

Abstract
With few exceptions, the almost 30,000 prostate cancer deaths annually in the United States are due to failure of androgen deprivation therapy. Androgen deprivation therapy prevents ligand-activation of the androgen receptor. Despite initial remission after androgen deprivation therapy, prostate cancer almost invariably progresses while continuing to rely on androgen receptor action. Androgen receptor’s transcriptional output, which ultimately controls prostate cancer behavior, is an alternative therapeutic target, but its molecular regulation is poorly understood. Recent insights in the molecular mechanisms by which the androgen receptor controls transcription of its target genes are uncovering gene-specificity as well as context-dependency. Heterogeneity in the androgen receptor’s transcriptional output is reflected both in its recruitment to diverse cognate DNA binding motifs and in its preferential interaction with associated pioneering factors, other secondary transcription factors and coregulators at those sites. This variability suggests that multiple, distinct modes of androgen receptor action that regulate diverse aspects of prostate cancer biology and contribute differentially to prostate cancer’s clinical progression are active simultaneously in prostate cancer cells. Recent progress in the development of peptidomimetics and small molecules, and application of Chem-Seq approaches indicate the feasibility for selective disruption of critical protein-protein and protein-DNA interactions in transcriptional complexes. Here, we review the recent literature on the different molecular mechanisms by which the androgen receptor transcriptionally controls prostate cancer progression, and we explore the potential to translate these insights into novel, more selective forms of therapies that may bypass prostate cancer’s resistance to conventional androgen deprivation therapy.
Keywords: androgen receptor, prostate cancer, nuclear receptor, coregulator, transcription, castration, hormonal therapy
Introduction
The pivotal role of the androgen-activated androgen receptor (AR) as a driver of prostate cancer (CaP) progression has long been recognized (Denmeade and Isaacs 2002; Schmidt and Tindall 2013). The first clinical androgen deprivation studies, published in 1941, indicated already that surgical or medical castration induces remission of, but does not cure, metastatic CaP (Huggins and Hodges 1941; Huggins 1941). Today, despite the development of an ever increasing and more refined repertoire of drugs and treatment regimens to achieve androgen deprivation therapy (ADT) (Attard, et al. 2016; Magnan, et al. 2015; Nguyen, et al. 2014; Valenca, et al. 2015), failure of ADT is still a clinical reality that contributes directly to the ~30,000 CaP deaths in the US annually (Siegel, et al. 2017). Aberrant re-activation of AR has been identified as the main culprit for CaP’s acquired resistance to ADT. The selective pressure of ADT induces amplification, somatic mutations, and ligand-independent activation of AR, stimulates intra-CaP production of AR-activating androgens, turns ADT drugs into partial AR agonists, and/or modifies the activity of key AR-co-operating proteins (Attard, et al. 2009; Dai, et al. 2017; Heemers and Tindall 2005; Heemers and Tindall 2010; Karantanos, et al. 2015; Knudsen and Penning 2010; Li, et al. 2016; Li, et al. 2015). All these mechanisms, which have been the topic of several excellent reviews (see above), lead to persistent AR action and have been shown to favor CaP cell growth under ADT in preclinical cell line and animal model systems. Whole exome and whole genome sequencing efforts on clinical castration-recurrent (CR) CaP specimens confirmed the presence of somatic alterations that contribute to AR re-activation in at least 60% of patients (Beltran, et al. 2013; Grasso, et al. 2012; Robinson, et al. 2015). Targeted deep sequencing of the AR gene locus on metastatic CR-CaP samples obtained at rapid autopsy and on circulating tumor DNA from CR-CaP patient serum have isolated additional genomic structural alterations in the AR gene that give rise to androgen-independent AR forms in 30%-50% of patients (De Laere, et al. 2017; Henzler, et al. 2016). The latter findings indicate that the actual fraction of patients whose CR-CaP harbors AR alterations is likely even higher.
