

Abstract
Tuberous sclerosis complex (TSC) is a pediatric disorder of dysregulated growth and differentiation caused by loss of function mutations in either the TSC1 or TSC2 genes, which regulate mTOR kinase activity. To study aberrations of early development in TSC, we generated induced pluripotent stem cells using dermal fibroblasts obtained from patients with TSC. During validation, we found that stem cells generated from TSC patients had a very high rate of integration of the reprogramming plasmid containing a shRNA against TP53. We also found that loss of one allele of TSC2 in human fibroblasts is sufficient to increase p53 levels and impair stem cell reprogramming. Increased p53 was also observed in TSC2 heterozygous and homozygous mutant human stem cells, suggesting that the interactions between TSC2 and p53 are consistent across cell types and gene dosage. These results support important contributions of TSC2 heterozygous and homozygous mutant cells to the pathogenesis of TSC and the important role of p53 during reprogramming.
Introduction
Tuberous sclerosis complex (TSC, OMIM #613254) is a genetic disorder of pediatric onset characterized by benign tumor growths (hamartomas) in multiple organ systems including the brain, kidney, heart, skin, and lungs (1). The most debilitating symptoms in TSC are a consequence of brain involvement, including a high rate of epilepsy, autism spectrum disorder, and learning disabilities (1). TSC is caused by a loss of function mutation in either the TSC1 or TSC2 genes, which encode the proteins hamartin and tuberin, respectively (2,3). These proteins bind together and also associate with TBC1D7 to negatively regulate mTOR (mammalian/mechanistic target of rapamycin) kinase complex 1 (mTORC1). Genetic loss of TSC1 or TSC2 leads to elevated and constitutively active mTORC1 signaling that impacts multiple processes including cell growth and protein translation. The discovery that hamartin/tuberin regulates mTORC1 kinase activity led to the use of mTORC1 inhibitors for treatment of various aspects of TSC. Although mTORC1 inhibitors effectively impact some aspects of the disease (e.g. subependymal giant cell astrocytomas [SEGA], angiomyolipomas [AML], and lymphangioleiomyomatosis [LAM]), reversal of growth is usually static and is restricted to the treatment period, pointing to the need for a better understanding of mechanisms controlling cell growth and differentiation (4–8).
Hamartomas in TSC are generally thought to arise from a ‘second-hit’ somatic mutation in the other allele of TSC1 or TSC2, resulting in unhindered growth but leaving the surrounding heterozygous tissue functionally normal. While this model is supported by existing data in kidney and lung, molecular analyses of cortical hamartomas in the brain (‘tubers’) demonstrate few if any cells with loss of heterozygosity (9–12). Furthermore, imaging and pathologic data indicate abnormalities also exist in non-hamartomatous regions including decreased gray matter volume, abnormal white matter tracts, focal dyslamination, and dysplastic neurons (13–17). Recent studies using human embryonic and induced pluripotent stem cells also provide support for a haploinsufficient phenotype (18,19). These results indicate an important role for heterozygous mutations of the TSC1 and TSC2 genes during human development.
Developmental brain abnormalities have been detected in TSC patients as early as 20 weeks of gestation suggesting an important role for hamartin and tuberin in early prenatal development (20–23). To better understand the impact of TSC2 loss on development, we generated induced pluripotent stem cells (iPSCs) using human dermal fibroblasts obtained from TSC patients and control volunteers. We reprogrammed primary fibroblasts using established episomal methods employing three plasmids expressing OCT4, KLF4, SOX2, L-MYC, LIN28, and a shRNA knockdown targeted to TP53 (24). Knocking down p53 enhances reprogramming efficiency and survival; however, the mTOR pathway is also known to interact with p53. p53 inhibits mTORC1 signaling, notably upregulating TSC2, but the role of mTORC1 in the regulation of p53 is less clear (25–30). Existing studies using cells with loss of TSC1 or TSC2, including TSC patient AML samples, found mTORC1 activation of p53 with increased apoptosis or senescence (31–36). As most studies were done in the context of homozygous loss of TSC1 or TSC2, it is unclear how heterozygous loss of TSC2 in patient fibroblasts might affect p53 and stem cell reprogramming. We now show that loss of one copy of TSC2 is sufficient to increase p53 levels and inhibit reprogramming to iPSCs. These results indicate critical interactions between hamartin/tuberin, mTOR, and p53 to regulate stem cell reprogramming, cell maintenance, and cell death.
Results
TSC patient fibroblasts harbor heterozygous mutations leading to nonsense mediated decay of TSC2
To develop a human in vitro model of TSC, we derived primary dermal fibroblast cultures from multiple patients with TSC. The diagnosis of TSC was based on clinical presentation, genetic testing, and imaging studies (Table 1). Fibroblasts were isolated from patients’ normal-appearing skin or from TSC associated skin lesions Shagreen patches or hypopigmented macules, (also known as ‘ash leaf spots’). The original attempt was to acquire fibroblasts with either heterozygous or homozygous genetic mutations in TSC1 or TSC2 reasoning that normal appearing skin had a germline mutation whereas TSC associated skin lesions also had a second somatic mutation. These Tuberous Sclerosis Patient (TSP) cells were later sequenced using highly redundant exome sequencing to identify pathogenic mutations in either TSC1 or TSC2. For all TSP lines, we identified only heterozygous mutations of either TSC1 or TSC2 regardless of whether the fibroblasts were obtained from normal appearing skin or skin lesions (data not shown). Thirty-three percent of the tested lines contained no identifiable mutation in TSC1 or TSC2, broadly consistent with clinical testing for patients with TSC (37). As mutations in TSC2 are more common than TSC1 and patients with mutations in TSC2 tend to have more severe disease (38–41), we selected three well characterized patient lines with nonsense mutations in TSC2 (denoted as TSP20, TSP23, TSP31) (Table 1). As premature stop codon mutations usually lead to nonsense mediated decay of RNA transcripts, we sequenced mRNA from these patient lines. Only the wild-type mRNA was detectable by sequencing in TSP20, TSP23, and TSP31 fibroblast lines (Fig. 1A). Consistent with these sequencing results, mRNA levels of TSC2 and expression of tuberin protein in TSC2 heterozygous mutant fibroblast lines were both reduced by approximately one-half compared with control fibroblasts (Fig. 1B and C, P = 0.039 and P = 0.016). As tuberin is an important negative regulator of mTORC1 we next measured phospho-S6, a downstream target of mTORC1. We did not observe increased mTORC1 activity in fibroblasts with or without nutrient starvation (data not shown).
Table 1.
TSC patient clinical history, biopsy information, and genotyping
| Patient | Gene | DNA (NG_005895.1) | Protein (NP_000539.2) | Exon | Age of Epilepsy Onset | Autism | Intellec Disability | Age at dermal biopsy | Biopsy skin lesion |
|---|---|---|---|---|---|---|---|---|---|
| TSP20 | TSC2 | g.11900T>A | p.Cys203Ter | 6/41 | 6 months | Y | Y | 6 years | Hypopigmented macule |
| TSP23 | TSC2 | g.34854C>T | p.Arg1032Ter | 26/41 | 6 months | Y | Y | 2 years | Hypopigmented macule |
| TSP31 | TSC2 | g.40172C>T | p.Gln1419Ter | 33/41 | 2 months | Y | Y | 12 years | Shagreen patch |
Figure 1.

Heterozygous nonsense mutations in TSC2 result in reduced tuberin mRNA levels and protein. (A) Sequencing confirms single nucleotide changes causes premature stop codons in TSC2+/− fibroblasts. DNA sequencing reveals wild type and mutant allele but only wild-type allele was detected in mRNA. (B) TSC1 and TSC2 mRNA quantified by qPCR in control and TSC patient (TSP) fibroblasts (n = 3 average of four experimental replicates; P = 0.039, t-test). (C) Tuberin and hamartin protein quantified by immunoblot (n = 3; P = 0.016, t-test).
