

Abstract
Understanding the relationship between dendritic excitability and synaptic plasticity is vital for determining how dendrites regulate the input–output function of the neuron. Dendritic calcium spikes have been associated with the induction of long-term changes in synaptic efficacy. Here we use direct recordings from cerebellar Purkinje cell dendrites to show that synaptically activated local dendritic calcium spikes are potent triggers of cannabinoid release, producing a profound and short-term reduction in synaptic efficacy at parallel fiber synapses. Enhancing dendritic excitability by modulating dendritic large-conductance calcium-activated potassium (BK) channels improves the spread of dendritic calcium spikes and enhances cannabinoid release at the expense of spatial specificity. Our findings reveal that dendritic calcium spikes provide a local and tunable coincidence detection mechanism that readjusts synaptic gain when synchronous activity reaches a threshold, and they reveal a tight link between the regulation of dendritic excitability and the induction of synaptic plasticity.
Keywords: dendritic spike, endocannabinoid, BK channel, DSE, Purkinje cell, cerebellum
Introduction
The dendrites of mammalian neurons have been shown to express a variety of voltage-gated ion channels (Migliore and Shepherd, 2002), and, as a consequence, dendrites exhibit a range of excitable behavior (Häusser et al., 2000). One of the most prominent forms of dendritic electrogenesis is the dendritic calcium spike. Dendritic calcium spikes were originally described in cerebellar Purkinje cells both in vivo (Llinas et al., 1968) and in vitro (Llinas and Sugimori, 1980). They also have been reported in hippocampal (Wong et al., 1979) and neocortical pyramidal cells (Amitai et al., 1993; Schiller et al., 1997), where they have been shown to be triggered by sensory stimulation (Helmchen et al., 1999; Larkum and Zhu, 2002). Dendritic calcium spikes generally do not propagate faithfully to the soma (Llinas and Sugimori, 1980; Schiller et al., 1997) and do not trigger axonal AP generation reliably (Schiller et al., 1997; Larkum et al., 2001), suggesting that there may exist more local forms of readout.
Given that dendritic calcium spikes cause significant postsynaptic calcium influx (Ross and Werman, 1987; Miyakawa et al., 1992; Yuste et al., 1994; Schiller et al., 1997), one attractive possibility is that dendritic calcium spikes could act as triggers for local synaptic plasticity, which is thought to require significant elevations in postsynaptic calcium. In pyramidal cells the initiation of calcium spikes by strong synaptic stimulation has been linked to the induction of non-Hebbian long-term potentiation (Golding et al., 2002) or depression (Holthoff et al., 2004). Recently, a novel form of short-term plasticity at excitatory synapses, termed depolarization-induced suppression of excitation (DSE), has been demonstrated at parallel fiber (PF) synapses in cerebellar Purkinje cells (Kreitzer and Regehr, 2001). DSE induction depends on dendritic cannabinoid release, which appears to require high postsynaptic calcium elevations (Brenowitz and Regehr, 2003), and can be triggered by bursts of PF inputs that are associated with large local calcium signals in Purkinje cell dendrites (Brown et al., 2003). This raises the possibility that physiological patterns of stimulation may cause DSE via the triggering of dendritic calcium spikes. We therefore made direct patch-clamp recordings from Purkinje cell dendrites in conjunction with calcium imaging to investigate the link between dendritic calcium spikes and synaptic efficacy at PF synapses.
Materials and Methods
Standard techniques were used to prepare 250-μm-thick sagittal brain slices from the cerebellum of 18–25 d postnatal rats (Wang et al., 2000). Artificial CSF (ACSF) for slicing and recording contained the following (in mm): 125 NaCl, 26 NaHCO3, 25 glucose, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, and 1 MgCl2 (305 mOsm). All recordings were performed at 34 ± 1°C in the presence of 10 μm SR95531.
Simultaneous somatic and dendritic whole-cell patch-clamp recordings were made from Purkinje neurons under visual control with the use of differential interference contrast optics (DIC) (Stuart and Häusser, 1994). Dendritic recordings were made at distances of 90–218 μm from the soma, measured along the dendritic branches. Patch electrodes were made from borosilicate glass and pulled to a resistance of 3–6 MΩ (soma) and 6–10 MΩ (dendrite). Intracellular patch solution contained the following (in mm): 130 methanesulfonic acid, 7 KCl, 2 Na2ATP, 2 Mg-ATP, 0.5 Na2GTP, 0.05 mm EGTA, and 0.4% biocytin, pH-adjusted to 7.30 with KOH (285 mOsm). For the imaging experiments 200 μm fluo-5F and 1 μm Alexa 594 (both from Invitrogen, Carlsbad, CA) were added routinely to the intracellular solution. The cells were loaded through a somatic electrode (3–5 MΩ) for at least 30 min before it was removed. Then the dendrite was patched with a patch electrode (6–8 MΩ) containing the same intracellular solution, and at least 10 min was allowed for the dyes to reach a stable concentration before the experiments were begun. Double and triple whole-cell current-clamp recordings were made by using dual Multiclamp 700A and 700B amplifiers (Molecular Devices, Union City, CA). PF inputs were stimulated every 2 s (10–90 V; 0.1–0.2 ms) with ACSF-filled patch pipettes placed under visual control beneath the dendritic arbor, immediately adjacent to the most distal dendritic recording site. To monitor EPSP amplitude during DSE experiments, we stimulated PF inputs at 0.5 Hz before and after the induction protocol (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Climbing fiber (CF) input was stimulated by placing the stimulating electrode in the granule cell layer beneath the Purkinje cell body; the all-or-none nature of the CF response allowed stimulation intensity to be adjusted carefully to permit selective activation of the CF. Recordings were low-pass filtered at 8 kHz and sampled at 20–50 kHz with the use of an Instrutech (Port Washington, NY) ITC18 DAC board connected to an Apple G4 PowerMac running Axograph (Molecular Devices) and were analyzed with IgorPro (WaveMetrics, Lake Oswego, OR). The imaging experiments were performed using a Polychrome IV monochromator as a light source and an Imago-QE camera (both from TILL Photonics, Gräfelfing, Germany). Fluo-5F was excited at 492 nm, and Alexa 594 was excited at 570 nm. The excitation time was 25 ms, giving a sampling rate of 40 Hz. The data were acquired by using TILL VisION (TILL Photonics) on a PC and analyzed with ImageJ (http://rsb.info.nih.gov/ij/) and IgorPro. Methanesulfonic acid was obtained from Fluka BioChemika (Ronkonkoma, NY), penitrem A was obtained from Alomone Laboratories (Jerusalem, Israel), and other chemicals were obtained from Sigma (St. Louis, MO) or Tocris Bioscience (Ellisville, MO).
Figure 1.

