

Abstract
The addition of O-linked β-N-acetylglucosamine (O-GlcNAc) to serine/threonine residues of proteins is a ubiquitous post-translational modification found in all multicellular organisms. Like phosphorylation, O-GlcNAc glycosylation (O-GlcNAcylation) is inducible and regulates a myriad of physiological and pathological processes. However, understanding the diverse functions of O-GlcNAcylation is often challenging due to the difficulty of detecting and quantifying the modification. Thus, robust methods to study O-GlcNAcylation are essential to elucidate its key roles in the regulation of individual proteins, complex cellular processes, and disease. In this chapter, we describe a set of chemoenzymatic labeling methods to (1) detect O-GlcNAcylation on proteins of interest, (2) monitor changes in both the total levels of O-GlcNAcylation and its stoichiometry on proteins of interest, and (3) enable mapping of O-GlcNAc to specific serine/threonine residues within proteins to facilitate functional studies. First, we outline a procedure for the expression and purification of a multi-use mutant galactosyltransferase enzyme (Y289L GalT). We then describe the use of Y289L GalT to modify O-GlcNAc residues with a functional handle, N-azidoacetylgalactosamine (GalNAz). Finally, we discuss several applications of the copper-catalyzed azide-alkyne cycloaddition “click” reaction to attach various alkyne-containing chemical probes to GalNAz and demonstrate how this functionalization of O-GlcNAc-modified proteins can be used to realize (1) – (3) above. Overall, these methods, which utilize commercially available reagents and standard protein analytical tools, will serve to advance our understanding of the diverse and important functions of O-GlcNAcylation.
Keywords: O-linked β-N-acetylglucosamine (O-GlcNAc), chemoenzymatic labeling, O-GlcNAcylation, protein glycosylation, post-translational modification, copper-catalyzed azide-alkyne cycloaddition (CuAAC)
1. Introduction
O-linked β-N-acetylglucosamine glycosylation (O-GlcNAcylation) is a dynamic, inducible post-translational modification (PTM) found on thousands of intracellular proteins (Hart, Slawson, Ramirez-Correa, & Lagerlof, 2011). Only two are enzymes responsible for O-GlcNAc cycling in metazoans: O-GlcNAc transferase (OGT), which catalyzes the addition of O-GlcNAc, and O-GlcNAcase (OGA), which catalyzes its removal (Hart et al., 2011). The O-GlcNAc modification plays a key role in many important cellular functions, including transcription (Rexach et al., 2012), nutrient sensing (X. Yang et al., 2008), metabolism (Yi et al., 2012), mitochondrial dynamics (Pekkurnaz, Trinidad, Wang, Kong, & Schwarz, 2014), and autophagy (Guo et al., 2014). Furthermore, dysregulation of O-GlcNAcylation has been strongly implicated in a number of human diseases such as diabetes (Copeland, Bullen, & Hart, 2008), cancer (Slawson & Hart, 2011), and neurodegenerative disorders (A. C. Wang, Jensen, Rexach, Vinters, & Hsieh-Wilson, 2016; Yuzwa & Vocadlo, 2014). However, the regulatory nature of the modification (e.g., dynamic, low cellular abundance) and its unique chemical characteristics (e.g., low immunogenicity of the neutral sugar, chemically labile) represent a central challenge in its detection and study. These features have necessitated the development of customized methods to visualize and quantify this PTM (Hart et al., 2011).
One of the first techniques developed to detect O-GlcNAcylation used bovine milk galactosyltransferase to transfer [3H]-galactose to terminal GlcNAc residues on proteins (Whiteheart et al., 1989). Along with metabolic labeling using [3H]-glucosamine (Roquemore, Chou, & Hart, 1994), this method facilitated early studies of O-GlcNAcylated proteins. Another common detection method took advantage of the specificity of wheat-germ agglutinin (WGA) lectin, which binds to all terminal GlcNAc sugars as well as sialic acid (Kelly & Hart, 1989). Several antibodies have also been produced using various O-GlcNAcylated proteins and peptide fragments, including the C-terminal domain of RNA polymerase II (CTD110.6) (Comer, Vosseller, Wells, Accavitti, & Hart, 2001) and the pore complex-lamina fraction from rat liver nuclear envelopes (RL-2) (Holt, Snow, Senior, Haltiwanger, & Hart, 1987; Snow, Senior, & Gerace, 1987). A comprehensive list of O-GlcNAc-specific antibodies has been recently described in detail elsewhere (Ma & Hart, 2014). However, the relatively poor immunogenicity of the neutral O-GlcNAc sugar has limited the availability of highly selective antibodies, and in particular, site-specific O-GlcNAc antibodies. Finally, metabolic oligosaccharide engineering (MOE) has emerged as a powerful method to label cellular glycans, including O-GlcNAc (Chuh & Pratt, 2015; Palaniappan & Bertozzi, 2016). Here, a non-natural, peracetylated sugar such as N-azidoacetylglucosamine (Vocadlo, Hang, Kim, Hanover, & Bertozzi, 2003) is incubated with live cells and incorporated into O-GlcNAcylated proteins by the cell’s native biosynthetic machinery. The bioorthogonal azide group can then be used for downstream detection and enrichment.
Although many of these detection methods have been widely adopted, they have some key limitations. Labeling of proteins with bovine milk galactosyltransferase requires handling of expensive, radioactive materials and often necessitates exposure times of days to months (Khidekel et al., 2003). While antibodies offer a convenient and faster means of detection, they exhibit a preference for specific sequence or structural motifs and thus only detect a subset of the O-GlcNAcylated proteins (Ma & Hart, 2014). Moreover, like tritium-based methods, both antibody- and lectin-based techniques are often not sensitive enough to detect O-GlcNAc modifications in cases of low abundance or where sample is limiting. Additionally, the lack of specificity of some detection reagents like the CTD110.6 antibody can lead to misinterpreted results. For example, the increase in O-GlcNAc levels observed upon nutrient deprivation by Western blotting with CTD110.6 was later found to be due to an increase in truncated N-glycans (Isono, 2011; Reeves, Lee, Henry, & Zachara, 2014). O-GlcNAc detection using antibodies and lectins has also resulted in conflicting reports on the levels of total protein O-GlcNAcylation in similar systems. For example, an increase in O-GlcNAcylation in Alzheimer’s disease brain tissue was observed by immunoblotting with the CTD110.6 antibody (Förster et al., 2014), whereas the opposite finding was observed using the RL-2 antibody (Liu et al., 2009). MOE methods also suffer from specificity issues, with reports of N-azidoacetylglucosamine incorporation into cell-surface N- and O-linked glycans (B. W. Zaro, Yang, Hang, & Pratt, 2011) that have prompted efforts to develop more selective probes (Chuh, Zaro, Piller, Piller, & Pratt, 2014; Balyn W. Zaro, Batt, Chuh, Navarro, & Pratt, 2017). Furthermore, the addition of relatively high concentrations of monosaccharide precursors for MOE labeling may alter O-GlcNAc cycling and artifically change physiological levels of O-GlcNAcylation in cells. Finally, MOE cannot achieve complete labeling due to competition with natural sugars, which limits its detection sensitivity and hinders the quantification of O-GlcNAc stoichiometry. Nevertheless, all of these methods can be effectively used for O-GlcNAc detection and remain key tools in the study of O-GlcNAcylation.
Given the above limitations, methods that are (1) highly sensitive and specific, (2) unbiased towards certain subsets of proteins, (3) able to easily and accurately assess changes in O-GlcNAcylation levels, and (4) compatible with traditional techniques for protein analysis are paramount to expanding our understanding of O-GlcNAcylation dynamics and function. Herein, we describe a chemoenzymatic labeling approach that utilizes engineered bovine β-1,4-galactosyltransferase 1 (Y289L GalT) to tag O-GlcNAcylated proteins with an N-azidoacetylgalactosamine (GalNAz) group (Figure 1A). When combined with peptide:N-glycosidase F (PNGase F) treatment to remove GlcNAc-containing N-linked glycans (Whelan & Hart, 2006), this method allows for the specific, unbiased, and global labeling of O-GlcNAcylated proteins. The appended azide handle can subsequently be reacted with a wide variety of alkyne-modified chemical probes, facilitating multiple downstream analyses. Here, we describe four distinct workflows that take advantage of the copper-catalyzed azide-alkyne cycloaddition (CuAAC) “click” reaction to attach biotin, 5-carboxytetramethylrhodamine (TAMRA), high-molecular-weight poly(ethylene glycol) (PEG), or cleavable Dde-biotin probes to O-GlcNAcylated proteins (Figure 1A).
Figure 1.