Selective retention of reprogramming plasmid with shRNA against TP53
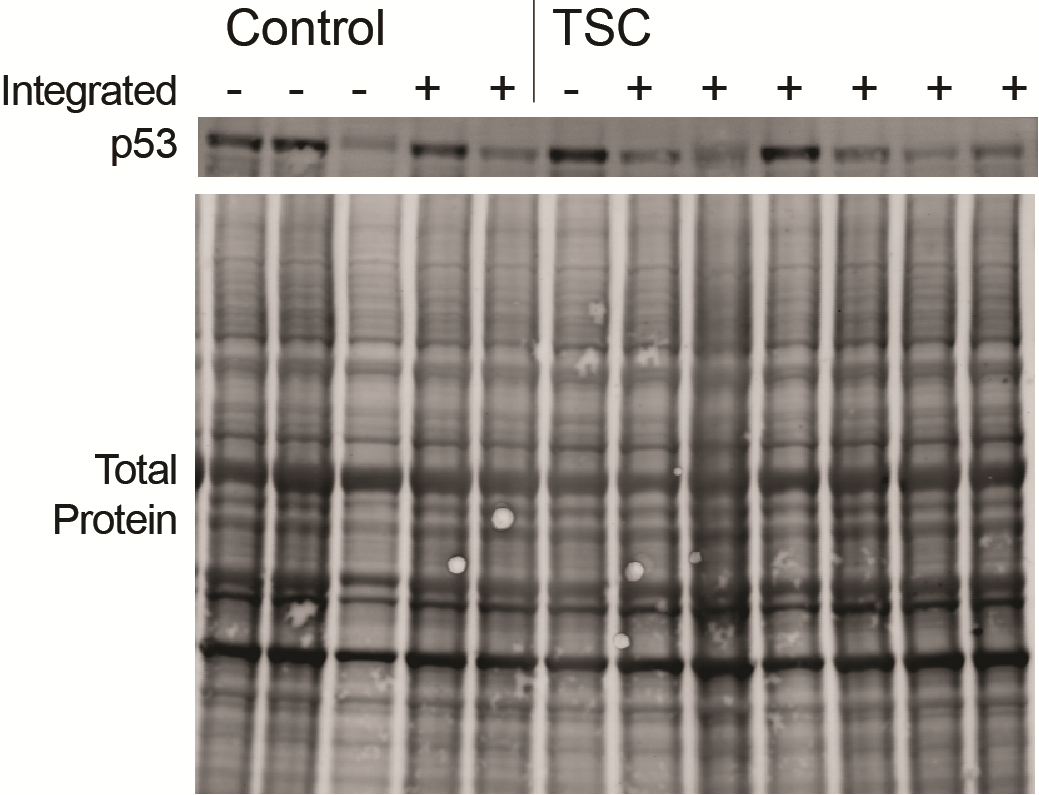
We next reprogrammed control (obtained from healthy unrelated volunteers) and TSP fibroblast lines to iPSCs by transfecting episomal plasmids expressing the reprogramming genes OCT4, KLF4, SOX2, L-MYC, LIN28, as well as a shRNA knockdown targeted to TP53 (24). All TSP and control iPSC lines were validated as pluripotent both by expression of established stem cell markers and the ability to differentiate to all three germ layers (Supplementary Material, Fig. S1). The reprogramming plasmids used are expected to remain episomal in the cytoplasm and then be lost from emerging iPSCs through the repeated cell division and passaging required for iPSC generation. As the original publication from the Yamanaka laboratory reported occasional genomic integration of the reprogramming plasmids (24), all iPSC lines were checked for plasmid integration using a PCR assay. Although control lines showed the expected low rate of integration (20%) (24,42), TSP lines showed a marked increase in the incidence of integration of one or more of the plasmids (80%, Fig. 2A, P = 0.023). To determine whether this was consistent across multiple rounds of reprogramming, we also included several additional iPSC lines from controls and patients with TSC that were not as fully characterized (Supplementary Material, Table S3). The high rate of plasmid retention was an unexpected finding that has not been observed while reprogramming any of the other genetic disorders modeled with iPSCs in our laboratory or those of collaborators (>100 independent iPSC lines made to date, data not shown). In TSP lines, this could be explained by an increased tendency to integrate any exogenous DNA or may reflect a requirement for sustained expression from one or more of the plasmid-encoded genes to drive cellular proliferation and survival during the stress of reprogramming. To distinguish these possibilities, we designed PCR primer sequences to unambiguously identify each plasmid used for reprogramming. In the control-integrated lines, one of the two integrated control lines integrated only the OCT4/shTP53 plasmid and the other had integrated more than one plasmid. In contrast, all of the TSP iPSC lines integrated only a single plasmid, the one that expresses both OCT4 and the shRNA directed against TP53 (Fig. 2B). Studies from our group and others supporting a complex interaction between tuberin, mTOR, and p53 led us to hypothesize that p53 signaling would be altered in cells from patients with TSC (31–36). To determine whether the incorporated shRNA to TP53 was expressed and functioning, we measured p53 protein levels in non-integrated control, integrated control, non-integrated TSP and integrated TSP iPSC lines. For the integrated TSP lines, p53 protein levels trended down but non-significantly reduced to 65% of control non-integrated iPSC levels (Fig. 2C, P = 0.259). Furthermore, integrated iPSCs displayed increased proliferation and survival following single-cell suspension, which is consistent with decreased p53 levels (data not shown). Given a recent report showing that human stem cells acquire mutations in p53 in culture, we further sequenced 16 identified ‘hot spots’ in our original fibroblasts as well as several iPSC lines. We did not identify any mutations in p53 that might explain the increased rate of integration in the TSC patient lines (43).
Figure 2.

Increased integration of reprogramming plasmids in TSP iPSC lines. (A) PCR for continued presence of the reprogramming plasmids at passage ≥10. Multiple iPSC clones generated from five individual patients and five individual controls were analysed. Plasmid bands were detectable in eight of ten TSC2 heterozygous mutant clones analysed and two of ten control clones from control volunteers (P = 0.023, Fisher‘s exact test). (B) PCR to distinguish between the three reprogramming plasmids (C) Protein levels of p53 were measured by immunoblot in non-integrated or integrated control and TSP iPSC lines (average of 2–3 experimental replicates; P = 0.259, t-test). See also Supplementary Material, Figure S2.
Increased levels of p53 in TSC2 heterozygous mutant fibroblasts
In normally growing ‘unstressed’ cells, p53 is a transcription factor that is rapidly ubiquitinated by MDM2 and degraded. However, following DNA stress, p53 is stabilized in the nucleus where it regulates gene transcription. We examined p53 levels and found expression in both TSP and control fibroblasts were barely detectable by immunoblotting and immunofluorescence. We tested whether p53 activity was increased in TSP fibroblasts after DNA damage. Control and heterozygous TSC patient fibroblasts were exposed to UV light, and 24 h later nuclear p53 was measured by immunofluorescence. The mean fluorescent intensity of p53 was significantly higher in TSC2 heterozygous mutant fibroblasts compared with controls (Fig. 3A, P = 0.012). To test whether the increase in p53 was due to increased mTORC1 activity, we treated fibroblasts with the mTORC1 inhibitor rapamycin beginning 24 h before and continuing 24 h after UV challenge. Rapamycin treatment at concentrations near the IC50 (0.2 nM) decreased nuclear p53 levels in both control and TSP cells (Fig. 3A, P = 0.008). With rapamycin treatment, the p53 signal decreased in both wildtype and TSP cells, and in TSP cells was reduced to the level of untreated control cells (Fig. 3A). This pattern is consistent with an interaction between TSC2, mTORC1 and p53.
Figure 3.

TSC patient fibroblasts display increased p53 in response to UV light. (A) Three control and two TSC patient fibroblast lines were challenged with UV light and then immunostained for p53. Cells were treated with 0.2 nM rapamycin for 24 h before through 24 h after UV challenge. Mean fluorescent intensity of p53 was quantified in nuclei defined by Hoechst staining. Nuclear p53 was analysed by two-way ANOVA in UV-treated cells (n = 2-3; interaction P = 0.532, genotype P = 0.012, rapamycin treatment P = 0.008; Bonferroni post-test control UV vs TSP UV P < 0.01). (B) Fibroblast lines were exposed to UV light and protein isolated at 0, 8 16, and 24 h post-exposure was analysed by immunoblot. (n = 3, average of 3–4 experimental replicates; two-way ANOVA). See also Supplementary Material, Figure S3.