Persistent AR action that controls CaP progression while CaP no longer responds to ADT represents a critical stalemate that prevents much-needed improvements in the care and survival rates for patients suffering from CR-CaP. An alternative approach to target for therapy aberrant AR action in CR-CaP may be found by taking a fresh look at a manner by which activated AR affects the behavior of CaP cells and disease progression (Figure 1). AR is a member of the ligand-activated nuclear receptor family of transcription factors (Heemers and Tindall 2007; Heinlein and Chang 2004). AR is activated by direct binding of androgens, which causes an inactive cytoplasmic AR to undergo conformational changes, alter its association with chaperones and translocate to the nucleus. Inside the nucleus, ligand-bound AR binds to DNA recognition motifs known as Androgen Response Elements (AREs) and recruits from a large array of coregulators and cofactors to control transcription of ARE-driven AR target genes. For more than 7 decades, the main approach to prevent AR activation, and the current mainstay of ADT, has been to deprive AR of its ligand (Denmeade and Isaacs 2002; Schmidt and Tindall 2013). This has been accomplished by preventing the production from the most bioactive androgen dihydrotestosterone (DHT) from precursors that are derived from gonads or adrenals or are produced intra-prostatically, or by administration of anti-androgens that complete with DHT for AR binding. Other targets in the pathways leading to aberrant AR activation such as interference with the chaperone proteins clusterin or Sigma-1 (Thomas, et al. 2017; Yamamoto, et al. 2015), or with the signaling cascade by which cytokines or growth factors activate AR activity (Culig 2004), to name but a few, have been explored also for therapeutic intervention. Parallel efforts have focused on targeting for therapy AR moieties other than its ligand-binding domain, such as its constitutively active N-terminal transactivation domain (Andersen, et al. 2010; Myung, et al. 2013) or its DNA binding-domain (Dalal, et al. 2014). The latter approaches have either not been tested yet in clinical trials or the results of such trials are not yet available.
Figure 1. Basic mechanism of AR-mediated transcription.
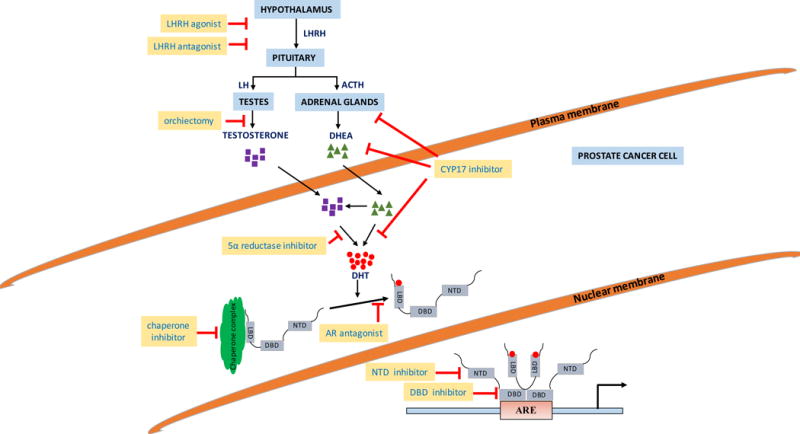
The hypothalamus produces LHRH which induces the production of LH and ACTH by the anterior pituitary. LH and ACTH stimulated secretion of testosterone and DHEA by the testes and adrenal glands, respectively. Testosterone and DHEA are converted into the bioactive androgen DHT in target cells. DHT binds to inactive AR in the cytoplasm, triggering a conformational change and nuclear translocation of AR. Inside the nucleus, DHT-bound AR homodimerizes and binds to AREs to drive the expression of AR target genes. Abbreviations: LHRH, luteinizing hormone-releasing hormone; LH, luteinizing hormone; ACTH, adrenocorticotropic hormone; DHEA, dehydroepiandrosterone; DHT, dihydrotestosterone; AR, androgen receptor; NTD, N-terminal domain; LBD, ligand-binding domain; DBD, DNA binding domain; ARE, androgen response element. Red lines mark current therapeutic targets to inhibit AR signaling.
Meanwhile, the technological advances that led to the genomic analyses of clinical CaP specimens have allowed also to map and characterize AREs, AR-dependent gene transcription, and the contribution of AR-associated proteins to AR target gene transcription in preclinical CaP models and in clinical specimens. The renewed insights in genome-wide DNA AR binding sites and patterns, and the dependence of AR’s transcriptional output on other transcriptional regulators indicate that AR’s regulation of target gene transcription does not follow a uniform pattern. The goal of this review is to summarize the knowledge that has been gained in terms of the distinct molecular mechanisms by AR target gene transcription occurs, and to explore whether this knowledge can be exploited to develop novel therapies that block specifically the action of androgen-activated AR that controls aggressive CaP cell behavior.
Diversity in AR-DNA interactions
Many efforts have been directed towards resolving the manner in which ligand-activated AR interacts with AREs. Initially, studies focused on distinguishing between the mechanisms by which AR and related nuclear receptors, such as the glucocorticoid receptor (GR), which are recruited to highly similar DNA motifs using structurally similar protein domains, bind selectively to their respective cognate genomic binding site. Although still far from understood completely, the current consensus is that AR binds as a dimer in a head-to-head conformation to inverted repeats of a 5′-AGAACA-3′ hexamer that are separated by 3 bases (Claessens, et al. 2017). We will refer to this 15-mer sequence as the canonical ARE motif. Domains other than AR’s DNA-binding domain, for instance its N-terminal domain, have been recognized to exert allosteric effects on the interaction between AR and ARE (Callewaert, et al. 2003; Tadokoro-Cuccaro, et al. 2014).