Local calcium spikes in Purkinje cell dendrites triggered by PF stimulation. A, Simultaneous triple whole-cell dendritic and somatic recording from a Purkinje cell. An image of the biocytin-filled cell is superimposed on the DIC image of the slice taken at the end of the recording. The recording electrodes and the corresponding traces are color-coded: red for the distal dendrite (218 μm from the soma), blue for the more proximal dendrite (164 μm), and black for the soma. The stimulating electrode (stim; gray) was buried in the slice directly underneath the most distal dendritic recording electrode. B, Traces from the recording shown in A; raising the PF stimulation (arrow) intensity from 30 V (dashed lines) to 40 V (solid lines) resulted in the initiation of a dendritic calcium spike (asterisk) near the distal dendritic recording site. Note the small amplitude of backpropagating APs at the dendritic recording sites. C, Suprathreshold stimulation evoked calcium spikes at rest (spontaneous firing with no holding current, solid lines) that are blocked when the cell is hyperpolarized (dashed lines). D, Graph showing the nonlinear relationship between stimulation intensity and peak depolarization evoked by the PF stimulation. The depolarization was measured at both dendritic locations (red, distal; blue, proximal) as the difference between a 5 ms baseline before the stimulation and the peak voltage reached in a 10 ms window after the stimulation. Note the step-like increase in peak depolarization at the distal recording site associated with initiation of the dendritic calcium spike (^) and the strong attenuation of the calcium spike at the more proximal recording site (54 μm away from the distal dendritic recording).
The apparent voltage threshold for calcium spikes was determined by taking the second derivative of the dendritic voltage trace over a time window 10 ms after the stimulus. The voltage threshold was defined as described previously (Gasparini et al., 2004). Briefly, we took the second derivative of the dendritic voltage trace and measured the voltage value at the 20% point of the second positive peak that followed the stimulus (see Fig. 7H). The amplitude of backpropagating APs at distal dendritic recording sites was <5 mV (Stuart and Häusser, 1994), and backpropagating APs were excluded from analysis by inspection of the somatic trace. We measured the amplitude of EPSPs at −70 mV by injecting hyperpolarizing current. In some cases EPSPs were mimicked by injection of current waveforms shaped like EPSCs, consisting of a double-exponential function with τrise = 0.6 ms and τdecay = 6 ms. To mimic physiological PF trains, we based the dynamics of the EPSC amplitudes on the facilitation–depression model of Dittman and colleagues (Dittman et al., 2000). When the induction parameters were varied during the recording, trials were randomized. Significance was assessed by Student’s t test, and all data are given as average ± SEM.
Figure 7.
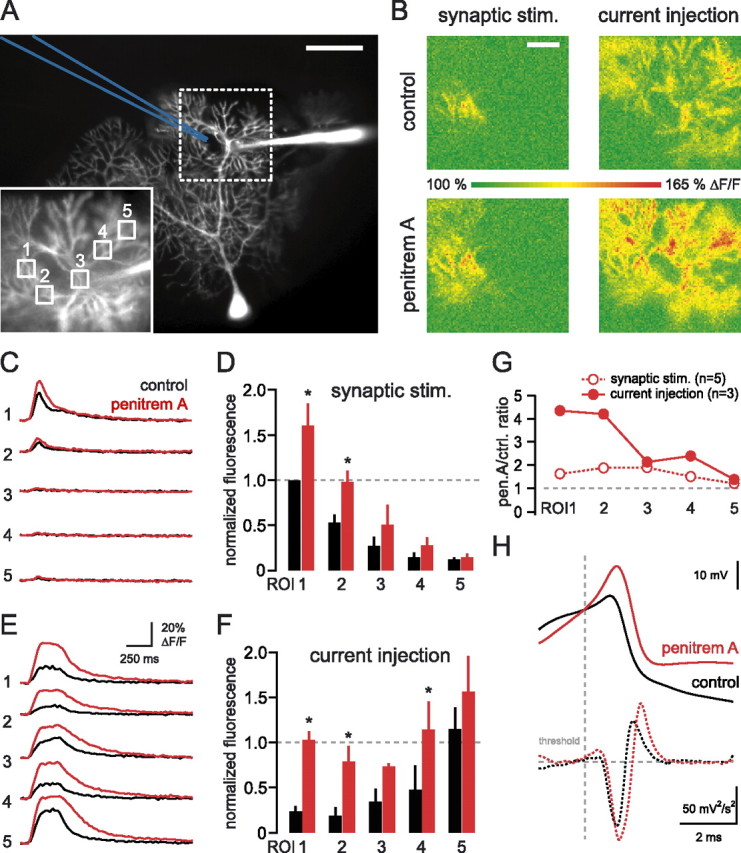
BK channels control dendritic calcium spike propagation. A, Dendritic recording from a Purkinje cell filled with Alexa 594 and fluo-5F, recorded 140 μm from the soma. The image was taken with Alexa fluorescence. The stimulating electrode is drawn in blue. Scale bar, 50 μm. Inset, High-magnification image of the area of interest (dotted box) with five ROIs indicated. B, Maximum intensity projections of image stacks acquired during synaptically or current injection-evoked calcium spikes in control ACSF and 100 nm penitrem A. Scale bar, 20 μm. C, Calcium transients recorded from the five respective ROIs in the inset of A during synaptically evoked calcium spikes before (black) and after the BK channels (red) were blocked. D, The similarly selected five ROIs were averaged over three cells and normalized to the synaptic maximum in control ACSF. E, Calcium transients recorded from the five respective ROIs in the inset of A during current injection-evoked calcium spikes before (black) and after (red) the BK channels (were blocked. F, The similarly selected five ROIs were averaged over several cells and normalized to the synaptic maximum in control ACSF. Blocking the BK channels clearly improved the spatial spread of calcium spikes. Asterisks (in D, F) denote statistical significance (p < 0.05); error bars indicate SEM. G, The change in the fluorescence signal after penitrem A application was quantified in every ROI under control conditions (open circles) and BK channel block (filled circles). H, Dendritic calcium spikes (aligned at threshold and averaged) evoked with current injection before (black) and after penitrem A application (red). The bottom panel shows the second derivative of the calcium spikes, with a dotted vertical gray line indicating the threshold (see Materials and Methods).
Results
Local dendritic calcium spikes activated by PF input
Simultaneous double or triple whole-cell patch recordings were made from the soma and dendrites of Purkinje cells in cerebellar slices (Fig. 1A). Double dendritic recording electrodes were placed on the same dendritic branch, separated by <75 μm (n = 5). PF synaptic input during spontaneous firing of Purkinje cells (Häusser and Clark, 1997) was activated by using a stimulation electrode located underneath the dendritic arbor, adjacent to the more distal dendritic recording electrode. Increasing the intensity of the stimulus produced a graded increase in the PF EPSP amplitude until a dendritic spike was triggered on top of the EPSP, identified by positive, regenerative deflections in membrane potential (see Materials and Methods). These events had a clear stimulation intensity threshold (Fig. 1B,D) and were abolished when the cell was hyperpolarized (Fig. 1C). The apparent voltage threshold of these dendritic spikes was –29 ± 2 mV (n = 14 cells), and the threshold amplitude of single dendritic EPSPs for triggering the dendritic spikes was 19.1 ± 2.7 mV (n = 10 cells; EPSPs measured at the dendritic recording site at –70 mV). To confirm that these dendritic spikes are calcium spikes, i.e., mediated predominantly by activation of voltage-gated calcium channels, we demonstrated that similar dendritic spikes evoked by using dendritic injection of EPSC-like waveforms were blocked by 200 μm Cd2+ (n = 5) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) (cf. Llinas and Sugimori, 1980).
Figure 2.

The amount of DSE depends on the number of dendritic calcium spikes. A, Dendritic recording 134 μm from the soma during DSE induction. Different stimulation intensities were used (black, 30 V; blue, 40 V; red, 50 V) during a train of 10 PF stimuli (arrowheads) at 100 Hz. Note the calcium spikes (asterisks) evoked at the higher stimulus intensities. B, Time course of changes in normalized dendritic PF EPSP amplitude (black, n = 4 trials; blue, n = 5 trials; red, n = 3 trials). All data are from the same cell as in A; error bars indicate SEM. C, Relationship between the number of dendritic calcium spikes during the train and the amount of DSE averaged across 12 cells. Open circles show the block of DSE by the CB1 antagonist AM251 (1 μm; n = 4 cells). The control data points were fit with a sigmoidal curve with baseline constrained to 1.