To determine the impact of TSC2 heterozygous mutations on the response to DNA damage, p53 levels in control and TSP fibroblasts were measured at 0, 8, 16, and 24 h following UV challenge. p53 levels rapidly increased with TSP fibroblasts upregulating p53 significantly more relative to controls (Fig. 3B, P = 0.035). Tuberin protein levels were also significantly increased in response to UV challenge, consistent with previous studies (Fig. 3B, P < 0.0001, time) (28,29). However, the relative response was similar in both control and TSP fibroblasts, suggesting the wild-type copy of TSC2 is appropriately upregulated in TSC2 heterozygous mutant fibroblasts following UV exposure. To further assess p53 activity and function, we measured p21, a cyclin-dependent kinase inhibitor whose levels are regulated by p53. Levels of p21 increased post-UV challenge and TSP lines displayed increased p21 relative to controls (Fig. 3B, P = 0.01). mTORC1 signaling, as measured by phospho-S6 levels, showed a small initial increase at 8 h before normalizing to pre-challenge levels; however, no difference between control and TSP lines was observed (Fig. 3B). The activity of mTORC2, as measured by pAktSer473, decreased following UV challenge but there was, again, no difference between control and TSP fibroblasts for genotype but there was for time (Fig. 3B, P = 0.836 and P = 0.001).
Impaired reprogramming and cell death in TSC2 heterozygous mutant cells
p53 has been reported to be increased during cellular reprogramming, likely via the DNA damage pathway, and knocking down p53 enhances reprogramming efficiency (24,44–47). We hypothesized that the increased p53 levels seen in TSC2 heterozygous mutant fibroblasts would impair reprogramming efficiency. To test this, we embarked on further rounds of reprogramming using two control and three TSP fibroblast lines with either the same three programming plasmids used previously or by using the same genes except omitting the shRNA cassette directed against TP53. Three weeks following plasmid reprogramming, we stained the resulting iPSC colonies for alkaline phosphatase (AP) as a marker of pluripotency and quantified the number of positive AP colonies (Fig. 4A). Reprogramming efficiency was significantly reduced in TSC2 heterozygous mutant fibroblasts compared with controls reprogrammed with shTP53 (Fig. 4A, P = 0.015). We found reprogramming efficiency correlated inversely with the expected p53 levels: the number of AP+ colonies was highest in control fibroblasts reprogrammed with the shTP53 and lowest in TSC2 heterozygous mutant fibroblasts reprogrammed without the shTP53 plasmid. As AP is an early marker of pluripotency, we also picked and expanded several iPSC colonies from each experiment and stained them for the more mature stem cell marker TRA-1-60. We found TRA-1-60 positive colonies from all control and TSP lines reprogrammed with or without shTP53 (data not shown). Given the known role for p53 in regulating apoptosis, we next hypothesized that increased p53 in our TSP fibroblasts would lead to an increased rate of apoptosis during reprogramming. We first measured apoptosis in fibroblasts by Annexin V staining 24 h after UV challenge. We found a significant increase in the percentage of Annexin V positive apoptotic cells in TSP fibroblast lines (Fig. 4B, P < 0.05). To determine whether mTORC1 signaling contributes to this finding, we next treated fibroblasts with rapamycin for a total of 48 h, starting 24 h before UV challenge and 24 h after. There was a significant interaction between rapamycin treatment and genotype (Fig. 4C, P = 0.012). Rapamycin had divergent effects on apoptosis in control and TSP fibroblasts. While rapamycin significantly increased apoptosis in control fibroblasts, there was a trend towards decreased apoptosis in TSP fibroblasts (Fig. 4C, P < 0.05).
Figure 4.

Impaired reprogramming and increased apoptosis in TSC patient fibroblasts. (A) Control (CA, CC) and patient (T20, T31, T23) fibroblasts were reprogrammed with the three plasmids including either the Oct4 plasmid containing shRNA to TP53 or the Oct4 plasmid alone. Pluripotent colonies were stained for alkaline phosphatase. Alkaline phosphatase positive colonies were counted using gray scale images in ImageJ (n = 2 controls and 3 patients, average of 1–2 wells per individual; P = 0.015, t-test). (B) Fibroblast lines were challenged with UV light and apoptosis measured by Annexin V staining by flow cytometry. Annexin V versus propidium iodide is plotted for untreated and UV challenged fibroblasts; control (blue) and TSP (green) samples have been combined for display purposes. (n = 3 controls and 2 TSP, average of 2 experimental replicates; interaction P = 0.048, genotype P = 0.189, UV treatment P = 0.007; Bonferroni post-test control UV vs TSP UV P < 0.05). (C) Fibroblasts were treated with 0.2 nM rapamycin or DMSO for a total of 48 h, starting 24 h before UV challenge and ending 24 h after. Apoptosis was measured by Annexin V staining by flow cytometry. (n = 3, average of 2 experimental replicates; interaction P = 0.012, genotype P = 0.678, rapamycin treatment P = 0.032; Bonferroni post-test Control Vehicle vs Rapa P < 0.05).
Possible mechanisms of increased levels of p53 in TSC2 heterozygous mutant cells
We next sought to determine the molecular mechanism of increased p53 protein in TSC2 heterozygous mutant fibroblasts. We first measured TP53 mRNA levels and found no increase in TP53 mRNA at baseline or following challenge with UV light (Fig. 5A and B). We instead observed decreased TP53 mRNA in TSP cells following challenge with UV light (Fig. 5B, P = 0.025). As alternative splicing results in multiple TP53 isoforms (48), we designed primers for both the 3’ end (Fig. 5A and B) and the 5’ end (Supplementary Material, Fig. S4) of the full-length transcript and found similar results. We next used the protease inhibitor MG132 to inhibit degradation of p53 in order to isolate the contribution of translation to increased p53 protein levels. Control and TSP fibroblasts were treated with MG132 for 6 h with or without UV light challenge. There was, again, no difference in p53 protein levels between patient and control fibroblasts after 6 h of MG132, with or without UV light challenge (Fig. 5C). Degradation of p53 protein is tightly regulated by ubiquitination by MDM2, which is activated by phosphorylation at serine residue 166. There were no differences seen in pMDM2Ser166 levels and it was appropriately upregulated in response to increased p53 levels in both controls and TSP lines (Fig. 3B). We next tested the stability of p53 protein by treating cells with the translation inhibitor cycloheximide. Fibroblasts were challenged with UV light 24 h prior to treatment with cycloheximide and protein samples were taken before and after 1 h of cycloheximide treatment. There was no difference in the relative protein remaining following treatment supporting a similar degradation rate of p53 protein in control and TSP fibroblasts (Fig. 5D).
Figure 5.