Today’s insights in the AR cistrome are derived from an array of ChIP-chip, ChIP-Seq and, more recently, ChIP-exo studies on diverse CaP model systems after treatment with different ligands for variable durations (Horie-Inoue and Inoue 2013; Jin, et al. 2013). Because of these efforts, thousands of AR binding sites, which contain AREs and are located preferentially in enhancer and intergenic regions, have been isolated. Owing to these data, evidence is emerging that even within the same cell AR can bind different classes of AREs, only a minority of which conform to the canonical ARE motif (Figure 2, discussed below). Diversity in the sequence composition of the AREs that regulate AR target gene expression should perhaps not be surprising. After all, even very early after the discovery of the first AREs in genes encoding for instance secretory component (SC), sex limiting protein (SLP), or prostate-specific antigen (PSA), the existence of 2 classes of AREs was proposed. In this model, some AREs were bound by AR alone (so-called selective AREs), whereas others could be recognized by both AR and GR (classical AREs) (Verrijdt, et al. 2006). It is now clear that the selective modes of AR-ARE interaction are not due to different dimerization patterns of AR at AREs (head-to-head versus head-to-tail (Shaffer, et al. 2004)) as originally thought. Rather, recent ChIP-Seq data have clarified that AR binds to selective AREs because of less stringent sequence requirements for the 3′ hexamer (Sahu, et al. 2014). Recent bioinformatics-guided modeling of the ARE motifs derived from similar high-throughput studies indicated that only a minority of the AREs fit perfectly the consensus AGAACAnnnTGTTCT 15-mer motif (Wilson, et al. 2016). The majority of AREs corresponded to “imperfect” AREs in which mismatches of 1 or more basepairs from the consensus motif occur. Consistent with other reports (see for instance (Massie, et al. 2007; Wang, et al. 2007)), for a significant number of AR binding sites only one ARE half site that corresponds to a perfect hexamer was present. Increased levels of degeneracy of the ARE have been linked with decreased level of transcriptional output of those sites (Wilson et al. 2016). As technology became more sensitive and allowed for higher resolution scanning of the ARE motif and its surrounding sequence, more details could be derived on the genomic regions occupied by AR. A recent ChIP-exo study that more precisely mapped the boundaries of AR-occupied DNA, has reported 4 different classes of agonist-bound AR binding elements based on the sequence composition of the ARE (hexamer) and border patterns that were protected from exonuclease digestion (Chen, et al. 2015) (Figure 2). Strikingly, the latter study also identified a previously unrecognized type of motif, 5′-NCHKGNnndDCHDGN-3′, to which antagonist- but not agonist-liganded AR is recruited. Some of these antagonist-specific motifs could be occupied by bicalutamide- as well as enzalutamide-liganded AR. Others were bound only by bicalutamide- or enzalutamide-liganded AR, indicating specificity of some of these sites for specific antagonist-AR interactions.
Figure 2. Schematic representation of AREs derived from ChIP-Seq and ChIP-exo analyses.
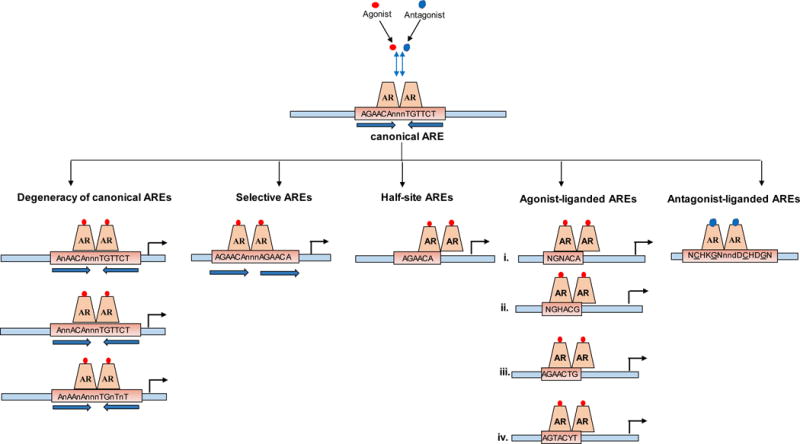
Top panel, consensus sequence for a canonical ARE, which reflects the ideal 15-mer motif of two hexamers arranged in inverted repeat with a 3-mer spacer. Bottom panels, Deviations from this conventional AREs, which include [from left to right]: degenerate AREs in which mismatches of 1 or more basepairs from the consensus canonical ARE motif occur, selective AREs which are composed of direct repeat of 6-mer half-sites, 6-mer half-site ARE sequences found to be enriched in AR binding sites derived from ChIP-based approaches, 4 types of ChIP-exo protected agonist-liganded AREs, ChIP-exo protected antagonist-liganded AREs. Note that, until proven conclusively otherwise, AR is assumed to bind as a dimer to all ARE versions.