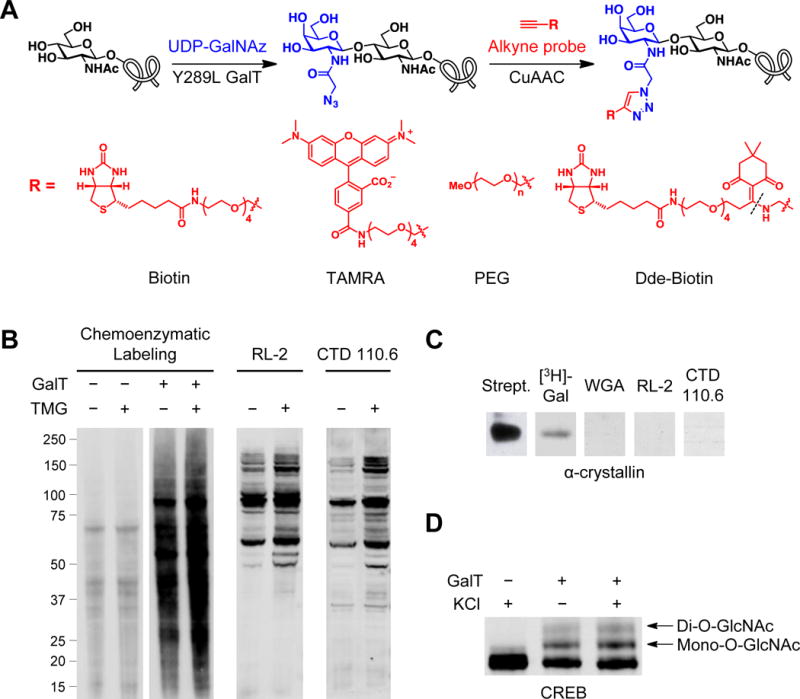
Chemoenzymatic labeling strategies for the study of O-GlcNAcylated proteins. (A) Incubation of O-GlcNAcylated proteins with Y289L GalT and UDP-GalNAz installs a chemical handle that can be further functionalized with alkyne-containing biotin, TAMRA, PEG, or Dde-biotin probes using CuAAC. The Dde-biotin probe is cleaved by hydrazine to provide a positively charged amine at the site indicated (dotted line). (B) Attachment of either an alkynyl biotin or alkynyl TAMRA probe allows for the assessment of total O-GlcNAcylation dynamics in response to cellular stimuli. HEK 293T cells were treated for 6 hours with 50 μM of the OGA inhibitor thiamet-G (TMG) or vehicle and subjected to the workflow in Sections 3 and 4. Total O-GlcNAcylation levels were visualized by blotting with AlexaFluor 680-conjugated streptavidin. Robust, unbiased labeling of proteins is observed by chemoenzymatic labeling. Note the marked difference in the number and molecular weight of O-GlcNAcylated proteins detected as compared to the anti-O-GlcNAc antibodies RL-2 and CTD110.6. (C) Attempts to detect O-GlcNAcylated α-crystallin by various methods show the sensitivity advantage afforded by functionalization with biotin-PEG4-alkyne. In all cases, 0.75 μg of bovine lens α-crystallin was resolved on SDS-PAGE (after chemoenzymatic labeling, as described in Section 3, or tritium labeling, as described in (Khidekel et al., 2003)) and detected as indicated. (D) Labeling proteins with alkynyl PEG (2-kDa) allows for the observation of multiple O-GlcNAcylation states (mono-, di-O-GlcNAcylated, etc.) and their corresponding stoichiometries. Mouse primary cortical neurons were treated with 60 mM KCl for 2 hours and subjected to the workflow in Sections 3 and 5. CREB O-GlcNAcylation was detected by immunoblotting with an anti-CREB antibody.
Biotinylation or TAMRA-labeling enables rapid, sensitive detection of O-GlcNAcylated proteins by Western/streptavidin blotting or direct in-gel fluorescence (Clark et al., 2008; Yi et al., 2012). Importantly, these approaches allow for unbiased protein detection (Figure 1B) and a significant enhancement in detection sensitivity (>380-fold relative to tritium labeling in the case of α-crystallin) compared to traditional methods (Figure 1C) (Clark et al., 2008; Khidekel et al., 2003). Thus, detection of novel O-GlcNAcylated proteins is more readily possible, such as CREB (Rexach et al., 2012) and PFK1 (Yi et al., 2012). The biotin and TAMRA tags also allow for the selective enrichment of O-GlcNAcylated proteins by using streptavidin or anti-TAMRA antibody affinity chromatography. Such an enrichment step can greatly improve the detection of O-GlcNAcylated peptides by mass spectrometry (MS) as the presence of unmodified peptides often suppresses the ionization of their O-GlcNAcylated counterparts (Z. Wang et al., 2010). To improve further the efficiency of enrichment and detection, we developed a chemically cleavable Dde-biotin probe (Figure 1A) (Griffin et al., 2016). This probe allows for the mild, quantitative elution of O-GlcNAcylated proteins from streptavidin beads and produces a positively charged amine upon cleavage to enhance ionization/MS-based detection.
Biotin or defined PEG mass tags can be used to monitor O-GlcNAcylation dynamics and stoichiometry by quantitative Western blotting. The ability to study changes in O-GlcNAcylation in response to cellular stimuli or across disease states is critical to understanding the specific functions of the O-GlcNAc modification. Chemoenzymatic labeling with biotin enables the rapid, global assessment of O-GlcNAcylation and has the advantage of detecting many more proteins than antibody-based methods (Figure 1B). Thus, it can be readily used to monitor changes in O-GlcNAcylation on either specific proteins or the total O-GlcNAcylated protein population in response to cellular stimuli. On the other hand, labeling with PEG tags of defined molecular mass provides the powerful ability to reveal O-GlcNAcylation states (i.e., mono-, di-O-GlcNAcylated, etc.) and quantify in vivo O-GlcNAcylation stoichiometries on proteins of interest (Figure 1D). Such information cannot be readily obtained using other methods. When combined with site-directed mutagenesis, the PEG tagging approach can also determine O-GlcNAc occupancy levels and cycling at specific serine/threonine sites in a protein (Rexach et al., 2012; Rexach et al., 2010; Yi et al., 2012). Quantitative comparisons of O-GlcNAcylation states and their stoichiometries following cellular stimulation or during disease progression are important for identifying the most physiologically relevant O-GlcNAcylation events.
We begin this chapter with an updated method for the purification of Y289L GalT (Section 2). We next describe how to use this enzyme to label O-GlcNAcylated proteins efficiently with GalNAz in a complex cellular lysate (Section 3). After attaching this azide handle, we outline a general method to install a variety of functional tags through CuAAC and detail how the four representative chemical probes can be used to detect, quantify, and measure the dynamics of O-GlcNAcylation both on individual proteins and on the total O-GlcNAcylated proteome (Sections 4, 5, and 6). Lastly, we briefly discuss methods to localize the O-GlcNAc modification to particular serine/threonine residues of proteins to facilitate in-depth functional analyses (Section 7).
2. Expression and Purification of Y289L GalT
The first step of the chemoenzymatic approach is the selective labeling of O-GlcNAcylated proteins with GalNAz. This is achieved using Y289L GalT, which is robustly expressed in E. coli, purified from inclusion bodies, and refolded to obtain active enzyme (Ramakrishnan & Qasba, 2013). The below procedure is a streamlined protocol for making large quantities of Y289L GalT. The enzyme can be stored for up to 1 year as the active protein or for several years in the unfolded form or cell pellet. We also describe procedures to test the purity and activity of the enzyme prior to its use. These checks are necessary before proceeding with the remaining techniques in this chapter.
2.1. Equipment
37°C incubator with shaker
UV-Vis spectrophotometer
Refrigerated centrifuge
Sonicator (Vibra-Cell VCX 130; Sonics & Materials)
Centricon Plus-70 Centrifugal Filter, 10-kDa nominal molecular weight limit (NMWL) (UFC700308, EMD Millipore)
Spectra/Por 4 Standard RC Dialysis Tubing, 12-14-kDa molecular weight cut-off (132706, Spectrum Labs)
Standard equipment for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie staining
- Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) MS instrumentation
- Other MS methods such as liquid chromatography-electrospray ionization MS (Khidekel et al., 2003) also work well.
2.2. Materials
Y289L GalT expressing plasmid (Ramakrishnan & Qasba, 2002)
- 2-Nitro-5-(Sulfothio)-Benzoate (NTSB) (Thannhauser, Konishi, & Scheraga, 1984)
- To prepare 10 mL of NTSB, dissolve 0.1 g of 5,5′-dithiobis(2-nitrobenzoic acid) in 10 mL of 1 M Na2SO3 at 37°C. Adjust the pH to 7.5 with NaOH and bubble air through the solution until the color changes from deep orange-red to pale yellow (~1-2 hours).
- Make fresh or store at −20°C immediately after production.
Electro- or chemically competent BL21(DE3) E. coli
Luria-Bertani agar (LA) plates containing 100 μg/mL of ampicillin
Luria-Bertani (LB) broth containing 100 μg/mL of ampicillin
100 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) in H2O
Phosphate-buffered saline (PBS) pH 7.4 (10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl), 1 mM ethylenediaminetetraacetic acid (EDTA)
25% (w/v) Sucrose, 0.1% Triton-X100, and 1 mM EDTA in PBS pH 7.4
5 M Guanidine hydrochloride in H2O*
5 M Guanidine hydrochloride and 0.3 M sodium sulfite in H2O*
Refolding buffer: 0.5 M L-arginine, 50 mM tris(hydroxymethyl)aminomethane HCl (Tris) pH 8, 5 mM EDTA, 4 mM cysteamine, and 2 mM cystamine*
Dialysis buffer: 50 mM Tris pH 8, 5 mM EDTA, 4 mM cysteamine, and 2 mM cystamine*
100 pmol/μL of Click-iT O-GlcNAc Peptide LC/MS Standard (Click-iT peptide) (C33374, Invitrogen)
100 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.9
- 10 mM uridine 5′-diphospho-2-acetonyl-2-deoxy-α-D-galactose (UDP-ketogal) (Khidekel et al., 2003) or 10 mM uridine 5′-diphospho-2-azidoacetylamino-2-deoxy-α-D-galactose (UDP-GalNAz) (Hang, Yu, Pratt, & Bertozzi, 2004) in H2O
- Store at −80°C and thaw on ice just before use.
100 mM MnCl2
C18 ZipTips (ZTC18M096, EMD Millipore)
MS grade acetonitrile (ACN)
MS grade H2O
1% trifluoroacetic acid (TFA) in H2O
-
Super-DHB (sDHB) MALDI matrix (50862, Sigma-Aldrich)
* Make fresh on the day of use.
2.3. Procedure
Transform 50 μL of electro- or chemically competent BL21(DE3) E. coli with 1 μL of Y289L GalT expressing plasmid (10 ng/μL), plate onto LA plates containing 100 μg/mL of ampicillin, and incubate overnight at 37°C.
Pick two colonies, add individually to 100 mL of LB containing 100 μg/mL of ampicillin, and incubate overnight with shaking (250 RPM) at 37°C.
Distribute the two 100-mL flasks equally between six 1-L volumes of LB containing 100 μg/mL of ampicillin.
Incubate with shaking (250 RPM) at 37°C until the optical density at 600 nm reaches 0.7.
Add IPTG to a final concentration of 1 mM to induce protein production.
Incubate with shaking (250 RPM) for another 4 hours at 37°C.
- Harvest the cells by centrifugation at 8000 × g for 10 minutes.
- The bacterial pellets can be frozen at −80°C for at least 2 years.
- Resuspend bacteria from 1 L of culture medium in 10 mL of PBS containing 1 mM EDTA and sonicate thoroughly (10-15 × 30 seconds, with 10-second rest) at 40% amplitude on ice.
- From now on, the volumes correspond to the purification of Y289L GalT from 1 L of bacterial culture; we generally purify Y289L GalT from 1-2 L of bacterial culture at a time and save the remaining pellets for future purification. Alternatively, the purification can be performed up to step 2.3.19 below and frozen at −80°C for at least 2 years.