Stability, mRNA and translation of p53 in TSC2+/− fibroblasts. (A) mRNA levels of TP53 in fibroblasts were measured by qPCR at baseline using primers directed to the 3’ end of the gene. Samples were normalized to ACTIN and PGK1. (n = 3, 4 technical replicates; P = 0.334, t-test) (B) Fibroblasts were challenged with UV light. Levels of TP53 mRNA were measured 24 h later by qPCR using primers directed to the 3’ end (n = 3, 4 technical replicates; P = 0.023, t-test) (C) Fibroblasts were treated with the proteasome inhibitor MG132 for 6 h with or without UV challenge. p53 protein levels were analysed by immunoblot. (D) Fibroblasts were challenged with UV light and 15 h later treated with the translation inhibitor cycloheximide (100 μM) for 2 h. P53 protein levels were analysed by immunoblot. See also Supplementary Material, Figure S4.
p53 is increased in TSC2 heterozygous and homozygous mutant iPSCs
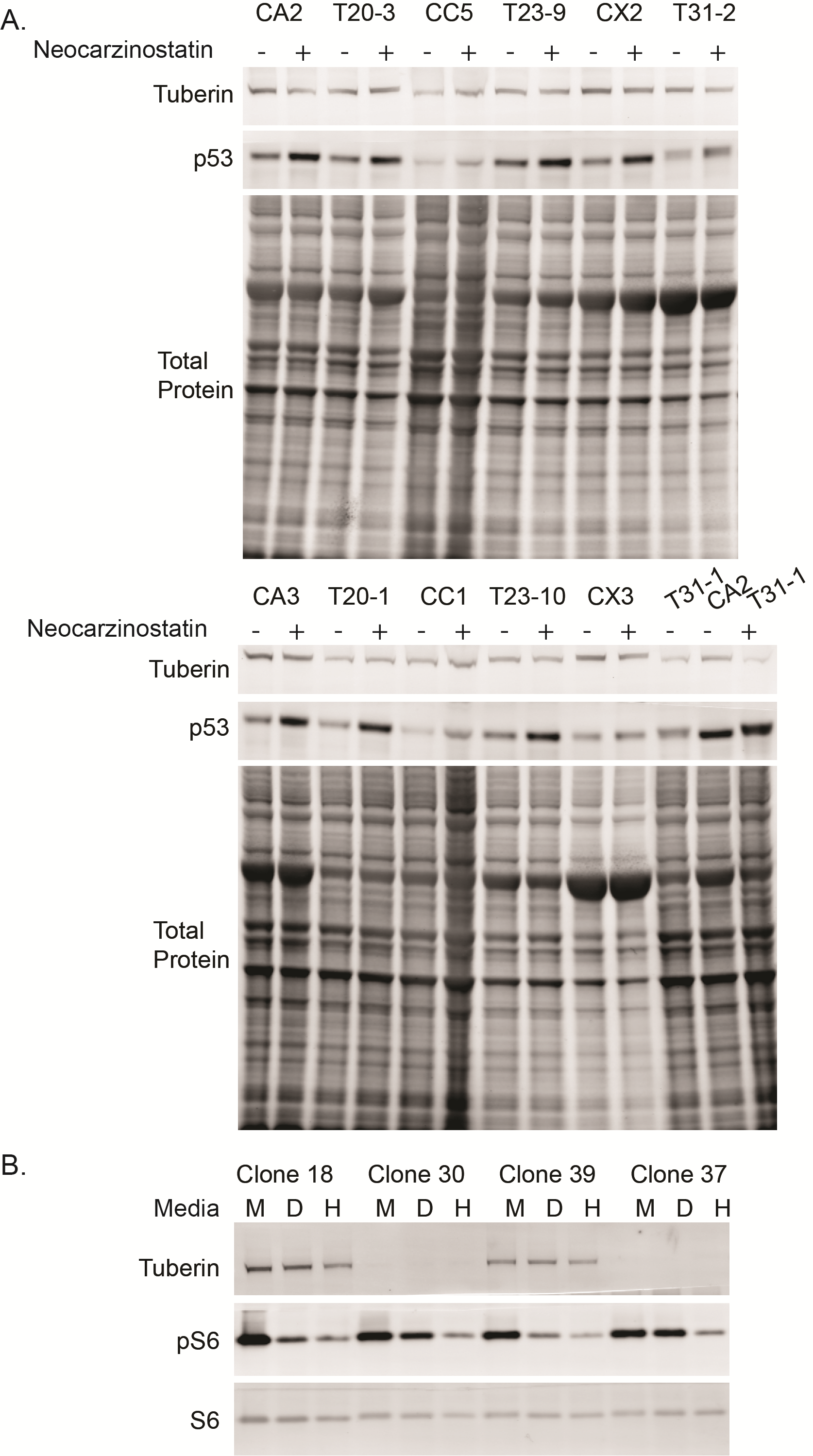
To confirm the interaction between tuberin and p53 was not limited to one cell type, we also measured p53 levels in additional TSC2 heterozygous iPSC lines generated using non-integrating Sendai viruses. Consistent with our results in heterozygous fibroblasts, mTORC1 activity was not elevated in TSC2 heterozygous TSP iPSCs with or without nutrient starvation (data not shown). Following 1 h of treatment with the DNA damaging agent neocarzinostatin, p53 was significantly elevated in TSC2 heterozygous mutant cells (Fig. 6A, P = 0.04). To further analyse the interaction between tuberin and p53, we also generated TSC2 homozygous mutant human iPSCs using CRISPR-Cas9 technology (49). At baseline, p53 is significantly elevated in TSC2 homozygous mutant iPSCs compared with isogenic control lines (Fig. 6B, P = 0.03). TSC2 homozygous mutant lines at baseline have unchanged phospho-S6, phospho-4EBP1, and phospho-AktSer473 levels (Fig. 6B). However, as iPSCs are maintained in mTeSR media that is nutrient-rich, these conditions likely do not reflect cell responses to changing stresses and metabolic demands. To address this point, we nutrient starved cells for 2.5 h in DMEM without glucose or serum. Wildtype cells decreased mTORC1 signaling appropriately, reflecting a switch in cellular metabolism from anabolic to catabolic. In contrast, TSC2 homozygous cells appeared unable to inhibit mTORC1 signaling with pS6 levels remaining high (Fig. 6C, P = 0.0002) However, when cells were starved in media without amino acids (HBSS), which signal to mTORC1 through Rag GTPases rather than tuberin/hamartin (50), both wildtype and TSC2 homozygous mutant cells appropriately inhibited mTORC1 signaling (Fig. 6C). We again measured p53 stability but found no difference between isogenic controls and TSC2 homozygous iPSC lines (data not shown). Treatment with the protease inhibitor MG132 to isolate the contribution of transcription and translation to the p53 phenotype also did not reveal a difference between isogenic controls and TSC2 homozygous iPSC lines (data not shown). Treatment of the iPSCs with the DNA damaging agent neocarzinostatin for 1–2 h, increased p53 levels in both control and mutant lines. Homozygous mutant TSC2 homozygous iPSCs also displayed increased p53 levels relative to control lines (data not shown).
Figure 6.

Increased p53 in heterozygous and homozygous TSC2 mutant stem cells. (A) Non-integrated iPSC lines generated from TSC2 heterozygous patient fibroblast lines treated with the DNA damaging agent neocarzinostatin for 1 h show increased p53 relative to controls. (n = 6, 1–3 experimental repeats per sample; P = 0.04 t-test) (B) Protein levels of p53 and mTORC1/mTORC2-related proteins were measured at baseline in homozygous knockout and control stem cells (n = 14, 2 clones with 7 experimental repeats; P = 0.03 t-test). (C) Homozygous TSC2−/− and isogenic control stem cells were nutrient starved for 2.5 h in DMEM without glucose or HBSS media. Protein levels of pS6 were elevated in TSC2−/− iPSCs following nutrient starvation. (n = 4, 2 clones with 2 experimental repeats; P = 0.0002 t-test) See also Supplementary Material, Figure S5.
Discussion
Our initial intent was to generate heterozygous and homozygous TSC2 mutant iPSCs from TSC patient fibroblasts to study the impact of gene dosage on somatic cell reprogramming and subsequent differentiation to various lineages impacted in TSC. This approach was designed to study the current favored ‘two hit’ model of TSC where the initial genetic event is a germline mutation in either TSC1 or TSC2 followed by a second somatic mutation in the other allele of TSC1 or TSC2. This model predicts that normal appearing tissues in patients with TSC to be heterozygous whereas any TSC associated focal abnormalities including skin lesions to be due to homozygous mutations. Despite deep sequencing of multiple primary lines, we were unable to find any homozygous mutant fibroblasts; all lines studied had only heterozygous mutations of TSC2 and tuberin was clearly present albeit reduced. Consistent with genotyping results, no difference in mTORC1 signaling was observed in patient fibroblast cells. This suggests that homozygous fibroblasts do not survive during the transition to fibroblast culture or do not contribute significantly to hypopigmented macules or Shagreen patches. A recent publication reported that focal areas of hypopigmentation in patients with TSC result from loss of heterozygosity in melanocytes rather than fibroblasts (51).