To determine the spatial spread of the calcium spikes in the dendritic tree, we compared the amplitude of synaptically evoked calcium spikes at the two dendritic recording sites. On average, the peak of calcium spikes decreased by 69 ± 3% over the average distance of 54 ± 11 μm (n = 5). By comparison, the peak amplitude of EPSPs (subthreshold for calcium spike generation) decayed by only 41 ± 3%, significantly less than attenuation of the calcium spikes (p < 0.005) over the same distance. These findings indicate that the dendritic calcium spikes activated by PF synaptic input are localized to near the site of synaptic stimulation.
Stimulus dependence of dendritic calcium spikes and DSE induction
We next investigated the dendritic voltage signals associated with induction of DSE at PF synapses. Previous work has shown that a train of PF stimuli can produce DSE of PF inputs (Brown et al., 2003). We made simultaneous somatic and dendritic recordings from spontaneously firing Purkinje cells during DSE induction (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), triggered by 10 PF stimuli delivered at 100 Hz (Brown et al., 2003). These experiments revealed that there was a stimulus-dependent threshold for the induction of DSE, which matched that observed for initiation of dendritic calcium spikes during the induction protocol. At low stimulation intensities no dendritic calcium spikes were evoked, and no DSE was observed (n = 12 cells) (Fig. 2A,B). However, when stimulation intensity was raised sufficiently to generate dendritic calcium spikes (Fig. 2A), DSE was always observed (n = 12) (Fig. 2B).
To demonstrate the link between dendritic calcium spike initiation and DSE induction, we plotted the number of calcium spikes detected in the dendritic recordings during the induction period against the resulting DSE (measured as the minimum normalized EPSP amplitude after induction) (Fig. 2C). When no dendritic calcium spikes were evoked, no DSE was observed (EPSP amplitude, 105 ± 3% of control). As more dendritic calcium spikes were recruited with increasing stimulus intensities, the degree of DSE was correlated with the number of calcium spikes evoked during induction in all cells (n = 12) (Fig. 2C, filled circles). This relationship could be described by a sigmoidal curve, with saturation of DSE being observed after five calcium spikes (no significant difference in the DSE value with larger numbers of calcium spikes; p0–2, 2–3, 4–5 < 0.05; p3–4, 5–6, 6–7 > 0.2). To assess the involvement of cannabinoid release in DSE triggered by calcium spikes, we blocked CB1 cannabinoid receptors by using N-(piperidin-1-yl)-5-(4- iodophenyl)-1-(2,4-dichlorophenyl)-4- methyl-1H-pyrazole-3-carboxamide (AM251; 1 μm). Under these conditions no change in EPSP amplitude was observed even when up to five dendritic calcium spikes were evoked by synaptic stimulation (EPSP amplitude, 101 ± 2% of control; n = 6; p = 0.6) (Fig. 2C). This indicates that dendritic calcium spikes cause DSE by triggering cannabinoid release.
Blocking dendritic calcium spikes by hyperpolarization prevents DSE
The activation of metabotropic glutamate receptors (mGluRs) has been shown to contribute to DSE activated by PF trains (Maejima et al., 2001; Brown et al., 2003). To separate the contributions of dendritic calcium spikes and mGluR activation during PF trains, we took advantage of the membrane potential dependence of dendritic calcium spikes. We used a stimulation strength for PF trains that was sufficient to trigger dendritic calcium spikes and cause DSE during spontaneous firing of the Purkinje cell (dendritic amplitude of the EPSP was 2.3 ± 0.2 mV at –70 mV) (Fig. 3A). We then hyperpolarized the cell with steady injected current. Hyperpolarization blocked the induction of calcium spikes and prevented the induction of DSE (Fig. 3B). On average, there was no significant DSE present at holding potentials of –70 and –90 mV (Fig. 3B). When the initiation of dendritic calcium spikes was titrated by carefully adjusting the membrane potential with injected current, the amount of DSE was dependent on the number of calcium spikes evoked during induction, with the relationship described by a sigmoidal curve (n = 7) (Fig. 3C).
Figure 3.

DSE and dendritic calcium spikes have a similar voltage threshold. A, Dendritic recording 108 μm from the soma. The same stimulation intensity was used to evoke 10 PF EPSPs at 100 Hz (arrowheads) when the dendrite was held at different membrane potentials (black, −90 mV; blue, −70 mV; red, no holding current, approximately −50 mV). Note that calcium spikes (asterisks) were observed only at the most depolarized potentials. B, Time course of changes in normalized dendritic PF EPSP amplitude (black, n = 3 trials; blue, n = 3 trials; red, n = 3 trials). All data are from the same cell as in A; error bars indicate SEM. C, Relationship between the number of calcium spikes during the train and the amount of DSE averaged across seven cells. The data points were fit with a sigmoidal curve with the baseline constrained to 1.
These findings indicate that the level of synaptic activation required for calcium spike induction is insufficient to trigger DSE via the mGluR pathway alone. The contribution of mGluRs to DSE induction could be revealed by increasing stimulus strength because of the strong dependence of mGluR activation on stimulus intensity (Batchelor and Garthwaite, 1997; Reichelt and Knöpfel, 2002). For stimuli generating a dendritic EPSP of 6.0 ± 0.9 mV at −70 mV, the selective and potent mGluR1 antagonist 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt; 100 μm) (Maejima et al., 2001; Brown et al., 2003) reduced the extent of DSE (29 ± 6% in control and 57 ± 13% in CPCCOEt; p < 0.009; n = 8). However, substantial DSE still remained in the presence of CPCCOEt, while dendritic calcium spikes were unaffected (6.9 ± 0.9 spikes in control and 6.9 ± 1.1 in CPCCOEt; p = 0.9). These findings indicate that the calcium spikes evoked by synaptic stimulation do not require mGluR1 activation to produce DSE and, thus, that synaptically evoked calcium spikes are sufficient to trigger DSE.
DSE induction is specific to dendritic calcium spikes triggered by PF input
These experiments indicate that synaptically evoked dendritic calcium spikes are necessary and sufficient for DSE induction by trains of PF inputs. To determine whether DSE is specific to dendritic calcium spikes triggered by PF input to the spiny branchlets, we examined whether calcium spikes activated by postsynaptic depolarization of the main dendrite alone also can trigger DSE. Dendritic calcium spikes were evoked in the absence of synaptic stimulation by injecting a train of EPSC waveforms via a dendritic pipette located on a main dendrite close to the synaptically activated spiny branchlets (Fig. 4A). Activation of a large number of such current-evoked dendritic calcium spikes (5.3 ± 0.5 spikes) did not change the amplitude of PF EPSPs activated by synaptic stimulation close to the dendritic recording site (98 ± 2% of control; p = 0.4; n = 6) (Fig. 4C). However, when the same synaptic input was used to activate dendritic calcium spikes (4.6 ± 0.4 spikes) (Fig. 4B), robust DSE was obtained (35 ± 9% of control; p < 0.0005; n = 6) (Fig. 4C). This indicates that dendritic calcium spikes triggered by PF input to a spiny branchlet under normal conditions are far more effective at triggering DSE than calcium spikes elicited by current injection on an adjacent main branch.