Recently, versions of AR that lack its ligand binding-domain but retain AR’s DNA-binding properties have been recognized to emerge under ADT, most often in the presence of full-length AR. More than 20 AR variants that arise either via alternative splicing or genomic rearrangements in the AR gene locus have been isolated (Dehm, et al. 2008; Henzler et al. 2016; Hu, et al. 2011). These variants, the best studied of which are ARV-7 and ARv567es, have been shown to facilitate nuclear localization of full-length AR under ADT and to form homodimers as well as heterodimers with other AR variants or with full-length AR (Cao, et al. 2014; Xu, et al. 2015; Zhan, et al. 2017). Whether these AR variants occupy the same AR binding sites or AREs as full-length AR is an important question, which has proven difficult to answer. Co-occupancy of canonical AR target genes by ARV-7 and full-length AR in a mutually dependent manner has been observed, whereas ARV-7 homodimers but not full-length AR were recruited to other, AR variant-specific genes (Cao et al. 2014). A ChIP-Seq study in cells that were engineered to express either full-length AR or ARv567es indicated that ARv567es recognizes the same target genes and canonical AREs as full-length AR, but with lower affinity (Chan, et al. 2015). Moving forward, the use of CaP model systems in which expression levels of different AR species can be controlled tightly will help resolve their precise DNA binding patterns and the degree to which these patterns overlap.
In addition to variability in the sequence of the AREs that are present, diversity has been observed also in the DNA motifs adjacent to AREs that correspond to binding sites for other transcription factors. Soon after the identification of the same, first AREs that are discussed above, it became obvious that other transcription factors contribute differentially to androgen regulation of reporter gene activity under their control (Haelens, et al. 1999; Scarlett and Robins 1995; Zhang, et al. 1997). At that time, the concept of the Androgen Response Unit (ARU) was proposed, which entailed that AREs are embedded in genomic domains containing also binding sites for other transcription factors that help to fine-tune the androgen response in a gene-specific manner (Celis, et al. 1993). Several of these factors will be discussed in more detail in the following section. These initial indications have been confirmed in ChIP-based approaches that extended knowledge beyond that obtained for a few known AR target genes (e.g. (Massie et al. 2007; Wang et al. 2007; Wang, et al. 2009)). Selective enrichment of binding motifs for other transactivating factors suggest that AR’s binding partners at a given AREs are different, and that molecular regulation of AR’s transcriptional output at different AREs varies. The relevance of such interactions is underscored further following the recent characterization of the AR cistrome at different phases of the CaP cell cycle progression (McNair, et al. 2017). Previous AR ChIP-chip, ChIP-Seq or ChIP-exo datasets were derived from cells that were androgen deprived prior to androgen stimulation, which means from cells that were stuck in G0 phase. AR ChIP-sequencing on cells that were arrested in specific stage of the cell cycle has identified the full complement of AR binding sites in mitotically active cells. This work has uncovered a large set of previously unknown AR binding sites. Some of these sites were present throughout cell cycle progression, and overlapped with the already isolated AR cistrome. Others, however, were specific for select stages of the cell cycle. Motif analyses of the specific AR binding sequences derived from each step of the cell cycle revealed strikingly selective enrichment for recognition motifs for other transcription factors. These data suggested that distinct interactions between AR and other transcriptional regulators control androgen-dependent progression through different stages of cell cycle progression.
Specific protein-protein interactions may contribute also to the differences in AR binding site patterns that are found between clinical CaP specimens and (synchronized) cell lines, and among prostate (cancer) samples obtained at different stages of disease progression. A shift in the AR cistrome between normal prostate tissues and localized ADT-naïve CaP specimens is accompanied by differential enrichment for binding sites for FoxA1 and HoxB13 in CaP-associated AR binding sites (Pomerantz, et al. 2015). Overexpression of either of these 2 transcription factors in AR-expressing normal prostate epithelial cells induced neoplastic transformation and a “benign-to-malignant” shift in the AR cistrome. In CR-CaP, a novel set of AR binding sites emerges that is not present in ADT-naïve CaP or in CaP cell line models. This CR-specific set of AR binding regions was enriched in binding motifs for factors such as STATs and IRFs (Sharma, et al. 2013). The relevance of specific AR-transcription factor interactions for the composition of the AR cistrome was underscored further in studies in which expression of the transcription factor FoxA1 was silenced. After silencing, some of the AR binding sites that were present before the loss of FoxA1 were gone, some were maintained, and a large number of novel AR binding sites was gained. Motif analysis of these regions indicated that gained AR binding sites harbor a classical 15-mer ARE consensus motif whereas the sites that were lost tended to be composed of a half-site ARE and a FoxA1 binding motif (Wang, et al. 2011a). These data indicate that AR-interacting proteins may restrict AR binding to some genomic sites or direct AR binding to others. Similar behavior has been noted also for the AR-associated transcription factor GATA2 (Zhao, et al. 2016).