Unexpectedly, by reprogramming TSC2 patient derived fibroblasts to make iPSCs, we also found strong evidence for an important role for p53. This supports a model of TSC2 haploinsufficiency contributing to cell dysfunction and clinical manifestations. Previous reports support a connection between loss of TSC1 or TSC2 and increased p53, but the nature of this interaction remains unclear. Potential mechanisms include S6 kinase mediated phosphorylation and inhibition of MDM2, decreased Akt mediated phosphorylation and activation of MDM2, increased translation of MDM2-inhibitor ARF, or increased p53 translation or transcription (32,36,52–54). The mTOR/p53 pathways may act together within a negative feedback loop where increased mTORC1 leads to increased p53 and sensitivity to DNA damage in proliferating cells. Completing this potential feedback loop, increased p53 activity in response to DNA damage or other cellular stress may inhibit mTORC1 to decrease the potential for excessive proliferation. Fibroblasts increase tuberin levels in response to DNA damaging agents; however, in the presence of a heterozygous mutation in TSC2, p53 levels increased above the usual response seen in control cells, suggesting that this critical feedback loop is disrupted. This may then contribute to one paradox seen in TSC, that despite having mutations in the mTORC1 pathway, cancer is very rarely seen.
The mTORC1 inhibitor, rapamycin, inhibited p53 accumulation following DNA damage, supporting an interaction between these two pathways. However, no difference in mTORC1 signaling was observed in heterozygous fibroblasts; such differences may be below the level of current detection, require environmental/nutrient stresses or mTORC1-independent mechanisms may further contribute to heterozygous phenotypes. Interestingly, rapamycin had a diverging effect on apoptosis in control and TSP fibroblasts. There was a trend towards enhancing apoptosis in control fibroblasts and inhibiting apoptosis in TSP fibroblasts. Although loss of TSC2 or TSC1 sensitizes cells to cell death (31–34,36), the effect of rapamycin on apoptosis and p53 is more complicated and appears to be cell type specific [reviewed in (55)]. Our results suggest that rapamycin sensitizes cells to apoptosis in wildtype fibroblasts but in the setting of heterozygous loss of TSC2, which have increased p53 and apoptosis, the dominant effect of rapamycin is to correct the phenotype and inhibit apoptosis.
Tuberin and p53 may intersect in multiple ways; however, we did not find a significant increase in p53 stability, phospho-MDM2 protein, p53 transcription/translation, or TP53 mRNA levels in TSC2 heterozygous mutant cells. Some of these changes may simply be below the sensitivity of current methods of detection. As treatment with MG132 induces other stress pathways and TSC2 homozygous mutant cells in general appear to be more sensitive to ER stress via proteasome inhibition (56), it is possible that these other functions are obscuring any difference in p53 transcription or translation between controls and TSC patient cells. In the future, translation can be measured more directly using polysome profiling or radioactive labeling of proteins. Interestingly, we observed a decrease in TP53 mRNA levels in TSC2 heterozygous mutant fibroblasts following UV exposure. There is conflicting evidence for p53 autoregulating its own transcription either positively or negatively (57–60). Our observation of inversely correlated patterns of TP53 mRNA and protein levels with heterozygous loss of TSC2 is most consistent with negative autoregulation.
While a circuitous route was required to finally generate them, we now report the first generation of isogenic iPSCs with homozygous knock-out of the TSC2 gene. Using CRISPR-Cas to generate isogenic lines, we were able to confirm that loss of tuberin leads to increased p53, both at baseline and in response to DNA damage. Further, we used a non-integrating Sendai viral method to reprogram heterozygous TSP fibroblasts to iPSCs. Using heterozygous TSP Sendai-generated iPSCs, we were also able to confirm that the increased p53 seen in patient fibroblasts in response to DNA damage also applied to the pluripotent stem cell state.
Relative levels of p53 during reprogramming tightly correlate with reprogramming efficiency; knocking down or knocking out p53 enhances formation of pluripotent colonies while activating p53 with the pharmacological agent Nutlin inhibits reprogramming (44,45). However, permanent loss of p53 enhances reprogramming at the expense of later genomic instability (46). The addition of shRNA to TP53 to the episomal plasmids attempts to circumvent this issue by transiently knocking down p53 only during reprogramming. Then, with the expected loss of episomal plasmids during cell division, p53 levels are restored to normal levels following the reprogramming process. Reprogramming efficiency we found correlated inversely with the theoretical levels of p53 in our fibroblasts, which was consistent with previous publications demonstrating increased reprogramming efficiency with knock-down or mutation of p53. However, the shRNA against p53 was not sufficient to completely rescue the TSC reprogramming phenotype. The transition of fibroblasts from partially reprogrammed cell to stable stem cell is regulated by a complicated network of changes and there likely are p53-independent alterations in the reprogramming process resulting from the heterozygous loss of TSC2. Future studies will dissect the contribution of TSC2 to each stage of reprogramming and stem cell maintenance. One previous study observed a tendency of integrated iPSC lines to integrate the OCT4/shTP53 plasmid preferentially, presumably due to a growth advantage conferred by continued inhibition of p53 (42). We hypothesize that a similar mechanism is occurring in heterozygous cells but at a higher rate due to increased p53 in TSC patient derived cells allowing continual p53 knockdown during the reprogramming process.
Finally, our results suggest that loss of one copy of TSC2 is sufficient to alter the cellular response to DNA damage and stress during reprogramming. The increased apoptosis in TSP derived lines then may further explain why cancer is quite rare in TSC and why any malignant transformation is usually associated with p53 deletion (34,61). We further speculate that the increased rate of apoptosis may also clarify why neural cells with second hits are either very rare or undetectable in the cortical lesions, (tubers) of patients with TSC. p53 also affects neural stem cell differentiation and self-renewal (62–66) and we further hypothesize that even small disruptions in p53 pathways, like those seen in our heterozygous cells, could alter neural development. Future studies will address regulation of cell death and neural development by gene dosage and non-cell-autonomous effects using control, heterozygous and now homozygous mutant TSC2 stem cell lines.
Materials and Methods
Cell culture and stem cell reprogramming
Fibroblasts were isolated from control volunteers (male, age 24 years, CA; female, age 18 years, CC; male, age 25 years, CX) or TSC patients (ages 2–12 years, female Table 1) from normal-appearing skin and TSC associated skin lesions (Shagreen patches or hypopigmented macules). Fibroblasts were cultured in DMEM with 10% FBS, 1% non-essential amino acids, and 1% pen-strep.
Fibroblasts were reprogrammed by transfection of cells using the Neon system (Invitrogen) with plasmids expressing KLF4, SOX2, OCT4, L-MYC and LIN28 with or without shRNA to TP53 (Addgene #27076, 27077, 27078, 27080) according to previously published protocols (24). Cells were switched to TeSR-E7 media (StemCell Technologies) 48 h post-transfection. Three weeks post-transfection, some cultures were stained for alkaline phosphatase expression following fixation with 2% paraformaldehyde (Millipore SCR004). Pluripotent colonies positive for alkaline phosphatase were quantified in ImageJ (ver1.50e) by analysing grayscale images, setting a threshold for positive staining, and counting the number of colonies with area ≥2 pixels (67). Concurrently colonies from parallel plates were picked and transferred to Matrigel treated plates and grown in mTeSR1 media (Corning; StemCell Technologies).
Sendai reprogramming was performed using the CytoTune iPS 2.0 Sendai Kit (ThermoFisher) in feeder-free conditions according to manufacturer‘s instructions. Nascent stem cells were passed to Matrigel and grown in mTeSR after one week.
Protein stability and translation
For p53 stability and translation analyses, cells were treated with 100 μM cycloheximide (Sigma 7698) or 1 μM MG132 (Millipore 474790). Appropriate doses were determined by in-cell Western assay to measure the dose at which puromycin incorporation into the nascent transcript was inhibited or p53 was stabilized.