- Unless otherwise noted, all steps from this point onward should be performed on ice or at 4°C, and all reagents should be ice cold.
Dilute the bacterial lysate to 80 mL with PBS containing 1 mM EDTA and centrifuge at 14,000 × g for 30 minutes.
Discard the supernatant and resuspend the pellet in 25% (w/v) sucrose in PBS containing 1 mM EDTA and 0.1% Triton X-100.
- Repeat the previous step 5 times.
- It is important to ensure that the pellet is completely resuspended with each wash step to ensure efficient purification of Y289L GalT. With repeated washes, the color of the pellet should change from yellow to ivory white.
- If necessary, the pellet can be stored overnight at 4°C at any point.
After the last centrifugation, resuspend the pellet in 50 mL of PBS containing 1 mM EDTA and centrifuge for 30 minutes at 14,000 × g.
Discard the supernatant, and repeat this step once more to remove any residual detergent.
Discard the supernatant, and resuspend the pellet in 20 mL of 5 M guanidine hydrochloride with 0.3 M sodium sulfite at room temperature.
- Add 2 mL of S-sulfonating agent, NTSB, and stir the reaction vigorously until it has turned pale yellow in color.
- When NTSB (pale yellow) is added to the mixture, the solution should turn red-orange. The reaction is complete when the solution has turned back to pale yellow, which usually takes approximately 45 minutes.
Add 180 mL of ice-cold H2O to precipitate the protein and centrifuge at 10,000 × g for 10 minutes.
Discard the supernatant, resuspend the pellet in 10 mL of H2O, and centrifuge the sample at 10,000 × g for 10 minutes.
Repeat two more times to remove any remaining sulfonating agent.
- Resuspend the pellet in 5 M guanidine hydrochloride to a protein concentration of 1 mg/mL.
- We typically use absorbance at 280 nm on a NanoDrop® 2000 UV-Vis Spectrophotometer (ThermoFisher Scientific) to determine the protein concentration.
- If desired, the unfolded, sulfonated protein can be frozen and stored at −80°C for at least 2 years.
- Dilute the protein solution 10-fold over the course of 15 minutes in refolding buffer.
- Add the refolding buffer in 10 portions over the course of 15 minutes while mixing the solution (by hand or with an orbital shaker).
- Some protein will precipitate as the refolding buffer is added.
- Dialyze the solution 3 × 12 hours with 4 L of dialysis buffer.
- A large amount of protein will precipitate during the dialysis process.
After dialysis, remove the precipitated protein by centrifugation at 10,000 × g for 15 minutes.
- Concentrate the Y289L GalT to 2 mg/mL (determined as previously described) using Centricon Plus-70 10-kDa NMWL Centrifugal Filter Units and store at 4°C.
- This typically requires more than 100-fold concentration.
- Protein can be stored for at least 1 year at 4°C; use the assay described below to ensure activity of older protein stocks before use.
Check the quality of the Y289L GalT by performing SDS-PAGE followed by staining with Coomassie blue (Figure 2A).
- Check that the purified Y289L GalT labels an O-GlcNAcylated peptide using UDP-ketogal or UDP-GalNAz. Set up a dilution series of enzyme as follows:
- Add 0.75 μL of 100 mM MnCl2; 1.5 μL of 100 mM HEPES (pH 7.9); 0, 0.375, or 0.75 μL of 2 mg/mL Y289L GalT; 0.75 μL of 10 mM UDP-ketogal or UDP-GalNAz; and 1.5 μL of 100 pmol/μL Click-iT peptide to 10.5, 10.125, or 9.75 μL of H2O (pipetting up and down after each condition to mix). The final reaction conditions are outlined in Table 1.
- Incubate the above reactions at 4°C for 16 hours.
After incubating for 16 hours, quench the reaction by adding 1.5 μL of 1% TFA and purify the peptides using ZipTips per the manufacturer’s instructions.
Analyze the peptides from each reaction by MALDI-TOF MS using a sDHB matrix (Tsarbopoulos et al., 1994).
- After MALDI-TOF analysis, look for the complete conversion of the unlabeled peptide to the labeled peptide to confirm quantitative labeling (Figure 2B).
- With UDP-ketogal, look for the labeled and unlabeled peptides at an m/z of 1320.5 and 1118.5 Da, respectively, in positive ionization mode. Often times their sodium adducts are also seen at 1342.5 and 1140.5 Da. If UDP-GalNAz is used, the labeled peptide will be observable at 1362.5 Da. A peak at [M-28], corresponding to the loss of N2, is also usually observed. Note that using reflector mode detection may complicate analysis when using UDP-GalNAz (Yejia Li, Hoskins, Sreerama, & Grayson, 2010).
Figure 2.
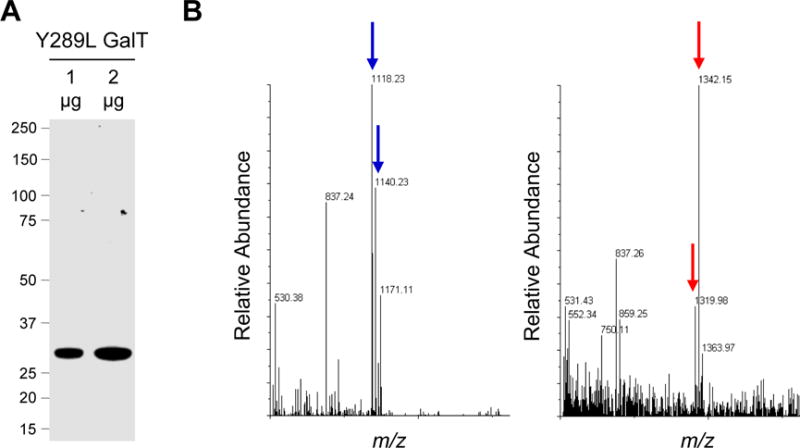
GalT Characterization. (A) Coomassie stained gel of purified Y289L GalT. (B) Representative MALDI-TOF spectra of the peptide labeling reaction with 0 mg/mL (left) or 0.1 mg/mL (right) Y289L GalT. The blue arrows indicate the unlabeled peptide (1118.23) and its sodium adduct (1140.23). The red arrows indicate the labeled peptide (1319.98) and its sodium adduct (1342.15).
Table 1.
Reaction conditions for testing Y289L GalT activity.
| Reaction Componentsa | Volume (μL) | Final Concentration |
|---|---|---|
| H2O | 10.5, 10.125, or 9.75 | |
| 100 mM MnCl2 | 0.75 | 5 mM |
| 100 mM HEPES pH 7.9 | 1.5 | 10 mM |
| 2 mg/mL Y289L GalT | 0, 0.375, or 0.75 | 0, 0.05, or 0.1 mg/mL |
| 10 mM UDP-ketogal or UDP-GalNAz | 0.75 | 500 μM |
| 100 pmol/μL Click-iT Peptide | 1.5 | 10 pmol/μL |
| Final Volume | 15 |
Flick tubes to mix and spin down briefly after each addition.
2.4. Notes
We usually obtain 5-10 mL of active Y289L GalT at a concentration of 2 mg/mL from the purification procedure described above. It is important to note that some protein will continue to precipitate over the first few days to weeks of storage. Although the precipitation of protein is usually not enough to significantly affect the concentration, it is important to re-check both the protein concentration and activity periodically to ensure consistent labeling in subsequent procedures. Overall, if stored properly at 4°C, the Y289L GalT should retain almost 100% of its activity for more than 1 year.
3. Labeling of O-GlcNAcylated Proteins with GalNAz
The Y289L GalT (hereafter referred to as ‘GalT’) produced in Section 2 can be used to label O-GlcNAcylated proteins with UDP-ketogal or UDP-GalNAz. Here, we focus on the use of UDP-GalNAz to add an azide handle to O-GlcNAcylated proteins in cell lysates for further functionalization with an alkyne-modified chemical probe by CuAAC (Sections 4–6).
3.1. Equipment
Centrifuge
Vortex mixer
End-over-end rotator
Spectrophotometer
Sonicator (Vibra-Cell VCX 130; Sonics & Materials)
3.2. Materials
20 mM Thiamet-G (TMG) stock in DMSO
50× cOmplete EDTA-free Protease Inhibitor Cocktail (11873580001, Sigma Aldrich) stock in H2O
- Lysate containing protein(s) of interest
- We generally use the following lysis buffer for most applications: 2% SDS, 100 mM Tris pH 8, 1× protease inhibitor cocktail, and 50 μM TMG.
- Other non-denaturing methods of lysis are also compatible with this protocol; however, it is important that OGA inhibitors, such as TMG (Yuzwa et al., 2008), be included at all steps until the protein is completely denatured to prevent the potential loss of protein O-GlcNAcylation.
- Purified protein obtained by recombinant expression or immunoprecipitated from cell lysate can also be used. In this case, reduce and alkylate the protein, and start at step 8 below.
Dithiothreitol (DTT)
Iodoacetamide (IAA)
IGEPAL CA-630 (I8896-50ML, Sigma-Aldrich)
Milli-Q (or equivalent) purified H2O
PNGase F (glycerol-free) (P0705S, New England Biolabs)
BCA Protein Assay Kit (23225, ThermoFisher Scientific)
Methanol
Chloroform
1% SDS in 20 mM HEPES pH 7.9
2.5× GalT labeling buffer: 50 mM HEPES pH 7.9, 125 mM NaCl, 5% IGEPAL CA-630*
100 mM MnCl2 in H2O*
- 500 μM UDP-GalNAz in 10 mM HEPES pH 7.9*
- Store at −80°C and thaw on ice just before use.
- 2 mg/mL GalT*
- Keep on ice.
* These materials are also commercially available as part of the Click-iT O-GlcNAc Enzymatic Labeling System (C33368, ThermoFisher Scientific).
3.3. Procedure
Lyse the cells in boiling lysis buffer, boil for 5 minutes, and sonicate the lysate 3 × 10 seconds to sheer DNA.
Centrifuge the sample for 5 minutes at 21,130 × g and transfer the supernatant to clean 1.5-mL microcentrifuge tubes.
Measure the protein concentration of the sample using the BCA assay and dilute the sample to 1-2 mg/mL prior to reduction and alkylation.
Add 25 mM (final concentration) of DTT, boil for 5 minutes, and allow the sample to return to room temperature.
