., Muscatelli F., Fainzilber M., Ouzounis C.A. (2012) Functional consequences of necdin nucleocytoplasmic localization. PloS One, 7, e33786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tcherpakov M., Bronfman F.C., Conticello S.G., Vaskovsky A., Levy Z., Niinobe M., Yoshikawa K., Arenas E., Fainzilber M. (2002) The p75 neurotrophin receptor interacts with multiple MAGE proteins. J. Biol. Chem., 277, 49101–49104. [DOI] [PubMed] [Google Scholar]
- 38. Lee A.K., Potts P.R. (2017) A comprehensive guide to the MAGE family of ubiquitin ligases. J. Mol. Biol., 429, 1114–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Laghmani K., Beck B.B., Yang S.-S., Seaayfan E., Wenzel A., Reusch B., Vitzthum H., Priem D., Demaretz S., Bergmann K.. et al. (2016) Polyhydramnios, transient Antenatal Bartter's syndrome, and MAGED2 mutations. N. Engl. J. Med., 374, 1853–1863. [DOI] [PubMed] [Google Scholar]
- 40. Choi Y., Chan A.P. (2015) PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics, 31, 2745–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Doyle J.M., Gao J., Wang J., Yang M., Potts P.R. (2010) MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol. Cell, 39, 963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hao Y.H., Doyle J.M., Ramanathan S., Gomez T.S., Jia D., Xu M., Chen Z.J., Billadeau D.D., Rosen M.K., Potts P.R. (2013) Regulation of WASH-dependent actin polymerization and protein trafficking by ubiquitination. Cell, 152, 1051–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hao Y.H., Fountain M.D. Jr., Fon Tacer K., Xia F., Bi W., Kang S.H., Patel A., Rosenfeld J.A., Le Caignec C., Isidor B.. et al. (2015) USP7 acts as a molecular rheostat to promote WASH-dependent endosomal protein recycling and is mutated in a human neurodevelopmental disorder. Mol. Cell, 59, 956–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roux K.J., Kim D.I., Burke B. (2013) BioID: a screen for protein-protein interactions. Curr. Protoc. Protein Sci., 74, Unit 19 23. [DOI] [PubMed] [Google Scholar]
- 45. Lambert J.P., Tucholska M., Go C., Knight J.D., Gingras A.C. (2015) Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J. Proteomics, 118, 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lievens S., Van der Heyden J., Masschaele D., De Ceuninck L., Petta I., Gupta S., De Puysseleyr V., Vauthier V., Lemmens I., De Clercq D.J.. et al. (2016) Proteome-scale binary interactomics in human cells. Mol. Cell Proteomics, 15, 3624–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Duan C., Li M., Rui L. (2004) SH2-B promotes insulin receptor substrate 1 (IRS1)- and IRS2-mediated activation of the phosphatidylinositol 3-kinase pathway in response to leptin. J. Biol. Chem., 279, 43684–43691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kozakova L., Vondrova L., Stejskal K., Charalabous P., Kolesar P., Lehmann A.R., Uldrijan S., Sanderson C.M., Zdrahal Z., Palecek J.J. (2015) The melanoma-associated antigen 1 (MAGEA1) protein stimulates the E3 ubiquitin-ligase activity of TRIM31 within a TRIM31-MAGEA1-NSE4 complex. Cell Cycle, 14, 920–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fujimoto I., Hasegawa K., Fujiwara K., Yamada M., Yoshikawa K. (2016) Necdin controls EGFR signaling linked to astrocyte differentiation in primary cortical progenitor cells. Cell Signal, 28, 94–107. [DOI] [PubMed] [Google Scholar]