Sequencing
Mutation screening of TSC fibroblast samples was performed using highly redundant exome sequencing in candidate genes. Genomic DNA was isolated and sheared, then exon-containing DNA captured using SureSelect XT (Agilent). Next generation sequencing was done on an Illumina HiSeq2000 (Solexa) through the Vanderbilt Genome Sciences Resource. Variants were submitted to the TSC2 Leiden Open Variant Database (http://www.LOVD.nl/TSC2). Confirmatory sequencing was performed on DNA or RNA isolated from fibroblast cultures using QuickExtract Buffer (epicentre) or Qiagen RNeasy or Invitrogen PureLink RNA kits. RNA was converted to cDNA using random hexamers and ThermoScientific Superscript First-Strand Synthesis Kit. Genomic or mRNA sequences were amplified using primers listed in Supplementary Material, Table S1. PCR amplicons were isolated using QiaQuick PCR Purification Kit.
Quantitative PCR
RNA was isolated from fibroblast cultures as above and converted to cDNA. Quantitative PCR was performed using Life technologies SYBR Green Master Mix and run on a QuantStudio real-time qPCR machine through the Vanderbilt VANTAGE Core. Samples were run in duplicate or quadruplicate technical replicates and normalized to levels of ACTIN and PGK1.
Stem cell validation
Integration was assessed in cell lines after ≥10 passages post-reprogramming using primers for the WPRE and EBNA segments of the Yamanaka reprogramming plasmids (24). Primers immediately flanking the shp53 segment were used to distinguish between the three plasmids; presence or absence of the shp53 plasmid was determined based on the size of the amplified fragment.
Pluripotency was assessed by the ability to differentiate to all three germline lineages using the hPSC Taqman Scorecard (ThermoFisher A15870). iPSCs were differentiated as embryoid bodies for one week in DMEM/F12 glutamax with 20% knockout serum replacement, non-essential amino acids and 55 μM β-mercaptoethanol. RNA was then isolated and converted to cDNA using the High-capacity Reverse Transcription Kit (ThermoFisher 4374966). Samples were analysed by qPCR on the Applied Biosystems 7900HT through the Vanderbilt VANTAGE Core.
Karyotype analysis was performed using standard techniques (Genetics Associates, Inc. Nashville, TN).
Immunoblotting
Protein was isolated by lysing and scraping cells in RIPA buffer plus protease inhibitor cocktails 2 and 3 and phosphatase inhibitor cocktail (Sigma P5726, P0044, P8340). Samples were rotated for 30 min at 4 °C, sonicated and then run on 4–12% NuPAGE BisTris gels in MOPS buffer. Gels were transferred to PVDF membrane at 95 V for 90 min using the Bio-Rad Mini Trans-Blot Cell. Membranes were incubated overnight in primary antibody diluted in 5% BSA. Membranes were then incubated for 1 h at room temperature in secondary antibody and imaged on an Odyssey Li-Cor machine. Samples were normalized to actin or total protein levels using Li-Cor REVERT stain.
Immunofluorescence
Cells were fixed in 4% paraformaldehyde for 20 min at room temperature, followed by 3×5 min PBS washes. For intracellular antibodies, cells were then permeabilized with 0.1% Triton in PBS for 20 min at room temperature and blocked in 0.1% Triton plus 5% normal goat serum for 1 h at room temperature. For extracellular antibodies, cells were blocked in 5% normal goat serum for 1 h at room temperature. Cells were then incubated in primary antibody rocking overnight at 4 °C. Following 5×10 min PBS washes, cells were incubated with secondary antibody for 3 h at room temperature.
Nuclear p53 was measured by immunofluorescence followed by analysis of images with ImageJ (67). A nuclear mask was generated using Hoechst staining and mean fluorescent intensity of p53 was measured using the outlines.
DNA damage
Primary fibroblasts were passed to 6 well plates at 100, 000 cells per well or 96 well plates at 5000 cells per well. Plates were then exposed to ∼24 J/m2 (30 s of UV light G51T8 bulb 4.9 Watts with wavelength 254 nm at a distance of approximately 0.706 meters). Analyses were conducted 24 h later unless otherwise noted.iPSC lines were treated with 50 ng/ml of neocarzinostatin for 1–2 h. Optimal neocarzinostatin doses were determined using a dose-response curve measured by in-cell Western assay for p53 response.
Flow cytometry
Single cell suspensions of fibroblasts were obtained by trypsinizing adherent cells and collecting supernatant, followed by filtration through a 40 μm filter. Cells were then counted, stained with 5 μl Annexin V (AF488, ThermoScientific A1320) and 0.5 μl of 1 mg/ml propidium iodide (Sigma P4864) per 105 cells and immediately analysed by flow cytometry. Cells were gated to exclude debris and doublets.
CRISPR
Doxycycline-inducible Cas9 iPSCs were generated and donated by the Conklin lab (49). Cells were treated with 2 μM doxycycline for 24 h prior to lipofection with Alt-R crRNA: trRNA complexes (IDTDNA). Guide sequences were designed using the online Zhang Lab CRISPR Design Tool (68). Guide RNA mixed with lipofectamine (1.5 μl 3 μM crRNA: trRNA, 0.3 μl lipofectamine 3000, 48.2 μl Opti-MEM) was added to a 96 well plate coated in Matrigel. Doxycycline-treated cells were isolated with Accutase and 40, 000 cells were then added to each well. Two days later cells were expanded to a 6 well plate for subsequent colony picking or DNA isolation for T7 endonuclease analysis.
Statistics
All statistical analyses were conducted using GraphPad PRISM. Individual patient fibroblast lines or iPSC clones are treated as independent n. All other experimental or technical replicates are considered not independent and are averaged to obtain the final value for each patient clone/line. Four isogenic clones (two TSC2+/+, two TSC2−/−) were generated using CRISPR-Cas9. For experiments involving isogenic lines, both experimental and clone replicates are considered independent repeats and are shown in the data.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank the Vanderbilt University Medical Center Flow Cytometry Shared Resource and the Vanderbilt VANTAGE Core provided technical assistance for this work.
Conflict of Interest statement. None declared.
Funding
National Institute of Neurological Disorders and Stroke (RO1 NS078289 to KCE), Public Health Service award T32 GM07347 from the National Institute of General Medical Studies for the Vanderbilt Medical Scientist Training Program (LCA). The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). VANTAGE is supported in part by CTSA Grant (5UL1 RR024975-03), the Vanderbilt Ingram Cancer Center (P30 CA68485), the Vanderbilt Vision Center (P30 EY08126), and National Institutes of Health/National Center for Research Resources (G20 RR030956).