Figure 4.

Dendritic spikes evoked by current injection or CF synaptic input fail to trigger DSE. A, Dendritic recording 112 μm from the soma. Dendritic calcium spikes (asterisks) were evoked by injecting a train of EPSC waveforms (shown below the dendritic recording; see Materials and Methods). B, Dendritic calcium spikes (asterisks) evoked in the same cell by stimulating PFs adjacent to the recording electrode. C, Comparison of the effect of calcium spikes evoked by current injection (red) or PF stimulation (black) on PF EPSP amplitudes averaged across six cells. D, Dendritic recording 114 μm from the soma. Stimulating CF input evoked a complex spike, which was associated with three dendritic calcium spikes (asterisks). E, Dendritic recording from the same cell showing a PF train that evoked dendritic calcium spikes (asterisks). F, Time course of PF EPSP amplitude before and after the two different induction protocols (blue, CF stimulation; black, PF stimulation) averaged across six cells. C, F, Error bars indicate SEM.
We also examined whether CF synaptic input is sufficient to induce DSE at PF synapses, because CF activation has been shown to trigger dendritic calcium spikes and widespread calcium entry in the Purkinje cell dendritic tree (Llinas and Sugimori, 1980; Ross and Werman, 1987; Miyakawa et al., 1992). When activation of the CF (triggering 2.4 ± 0.2 dendritic calcium spikes) was used instead of a PF train, no DSE was observed (98 ± 6% of control; p = 0.3; n = 6) (Fig. 4D,F). In contrast, activation of calcium spikes with the same PF synapses (triggering 3.2 ± 0.4 calcium spikes) produced robust DSE (53 ± 3% of control; p < 0.0001; n = 6) (Fig. 4E,F). These findings indicate that DSE activated by calcium spikes is specific to calcium spikes that are evoked directly by PF inputs.
To explain why DSE is specific to calcium spikes evoked by PF synaptic input, we examined two hypotheses in detail. First, it is possible that presynaptic activity is required for the physiological induction of DSE (potentially mediated by presynaptic NMDA receptors at PF synapses) (Casado et al., 2000; Sjöström et al., 2003). Alternatively, calcium spikes evoked by dendritic current injection might be triggered at a different location from those evoked synaptically such that there is spatial segregation of the activated synapses and the injected current-evoked calcium spikes. As a consequence, the resulting release of endocannabinoids may not reach the activated synapses because of their highly restricted range of action (Brown et al., 2003).
Calcium spikes evoked by current injection are insufficient to trigger DSE but can act synergistically with synaptic stimulation
To test whether presynaptic activity is required during the induction of DSE by dendritic calcium spikes, we paired calcium spikes evoked by dendritic current injection with different intensities of synaptic stimulation. The stimulus train during induction always was delivered with the same intensity as the stimuli testing for synaptic strength in order to activate the same pool of PFs. Dendritic current injection capable of activating many calcium spikes, which did not induce DSE on its own (103 ± 4% of control; n = 4; p = 0.6) (see above), was paired with three different intensities of synaptic stimulation. When the evoked EPSPs were <2 mV, they failed to trigger calcium spikes and evoke DSE (106 ± 7% of control; p = 0.7; n = 4) when delivered in a train of 10 stimuli at 100 Hz. Combining the stimulus train with calcium spikes evoked by depolarizing current injection (Fig. 5A) also failed to induce DSE (105 ± 8% of control; p = 0.4; n = 4). This result shows that pairing postsynaptic calcium spikes triggered by dendritic current injection with presynaptic activity cannot rescue the induction of DSE. When the synaptically triggered EPSPs were between 2 and 5 mV in amplitude, the stimulation on its own resulted in calcium spikes and a moderate degree of DSE (77 ± 2% of control; n = 4; p < 0.005). Combining this synaptic stimulus with current injection-evoked calcium spikes increased the amount of DSE significantly (57 ± 8% of control; n = 4; p < 0.007) (different from synaptic stimulation alone; p < 0.005). Thus the amount of DSE evoked by synaptic stimulation can be increased supralinearly by additional postsynaptic depolarization, but only if the synaptic stimulation alone was able to trigger some degree of DSE. Using stronger synaptic activation, which triggered EPSPs >5 mV (14.7 ± 3 mV) and more than six calcium spikes, resulted in robust DSE (21 ± 3% of control; n = 4; p < 0.001). The pairing of this stimulation with current injection was unable to increase the amount of DSE that was triggered (23 ± 4% of control; n = 4; p < 0.003) (compared with stimulation alone: p = 0.7). Thus DSE triggered by synaptically evoked calcium spikes can be saturated such that additional depolarization fails to increase the amount of DSE.
Figure 5.

Presynaptic activity during calcium spikes evoked by current injection does not rescue DSE. A, Dendritically recorded calcium spikes (150 μm from the soma) during synaptic stimulation (black) and combined current injection and synaptic stimulation (red). The current injection was adjusted to evoke several calcium spikes. Shown in the bottom panel is the time course of normalized synaptic strength of four cells. Weak synaptic stimulation during current injection-evoked calcium spikes failed to depress the stimulated synapses. B, When the current injection (same as in A) was paired with stronger synaptic stimulation, which alone evoked calcium spikes and some degree of DSE, the amount of depression was increased (bottom panel, pooled data from 4 cells; asterisks denote p < 0.05 between the two conditions). C, When we used synaptic stimuli evoking six or more calcium spikes, pairing with current injection (same as in A, B) failed to increase the degree of DSE (pooled data from 4 cells). EPSP amplitudes were measured at −70 mV. Error bars indicate SEM.
The inability of low-intensity stimulation paired with calcium spikes evoked by current injection to evoke DSE can indicate either that presynaptic activity is not required for DSE induction or that the current injection-evoked calcium spikes fail to propagate to the activated synapses. Nevertheless, the results demonstrate that calcium spikes triggered by postsynaptic depolarization can act in a synergistic manner to enhance DSE evoked by synaptic stimulation but do not appear to change its threshold or maximal value.
Spatial localization of calcium spikes evoked by synaptic stimulation and current injection
As discussed above, an alternative possibility to explain why calcium spikes evoked by current injection were unable to induce DSE is that these calcium spikes are highly localized and do not overlap sufficiently with the sites of synaptic stimulation. To test this hypothesis, we determined the spatial spread of calcium spikes by imaging dendritic calcium transients with a fast CCD camera to compare their location with respect to that of the activated synapses (see Materials and Methods).
Calcium spikes triggered by synaptic activation (10 PF stimuli at 100 Hz) were associated with calcium transients that were highly localized to the region close to the stimulation electrode (Fig. 6B), consistent with previous reports (Miyakawa et al., 1992; Hartell, 1996; Brown et al., 2003) and with our dendritic electrophysiological recordings (Fig. 1). The peak calcium signal during synaptic stimulation (ROISYN) was correlated strongly with the number of evoked calcium spikes (r = 0.98–0.99; five cells) and was reduced greatly when hyperpolarizing current was injected to prevent calcium spikes (19 ± 5% of control; n = 5; p < 0.02 for stimuli evoking 4.3 ± 1.2 calcium spikes), confirming that most of the dendritic calcium signal was attributable to the entry of calcium via voltage-gated calcium channel activation. In contrast, calcium spikes triggered by dendritic current injection were associated with more widespread but smaller calcium signals (Fig. 6C). Approximately twice the number of calcium spikes (10.1 ± 1.3 for current injection vs 4.8 ± 0.2 for synaptic stimulation; n = 5) was required to evoke calcium transients of similar magnitude (peak amplitude, 108 ± 22% of synaptically evoked transients).