Transcriptional regulation of gene expression results from an interplay between DNA sequence, transcription factor action, chromatin accessibility, and nucleosome assembly. It is, therefore, important to note that sequence analyses on data from nucleosome (re)positioning and DNase I hypersensitivity assays after androgen treatment of CaP cells support diversity in AR binding site composition and AR binding partners. Examination of the position of H3K4me2 marks, which are associated with transcriptionally active enhancers, have shown that androgen treatment leads to dismissal of a central nucleosome over the AR binding site and repositioning of flanking nucleosomes (He, et al. 2010). These nucleosome relocation patterns predicted the presence of binding sites for AR and AR-associated transcription factors such as FoxA1 and Oct1. Furthermore, a direct comparison of the DNA regions obtained by DNase I protection assays and AR-ChIP sequencing data from androgen-treated CaP cells revealed 3 different footprinting patterns that overlap with AR binding sites (Tewari, et al. 2012). Two of these patterns contain an ARE half-site and binding motifs for Fox family members, whereas the third harbors full length AREs and are specifically enriched for motifs recognized by NF1C.
Taken together, literature indicates that AR’s recruitment to a diversity of ARE motifs where it interacts selectively with other transcription factors that bind their own recognition motif leads to heterogeneity in AR target gene transcription. In addition, AR-interacting transcription factors may control AR’s access to genomic AR binding sites.
Heterogeneity in the interaction between AR and its associated transcriptional regulators at AREs
DNA-bound AR executes transcription of its target genes via interaction with a large number of functionally diverse proteins (DePriest, et al. 2016; Gottlieb, et al. 2012; Heemers and Tindall 2007). Not taken into account the general transcriptional machinery, these proteins fall into 2 major classes: transcription factors that bind their own consensus binding motifs nearby AREs and coregulators that associate with ARE-bound AR without binding DNA themselves. More than 300 proteins that belong to the latter 2 categories have been reported to contribute directly to AR-mediated transcription. In all likelihood that number is a significant underestimation considering the hundreds of proteins that have been identified as previously unrecognized coregulators via IP-mass spectrometry approaches, and the novel interaction modes among such proteins (Malovannaya, et al. 2011). Recent mapping of the genome-wide binding sites of some of these AR-associated proteins indicate variability in recruitment patterns to different AREs. Such diversity could signify the formation of differentially composed protein-protein complexes at AREs and thus selective control by these complexes over specific biological processes in CaP cells. This possibility is in line with findings from gene expression studies in which key AR-associated regulators were silenced or inhibited, which affected androgen regulation of subfractions only of the AR-transcriptome. In this section, we provide an overview of the mechanisms by which pioneer transcription factors, other transcription factors, and coregulators may contribute to a selective output from the AR transcriptional complex (Figure 3).
Figure 3. Schematic depiction of some of the mechanisms by which pioneer transcription factors, other transcription factors and coregulators selectively contribute to AR-dependent transcription.
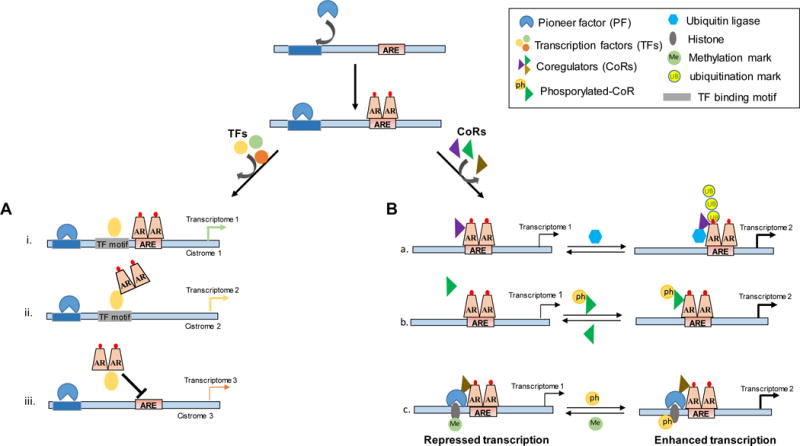
Top panel, pioneer factor opens up the chromatin environment to facilitate AR–ARE interactions and subsequent recruitment of other components of the AR transcriptional complex. Recruited components include other transcription factors (TFs) (A) and coregulators (B). A. Schematic representation of the manner in which a single TF (e.g. HoxB13) either increases (i. and ii.) or decreases (iii.) androgen regulation of select AR target genes. B) Schematic representation of the mechanisms which coregulators selectively control portions of AR’s transcriptional output. Coregulators can enhance the transcriptional activity of AR by degrading a transcriptionally-inactive pool of AR (Siah2, a), bind to AR only when it has undergone a specific post-translational modification (ph-Ezh2, b) and c) bind at specific AREs guided by histone modification patterns to act as coactivator or corepressor (LSD-1, c).