References
- 1. Sahin M., Henske E.P., Manning B.D., Ess K.C., Bissler J.J., Klann E., Kwiatkowski D.J., Roberds S.L., Silva A.J., Hillaire-Clarke C.S.. et al. (2016) Advances and Future Directions for Tuberous Sclerosis Complex Research: Recommendations From the 2015 Strategic Planning Conference. Pediatr. Neurol., 60, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. European. (1993) Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell, 75, 1305–1315. [DOI] [PubMed] [Google Scholar]
- 3. van Slegtenhorst M., de Hoogt R., Hermans C., Nellist M., Janssen B., Verhoef S., Lindhout D., van den Ouweland A., Halley D., Young J.. et al. (1997) Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science, 277, 805–808. [DOI] [PubMed] [Google Scholar]
- 4. Bissler J.J., McCormack F.X., Young L.R., Elwing J.M., Chuck G., Leonard J.M., Schmithorst V.J., Laor T., Brody A.S., Bean J.. et al. (2008) Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N. Engl. J. Med., 358, 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cardamone M., Flanagan D., Mowat D., Kennedy S.E., Chopra M., Lawson J.A. (2014) Mammalian target of rapamycin inhibitors for intractable epilepsy and subependymal giant cell astrocytomas in tuberous sclerosis complex. J. Pediatr., 164, 1195–1200. [DOI] [PubMed] [Google Scholar]
- 6. Franz D.N., Leonard J., Tudor C., Chuck G., Care M., Sethuraman G., Dinopoulos A., Thomas G., Crone K.R. (2006) Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann. Neurol., 59, 490–498. [DOI] [PubMed] [Google Scholar]
- 7. Sasongko T.H., Ismail N.F., Zabidi-Hussin Z. (2016) Rapamycin and rapalogs for tuberous sclerosis complex. Cochrane Database Syst. Rev., 7, CD011272.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McCormack F.X., Inoue Y., Moss J., Singer L.G., Strange C., Nakata K., Barker A.F., Chapman J.T., Brantly M.L., Stocks J.M.. et al. (2011) Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N. Engl. J. Med., 364, 1595–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Niida Y., Stemmer-Rachamimov A.O., Logrip M., Tapon D., Perez R., Kwiatkowski D.J., Sims K., MacCollin M., Louis D.N., Ramesh V. (2001) Survey of somatic mutations in tuberous sclerosis complex (TSC) hamartomas suggests different genetic mechanisms for pathogenesis of TSC lesions. Am. J. Hum. Genet., 69, 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Henske E.P., Scheithauer B.W., Short M.P., Wollmann R., Nahmias J., Hornigold N., van Slegtenhorst M., Welsh C.T., Kwiatkowski D.J. (1996) Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am. J. Hum. Genet., 59, 400–406. [PMC free article] [PubMed] [Google Scholar]
- 11. Qin W., Chan J.A., Vinters H.V., Mathern G.W., Franz D.N., Taillon B.E., Bouffard P., Kwiatkowski D.J. (2010) Analysis of TSC cortical tubers by deep sequencing of TSC1, TSC2 and KRAS demonstrates that small second-hit mutations in these genes are rare events. Brain Pathol., 20, 1096–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Martin K.R., Zhou W., Bowman M.J., Shih J., Au K.S., Dittenhafer-Reed K.E., Sisson K.A., Koeman J., Weisenberger D.J., Cottingham S.L.. et al. (2017) The genomic landscape of tuberous sclerosis complex. Nat. Commun., 8, 15816.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chandra P.S., Salamon N., Nguyen S.T., Chang J.W., Huynh M.N., Cepeda C., Leite J.P., Neder L., Koh S., Vinters H.V.. et al. (2007) Infantile spasm-associated microencephaly in tuberous sclerosis complex and cortical dysplasia. Neurology, 68, 438–445. [DOI] [PubMed] [Google Scholar]
- 14. Ridler K., Bullmore E.T., De Vries P.J., Suckling J., Barker G.J., Meara S.J., Williams S.C., Bolton P.F. (2001) Widespread anatomical abnormalities of grey and white matter structure in tuberous sclerosis. Psychol. Med., 31, 1437–1446. [DOI] [PubMed] [Google Scholar]
- 15. Lewis W.W., Sahin M., Scherrer B., Peters J.M., Suarez R.O., Vogel-Farley V.K., Jeste S.S., Gregas M.C., Prabhu S.P., Nelson C.A. 3rd. et al. (2013) Impaired language pathways in tuberous sclerosis complex patients with autism spectrum disorders. Cereb. Cortex, 23, 1526–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peters J.M., Sahin M., Vogel-Farley V.K., Jeste S.S., Nelson C.A. 3rd, Gregas M.C., Prabhu S.P., Scherrer B., Warfield S.K. (2012) Loss of white matter microstructural integrity is associated with adverse neurological outcome in tuberous sclerosis complex. Acad. Radiol., 19, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marcotte L., Aronica E., Baybis M., Crino P.B. (2012) Cytoarchitectural alterations are widespread in cerebral cortex in tuberous sclerosis complex. Acta Neuropathol., 123, 685–693. [DOI] [PubMed] [Google Scholar]
- 18. Costa V., Aigner S., Vukcevic M., Sauter E., Behr K., Ebeling M., Dunkley T., Friedlein A., Zoffmann S., Meyer C.A.. et al. (2016) mTORC1 inhibition corrects neurodevelopmental and synaptic alterations in a human stem cell model of tuberous sclerosis. Cell Rep., 15, 86–95. [DOI] [PubMed] [Google Scholar]
- 19. Li Y., Cao J., Chen M., Li J., Sun Y., Zhang Y., Zhu Y., Wang L., Zhang C. (2017) Abnormal neural progenitor cells differentiated from induced pluripotent stem cells partially mimicked development of TSC2 neurological abnormalities. Stem Cell Rep., 8, 883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsai V., Parker W.E., Orlova K.A., Baybis M., Chi A.W., Berg B.D., Birnbaum J.F., Estevez J., Okochi K., Sarnat H.B.. et al. (2014) Fetal brain mTOR signaling activation in tuberous sclerosis complex. Cereb. Cortex, 24, 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Prabowo A.S., Anink J.J., Lammens M., Nellist M., van den Ouweland A.M., Adle-Biassette H., Sarnat H.B., Flores-Sarnat L., Crino P.B., Aronica E. (2013) Fetal brain lesions in tuberous sclerosis complex: TORC1 activation and inflammation. Brain Pathol., 23, 45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park S.H., Pepkowitz S.H., Kerfoot C., De Rosa M.J., Poukens V., Wienecke R., DeClue J.E., Vinters H.V. (1997) Tuberous sclerosis in a 20-week gestation fetus: immunohistochemical study. Acta Neuropathol., 94, 180–186. [DOI] [PubMed] [Google Scholar]
- 23. Muhler M.R., Rake A., Schwabe M., Schmidt S., Kivelitz D., Chaoui R., Hamm B. (2007) Value of fetal cerebral MRI in sonographically proven cardiac rhabdomyoma. Pediatr. Radiol., 37, 467–474. [DOI] [PubMed] [Google Scholar]
- 24. Okita K., Matsumura Y., Sato Y., Okada A., Morizane A., Okamoto S., Hong H., Nakagawa M., Tanabe K., Tezuka K.. et al. (2011) A more efficient method to generate integration-free human iPS cells. Nat Methods, 8, 409–412. [DOI] [PubMed] [Google Scholar]
- 25. Cam M., Bid H.K., Xiao L., Zambetti G.P., Houghton P.J., Cam H. (2014) p53/TAp63 and AKT regulate mammalian target of rapamycin complex 1 (mTORC1) signaling through two independent parallel pathways in the presence of DNA damage. J. Biol. Chem., 289, 4083–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Budanov A.V., Karin M. (2008) p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell, 134, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stambolic V., MacPherson D., Sas D., Lin Y., Snow B., Jang Y., Benchimol S., Mak T.W. (2001) Regulation of PTEN transcription by p53. Mol. Cell, 8, 317–325. [DOI] [PubMed] [Google Scholar]
- 28. Feng Z., Zhang H., Levine A.J., Jin S. (2005) The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. U. S. A, 102, 8204–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Feng Z., Hu W., de Stanchina E., Teresky A.K., Jin S., Lowe S., Levine A.J. (2007) The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res., 67, 3043–3053. [DOI] [PubMed] [Google Scholar]
- 30. Horton L.E., Bushell M., Barth-Baus D., Tilleray V.J., Clemens M.J., Hensold J.O. (2002) p53 activation results in rapid dephosphorylation of the eIF4E-binding protein 4E-BP1, inhibition of ribosomal protein S6 kinase and inhibition of translation initiation. Oncogene, 21, 5325–5334. [DOI] [PubMed] [Google Scholar]