Figure 6.

Calcium spikes evoked by dendritic current injection are segregated spatially from synaptically evoked calcium spikes. A, Purkinje cell filled with Alexa 594 and fluo-5F (the image shows the Alexa fluorescence). The positions of the PF stimulation electrode (blue) and the dendritic recording electrode (red; 90 μm from the soma) are indicated. B, The area of interest (shown by the white rectangle in A) was imaged for calcium signals during synaptic stimulation, triggering calcium spikes. The image is a maximum intensity projection of a stack of 41 consecutive images acquired at 40 Hz. C, Maximum intensity projection of a stack recorded during calcium spikes evoked by current injection. Two ROIs outlining the branchlets showing calcium signals during synaptic stimulation (ROISYN) or current injection (ROIINJ) were selected. The time course of the fluorescent signal in the two ROIs is plotted below the images. The same number of calcium spikes was evoked by the synaptic stimulation and the current injection. Scale bars: B, C, 10 μm. D, Pooled data from eight cells showing the peak fluorescence change during synaptic stimulation-evoked calcium spikes at the two ROIs selected in the same way as in B, C. E, Pooled data from the same eight cells showing fluorescence changes during current injection-evoked calcium spikes.
Comparing the calcium transients triggered by synaptic stimulation and current injection revealed a spatial separation of calcium influx during the two different protocols. During current injection-evoked calcium spikes the peak at ROISYN was only 25 ± 4% (n = 8; p < 0.001) of the peak at ROIINJ. Conversely, during synaptic stimulation the calcium transient at the location of the peak calcium signal during dendritic current injection (ROIINJ) reached only 21 ± 4% (n = 8; p < 0.001) of the peak at the ROISYN (Fig. 6D). These experiments support the hypothesis that the peak calcium signal associated with calcium spikes triggered by current injection often is spatially separated from the activated synapses. The resulting lower calcium signal at these synapses thus prevents them from undergoing DSE, explaining why calcium spikes evoked by current injection are normally ineffective at triggering DSE.
Modulating the propagation of dendritic calcium spikes
Next we sought to improve the propagation of calcium spikes triggered by dendritic current injection to determine whether this could promote endocannabinoid release and short-term plasticity at the activated synapses. Purkinje cell dendrites express calcium-activated potassium channels that can be activated by calcium spikes (Womack et al., 2004) and that have been shown to limit the spread of regenerative dendritic events in cultured pyramidal neurons (Cai et al., 2004). We therefore examined the effect of the high-conductance calcium-activated potassium (BK) channel blocker penitrem A (Knaus et al., 1994) on the spread of calcium spikes by using calcium imaging. Because the peaks of synaptically evoked and current injection-evoked calcium signals were separated spatially (Fig. 6), we evaluated the spread of the calcium signals by using ROIs placed along the dendrite on the path between the two peak signals (Fig. 7A), with ROI1 corresponding to ROISYN, ROI3 to the location of the dendritic recording electrode, and ROI5 to ROIINJ. This allowed us to compare the spread of calcium signals systematically across different dendritic morphologies.
The dendritic calcium signals in control and in the presence of penitrem A were measured for similar numbers of dendritic calcium spikes (6.7 ± 0.6 vs 8.2 ± 0.7 and n = 5 for synaptically evoked calcium spikes; 11.7 ± 1.7 vs 14 ± 1.1 and n = 3 for current injection-evoked calcium spikes; p > 0.05 for both cases). The calcium signal evoked by synaptic stimulation at the site of the active synapses (ROISYN) was increased significantly by penitrem A (161 ± 24% of control; n = 5; p < 0.04). The peak current injection-evoked calcium signal (ROIINJ) also appeared to increase, although this was not statistically significant (135 ± 10% of control; n = 3; p = 0.11). Penitrem A enhanced the spread of calcium transients evoked by synaptic stimulation as determined by the normalized peak calcium signals at successive ROIs from ROISYN (Fig. 7C,D). The calcium signal associated with dendritic spikes evoked by current injection spread even more effectively from the location of the peak in the presence of penitrem A (Fig. 7E,F). As a consequence, the peak calcium transient reached during current injection-evoked dendritic spikes at the site of the activated synapses was enhanced dramatically (435 ± 32%; n = 3; p < 0.01) (Fig. 7G).
These results suggest that blocking BK channels improves the propagation of calcium spikes that are evoked by dendritic current injection. To verify this directly and electrophysiologically, we examined the properties of calcium spikes reaching the dendritic recording site, which presumably represent propagated spikes because the maximal calcium signal always was located away from this site. Synaptically evoked calcium spikes showed small increases in amplitude (6.4 ± 1.1 vs 5.1 ± 1.5 mV in control; n = 3 cells; p = 0.1) and half-width (1.02 ± 0.1 vs 0.89 ± 0.11 ms in control; n = 3; p = 0.1) in the presence of penitrem A. However, calcium spikes evoked by dendritic current injection (Fig. 7H) were enhanced dramatically both in amplitude (9.9 ± 3.8 vs 2.4 ± 2.2 mV in control; n = 3; p < 0.04) and half-width (0.71 ± 0.17 vs 0.38 ± 0.22 ms in control; n = 3; p < 0.03), consistent with the calcium-imaging data.
BK channel block enables current-evoked calcium spikes and CF input to trigger DSE
Next we tested whether the improvement in propagation of calcium spikes caused by BK channel block could enhance the ability of calcium spikes evoked by dendritic current injection to elicit DSE. Under control conditions calcium spikes triggered by dendritic current injection (Fig. 8A,B) did not generate DSE, even with up to 41 calcium spikes (n = 8) (Fig. 8C). When BK channels were blocked by penitrem A (100 nm), calcium spikes evoked by dendritic current injection were able to produce robust DSE (Fig. 8B,C). Interestingly, the relationship between the number of calcium spikes evoked by current injection and the amount of DSE followed an approximately sigmoidal relationship in penitrem A (n = 8) (compare Figs. 8C, 3C). To verify that DSE in the presence of penitrem A is also endocannabinoid-dependent, we conducted additional experiments by using the CB1 receptor antagonist AM251. In the presence of penitrem A synaptically evoked calcium spikes produced robust DSE, which was blocked by 1 μm AM251 (36 ± 6 vs 111 ± 10% of control, respectively; n = 6; p < 0.0001). In the same cells DSE evoked by calcium spikes triggered by current injection was blocked similarly by 1 μm AM251 (66 ± 1 vs 97 ± 3% of control; n = 6; p < 0.001). These results provide additional confirmation that presynaptic activity is not required for DSE induction via dendritic calcium spikes. BK channel block by penitrem A did not affect baseline EPSP amplitudes (101 ± 12% of control; n = 6; p = 0.4), paired pulse facilitation (99 ± 2% of control; 10 ms interval; n = 6; p = 0.4), or input resistance (99 ± 4% of control; n = 6; p = 0.8), ruling out effects of BK channel block on baseline synaptic transmission or postsynaptic properties.
Figure 8.