Pioneer transcription factors
The isolation of the AR cistrome via ChIP-based approaches has provided insights also in the manner in which AR is recruited to unoccupied AREs in a largely unaccessible chromatin environment after androgen stimulation. As for other transcription factors, AR recruitment is facilitated by, and dependent on, pioneer factors. These factors are able to bind to compact chromatin before AR does, open up the local chromatin environment, and make it competent for the AR to execute the transcription of its target genes (Jozwik and Carroll 2012; Zaret and Carroll 2011).
The first ChIP-chip study that defined the AR binding sites on chromosomes 21 and 22 reported co-occurrence of AREs (half-sites) with 3 significantly enriched DNA motifs, namely those to which Forkhead, GATA, and Oct transcription factors are recruited (Wang et al. 2007). These sites were subsequently shown to be occupied preferentially by FoxA1, GATA2, and Oct1, respectively. The 3 factors were recruited in various combinations, independently or simultaneously, to 80% of the AR binding sites tested, which indicated already gene selectivity. Strikingly, significant binding of each of the 3 factors to the AR binding regions was observed already in the absence of androgens, which led to their being labeled as potential pioneer factors that facilitate subsequent AR-dependent transcription. None-the-less, androgen treatment increased also the specific factor binding at specific sites. Interactions of FoxA1, GATA2 and Oct1 with AR at a few binding sites that were selected for further study (those present in PSA and TMPRSS2 genes) depended on their respective functional cis-acting motifs. AR formed independent complexes with each factor, and interacted with GATA2 and Oct1 in a hormone-dependent manner while its interaction with FoxA1 was not affected by androgens. Recruitment of AR to these select ARE-containing enhancers was significantly decreased following transfection with siRNAs targeting FoxA1 or GATA2, both in the absence of ligand and, to a greater extent, in the presence of hormone. In contrast, silencing of Oct1 did not affect AR binding on the PSA and TMPRSS2 enhancers in the absence and presence of hormone. Oct1 recruitment to the PSA and TMPRSS2 enhancers was similarly attenuated when GATA2 was silenced, whereas GATA2’s presence was not affected by Oct1 silencing. These data suggested that GATA2, FoxA1 and Oct1 play necessary roles both in the basal recruitment of AR to chromatin and in their recruitment following androgen stimulation. Interestingly, these data indicated also that GATA2 and Oct1 act at distinct steps in AR signaling, with GATA2 acting upstream of Oct1 recruitment.
Independent ChIP-Seq studies to define the genome-wide recruitment pattern of these factors have confirmed and expanded on the initial findings for candidate genes. It is now evident that more than 50%, 25% and 10% of AR recruitment sites overlap with binding sites for FoxA1, GATA2 and Oct1, respectively (He, et al. 2014; Obinata, et al. 2016; Sahu, et al. 2011; Sahu, et al. 2012; Wu, et al. 2014; Zhao et al. 2016). These numbers are rough estimates only as individual studies used variable treatment duration, different AR agonists, or were done in different cell lines. Some ARE-harboring genes contained binding peaks for the 3 factors, whereas others harbored only recruitment sites for one or two factors. Gene selectivity of these binding patterns was reflected also in corresponding gene expression studies, which showed that androgen-response of portions of the AR transcriptome only were altered after silencing of each factor individually (He et al. 2014; Obinata et al. 2016; Sahu et al. 2011; Wu et al. 2014; Zhao et al. 2016).
The role for pioneer factors in AR-dependent transcription is sustained, and evolves, during CaP progression. Direct comparisons between isogenic cell lines that represent ADT-naïve CaP or CR-CaP have shown higher occupancy of FoxA1 and GATA2 at representative CR-specific AR target genes such as UBE2C (Wang et al. 2009). Similarly, loss of FoxA1 and GATA2 reduced expression of these CR-CaP specific AR target genes only in CR-CaP cells, even though expression levels of FoxA1 and GATA2 were not elevated in these cells. Differential levels of gene occupancy for these factors correlated with differences in gene expression patterns also in clinical CaP specimens obtained before and after ADT and may, thus, contribute to AR transcriptome differences between ADT-naive and CR-CaP.