- 31. Zhang H., Cicchetti G., Onda H., Koon H.B., Asrican K., Bajraszewski N., Vazquez F., Carpenter C.L., Kwiatkowski D.J. (2003) Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J. Clin. Invest., 112, 1223–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miceli A.P., Saporita A.J., Weber J.D. (2012) Hypergrowth mTORC1 signals translationally activate the ARF tumor suppressor checkpoint. Mol. Cell. Biol., 32, 348–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pai G.M., Zielinski A., Koalick D., Ludwig K., Wang Z.Q., Borgmann K., Pospiech H., Rubio I. (2016) TSC loss distorts DNA replication programme and sensitises cells to genotoxic stress. Oncotarget, 7, 85365–85380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Habib S.L., Yadav A., Mahimainathan L., Valente A.J. (2011) Regulation of PI 3-K, PTEN, p53, and mTOR in Malignant and Benign Tumors Deficient in Tuberin. Genes Cancer, 2, 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wataya-Kaneda M., Kaneda Y., Hino O., Adachi H., Hirayama Y., Seyama K., Satou T., Yoshikawa K. (2001) Cells derived from tuberous sclerosis show a prolonged S phase of the cell cycle and increased apoptosis. Arch. Dermatol. Res., 293, 460–469. [DOI] [PubMed] [Google Scholar]
- 36. Lee C.H., Inoki K., Karbowniczek M., Petroulakis E., Sonenberg N., Henske E.P., Guan K.L. (2007) Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. embo J., 26, 4812–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rosset C., Netto C.B.O., Ashton-Prolla P. (2017) TSC1 and TSC2 gene mutations and their implications for treatment in Tuberous Sclerosis Complex: a review. Genet. Mol. Biol., 40, 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jansen F.E., Braams O., Vincken K.L., Algra A., Anbeek P., Jennekens-Schinkel A., Halley D., Zonnenberg B.A., van den Ouweland A., van Huffelen A.C.. et al. (2008) Overlapping neurologic and cognitive phenotypes in patients with TSC1 or TSC2 mutations. Neurology, 70, 908–915. [DOI] [PubMed] [Google Scholar]
- 39. Dabora S.L., Jozwiak S., Franz D.N., Roberts P.S., Nieto A., Chung J., Choy Y.S., Reeve M.P., Thiele E., Egelhoff J.C.. et al. (2001) Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet., 68, 64–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kothare S.V., Singh K., Chalifoux J.R., Staley B.A., Weiner H.L., Menzer K., Devinsky O. (2014) Severity of manifestations in tuberous sclerosis complex in relation to genotype. Epilepsia, 55, 1025–1029. [DOI] [PubMed] [Google Scholar]
- 41. Devlin L.A., Shepherd C.H., Crawford H., Morrison P.J. (2006) Tuberous sclerosis complex: clinical features, diagnosis, and prevalence within Northern Ireland. Dev. Med. Child Neurol., 48, 495–499. [DOI] [PubMed] [Google Scholar]
- 42. Schlaeger T.M., Daheron L., Brickler T.R., Entwisle S., Chan K., Cianci A., DeVine A., Ettenger A., Fitzgerald K., Godfrey M.. et al. (2015) A comparison of non-integrating reprogramming methods. Nat. Biotechnol., 33, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Merkle F.T., Ghosh S., Kamitaki N., Mitchell J., Avior Y., Mello C., Kashin S., Mekhoubad S., Ilic D., Charlton M.. et al. (2017) Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature, 545, 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kawamura T., Suzuki J., Wang Y.V., Menendez S., Morera L.B., Raya A., Wahl G.M., Izpisua Belmonte J.C. (2009) Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature, 460, 1140–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hong H., Takahashi K., Ichisaka T., Aoi T., Kanagawa O., Nakagawa M., Okita K., Yamanaka S. (2009) Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature, 460, 1132–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marion R.M., Strati K., Li H., Murga M., Blanco R., Ortega S., Fernandez-Capetillo O., Serrano M., Blasco M.A. (2009) A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature, 460, 1149–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li H., Collado M., Villasante A., Strati K., Ortega S., Canamero M., Blasco M.A., Serrano M. (2009) The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature, 460, 1136–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marcel V., Dichtel-Danjoy M.L., Sagne C., Hafsi H., Ma D., Ortiz-Cuaran S., Olivier M., Hall J., Mollereau B., Hainaut P.. et al. (2011) Biological functions of p53 isoforms through evolution: lessons from animal and cellular models. Cell Death Differ., 18, 1815–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mandegar M.A., Huebsch N., Frolov E.B., Shin E., Truong A., Olvera M.P., Chan A.H., Miyaoka Y., Holmes K., Spencer C.I.. et al. (2016) CRISPR interference efficiently induces specific and reversible gene silencing in human iPSCs. Cell Stem Cell, 18, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sancak Y., Peterson T.R., Shaul Y.D., Lindquist R.A., Thoreen C.C., Bar-Peled L., Sabatini D.M. (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science, 320, 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cao J., Tyburczy M.E., Moss J., Darling T.N., Widlund H.R., Kwiatkowski D.J. (2017) Tuberous sclerosis complex inactivation disrupts melanogenesis via mTORC1 activation. J. Clin. Invest., 127, 349–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lai K.P., Leong W.F., Chau J.F., Jia D., Zeng L., Liu H., He L., Hao A., Zhang H., Meek D.. et al. (2010) S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. embo J., 29, 2994–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Goudarzi K.M., Nister M., Lindstrom M.S. (2014) mTOR inhibitors blunt the p53 response to nucleolar stress by regulating RPL11 and MDM2 levels. Cancer Biol. Ther, 15, 1499–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ogawara Y., Kishishita S., Obata T., Isazawa Y., Suzuki T., Tanaka K., Masuyama N., Gotoh Y. (2002) Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem, 277, 21843–21850. [DOI] [PubMed] [Google Scholar]
- 55. Castedo M., Ferri K.F., Kroemer G. (2002) Mammalian target of rapamycin (mTOR): pro- and anti-apoptotic. Cell Death Differ, 9, 99–100. [DOI] [PubMed] [Google Scholar]
- 56. Kang Y.J., Lu M.K., Guan K.L. (2011) The TSC1 and TSC2 tumor suppressors are required for proper ER stress response and protect cells from ER stress-induced apoptosis. Cell Death Differ, 18, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Benoit V., Hellin A.C., Huygen S., Gielen J., Bours V., Merville M.P. (2000) Additive effect between NF-kappaB subunits and p53 protein for transcriptional activation of human p53 promoter. Oncogene, 19, 4787–4794. [DOI] [PubMed] [Google Scholar]
- 58. Ginsberg D., Mechta F., Yaniv M., Oren M. (1991) Wild-type p53 can down-modulate the activity of various promoters. Proc. Natl. Acad. Sci. U. S. A, 88, 9979–9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Deffie A., Wu H., Reinke V., Lozano G. (1993) The tumor suppressor p53 regulates its own transcription. Mol. Cell. Biol., 13, 3415–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hudson J.M., Frade R., Bar-Eli M. (1995) Wild-type p53 regulates its own transcription in a cell-type specific manner. DNA Cell Biol., 14, 759–766. [DOI] [PubMed] [Google Scholar]
- 61. Hayashi T., Koike K., Kumasaka T., Saito T., Mitani K., Terao Y., Ogishima D., Yao T., Takeda S., Takahashi K.. et al. (2012) Uterine angiosarcoma associated with lymphangioleiomyomatosis in a patient with tuberous sclerosis complex: an autopsy case report with immunohistochemical and genetic analysis. Hum. Pathol., 43, 1777–1784. [DOI] [PubMed] [Google Scholar]
- 62. Li Y., Stockton M.E., Bhuiyan I., Eisinger B.E., Gao Y., Miller J.L., Bhattacharyya A., Zhao X. (2016) MDM2 inhibition rescues neurogenic and cognitive deficits in a mouse model of fragile X syndrome. Sci. Trans. Med., 8, 336ra361.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tedeschi A., Di Giovanni S. (2009) The non-apoptotic role of p53 in neuronal biology: enlightening the dark side of the moon. EMBO Rep., 10, 576–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Meletis K., Wirta V., Hede S.M., Nister M., Lundeberg J., Frisen J. (2006) p53 suppresses the self-renewal of adult neural stem cells. Development, 133, 363–369. [DOI] [PubMed] [Google Scholar]
- 65. Mendrysa S.M., Ghassemifar S., Malek R. (2011) p53 in the CNS: Perspectives on development, stem cells, and cancer. Genes Cancer, 2, 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jacobs W.B., Kaplan D.R., Miller F.D. (2006) The p53 family in nervous system development and disease. J. Neurochem., 97, 1571–1584. [DOI] [PubMed] [Google Scholar]
- 67. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B.. et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O.. et al. (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol., 31, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.