Blocking BK channels enables calcium spikes evoked by current injection or CF activation to trigger DSE. A, Dendritic calcium spikes evoked by dendritic current injection in control ACSF (black trace) and 100 nm penitrem A (red trace), recorded 130 μm from the soma. The amplitude of the injected dendritic current waveform (blue traces) was adjusted to evoke similar numbers of calcium spikes under the two conditions. B, Time course of PF synaptic strength before and after the calcium spikes evoked by the current injection shown in A. The same number of calcium spikes, which were unable to trigger DSE in control ACSF, produced strong short-term depression of PF EPSPs when the BK channels were blocked. C, The amount of DSE was plotted versus the number of calcium spikes evoked by current injection. An increasing amount of DSE was triggered with an increasing number of calcium spikes but only when BK channels were blocked by 100 nm penitrem A. Pooled data are from six cells. D, The amount of DSE plotted against the number of synaptically evoked calcium spikes. Block of BK channels did not change the efficacy of synaptically evoked calcium spikes to evoke DSE. Pooled data are from four cells. E, Time course of PF synaptic strength shows prominent DSE after 20 CF stimuli when the BK channels were blocked, but not in control conditions. Single trials are from the same cell. F, The amount of DSE was plotted versus the number of CF stimuli delivered during the induction. Pooled data are from five cells. C F, Error bars indicate SEM, and asterisks denote p < 0.05.
We also tested whether BK channel block affected the ability of calcium spikes evoked by synaptic stimulation to evoke DSE. The relationship between the number of calcium spikes and the amount of DSE evoked during trains of PF stimuli was sigmoidal (Fig. 2), which was unchanged when BK channels were blocked by penitrem A (n = 5; p = 0.8) (Fig. 8D). This suggests that the block of BK channels did not change the ability of calcium spikes to trigger DSE at their initiation site (i.e., there was no change in synaptically evoked DSE) but instead may help calcium spikes evoked at other dendritic locations to spread to the test synapses and evoke DSE.
To test this idea further, we investigated activation of the CF, which produces widespread dendritic calcium entry in Purkinje cells (Miyakawa et al., 1992; Wang et al., 2000) but which, however, is normally insufficient to trigger DSE of PF input (Fig. 4D) (Brenowitz and Regehr, 2005). To test whether BK channel activation also limits the spread of the CF-triggered depolarization to PF synapses, we examined the effect of penitrem A on the ability of the CF to trigger DSE of PF inputs. Experiments were performed in the presence of the specific mGluR1 antagonist CPCCOEt to block the effect of glutamate spillover from the CF terminals to mGluR1 receptors present on PF synapses (Maejima et al., 2001; Brenowitz and Regehr, 2005). In control conditions 20 CF stimuli (at 50 Hz) were required to produce significant DSE (80 ± 7% of control; p < 0.025; n = 5) (Fig. 8E). However, when BK channels were blocked by penitrem A (100 nm), as few as three CF stimuli triggered significant DSE (85 ± 7% of control; p < 0.02; n = 5). For all numbers of CF stimuli (except single CF stimuli) DSE produced in the presence of penitrem A was significantly greater than in the control condition (p < 0.05; n = 5) (Fig. 8F). This suggests that the activation of BK channels limits the ability of CF input to modulate PF transmission via endocannabinoid release.
Discussion
Our findings provide direct evidence linking the initiation and spread of local dendritic calcium spikes with dendritic cannabinoid release and short-term regulation of synaptic efficacy in cerebellar Purkinje cells. By modulating dendritic excitability, we demonstrate that regulating the spread of dendritic calcium spikes can set the threshold for endocannabinoid-dependent regulation of synaptic strength. These findings reveal that dendritic voltage-gated channels can act as tunable coincidence detectors for local feedback regulation of synaptic strength at cerebellar PF synapses.
The link between local dendritic calcium spikes and synaptic plasticity
We have used four different approaches to link directly the initiation of dendritic calcium spikes with plasticity at PF synapses. First, we showed that the stimulus threshold for induction of dendritic calcium spikes is identical to that for induction of DSE: whenever dendritic calcium spikes were initiated by PF stimulation, DSE also followed; when calcium spikes were blocked, DSE induction also failed. This link is exemplified by the sigmoidal relationship between the number of calcium spikes and the degree of DSE, with significant DSE observed with only two dendritic calcium spikes and DSE saturating above five dendritic calcium spikes. Second, we showed that DSE induction and dendritic calcium spike initiation have a similar voltage threshold such that hyperpolarization blocks both dendritic calcium spikes and DSE induction. When the number of dendritic calcium spikes was titrated by adjusting membrane potential, the degree of resulting DSE tracked the number of calcium spikes that were elicited. Third, we demonstrated that DSE can be triggered by synaptically evoked calcium spikes in the presence of an mGluR1 antagonist, indicating that calcium spikes alone are sufficient to trigger DSE even in the absence of mGluR1 activation. Finally, we demonstrated that enhancing the spread of dendritic calcium spikes by blocking BK-type calcium-activated potassium channels also gates the induction of DSE by dendritic calcium spikes triggered by dendritic current injection or CF stimulation.
Together, these findings suggest that dendritic calcium spikes are both necessary and sufficient for triggering DSE with a train of PF stimuli under physiological conditions. Our results thus provide some of the strongest evidence to date linking dendritic calcium spikes with the induction of synaptic plasticity. Previous work has correlated the initiation of calcium spikes by strong synaptic stimulation with non-Hebbian long-term potentiation (Golding et al., 2002) or depression (Holthoff et al., 2004) in pyramidal neurons but relied on relatively indirect evidence, because in these studies dendritic recordings were not performed during the induction of plasticity. In contrast, we directly manipulated dendritic spike initiation and propagation via dendritic recordings, allowing us to provide a strong quantitative link with synaptic plasticity.
Implications for the regulation of plasticity
The tight link between dendritic calcium spikes and DSE induction has several implications for our understanding of this form of short-term synaptic plasticity. First, our results provide a biophysical basis for the specificity of DSE induction by PF input. We demonstrate that dendritic calcium spikes evoked by current injection in the absence of PF stimulation or by CF input were insufficient to trigger DSE at the PF synapses that were tested. This indicates that physiological activation of DSE is restricted to calcium spikes triggered by the PF synapses undergoing DSE. Such specificity is important, because otherwise continuous CF activity in vivo, occurring at mean rates of ∼1 Hz (Lang et al., 1999), would suppress the ∼200,000 PF synapses tonically. The specificity is attributable to the fact that the calcium spike must be sufficiently large at the site of the active synapses to deliver sufficient calcium to exceed the high-calcium threshold for cannabinoid release (Brenowitz and Regehr, 2003). As suggested by our calcium-imaging data, calcium spikes activated by current injection or via CF activation (Wang et al., 2000) do not reach this threshold. We also rule out that coincident presynaptic activity is required for DSE activated by PF input under physiological conditions. This is consistent with previous evidence that prolonged postsynaptic depolarizations to 0 mV when postsynaptic potassium channels have been blocked with internal Cs+, which presumably produces calcium influx far in excess of that generated via dendritic calcium spikes, can trigger DSE in the absence of presynaptic activity (Kreitzer and Regehr, 2001; Maejima et al., 2001; Brenowitz and Regehr, 2003).