- Add 50 mM (final concentration) of IAA and incubate with end-over-end rotation at room temperature in the dark for 60 minutes.
- Reduction and alkylation of the protein lysate is very important for preventing side reactions between free cysteine residues and the alkyne probes (Conte et al., 2011; Hoogenboom, 2010; Yiming Li, Pan, Li, Huang, & Guo, 2013). These side reactions produce nonspecific background and artificially increase the O-GlcNAcylation stoichiometry.
- Dilute cell lysate containing 150 μg of protein with H2O to a total volume of 200 μL.
- This amount is a good starting point for detecting specific O-GlcNAcylated proteins of interest from cell lysates; however, the reaction can be easily scaled depending on the application.
- Precipitate the protein to remove it from endogenous UDP-sugars in the cell lysate:
- Add 600 μL of methanol and vortex the sample.
- Add 200 μL of chloroform and vortex.
- Add 450 μL of H2O, vortex, and centrifuge at 21,130 × g for 5 minutes to separate the aqueous and organic phases.
- The protein will be a thin solid layer at the aqueous-organic interface.
- Remove the top, aqueous layer carefully without disturbing the pellet.
- Add 450 μL of methanol and invert gently to produce a single organic layer.
- Remove all solvent and resuspend the pellet gently in another 450 μL of methanol.
- Remove all solvent, and allow the pellet to air-dry until it is translucent (~10-20 minutes).
- Do not over-dry the pellet as it becomes much more difficult to redissolve.
- Resuspend the protein in 40 μL of 1% SDS, 20 mM HEPES pH 7.9.
- Usually the protein will redissolve completely after ~15-30 minutes; however, the samples can be boiled or sonicated for up to 5 minutes if necessary.
After the protein has redissolved, add (in order) 45 μL of H2O, 4 μL of 50× protease inhibitor cocktail, 80 μL of 2.5× GalT labeling buffer, and 11 μL of 100 mM MnCl2.
Vortex the sample to mix and transfer to ice.
- Add 10 μL of 500 μM UDP-GalNAz and 10 μL of 2 mg/mL GalT or H2O. The final reaction components are outlined in Table 2.
- Adding H2O in place of GalT is a suitable and important negative control for all of the workflows presented hereafter.
- If total O-GlcNAcylation levels or the O-GlcNAcylation level of a transmembrane protein is being measured, it is important to remove N-glycans as they may contain terminal GlcNAc residues that can be labeled by GalT. To achieve this for the reaction described above, add only 42 μL of H2O and 3 μL of 500 U/μL PNGase F after adding GalT.
- Ovalbumin can serve as a useful positive control for PNGase F treatment, see (Whelan & Hart, 2006).
Rotate the reaction end-over-end at 4°C for 16 hours.
- Precipitate the proteins using the methanol/chloroform/H2O method as described above and dry the pellet as before.
- If necessary, at this point, the pellets can be stored in methanol at −80°C for up to 6 months.
Table 2.
Reaction conditions for GalNAz labeling.
| Reaction Components | Volume (μL) | Final Concentration |
|---|---|---|
| Protein mixture (3.75 mg/mL in 1% SDS, 20 mM HEPES pH 7.9) | 40 | 0.75 mg/mL |
| H2Oa | 45 | |
| 50× protease inhibitor cocktail | 4 | 1× |
| 2.5× GalT Labeling bufferb | 80 | 1× |
| 100 mM MnCl2 | 11 | 5.5 mM |
| 500 μM UDP-GalNAz | 10 | 25 μM |
| 2 mg/mL GalTc | 10 | 0.1 mg/mL |
| Final Volume | 200 |
If using PNGase F, add only 42 μL, and add 3 μL PNGase F (500 U/μL) after GalT.
Vortex gently to mix after this step.
Use H2O for negative control.
4. CuAAC “Click” Reaction with Small Molecule Alkyne Probes
After labeling protein(s) with GalNAz, they are ready to be further functionalized with chemical probes and analyzed. In this section, we provide a workflow using CuAAC to append a biotin, TAMRA, or chemically cleavable biotin probe to GalNAz-labeled proteins. However, this general method can be used with a wide variety of alkyne-containing molecules with minimal modifications. Labeling of O-GlcNAcylated proteins with biotin or TAMRA provides a robust and sensitive method for quantifying the total levels of O-GlcNAcylation by streptavidin/Western blotting or direct in-gel fluorescence (Section 4.4). Moreover, biotin-labeled proteins of interest can be captured using streptavidin resin to determine the fraction of labeled proteins and approximate their O-GlcNAcylation stoichiometry (Section 6.3.1). TAMRA labeling followed by immunoprecipitation using an anti-TAMRA antibody can also be used for these same purposes (Section 6.3.2). Finally, labeling with Dde-biotin-alkyne provides a convenient method for the efficient capture and release of O-GlcNAcylated proteins under mild conditions; this may be especially useful for proteins whose O-GlcNAc sites have proven difficult to map (Section 6.3.3).
4.1. Equipment
Centrifuge
Vortex mixer
End-over-end rotator
Standard equipment for SDS-PAGE and Western blotting
4.2. Materials
GalNAz-labeled protein pellet (step 3.3.14)
1% SDS, 20 mM HEPES pH 7.9
50 mM CuSO4-5H2O in H2O*
- 100 mM sodium ascorbate (NaAsc) in H2O*
- Prepare fresh immediately before use and protect from light.
Milli-Q (or equivalent) purified H2O
Methanol
Chloroform
10 X PBS pH 7.4 (100 mM Na2HPO4, 18 mM KH2PO4, 1.37 M NaCl, 27 mM KCl)*
- Alkyne-functionalized chemical probe
- 5 mM Biotin-PEG4-alkyne in DMSO (TA105-25, Click Chemistry Tools)
- 5 mM Dde-biotin-alkyne in DMSO (1137-10, Click Chemistry Tools)
- 5 mM TAMRA alkyne in DMSO (TA108-5, Click Chemistry Tools)
10 mM Tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) in DMSO* (1010-100, Click Chemistry Tools)
SDS-PAGE loading buffer: 2% SDS, 50 mM Tris pH 6.8, 100 mM DTT, 0.1% (w/v) bromophenol blue, and 10% glycerol
* As an alternative, commercial kits are also available as the Click-iT Biotin or Tetramethylrhodamine (TAMRA) Protein Analysis Detection Kits (C33372 and C33370, ThermoFisher Scientific). Furthermore, the Click-iT Protein Buffer Kit (C10276, ThermoFisher Scientific) provides all necessary components except for the alkyne probe for reaction customization.
4.3. Procedure
- Redissolve the protein pellet with 37.5 μL of 1% SDS, 20 mM HEPES pH 7.9.
- As before, this step may take an extended amount of time and can be facilitated by boiling or sonication.
After the protein is completely dissolved, add (in order) 87 μL of H2O, 15 μL of 10× PBS, 3 μL of the 5 mM alkyne probe (if using other probes/volumes, adjust the volume of H2O accordingly), 1.5 μL of 10 mM THPTA, and 3 μL of 50 mM CuSO4 with vortex mixing after each addition. The final reaction conditions are outlined in Table 3.
Incubate the solution with end-over-end rotation for 1 hour at room temperature.
Precipitate the protein as previously described and wash three times with 1 mL of methanol to remove any unreacted alkyne probe.
- Dry the pellet as previously described and redissolve in 40 μL of 20 mM HEPES pH 7.9 containing 1% SDS.
- At this point, the redissolved, functionalized proteins can be frozen and stored for later analysis or subjected directly to SDS-PAGE (see Section 4.4 below). Labeled proteins can also be enriched (Section 6) for further analysis and quantification.
Table 3.
Reaction conditions for CuAAC with small molecule alkyne probes.
| Reaction Componentsa | Volume (μL) | Final Concentration |
|---|---|---|
| Protein mixture (4 mg/mL in 1% SDS, 20 mM HEPES pH 7.9) | 37.5 | 1 mg/mL |
| H2O | 87 | |
| 10× PBS | 15 | 1× |
| 5 mM alkyne probe | 3 | 100 μM |
| 100 mM sodium ascorbate | 3 | 2 mM |
| 10 mM THPTA | 1.5 | 100 μM |
| 50 mM CuSO4 | 3 | 1 mM |
| Final Volume | 150 |
Vortex briefly to mix after each addition.
4.4. Quantifying total O-GlcNAcylation levels
After labeling O-GlcNAcylated proteins from a cellular lysate with biotin or TAMRA, the total levels of protein O-GlcNAcylation can be quantified. Typically, we resolve the labeled proteins (along with a negative control subjected to the same workflow but without the addition of GalT) by SDS-PAGE, transfer the gel to a PVDF membrane, and blot with an anti-TAMRA antibody (A6397, ThermoFisher Scientific) or streptavidin-conjugated dye (S32358, ThermoFisher Scientific or 32230, LI-COR Biosciences). The total levels of O-GlcNAcylation can then be assessed using densitometry. Alternatively, TAMRA-labeled proteins can be resolved by SDS-PAGE and subjected to direct in-gel fluorescence analysis. These approaches are particularly useful for monitoring changes in protein O-GlcNAcylation in response to cellular stimuli (Figure 1B). Moreover, the captured O-GlcNAcylated proteins can be excised from the gel, subjected to proteolytic digestion, and identified by MS analysis (Clark et al., 2008). Together, these probes provide a robust, unbiased method for comparing total O-GlcNAcylation levels under various conditions and may avoid previously reported issues with O-GlcNAc antibodies.
5. CuAAC “Click” Reaction with PEG Alkyne
Using a similar procedure to that described in Section 4, it is also possible to add an alkyne-functionalized PEG tag of defined molecular mass (e.g., 2-kDa or 5-kDa) to O-GlcNAcylated proteins. The PEG-labeled lysates can be resolved by SDS-PAGE and immunoblotted for specific proteins of interest (Figure 1D). This provides a rapid, convenient way to monitor the O-GlcNAcylation state and stoichiometry across different conditions with minimal manipulation after labeling. Tagging O-GlcNAcylated proteins with alkynyl PEG is our method of choice for most applications, including: (a) monitoring O-GlcNAcylation in different tissues or in response to various treatments (Rexach et al., 2012; Yi et al., 2012), (b) confirming sites of O-GlcNAcylation (Rexach et al., 2012), and (c) detecting O-GlcNAc on proteins of interest in a more rapid, parallel fashion (Rexach et al., 2010).