- 50. Bronfman F.C., Tcherpakov M., Jovin T.M., Fainzilber M. (2003) Ligand-induced internalization of the p75 neurotrophin receptor: a slow route to the signaling endosome. J. Neurosci. Off. J. Soc. Neurosci., 23, 3209–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kuwako K., Taniura H., Yoshikawa K. (2004) Necdin-related MAGE proteins differentially interact with the E2F1 transcription factor and the p75 neurotrophin receptor. J. Biol. Chem., 279, 1703–1712. [DOI] [PubMed] [Google Scholar]
- 52. Kuwako K., Hosokawa A., Nishimura I., Uetsuki T., Yamada M., Nada S., Okada M., Yoshikawa K. (2005) Disruption of the paternal necdin gene diminishes TrkA signaling for sensory neuron survival. J. Neurosci. Off. J. Soci. Neurosci., 25, 7090–7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rieusset A., Schaller F., Unmehopa U., Matarazzo V., Watrin F., Linke M., Georges B., Bischof J., Dijkstra F., Bloemsma M.. et al. (2013) Stochastic loss of silencing of the imprinted Ndn/NDN allele, in a mouse model and humans with prader-willi syndrome, has functional consequences. PLoS Genet., 9, e1003752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Miller J.L., Lynn C.H., Driscoll D.C., Goldstone A.P., Gold J.A., Kimonis V., Dykens E., Butler M.G., Shuster J.J., Driscoll D.J. (2011) Nutritional phases in Prader-Willi syndrome. Am. J. Med. Genet. A, 155A, 1040–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kamaludin A.A., Smolarchuk C., Bischof J.M., Eggert R., Greer J.J., Ren J., Lee J.J., Yokota T., Berry F.B., Wevrick R. (2016) Muscle dysfunction caused by loss of Magel2 in a mouse model of Prader-Willi and Schaaf-Yang syndromes. Hum. Mol. Genet., 25, 3798–3809. [DOI] [PubMed] [Google Scholar]
- 56. Jacomin A.C., Bescond A., Soleilhac E., Gallet B., Schoehn G., Fauvarque M.O., Taillebourg E. (2015) The deubiquitinating enzyme UBPY is required for lysosomal biogenesis and productive autophagy in Drosophila. PloS One, 10, e0143078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ceriani M., Amigoni L., D'Aloia A., Berruti G., Martegani E. (2015) The deubiquitinating enzyme UBPy/USP8 interacts with TrkA and inhibits neuronal differentiation in PC12 cells. Exp. Cell Res., 333, 49–59. [DOI] [PubMed] [Google Scholar]
- 58. Smith G.A., Fearnley G.W., Abdul-Zani I., Wheatcroft S.B., Tomlinson D.C., Harrison M.A., Ponnambalam S. (2016) VEGFR2 trafficking, signaling and proteolysis is regulated by the ubiquitin isopeptidase USP8. Traffic, 17, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Berlin I., Schwartz H., Nash P.D. (2010) Regulation of epidermal growth factor receptor ubiquitination and trafficking by the USP8.STAM complex. J. Biol. Chem., 285, 34909–34921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ju H., Lee S., Kang S., Kim S.S., Ghil S. (2014) The alpha subunit of Go modulates cell proliferation and differentiation through interactions with Necdin. Cell Commun. Signal, 12, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ingraham C.A., Wertalik L., Schor N.F. (2011) Necdin and neurotrophin receptors: interactors of relevance for neuronal resistance to oxidant stress. Pediatr. Res., 69, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bush J.R., Wevrick R. (2010) Loss of Necdin impairs myosin activation and delays cell polarization. Genesis, 48, 540–553. [DOI] [PubMed] [Google Scholar]
- 63. Luck C., Vitaterna M.H., Wevrick R. (2016) Dopamine pathway imbalance in mice lacking Magel2, a Prader-Willi syndrome candidate gene. Behav. Neurosci., 130, 448–459. [DOI] [PubMed] [Google Scholar]
- 64. Meziane H., Schaller F., Bauer S., Villard C., Matarazzo V., Riet F., Guillon G., Lafitte D., Desarmenien M.G., Tauber M.. et al. (2015) An early postnatal oxytocin treatment prevents social and learning deficits in adult mice deficient for Magel2, a gene involved in Prader-Willi syndrome and autism. Biol. Psychiatry, 78, 85–94. [DOI] [PubMed] [Google Scholar]