Second, our results provide a deeper understanding of the cooperativity requirements for DSE induction by PF input (Brown et al., 2003; Marcaggi and Attwell, 2005). We demonstrate that both the activation of dendritic calcium spikes by PF input and the induction of DSE require a threshold level of synchronous PF activity. Thus calcium spikes and DSE induction read out the level of spatial and temporal synchrony in the PF population. This is presumably attributable to the highly nonlinear recruitment of voltage-gated calcium channels during the initiation of dendritic calcium spikes by synaptic input.
Initiation of dendritic calcium spikes and activation of mGluR receptors both require temporal and spatial clustering of PF input (Batchelor and Garthwaite, 1997; Reichelt and Knöpfel, 2002). The fact that DSE was prevented when calcium spikes were blocked by hyperpolarization indicates that the threshold for DSE induction via calcium spikes is lower than for via mGluR activation and that the relative contribution of calcium spikes and mGluR activation will depend on membrane potential. However, although mGluR activation is not required for DSE induction, there may exist additional cooperativity between postsynaptic mGluR activation and calcium entering via the dendritic calcium spike to cause supralinear calcium signals (Wang et al., 2000; Ohno-Shosaku et al., 2002) or to act synergistically via a calcium-independent mGluR pathway (Maejima et al., 2001). The involvement of phospholipase Cβ, which can integrate calcium and Gq-coupled receptor signals leading to DSI in the hippocampus (Hashimotodani et al., 2005), may contribute to the supralinear effect on DSE induction of combining synaptic stimulation and dendritic spikes evoked by current injection, as well as the cooperativity between PF and CF input (Brenowitz and Regehr, 2005). Although our understanding of endocannabinoid production and release remains limited (Freund et al., 2003; Piomelli, 2003), the local nature of both dendritic calcium spikes and mGluR activation, potentially acting in concert, may help to ensure the synapse specificity of the resulting synaptic plasticity.
Modulation of dendritic spikes influences plasticity
Our results provide direct evidence for the regulation of dendritic calcium spikes by BK channels in Purkinje cells. This supports earlier findings that BK channels play an important role in modulating the excitability of these neurons (Edgerton and Reinhart, 2003; Sausbier et al., 2004; Womack et al., 2004) and may be related to the deficits in motor learning and performance observed in a BK channel knock-out mouse (Sausbier et al., 2004). Our findings are consistent with the results of Golding and colleagues (1999) in CA1 pyramidal cells, who showed that BK channels are responsible in part for determining the duration of dendritic calcium spikes. We also demonstrate that BK channels are critical for regulating the spatial spread of calcium spikes, in contrast with cultured hippocampal pyramidal neurons in which SK-type calcium-activated potassium channels appear to play a dominant role (Cai et al., 2004).
We demonstrate that the modulation of dendritic calcium spikes by the block of BK channels can, in turn, influence endocannabinoid release and thus the induction of plasticity by dendritic calcium spikes. This provides the first direct evidence that modulation of dendritic excitability in turn can regulate the threshold for induction of synaptic plasticity. Interestingly, BK channels can be located presynaptically (Knaus et al., 1996), where they limit transmitter release from synaptic terminals (Robitaille et al., 1993; Raffaelli et al., 2004). Their postsynaptic localization in Purkinje cell dendrites could serve an analogous role. As we demonstrate that BK channel block extends the spatial range of action of dendritic calcium spikes evoked by dendritic current injection or CF activation, the modulation of BK channels also may influence the spatial specificity of DSE evoked by synaptic stimuli. Recent work has demonstrated that the induction of synaptic plasticity also can alter dendritic excitability in pyramidal neurons (Frick et al., 2004; Li et al., 2004). Our study thus “closes the loop” and shows that dendritic excitability may, in turn, regulate the induction of plasticity and be a substrate for metaplasticity (Frick and Johnston, 2005).
What are the physiological regulators of BK channels and thus DSE induction? BK channels are activated by the endogenous cannabinoid anandamide (Sade et al., 2006), providing a negative feedback loop for self-regulation of dendritic calcium spikes and subsequent endocannabinoid release. BK channels also are subject to modulation by numerous second messenger pathways via protein kinase C (PKC), protein phosphatases (Widmer et al., 2003), or phospholipase C (Clarke et al., 2002). These pathways, activated by noradrenaline and serotonin (Schweighofer et al., 2004), thus also could influence DSE. Interestingly, the induction of long-term depression (LTD) also is accompanied by activation of PKC and protein phosphatase pathways (Daniel et al., 1998). Because dendritic calcium spikes also may be involved in the induction of LTD (Wang et al., 2000), this might provide a form of metaplasticity whereby LTD in turn regulates the link between calcium spikes and DSE, setting the threshold at which subsequent patterns of activity are converted into long-term changes in synaptic strength.
Footnotes
This work was supported by grants from the Wellcome Trust, European Commission, and the Gatsby Foundation. E.A.R. held a Prize Studentship of the Wellcome Trust. We thank Jenny Davie, Arnd Roth, and Jesper Sjöström for helpful comments; Beverley Clark, Kazuo Kitamura, and Jesper Sjöström for help with imaging; and Latha Ramakrishnan for technical assistance.
References
- Amitai Y, Friedman A, Connors BW, Gutnick MJ (1993). Regenerative activity in apical dendrites of pyramidal cells in neocortex. Cereb Cortex 3:26–38. [DOI] [PubMed] [Google Scholar]
- Batchelor AM, Garthwaite J (1997). Frequency detection and temporally dispersed synaptic signal association through a metabotropic glutamate receptor pathway. Nature 385:74–77. [DOI] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG (2003). Calcium dependence of retrograde inhibition by endocannabinoids at synapses onto Purkinje cells. J Neurosci 23:6373–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG (2005). Associative short-term synaptic plasticity mediated by endocannabinoids. Neuron 45:419–431. [DOI] [PubMed] [Google Scholar]
- Brown SP, Brenowitz SD, Regehr WG (2003). Brief presynaptic bursts evoke synapse-specific retrograde inhibition mediated by endogenous cannabinoids. Nat Neurosci 6:1048–1057. [DOI] [PubMed] [Google Scholar]
- Cai X, Liang CW, Muralidharan S, Kao JP, Tang CM, Thompson SM (2004). Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron 44:351–364. [DOI] [PubMed] [Google Scholar]
- Casado M, Dieudonne S, Ascher P (2000). Presynaptic N-methyl-d-aspartate receptors at the parallel fiber–Purkinje cell synapse. Proc Natl Acad Sci USA 97:11593–11597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AL, Petrou S, Walsh JV Jr, Singer JJ (2002). Modulation of BKCa channel activity by fatty acids: structural requirements and mechanism of action. Am J Physiol Cell Physiol 283:C1441–C1453. [DOI] [PubMed] [Google Scholar]
- Daniel H, Levenes C, Crepel F (1998). Cellular mechanisms of cerebellar LTD. Trends Neurosci 21:401–407. [DOI] [PubMed] [Google Scholar]
- Dittman JS, Kreitzer AC, Regehr WG (2000). Interplay between facilitation, depression, and residual calcium at three presynaptic terminals. J Neurosci 20:1374–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgerton JR, Reinhart PH (2003). Distinct contributions of small and large conductance Ca2+-activated K+ channels to rat Purkinje neuron function. J Physiol (Lond) 548:53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D (2003). Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83:1017–1066. [DOI] [PubMed] [Google Scholar]