5.1. Equipment
Centrifuge
Vortex mixer
End-over-end rotator
37°C incubator
Standard equipment for SDS-PAGE and Western blotting
5.2. Materials
GalNAz labeled protein pellet (step 3.3.14)
1% SDS, 50 mM Tris pH 8
Milli-Q (or equivalent) purified H2O
Methanol
Chloroform
50 mM CuSO4 in H2O
- 100 mM Tris(2-carboxyethyl)phosphine (TCEP) in H2O
- Prepare fresh immediately before use and protect from light.
- 10 mM PEG alkyne in 200 mM Na2HPO4 pH 8
- We typically use 2-kDa or 5-kDa mPEG alkyne (PLS-2035 or PLS-2034, Creative PEGWorks).
- Prepare fresh immediately before use.
- For proteins with a very high molecular weight (MW), it may be possible to use a higher MW PEG (Deiters & Schultz, 2005). Polymers up to 30-kDa are available from Creative PEGWorks. However, we find that longer reaction times are necessary for efficient labeling with larger PEG molecules.
- Inspect the quality of any PEG compound prior to use by MALDI-TOF MS. High-quality PEG will present a narrow set of peaks centered on the reported MW.
500 mM Tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) in DMSO
SDS-PAGE loading buffer: 2% SDS, 50 mM Tris (pH 6.8), 100 mM DTT, 0.1% (w/v) bromophenol blue, and 10% glycerol
5.3. Procedure
- Redissolve the protein pellet in 50 μL of 1% SDS, 50 mM Tris pH 8.
- As before, this step may take an extended amount of time and can be facilitated by boiling or sonication.
Add 46 μL of H2O and vortex briefly.
- Pre-mix THPTA, CuSO4, and PEG stock solutions:
- Add 0.5 μL of 500 mM THPTA to 5 μL of 50 mM CuSO4, mix, and incubate for 1-2 minutes.
- Add 100 μL of 10 mM PEG alkyne to the CuSO4-THPTA mixture and incubate for 1 minute.
- Add (in order) 100 μL of the THPTA-CuSO4-PEG solution and 4 μL of 100 mM TCEP (vortex briefly to mix after each addition). The final reaction conditions are outlined in Table 4.
- The solution should turn bright yellow after vortex mixing, but will slowly turn clear over the next 1-2 minutes.
- We have found that TCEP works better than sodium ascorbate for click reactions incubated for extended periods of time. This is likely due to the presence of unwanted side reactions between proteins and sodium ascorbate decomposition products (Hong, Presolski, Ma, & Finn, 2009).
- Incubate the reaction in the dark at 37°C for 18-24 hours with end-over-end rotation.
- If using 5-kDa PEG, sometimes incubating for an additional 24 hours (48 hours total) may improve labeling efficiency. Furthermore, for the potential use of higher MW PEG, we have found that many proteins are stable for at least 60 hours under these conditions.
Precipitate the protein as previously described and wash three times with 1 mL of methanol to remove unreacted PEG.
Redissolve the protein in the desired volume of loading buffer for SDS-PAGE and analysis by Western blotting (see Section 5.4 below).
Table 4.
Reaction conditions for CuAAC with PEG alkyne.
| Reaction Componentsa | Volume (μL) | Final Concentration |
|---|---|---|
| Protein mixture (3 mg/mL in 1% SDS, 50 mM Tris pH 8) | 50 | 0.75 mg/mL |
|
| ||
| H2O | 46 | |
|
| ||
| PEG-CuSO4-THPTA mixtureb | 100 | |
| 0.5 μL 500 mM THPTA | 1.2 mM | |
| 5 μL 50 mM CuSO4 | 1.2 mM | |
| 100 μL 10 mM PEG alkyne | 4.75 mM | |
|
| ||
| 100 mM TCEP | 4 | 2 mM |
|
| ||
| Final Volume | 200 | |
Vortex briefly to mix after each addition.
Prepared according to instructions in text.
5.4. Quantification of protein O-GlcNAcylation stoichiometry
The lysates are resolved by SDS-PAGE and analyzed by Western blotting using antibodies against the protein(s) of interest. For standard cell lysates, we typically use 50 to 100 μg of total protein for analysis, depending on the abundance of the protein. For other applications, such as detecting O-GlcNAcylation of purified or overexpressed proteins, the amount needed depends on the O-GlcNAcylation stoichiometry and resolution capacity of the gel. Successful PEG labeling will reveal a mass-shifted band corresponding to the MW of the PEG tag for each O-GlcNAc group on the protein. The relative abundance of each O-GlcNAcylated form and the total O-GlcNAcylation stoichiometry can be obtained by quantifying the ratio of the signals for the mass shifted band(s) to the total, protein-specific band (Rexach et al., 2012; Rexach et al., 2010). See Figure 1D for typical results using an antibody against cAMP response element binding protein (CREB) (4820, Cell Signaling Technologies). In general, we have found that CREB serves as an excellent positive control that is expressed in many mammalian cell lines with an O-GlcNAcylation stoichiometry of approximately 20-30%. Although other well-characterized O-GlcNAcylated proteins can be used, we recommend against using highly O-GlcNAcylated proteins as a positive control because they often show a mass shift even when the labeling is incomplete. It is important that the labeling be complete for an accurate assessment of O-GlcNAcylation stoichiometry. Notably, this method can also be used to study the interplay between O-GlcNAcylation and other PTMs such as phosphorylation using phosphorylation state-specific antibodies (Rexach et al., 2012). Finally, in all of the above cases, it is important to employ a quantitative Western blotting system, such as the Odyssey Imaging System (LI-COR Biosciences). Overall, this method provides the only direct way to measure O-GlcNAcylation stoichiometries without the use of laborious MS methods. Moreover, this approach utilizes standard techniques for protein analysis without the need for advanced instrumentation or radiolabels.
6. Biotin/TAMRA Immunoprecipitation
This section describes our workflow for streptavidin capture or TAMRA immunoprecipitation of O-GlcNAcylated protein(s) from a complex cell lysate. Upon release, the O-GlcNAcylated protein(s) can be subjected to detection and quantification (Section 6.4) or MS analysis (Section 7). These methods are most useful for initial detection of O-GlcNAcylation on a protein of interest or for MS-based detection and site-mapping of O-GlcNAcylated peptides (Section 7).
6.1. Equipment
Centrifuge
Vortex mixer
End-over-end rotator
Magnetic separator
Standard equipment for SDS-PAGE and Western blotting
6.2. Materials
Biotin or TAMRA-labeled proteins (step 4.3.5)
Neutralization buffer: 6% IGEPAL CA-630, 100 mM Na2HPO4 pH 8, 150 mM NaCl
Dilution buffer: 100 mM Na2HPO4 pH 8, 150 mM NaCl
Streptavidin magnetic beads (Pierce, for biotin IP)
Protein A/G magnetic beads (Pierce, for TAMRA IP)
- TRITC polyclonal antibody (A-6397, ThermoFisher)
- Alternatively, a TAMRA monoclonal antibody (MA1-041, ThermoFisher) can also be used.
High Capacity NeutrAvidin agarose (Pierce, for cleavable biotin IP)
TAMRA wash buffer: 1% IGEPAL CA-630, 100 mM Na2HPO4 pH 8, 150 mM NaCl
- Biotin low salt wash buffer: 100 mM Na2HPO4 pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS
- While cleavage of the Dde-biotin-alkyne in buffers containing SDS has been previously reported (Y. Yang & Verhelst, 2013), we have found that it is stable in up to 1% SDS at room temperature (Griffin et al., 2016).
Biotin high salt wash buffer: 100 mM Na2HPO4 pH 7.5, 500 mM NaCl, 0.2% Triton X-100
Biotin elution buffer: 50 mM Tris pH 6.8, 2.5% SDS, 100 mM DTT, 10% glycerol, and 2 mM biotin (add 0.1% (w/v) bromophenol blue if to be used directly for Western blotting)
TAMRA wash buffer: 50 mM Tris pH 7.4, 150 mM NaCl, 1% Triton X-100
TAMRA elution buffer: 2% SDS, 50 mM Tris pH 6.8, 100 mM DTT, and 10% glycerol (add 0.1% (w/v) bromophenol blue if to be used directly for Western blotting)
PBS pH 7.4 (10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl)
- 2% (w/v) hydrazine monohydrate in H2O
- Make this solution fresh immediately before use.
Acetone
1% SDS in 20 mM HEPES pH 7.9
6.3. Procedure
Save 1/10 (4 μL) of the biotin or TAMRA-labeled protein solution as input to confirm effective enrichment or to quantify the O-GlcNAcylation stoichiometry of labeled proteins.
To avoid denaturing proteins on the affinity resins, neutralize the SDS in the protein sample with an equivalent volume of neutralization buffer.
Dilute the sample to 500 μL with dilution buffer.
- Wash the beads twice with 1 mL of wash buffer. Typically, we use the following amounts for the applications discussed:
- 40 μL of streptavidin magnetic beads (Section 6.3.1)
- 50 μL of Protein A/G magnetic beads (Section 6.3.2)
- 25 μL of high capacity NeutrAvidin agarose (Section 6.3.3)
Add the diluted protein solution to the washed beads.
6.3.1. Non-cleavable biotin
Rotate end-over-end for 2 hours at room temperature.
- After the precipitation is complete, remove the flowthrough, resuspend the beads in 1 mL of biotin low salt wash buffer, and rotate end-over-end for 5 minutes at room temperature
- A portion of the flowthrough can be retained and analyzed to ensure complete precipitation by the affinity resin.
- Remove the wash buffer and repeat four more times with 1 mL of low salt wash buffer and five times with 1 mL of high salt wash buffer to remove non-O-GlcNAcylated proteins.
- In applications where there tends to be high background (e.g., protein overexpression), the wash conditions can be made more stringent by replacing the low salt wash buffer with 100 mM Na2HPO4 pH 7.5, 150 mM NaCl, 1-3% SDS. However, this may elute some biotinylated proteins as well.
After the last wash, centrifuge the beads briefly (5-10 seconds) at 500 × g and remove any residual wash buffer.
Resuspend the beads with 30 μL of biotin elution buffer and boil for 15 minutes with occasional vortex mixing.
- Centrifuge the tube briefly and transfer the elution buffer to a new tube.
- The eluent can be safely stored indefinitely at −80°C.