- 65. Tauber M., Boulanouar K., Diene G., Cabal-Berthoumieu S., Ehlinger V., Fichaux-Bourin P., Molinas C., Faye S., Valette M., Pourrinet J.. et al. (2017) The use of oxytocin to improve feeding and social skills in infants with Prader-Willi syndrome. Pediatrics, 139, e20162976. [DOI] [PubMed] [Google Scholar]
- 66. Lee S., Walker C.L., Karten B., Kuny S.L., Tennese A.A., O'Neill M.A., Wevrick R. (2005) Essential role for the Prader-Willi syndrome protein necdin in axonal outgrowth. Hum. Mol. Genet., 14, 627–637. [DOI] [PubMed] [Google Scholar]
- 67. Seo S., Guo D.F., Bugge K., Morgan D.A., Rahmouni K., Sheffield V.C. (2009) Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum. Mol. Genet., 18, 1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jackson R.S., Creemers J.W., Ohagi S., Raffin-Sanson M.L., Sanders L., Montague C.T., Hutton J.C., O'Rahilly S. (1997) Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat. Genet., 16, 303–306. [DOI] [PubMed] [Google Scholar]
- 69. Tarpey P.S., Raymond F.L., O’Meara S., Edkins S., Teague J., Butler A., Dicks E., Stevens C., Tofts C., Avis T.. et al. (2007) Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am. J. Hum. Genet., 80, 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Webster E., Cho M.T., Alexander N., Desai S., Naidu S., Bekheirnia M.R., Lewis A., Retterer K., Juusola J., Chung W.K. (2016) De novo PHIP-predicted deleterious variants are associated with developmental delay, intellectual disability, obesity, and dysmorphic features. Cold Spring Harb. Mol. Case Stud., 2, a001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Seifert W., Kuhnisch J., Maritzen T., Horn D., Haucke V., Hennies H.C. (2011) Cohen syndrome-associated protein, COH1, is a novel, giant Golgi matrix protein required for Golgi integrity. J. Biol. Chem., 286, 37665–37675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lim K.L., Chew K.C., Tan J.M., Wang C., Chung K.K., Zhang Y., Tanaka Y., Smith W., Engelender S., Ross C.A.. et al. (2005) Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J. Neurosci. Off. J. Soc. Neurosci., 25, 2002–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Seiler C.Y., Park J.G., Sharma A., Hunter P., Surapaneni P., Sedillo C., Field J., Algar R., Price A., Steel J.. et al. (2014) DNASU plasmid and PSI: Biology-Materials repositories: resources to accelerate biological research. Nucleic Acids Res., 42, D1253–D1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Couzens A.L., Knight J.D., Kean M.J., Teo G., Weiss A., Dunham W.H., Lin Z.Y., Bagshaw R.D., Sicheri F., Pawson T.. et al. (2013) Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal, 6, rs15. [DOI] [PubMed] [Google Scholar]
- 75. Bovijn C., Desmet A.S., Uyttendaele I., Van Acker T., Tavernier J., Peelman F. (2013) Identification of binding sites for myeloid differentiation primary response gene 88 (MyD88) and Toll-like receptor 4 in MyD88 adapter-like (Mal). J. Biol. Chem., 288, 12054–12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rolland T., Taşan M., Charloteaux B., Pevzner S.J., Zhong Q., Sahni N., Yi S., Lemmens I., Fontanillo C., Mosca R.. et al. (2014) A proteome-scale map of the human interactome network. Cell, 159, 1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lievens S., Vanderroost N., Van der Heyden J., Gesellchen V., Vidal M., Tavernier J. (2009) Array MAPPIT: high-throughput interactome analysis in mammalian cells. J. Proteome Res., 8, 877–886. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.