Consistent with the findings for FoxA1 that are mentioned above, the composition of the AR cistrome is impacted also by the action of GATA2 and Oct1. Silencing of GATA2 and Oct1 led to loss of a fraction of AR binding sites while other sites were maintained. New AR binding sites were gained also (Obinata et al. 2016; Zhao et al. 2016). However, the number of these novel sites was significantly lower than that gained after loss of FoxA1 expression, and the gained sites did not show enrichment of 15mer ARE motifs. Supporting the notion of a higher order in the action of these transcription factors, silencing of FoxA1 led to redistribution of the GATA2 cistrome, whereas loss of GATA2 did not impact on the location of the FoxA1 binding sites (Zhao et al. 2016).
Taken together, these findings indicate that FoxA1 and GATA2, and perhaps to a lesser extent Oct1, function as pioneer transcription factors for AR, in a gene- and/or location-specific manner. Hierarchy exists in the action of these proteins for generation of the AR cistrome wherein FoxA1 can program also GATA2’s pioneering function and GATA2 acts upstream of Oct1 (Wang et al. 2007; Zhao et al. 2016). Evidence is emerging also for additional, non-pioneering roles for these 3 factors in AR-dependent transcription, for instance control over AR expression levels (He et al. 2014) and restricting AR access from select AREs (Sahu et al. 2011; Wang et al. 2011a).
Non-pioneer transcription factors
Analyses similar to those that isolated the FKHD, GATA and Oct consensus motifs in AR binding sites have been done for most, if not all, ChIP-based genomic AR mapping efforts. These studies have recognized that consensus binding motifs for transcription factors that do not fit a pioneering role are also significantly enriched near the AR cistrome. Examples include recognition sites for CREB and Nkx3.1 (He et al. 2010; Massie et al. 2007; Tan, et al. 2012). As indicated above, several of these transcription factors, for instance HoxB13 and IRFs (Pomerantz et al. 2015; Sharma et al. 2013), may contribute in a stage-specific manner to AR cistrome programming that occurs during clinical CaP progression. Others, such as ETS transcription factors, have been implicated in aggressive CaP cell behavior in preclinical model systems (Klezovitch, et al. 2008; Tomlins, et al. 2008; Wang, et al. 2008a). Although androgen-dependent cistromes for all the transcription factors for which a motif has been isolated near AR binding sites are not yet available, the DNA binding patterns for the factors that have been examined do suggest contribution to a subset of AR target genes and the control over distinct CaP cell biology. Here, we highlight a few of these factors, the manner in which they interact with DNA-bound AR, and their role in specific AR target gene expression patterns that are relevant to CaP progression and survival.
i. ETS transcription factor family members
A pioneering AR ChIP-chip study on 24,275 promoter-only gene regions yielded 1,532 promoter domains to which AR bound after androgen stimulation of LNCaP cells (Massie et al. 2007). When analyzing the corresponding DNA sequences, the canonical 15-mer ARE was found in less than 10% of AR binding sites. Instead, the most frequently occurring motif was that of an ARE half site, which was present in almost 80% of promoter regions. This motif was closely followed in frequency by a consensus binding sequence for the avian erythroblastosis virus E26 homologue (ETS) family of transcription factors, which was found in 70% of AR binding sites. Validation experiments confirmed that the ETS1 family member can bind to AREs. ETS1 was present also in AR immunocomplexes and selectively contributed to androgen regulation of ARE-driven genes that contain also the 6-mer ETS motif, but not genes that do not harbor such a motif. Strikingly, ETS1 was recruited to AR binding sites in an androgen-dependent manner, which did not fit the behavior of a pioneer factor. These findings thus uncovered a novel, selective, role for ETS family members in AR-dependent transcription.
ETS1 belongs to the same family of transcription factors as ERG. ERG’s function in CaP has become a hot topic since the identification of the TMPRSS2-ERG gene fusion, which has been linked to CaP cell invasion in preclinical models and to induction of CaP precursor-like lesions in mice (Klezovitch et al. 2008; Tomlins et al. 2008; Tomlins, et al. 2005; Wang et al. 2008a).