- For endogenous proteins, we typically resolve the entire eluent in a single gel lane. However, for overexpressed proteins, it is usually sufficient to run as little as 1/10 of the eluent.
6.3.2. TAMRA
Add 10 μL of TRITC polyclonal antibody to the mixture.
Rotate end-over-end overnight at 4°C.
Remove the flow-through and wash the beads by rotating end-over-end for 5 minutes at 4°C with 1 mL of TAMRA wash buffer.
Remove the wash buffer and repeat four additional times.
- After the final wash, elute TAMRA-labeled proteins by boiling with 30 μL of TAMRA elution buffer for 15 minutes and proceed as described for the biotin samples.
- These conditions are used to quantitatively release TAMRA-labeled proteins, but they will also elute the antibody, which can interfere with Western blot quantification. We suggest the use of a conformation-specific secondary antibody for detection during Western blotting (e.g., 3678S, Cell Signaling Technology).
- Alternatively, 100 mM glycine pH 2.5 can be used to elute the protein in cases where co-elution of TAMRA antibody is problematic and/or stoichiometric elution is not required.
6.3.3. Cleavable Dde-biotin
Rotate end-over-end for 2 hours at room temperature.
Transfer the beads to a spin filter and wash the beads as described for the non-cleavable biotin.
Wash the beads twice more with 0.5 mL of PBS.
Resuspend the beads with 50 μL of 2% (w/v) hydrazine monohydrate and incubate with end-over-rotation for 1 hour at room temperature.
Collect the eluent via centrifugation at 2,000 × g for 30 seconds.
Resuspend the beads in 50 μL of PBS and incubate with end-over-end rotation for 5 minutes at room temperature.
Collect this wash by centrifugation as before and combine with the eluent.
- Precipitate the eluted proteins by the addition of 4 volumes (400 μL) of −20°C acetone and incubate for 2 hours to overnight at −20°C.
- Do not use the methanol/chloroform/H2O method here as it is extremely difficult to form a pellet with low quantities of protein. The precipitated proteins can be stored for up to 6 months in acetone at −80°C.
Centrifuge the precipitated proteins at 21,130 × g for 10 minutes at 4°C, remove the acetone, and dry the pellet as before.
- For analysis by SDS-PAGE, resuspend the pellet in 40 μL of SDS-PAGE loading buffer.
- For direct analysis by MS, redissolve the proteins in a buffer compatible with an in-solution digestion method, such as 8 M urea, 50 mM Tris pH 8.
6.4. Detection and quantification of enriched O-GlcNAcylated proteins
Western blotting of the eluted solution can provide putative evidence for O-GlcNAcylation of specific proteins. It is important to run a negative control simultaneously in which GalT has been omitted from the GalNAz labeling step (as described earlier) to corroborate these results. Approximate O-GlcNAcylation stoichiometries can be determined following biotin labeling by resolving the eluted sample (step 6.3.1.6) by SDS-PAGE along with a portion of the retained input sample (step 6.3.1) and calculating the fraction of eluted O-GlcNAcylated protein to the total protein signal (Yi et al., 2012). Furthermore, the method can be used to monitor changes in the O-GlcNAcylation levels of individual proteins across different physiological conditions. Although this method cannot reveal the presence of multiple O-GlcNAcylation sites, it does have some advantages over labeling proteins with PEG alkyne. Specifically, (1) enrichment allows for the detection of low O-GlcNAcylation stoichiometries, (2) O-GlcNAcylation of high MW proteins can be readily measured without relying on a mass shift, and (3) some antibodies may not recognize mass-shifted proteins. Regardless, after observing that a given protein is likely O-GlcNAcylated, MS-based methods can be used as further, more definitive verification and to identify the exact site(s) of O-GlcNAcylation.
7. O-GlcNAc Site Identification
The identification of specific serine/threonine residues that are modified by O-GlcNAc is often critical to interrogate its functional relevance. Accordingly, several techniques have been developed for this purpose. Originally, O-GlcNAcylated peptides were labeled with [3H]-galactose and sequenced by Edman degradation (Whelan & Hart, 2006). However, with the rise of powerful MS-based methods for peptide/protein sequencing, many techniques have been developed to map O-GlcNAc sites on proteins (Alfaro et al., 2012; Griffin et al., 2016; Haynes & Aebersold, 2000; Khidekel, Ficarro, Peters, & Hsieh-Wilson, 2004; Trinidad et al., 2012; Vosseller et al., 2006; Z. Wang et al., 2010); a thorough review of current methods for O-GlcNAc site mapping can be found elsewhere (Ma & Hart, 2014). Here we describe a general approach for O-GlcNAc site mapping and its enhancement using chemoenzymatic labeling. Unlike MS analysis of immunoprecipitated protein alone, chemoenzymatic labeling has the specific advantages of providing 1) an effective method for enrichment of O-GlcNAcylated peptides/proteins to greatly improve the detection sensitivity, 2) a positively charged amine left by the cleavable biotin tag to facilitate O-GlcNAc site mapping, and 3) a unique ion fragment signature upon MS/MS analysis for conclusive identification of the O-GlcNAc modification. Once modification sites are discovered, the tools outlined in the previous sections can be used to investigate how O-GlcNAcylation at individual sites changes in response to stimuli.
7.1. Mapping of O-GlcNAc sites using MS
To map the O-GlcNAc sites on a protein of interest, a tagged form of the protein can be overexpressed in HEK 293T cells (or any other easily transfectable cell line). TMG can be added to the cells to enhance the overall levels of O-GlcNAcylation. The protein of interest is then immunoprecipitated, subjected to SDS-PAGE, and excised from the gel. After reduction, alkylation, and proteolytic digestion in gel, the extracted peptides are analyzed by MS (Shevchenko, Tomas, Havli, Olsen, & Mann, 2007). The use of electron-transfer dissociation tandem MS (ETD-MS/MS) has become a powerful technique for mapping O-GlcNAc sites due to its ability to fragment peptides without breaking the glycosidic linkage between serine/threonine residues and GlcNAc (Ma & Hart, 2014). Indeed, we and others have used this workflow to successfully map O-GlcNAc sites on multiple occasions (Khidekel et al., 2007; Pekkurnaz et al., 2014; Rexach et al., 2012; Yi et al., 2012).
Recently, we used a similar workflow in conjunction with a cleavable Dde-biotin-alkyne probe to identify two known and four novel O-GlcNAcylation sites on OGT (Griffin et al., 2016). The Dde-biotin moiety allows for efficient capture and release of O-GlcNAcylated peptides and generates a positively charged amine functionality on the glycopeptide to improve its ionization efficiency (Figure 1A). We believe that this new probe has significant potential to facilitate the identification of O-GlcNAc sites. In this case, overexpressed, immunoprecipitated protein could be subjected to the workflow described in Section 6.3 using Dde-biotin-alkyne as the azide-reactive probe. The eluted O-GlcNAcylated protein is then purified by SDS-PAGE and processed as outlined above. Alternatively, the eluent can be processed directly by in-solution digestion or with filter-aided sample preparation (Wiśniewski, Zougman, Nagaraj, & Mann, 2009).
Labeled O-GlcNAcylated peptides can be easily identified using higher-energy collisional dissociation (HCD) MS/MS fragmentation by the presence of up to three fragmented linker ions at 300.1, 318.1, and 503.2 m/z. These fragment ions correspond to cleavage of the glycosidic bond between GlcNAc and GalNAz and its water adduct (300.1 and 318.1 m/z, respectively) and cleavage between GlcNAc and the serine/threonine residue (503.2 m/z). For subsequent ETD-MS/MS analysis, we typically use LTQ-Velos, Orbitrap Elite, and/or Orbitrap Fusion instruments (ThermoFisher Scientific), but any instrument capable of performing ETD-MS/MS would be sufficient. Regardless, it is important to account for the variable addition of the mass tag (502.202341 Daltons) to serine and threonine residues in the analysis (Griffin et al., 2016). In summary, we believe that the procedure outlined may aid in the detection of low-abundance or otherwise hard to identify O-GlcNAc sites.
8. Integrated Workflow for Discovery and Biological Assessment of Novel O-GlcNAcylated Proteins
Together, chemoenzymatic tagging methods provide a powerful approach for the detection and analysis of novel O-GlcNAcylated proteins. In our lab, we follow an integrated workflow when assessing the importance of a putatively modified protein. First, we use biotin or TAMRA labeling followed by immunoprecipitation and Western blot detection (Sections 4 and 6.3.1 or 6.3.2) to provide direct evidence of protein O-GlcNAcylation. Importantly, we perform a negative control experiment without adding GalT to ensure that the observed signal is specific for the O-GlcNAcylated structure. Next, we map the modification site(s) by either direct MS analysis of a FLAG-tagged, immunoprecipitated protein of interest (Section 7.1) or labeling and enrichment with the Dde-biotin-alkyne probe prior to MS analysis (Sections 4 and 6.3.3). If the protein has multiple sites, expression of alanine mutants lacking specific O-GlcNAcylation sites in cells, followed by analysis of their stoichiometries using PEG or biotin labeling, can sometimes be used to determine the major sites (Sections 5 and 6). Moreover, these mutants can be expressed in the presence of TMG or other stimuli to uncover which sites are dynamic or rapidly cycled. Finally, cells with the endogenous protein or overexpressed mutants can be subjected to physiologically relevant treatments such as serum starvation, hypoxia, or KCl-mediated depolarization followed by PEG labeling to measure changes in the levels of each O-GlcNAcylation state in response to biological stimuli. Again, we have found CREB to be an excellent positive control for the strategies outlined in this section. As an example, the increase in O-GlcNAcylation of CREB stimulated by TMG treatment can be seen in Figure 3. Using PEG labeling, mono-O-GlcNAcylation of CREB was also found to increase in neurons after KCl-induced depolarization (Figure 1D). Ultimately, the collective results of this integrated workflow will provide strong evidence for novel O-GlcNAcylation events and guide subsequent functional studies.
Figure 3.