- Frick A, Johnston D (2005). Plasticity of dendritic excitability. J Neurobiol 64:100–115. [DOI] [PubMed] [Google Scholar]
- Frick A, Magee J, Johnston D (2004). LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat Neurosci 7:126–135. [DOI] [PubMed] [Google Scholar]
- Gasparini S, Migliore M, Magee JC (2004). On the initiation and propagation of dendritic spikes in CA1 pyramidal neurons. J Neurosci 24:11046–11056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding NL, Jung HY, Mickus T, Spruston N (1999). Dendritic calcium spike initiation and repolarization are controlled by distinct potassium channel subtypes in CA1 pyramidal neurons. J Neurosci 19:8789–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding NL, Staff NP, Spruston N (2002). Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature 418:326–331. [DOI] [PubMed] [Google Scholar]
- Hartell NA (1996). Strong activation of parallel fibers produces localized calcium transients and a form of LTD that spreads to distant synapses. Neuron 16:601–610. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi K, Shin HS, Kano M (2005). Phospholipase Cβ serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron 45:257–268. [DOI] [PubMed] [Google Scholar]
- Häusser M, Clark BA (1997). Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron 19:665–678. [DOI] [PubMed] [Google Scholar]
- Häusser M, Spruston N, Stuart GJ (2000). Diversity and dynamics of dendritic signaling. Science 290:739–744. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Svoboda K, Denk W, Tank DW (1999). In vivo dendritic calcium dynamics in deep-layer cortical pyramidal neurons. Nat Neurosci 2:989–996. [DOI] [PubMed] [Google Scholar]
- Holthoff K, Kovalchuk Y, Yuste R, Konnerth A (2004). A single-shock LTD by local dendritic spikes in pyramidal neurons of mouse visual cortex. J Physiol (Lond) 560:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus HG, McManus OB, Lee SH, Schmalhofer WA, Garcia-Calvo M, Helms LM, Sanchez M, Giangiacomo K, Reuben JP, Smith 3rd AB (1994). Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry 33:5819–5828. [DOI] [PubMed] [Google Scholar]
- Knaus HG, Schwarzer C, Koch RO, Eberhart A, Kaczorowski GJ, Glossmann H, Wunder F, Pongs O, Garcia ML, Sperk G (1996). Distribution of high-conductance Ca2+-activated K+ channels in rat brain: targeting to axons and nerve terminals. J Neurosci 16:955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001). Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29:717–727. [DOI] [PubMed] [Google Scholar]
- Lang EJ, Sugihara I, Welsh JP, Llinas R (1999). Patterns of spontaneous Purkinje cell complex spike activity in the awake rat. J Neurosci 19:2728–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ (2002). Signaling of layer 1 and whisker-evoked Ca2+ and Na+ action potentials in distal and terminal dendrites of rat neocortical pyramidal neurons in vitro and in vivo. J Neurosci 22:6991–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ, Sakmann B (2001). Dendritic mechanisms underlying the coupling of the dendritic with the axonal action potential initiation zone of adult rat layer 5 pyramidal neurons. J Physiol (Lond) 533:447–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CY, Lu JT, Wu CP, Duan SM, Poo MM (2004). Bidirectional modification of presynaptic neuronal excitability accompanying spike timing-dependent synaptic plasticity. Neuron 41:257–268. [DOI] [PubMed] [Google Scholar]
- Llinas R, Sugimori M (1980). Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. J Physiol (Lond) 305:197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Nicholson C, Freeman JA, Hillman DE (1968). Dendritic spikes and their inhibition in alligator Purkinje cells. Science 160:1132–1135. [DOI] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M (2001). Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31:463–475. [DOI] [PubMed] [Google Scholar]
- Marcaggi P, Attwell D (2005). Endocannabinoid signaling depends on the spatial pattern of synapse activation. Nat Neurosci 8:776–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliore M, Shepherd GM (2002). Emerging rules for the distributions of active dendritic conductances. Nat Rev Neurosci 3:362–370. [DOI] [PubMed] [Google Scholar]
- Miyakawa H, Lev-Ram V, Lasser-Ross N, Ross WN (1992). Calcium transients evoked by climbing fiber and parallel fiber synaptic inputs in guinea pig cerebellar Purkinje neurons. J Neurophysiol 68:1178–1189. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Shosaku J, Tsubokawa H, Kano M (2002). Cooperative endocannabinoid production by neuronal depolarization and group I metabotropic glutamate receptor activation. Eur J Neurosci 15:953–961. [DOI] [PubMed] [Google Scholar]
- Piomelli D (2003). The molecular logic of endocannabinoid signaling. Nat Rev Neurosci 4:873–884. [DOI] [PubMed] [Google Scholar]
- Raffaelli G, Saviane C, Mohajerani MH, Pedarzani P, Cherubini E (2004). BK potassium channels control transmitter release at CA3–CA3 synapses in the rat hippocampus. J Physiol (Lond) 557:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichelt W, Knöpfel T (2002). Glutamate uptake controls expression of a slow postsynaptic current mediated by mGluRs in cerebellar Purkinje cells. J Neurophysiol 87:1974–1980. [DOI] [PubMed] [Google Scholar]
- Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP (1993). Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron 11:645–655. [DOI] [PubMed] [Google Scholar]
- Ross WN, Werman R (1987). Mapping calcium transients in the dendrites of Purkinje cells from the guinea-pig cerebellum in vitro. J Physiol (Lond) 389:319–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade H, Muraki K, Ohya S, Hatano N, Imaizumi Y (2006). Activation of large-conductance Ca2+-activated K+ channels by cannabinoids. Am J Physiol Cell Physiol 290:C77–C86. [DOI] [PubMed] [Google Scholar]
- Sausbier M, Hu H, Arntz C, Feil S, Kamm S, Adelsberger H, Sausbier U, Sailer CA, Feil R, Hofmann F, Korth M, Shipston MJ, Knaus HG, Wolfer DP, Pedroarena CM, Storm JF, Ruth P (2004). Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci USA 101:9474–9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Stuart G, Sakmann B (1997). Calcium action potentials restricted to distal apical dendrites of rat neocortical pyramidal neurons. J Physiol (Lond) 505:[Pt 3]605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweighofer N, Doya K, Kuroda S (2004). Cerebellar aminergic neuromodulation: towards a functional understanding. Brain Res Brain Res Rev 44:103–116. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB (2003). Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39:641–654. [DOI] [PubMed] [Google Scholar]
- Stuart G, Häusser M (1994). Initiation and spread of sodium action potentials in cerebellar Purkinje cells. Neuron 13:703–712. [DOI] [PubMed] [Google Scholar]
- Wang SS, Denk W, Häusser M (2000). Coincidence detection in single dendritic spines mediated by calcium release. Nat Neurosci 3:1266–1273. [DOI] [PubMed] [Google Scholar]
- Widmer HA, Rowe IC, Shipston MJ (2003). Conditional protein phosphorylation regulates BK channel activity in rat cerebellar Purkinje neurons. J Physiol (Lond) 552:379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Chevez C, Khodakhah K (2004). Calcium-activated potassium channels are selectively coupled to P/Q-type calcium channels in cerebellar Purkinje neurons. J Neurosci 24:8818–8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RK, Prince DA, Basbaum AI (1979). Intradendritic recordings from hippocampal neurons. Proc Natl Acad Sci USA 76:986–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Gutnick MJ, Saar D, Delaney KR, Tank DW (1994). Ca2+ accumulations in dendrites of neocortical pyramidal neurons: an apical band and evidence for two functional compartments. Neuron 13:23–43. [DOI] [PubMed] [Google Scholar]