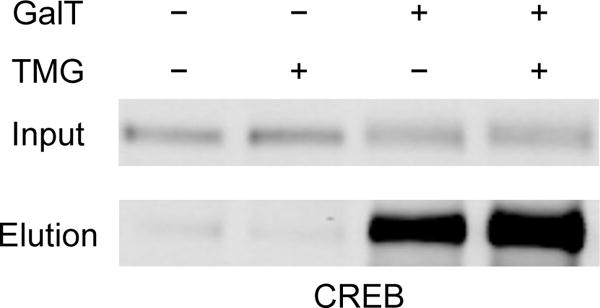
Streptavidin capture of O-GlcNAcylated proteins labeled with biotin can allow for the detection and dynamic monitoring of novel O-GlcNAcylated proteins. HEK 293T cells were treated with 50 μM TMG or vehicle for 6 hours and subjected to the workflow outlined in Sections 3 and 6. Immunoblotting with an anti-CREB antibody revealed a robust increase in O-GlcNAcylation of CREB upon TMG treatment.
9. Conclusions and Future Directions
The O-GlcNAcylation of intracellular proteins is emerging as a major regulator of key cellular processes in both health and disease. Effective methods to characterize the presence and dynamics of this post-translational modification will greatly propel studies of its functional significance. Traditional methods such as tritium labeling and the use of certain O-GlcNAc-specific antibodies suffer from a lack of sensitivity and specificity. Moreover, using such methods to detect changes in O-GlcNAcylation levels and stoichiometry reliably in a complex lysate can be very difficult or in some cases impossible. Although the rapid proliferation of MS-based techniques has alleviated some of these concerns, MS-based approaches require specialized expertise and access to expensive instrumentation. Thus, highly accessible, cost-effective approaches that can be readily performed in most laboratories on a routine basis remain vital to elucidate the physiological importance of O-GlcNAcylation.
The protocols above provide a practical platform that is easy to implement in many research settings. GalT labeling and the CuAAC reaction provide an efficient means to install a variety of functional tags for diverse downstream applications. The commercial availability of the reagents and the simplicity of the experimental approach should facilitate adoption of this methodology by non-specialists. In the future, further development of these techniques in conjunction with MS will pave the way for more quantitative, high-throughput methods to study O-GlcNAcylation dynamics on individual proteins as well as on a global scale. Combining the techniques with established methods such as stable isotope labeling by amino acids in cell culture (SILAC) (Ong et al., 2002), isobaric tag for relative and absolute quantitation (iTRAQ) (Ross et al., 2004), or tandem mass tags (TMT) (Thompson et al., 2003) should allow for direct comparison of the O-GlcNAcome across diverse physiological conditions. Together, we hope that these advances will illuminate the broad importance of O-GlcNAcylation and lead to new discoveries at the frontiers of biology and human health.
Acknowledgments
This work was supported by the National Institutes of General Medical Sciences (NIGMS) grant R01-GM084724 (L. H. W.), NIGMS training grants T32-GM008042 and T32-GM07616 (J. W. T.), the UCLA-Caltech Medical Scientist Training Program (J. W. T.), and the National Science Foundation Graduate Research Fellowship DGE-1144469 (M. E. G.). We thank Dr. P. Qasba (National Cancer Institute at Frederick) for generously providing the Y289L GalT construct and Dr. M. Shahgholi (Caltech CCE Multiuser Mass Spectrometry Laboratory) for assistance with MALDI-TOF MS analysis of O-GlcNAcylated peptides. We also thank Drs. E. Peters and D. Mason (Genomics Research Institute of the Novartis Research Foundation) for their suggestions, feedback, and critical discussions about O-GlcNAc site mapping.
References
- Alfaro JF, Gong CX, Monroe ME, Aldrich JT, Clauss TR, Purvine SO, Smith RD. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(19):7280–7285. doi: 10.1073/pnas.1200425109. http://dx.doi.org/10.1073/pnas.1200425109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuh KN, Pratt MR. Chemical methods for the proteome-wide identification of posttranslationally modified proteins. Current Opinion in Chemical Biology. 2015;24:27–37. doi: 10.1016/j.cbpa.2014.10.020. http://doi.org/10.1016/j.cbpa.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuh KN, Zaro BW, Piller F, Piller V, Pratt MR. Changes in metabolic chemical reporter structure yield a selective probe of O-GlcNAc modification. Journal of the American Chemical Society. 2014;136(35):12283–12295. doi: 10.1021/ja504063c. http://dx.doi.org/10.1021/ja504063c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark PM, Dweck JF, Mason DE, Hart CR, Buck SB, Peters EC, Hsieh-Wilson LC. Direct in-gel fluorescence detection and cellular imaging of O-GlcNAc-modified proteins. Journal of the American Chemical Society. 2008;130(35):11576–11577. doi: 10.1021/ja8030467. http://dx.doi.org/10.1021/ja8030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer FI, Vosseller K, Wells L, Accavitti MA, Hart GW. Characterization of a mouse monoclonal antibody apecific for O-linked N-acetylglucosamine. Analytical Biochemistry. 2001;293(2):169–177. doi: 10.1006/abio.2001.5132. http://dx.doi.org/10.1006/abio.2001.5132. [DOI] [PubMed] [Google Scholar]
- Conte ML, Staderini S, Marra A, Sanchez-Navarro M, Davis BG, Dondoni A. Multi-molecule reaction of serum albumin can occur through thiol-yne coupling. Chemical Communications. 2011;47(39):11086. doi: 10.1039/c1cc14402b. http://dx.doi.org/10.1039/c1cc14402b. [DOI] [PubMed] [Google Scholar]
- Copeland RJ, Bullen JW, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: roles in insulin resistance and glucose toxicity. American Journal of Physiology. Endocrinology and Metabolism. 2008;295(1):E17–28. doi: 10.1152/ajpendo.90281.2008. http://dx.doi.org/10.1152/ajpendo.90281.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiters A, Schultz PG. In vivo incorporation of an alkyne into proteins in Escherichia coli. Bioorganic & Medicinal Chemistry Letters. 2005;15(5):1521–1524. doi: 10.1016/j.bmcl.2004.12.065. http://dx.doi.org/10.1016/j.bmcl.2004.12.065. [DOI] [PubMed] [Google Scholar]
- Förster S, Welleford AS, Triplett JC, Sultana R, Schmitz B, Butterfield DA. Increased O-GlcNAc levels correlate with decreased O-GlcNAcase levels in Alzheimer disease brain. Biochimica et Biophysica Acta. 2014;1842(9):1333–1339. doi: 10.1016/j.bbadis.2014.05.014. http://dx.doi.org/10.1016/j.bbadis.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin ME, Jensen EH, Mason DE, Jenkins CL, Stone SE, Peters EC, Hsieh-Wilson LC. Comprehensive mapping of O-GlcNAc modification sites using a chemically cleavable tag. Molecular bioSystems. 2016 doi: 10.1039/c6mb00138f. http://dx.doi.org/10.1039/c6mb00138f. [DOI] [PMC free article] [PubMed]
- Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P, Zhang H. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nature Cell Biology. 2014;16(12):1215–1226. doi: 10.1038/ncb3066. http://dx.doi.org/10.1038/ncb3066. [DOI] [PubMed] [Google Scholar]
- Hang HC, Yu C, Pratt MR, Bertozzi CR. Probing glycosyltransferase activities with the Staudinger ligation. Journal of the American Chemical Society. 2004;126(1):6–7. doi: 10.1021/ja037692m. http://dx.doi.org/10.1021/ja037692m. [DOI] [PubMed] [Google Scholar]
- Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annual Review of Biochemistry. 2011;80:825–858. doi: 10.1146/annurev-biochem-060608-102511. http://dx.doi.org/10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes PA, Aebersold R. Simultaneous detection and identification of O-GlcNAc-modified glycoproteins using liquid chromatography-tandem mass spectrometry. Analytical Chemistry. 2000;72(21):5402–5410. doi: 10.1021/ac000512w. http://dx.doi.org/10.1021/ac000512w. [DOI] [PubMed] [Google Scholar]
- Holt GD, Snow CM, Senior A, Haltiwanger RS, Hart GW. Nuclear pore complex glycoproteins contain cytoplasmically disposed O-linked N-acetylglucosamine. The Journal of Cell Biology. 1987;104(5):1157–1164. doi: 10.1083/jcb.104.5.1157. http://dx.doi.org/10.1083/jcb.104.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong V, Presolski S, Ma C, Finn MG. Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angewandte Chemie (International ed in English) 2009;48(52):9879–9883. doi: 10.1002/anie.200905087. http://dx.doi.org/10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenboom R. Thiol-yne chemistry: a powerful tool for creating highly functional materials. Angewandte Chemie (International ed in English) 2010;49(20):3415–3417. doi: 10.1002/anie.201000401. http://dx.doi.org/10.1002/anie.201000401. [DOI] [PubMed] [Google Scholar]
- Isono T. O-GlcNAc-specific antibody CTD110.6 cross-reacts with N-GlcNAc2-modified proteins induced under glucose deprivation. PLoS ONE. 2011;6(4):e18959. doi: 10.1371/journal.pone.0018959. http://dx.doi.org/10.1371/journal.pone.0018959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly WG, Hart GW. Glycosylation of chromosomal proteins: localization of O-linked N-acetylglucosamine in Drosophila chromatin. Cell. 1989;57(2):243–251. doi: 10.1016/0092-8674(89)90962-8. http://dx.doi.org/10.1016/0092-8674(89)90962-8. [DOI] [PubMed] [Google Scholar]
- Khidekel N, Arndt S, Lamarre-Vincent N, Lippert A, Poulin-Kerstien KG, Ramakrishnan B, Hsieh-Wilson LC. A chemoenzymatic approach toward the rapid and sensitive detection of O-GlcNAc posttranslational modifications. Journal of the American Chemical Society. 2003;125(52):16162–16163. doi: 10.1021/ja038545r. http://dx.doi.org/10.1021/ja038545r. [DOI] [PubMed] [Google Scholar]
- Khidekel N, Ficarro SB, Clark PM, Bryan MC, Swaney DL, Rexach JE, Hsieh-Wilson LC. Probing the dynamics of O-GlcNAc glycosylation in the brain using quantitative proteomics. Nat Chem Biol. 2007;3(6):339–348. doi: 10.1038/nchembio881. http://dx.doi.org/10.1038/nchembio881. [DOI] [PubMed] [Google Scholar]
- Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC. Exploring the O-GlcNAc proteome: direct identification of O-GlcNAc-modified proteins from the brain. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(36):13132–13137. doi: 10.1073/pnas.0403471101. http://dx.doi.org/10.1073/pnas.0403471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hoskins JN, Sreerama SG, Grayson SM. MALDI−TOF mass spectral characterization of polymers containing an azide group: evidence of metastable ions. Macromolecules. 2010;43(14):6225–6228. doi: 10.1021/ma100599n. http://dx.doi.org/10.1021/ma100599n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Pan M, Li Y, Huang Y, Guo Q. Thiol-yne radical reaction mediated site-specific protein labeling via genetic incorporation of an alkynyl-L-lysine analogue. Organic & Biomolecular Chemistry. 2013;11(16):2624. doi: 10.1039/c3ob27116a. http://dx.doi.org/10.1039/c3ob27116a. [DOI] [PubMed] [Google Scholar]
- Liu F, Shi J, Tanimukai H, Gu J, Gu J, Grundke-Iqbal I, Gong CX. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain. 2009;132(Pt 7):1820–1832. doi: 10.1093/brain/awp099. http://dx.doi.org/10.1093/brain/awp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Hart GW. O-GlcNAc profiling: from proteins to proteomes. Clinical Proteomics. 2014;11(1):8. doi: 10.1186/1559-0275-11-8. http://dx.doi.org/10.1186/1559-0275-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Molecular & Cellular Proteomics. 2002;1(5):376–386. doi: 10.1074/mcp.m200025-mcp200. http://dx.doi.org/10.1074/mcp.M200025-MCP200. [DOI] [PubMed] [Google Scholar]
- Palaniappan KK, Bertozzi CR. Chemical glycoproteomics. Chemical Reviews. 2016;116(23):14277–14306. doi: 10.1021/acs.chemrev.6b00023. http://dx.doi.org/10.1021/acs.chemrev.6b00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekkurnaz G, Trinidad JC, Wang X, Kong D, Schwarz TL. Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell. 2014;158(1):54–68. doi: 10.1016/j.cell.2014.06.007. http://dx.doi.org/10.1016/j.cell.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan B, Qasba PK. Structure-based design of 1,4-galactosyltransferase I (4Gal-T1) with equally efficient N-acetylgalactosaminyltransferase activity. Journal of Biological Chemistry. 2002;277(23):20833–20839. doi: 10.1074/jbc.M111183200. http://dx.doi.org/10.1074/jbc.M111183200. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan B, Qasba PK. In vitro folding of beta-1,4galactosyltransferase and polypeptide-alpha-N-acetylgalactosaminyltransferase from the inclusion bodies. Methods in Molecular Biology. 2013;1022:321–333. doi: 10.1007/978-1-62703-465-4_24. http://dx.doi.org/10.1007/978-1-62703-465-4_24. [DOI] [PubMed] [Google Scholar]
- Reeves RA, Lee A, Henry R, Zachara NE. Characterization of the specificity of O-GlcNAc reactive antibodies under conditions of starvation and stress. Analytical Biochemistry. 2014;457:8–18. doi: 10.1016/j.ab.2014.04.008. http://dx.doi.org/10.1016/j.ab.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexach JE, Clark PM, Mason DE, Neve RL, Peters EC, Hsieh-Wilson LC. Dynamic O-GlcNAc modification regulates CREB-mediated gene expression and memory formation. Nature Chemical Biology. 2012;8(3):253–261. doi: 10.1038/nchembio.770. http://dx.doi.org/10.1038/nchembio.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexach JE, Rogers CJ, Yu SH, Tao J, Sun YE, Hsieh-Wilson LC. Quantification of O-glycosylation stoichiometry and dynamics using resolvable mass tags. Nature Chemical Biology. 2010;6(9):645–651. doi: 10.1038/nchembio.412. http://dx.doi.org/10.1038/nchembio.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roquemore EP, Chou TY, Hart GW. Detection of O-linked N-acetylglucosamine (O-GlcNAc) on cytoplasmic and nuclear proteins. Methods in Enzymology. 1994;230:443–460. doi: 10.1016/0076-6879(94)30028-3. http://dx.doi.org/10.1016/0076-6879(94)30028-3. [DOI] [PubMed] [Google Scholar]
- Ross PL, Huang YN, Marchese B, Williamson B, Parker K, Hattan S, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Molecular & Cellular Proteomics. 2004;3(12):1154–1169. doi: 10.1074/mcp.M400129-MCP200. http://dx.doi.org/10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havli J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nature Protocols. 2007;1(6):2856–2860. doi: 10.1038/nprot.2006.468. http://dx.doi.org/10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Slawson C, Hart GW. O-GlcNAc signalling: implications for cancer cell biology. Nature Reviews Cancer. 2011;11(9):678–684. doi: 10.1038/nrc3114. http://dx.doi.org/10.1038/nrc3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow CM, Senior A, Gerace L. Monoclonal antibodies identify a group of nuclear pore complex glycoproteins. The Journal of Cell Biology. 1987;104(5):1143–1156. doi: 10.1083/jcb.104.5.1143. http://dx.doi.org/10.1083/jcb.104.5.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannhauser TW, Konishi Y, Scheraga HA. Sensitive quantitative analysis of disulfide bonds in polypeptides and proteins. Analytical Biochemistry. 1984;138(1):181–188. doi: 10.1016/0003-2697(84)90786-3. http://dx.doi.org/10.1016/0003-2697(84)90786-3. [DOI] [PubMed] [Google Scholar]
- Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Analytical Chemistry. 2003;75(8):1895–1904. doi: 10.1021/ac0262560. http://dx.doi.org/10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- Trinidad JC, Barkan DT, Gulledge BF, Thalhammer A, Sali A, Schoepfer R, Burlingame AL. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Molecular & Cellular Proteomics. 2012;11(8):215–229. doi: 10.1074/mcp.O112.018366. http://dx.doi.org/10.1074/mcp.O112.018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsarbopoulos A, Karas M, Strupat K, Pramanik BN, Nagabhushan TL, Hillenkamp F. Comparative mapping of recombinant proteins and glycoproteins by plasma desorption and matrix-assisted laser desorption/ionization mass spectrometry. Analytical Chemistry. 1994;66(13):2062–2070. doi: 10.1021/ac00085a022. http://dx.doi.org/10.1021/ac00085a022. [DOI] [PubMed] [Google Scholar]
- Vocadlo DJ, Hang HC, Kim EJ, Hanover JA, Bertozzi CR. A chemical approach for identifying O-GlcNAc-modified proteins in cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(16):9116–9121. doi: 10.1073/pnas.1632821100. http://dx.doi.org/10.1073/pnas.1632821100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosseller K, Trinidad JC, Chalkley RJ, Specht CG, Thalhammer A, Lynn AJ, Burlingame AL. O-linked N-acetylglucosamine proteomics of postsynaptic density preparations using lectin weak affinity chromatography and mass spectrometry. Molecular & cellular proteomics: MCP. 2006;5(5):923–934. doi: 10.1074/mcp.T500040-MCP200. http://dx.doi.org/10.1074/mcp.T500040-MCP200. [DOI] [PubMed] [Google Scholar]
- Wang AC, Jensen EH, Rexach JE, Vinters HV, Hsieh-Wilson LC. Loss of O-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(52):15120–15125. doi: 10.1073/pnas.1606899113. http://dx.doi.org/10.1073/pnas.1606899113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Udeshi ND, O’Malley M, Shabanowitz J, Hunt DF, Hart GW. Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry. Molecular & cellular proteomics: MCP. 2010;9(1):153–160. doi: 10.1074/mcp.M900268-MCP200. http://dx.doi.org/10.1074/mcp.M900268-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan SA, Hart GW. Identification of O‐GlcNAc sites on proteins. Methods in Enzymology. 2006;415:113–133. doi: 10.1016/S0076-6879(06)15008-9. http://dx.doi.org/10.1016/s0076-6879(06)15008-9. [DOI] [PubMed] [Google Scholar]
- Whiteheart SW, Passaniti A, Reichner JS, Holt GD, Haltiwanger RS, Hart GW. Glycosyltransferase probes. Methods in Enzymology. 1989;179:82–95. doi: 10.1016/0076-6879(89)79116-3. http://dx.doi.org/10.1016/0076-6879(89)79116-3. [DOI] [PubMed] [Google Scholar]
- Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nature Methods. 2009;6(5):359–362. doi: 10.1038/nmeth.1322. http://dx.doi.org/10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Yang X, Ongusaha PP, Miles PD, Havstad JC, Zhang F, So WV, Evans RM. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature. 2008;451(7181):964–969. doi: 10.1038/nature06668. http://dx.doi.org/10.1038/nature06668. [DOI] [PubMed] [Google Scholar]
- Yang Y, Verhelst SHL. Cleavable trifunctional biotin reagents for protein labelling, capture and release. Chemical Communications. 2013;49(47):5366. doi: 10.1039/c3cc42076k. http://dx.doi.org/10.1039/c3cc42076k. [DOI] [PubMed] [Google Scholar]
- Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, 3rd, Hsieh-Wilson LC. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. 2012;337(6097):975–980. doi: 10.1126/science.1222278. http://dx.doi.org/10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Vocadlo DJ. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nature Chemical Biology. 2008;4(8):483–490. doi: 10.1038/nchembio.96. http://dx.doi.org/10.1038/nchembio.96. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Vocadlo DJ. O-GlcNAc and neurodegeneration: biochemical mechanisms and potential roles in Alzheimer’s disease and beyond. Chemical Society Reviews. 2014;43(19):6839–6858. doi: 10.1039/c4cs00038b. http://dx.doi.org/10.1039/c4cs00038b. [DOI] [PubMed] [Google Scholar]
- Zaro BW, Batt AR, Chuh KN, Navarro MX, Pratt MR. The small molecule 2-Azido-2-deoxy-glucose is a metabolic chemical reporter of O-GlcNAc modifications in mammalian cells, revealing an unexpected promiscuity of O-GlcNAc transferase. ACS Chemical Biology. 2017 doi: 10.1021/acschembio.6b00877. http://dx.doi.org/10.1021/acschembio.6b00877. [DOI] [PMC free article] [PubMed]
- Zaro BW, Yang YY, Hang HC, Pratt MR. Chemical reporters for fluorescent detection and identification of O-GlcNAc-modified proteins reveal glycosylation of the ubiquitin ligase NEDD4-1. Proc Natl Acad Sci U S A. 2011;108(20):8146–8151. doi: 10.1073/pnas.1102458108. http://dx.doi.org/10.1073/pnas.1102458108. [DOI] [PMC free article] [PubMed] [Google Scholar]