The TMPRSS2-ERG gene fusion is present in about half of localized CaP cases, which makes it the most frequent gene-specific alteration in organ-confined treatment-naive CaP (Cancer Genome Atlas Research Network. Electronic address and Cancer Genome Atlas Research 2015). Although these gene fusions have been associated with the early onset of CaP (Steurer, et al. 2014; Weischenfeldt, et al. 2013), their clinical utility as a prognostic or predictive biomarker for CaP progression is still unclear. Many studies have evaluated their role in patients’ outcome after radical prostatectomy, risk for biochemical recurrence, response to ADT, and lethal CaP development (reviewed in (Bostrom, et al. 2015)) and have reached different conclusions. The reason for the discrepancies between these studies is not entirely clear, but is likely related at least in part to differences in TMPRSS2-ERG fusion status among and within intraprostatic foci of localized CaP (Boutros, et al. 2015; Svensson, et al. 2011; Wei, et al. 2016). Inconsistent association between TMPRSS2-ERG status and post-operative risk for CaP progression may, thus, be due to inadequate tissue sampling.
The ERG and AR cistromes have been defined in VCaP cells, which harbor the TMPRSS2-ERG fusion (Yu, et al. 2010). ChIP-Seq analysis was done, which allowed to assess also occupancy of enhancer and intergenic regions. In this work, the canonical 15-mer ARE was isolated as the most enriched motif in AR binding peaks and the ETS family motif ranked as the second enriched motif. Notably, molecular concept mapping of AR-bound gene sets also reported an enrichment in ERG-regulated genes. 44% of the AR-bound gene regions in VCaP cells overlapped with ERG binding peaks, and, strikingly, AR and ERG ChIP-Seq analysis of an ERG-TMPRSS2 positive clinical CaP specimen also identified a 44% overlap in AR- and ERG-enriched genomic regions. In addition, the biology of genes bound by AR and ERG in vivo reflected AR and ERG function in in vitro LNCaP and VCaP cell lines. Mechanistically, ERG was found to repress AR function in part by binding the promoter of the AR gene and decreasing AR expression. ERG overexpression correlated with repression of AR target genes in vivo and decreased androgen regulation of a lineage-specific differentiation program. Gradual overexpression of ERG in ERG-negative LNCaP cells also gradually increased the ERG binding but decreased AR binding at gene-specific loci. Conversely, ChIP-Seq on VCaP cells in which ERG had been silenced led to 24% decrease in ERG binding sites and a 26% increase in AR binding sites. This molecular regulation of the AR-dependent cistrome and transcriptome by ERG indicated selective effects on AR action that inhibit specifically AR-dependent pro-differentiation potentiating pathways and result in CaP dedifferentiation.
In addition to ETS1 and ERG, other ETS family members such as ETV1 and GABPα have been shown also to modulate AR signaling in CaP cells (Baena, et al. 2013; Sharma, et al. 2014). Analyses on the cistromes of these 4 ETS factors have indicated that both specificity and synergy may exist in their contribution to AR action and CaP biology (Sharma et al. 2014).
ii. HoxB13
HoxB13 is a member of the homeodomain family of sequence-specific transcription factors. During embryogenesis, HoxB13 is expressed prominently in the urogenital sinus and plays a critical role in the development of the prostate (Huang, et al. 2007; Zeltser, et al. 1996). HoxB13 was identified as an AR-interacting protein via a T7 phage display (Norris, et al. 2009b). Although reported before to repress androgen-dependent reporter gene activity (Jung, et al. 2004), HoxB13 was shown to selectively induce or repress transcription of androgen-responsive genes (Norris, et al. 2009a). These differential effects were linked to its ability to increase or decrease recruitment of AR and AR-associated coregulators to genomic AR and/or HoxB13 binding sites in the affected genes. Specifically, at genes for which HoxB13 acts as a transcriptional repressor, loss of HoxB13 increased binding of AR and coregulators such as TRAP220, p300 and SRC1. Conversely, at genes for which HoxB13 acts as a transcriptional activator, silencing of HoxB13 led to a decrease in both AR and coregulatory occupancy, or maintenance of AR but loss of AR-associated coregulators. These insights were derived from studying a set of 6 ARE-driven genes for which AR and HoxB13 binding sites and recruitment modes have been thoroughly characterized (Norris et al. 2009a). Elegant studies in which AR and/or HoxB13 DNA binding motifs were mutated or in which the DNA binding domain of AR or HoxB13 underwent mutations to prevent DNA binding uncovered 3 molecular mechanisms of HoxB13 action at these AR-recruiting genes. For AR target genes for which HoxB13 acts as an activator, 2 models were identified. In genes for which the collaborative model applies, DNA binding sites for AR and HoxB13 were present and both AR and HoxB13 bound. In the alternative, tethering model, genes did not harbor canonical AREs but AR was recruited via HoxB13 that was bound to its own consensus motif. A third repressor model applied to genes for which HoxB13 acts as a repressor. Genes whose androgen regulation follows the latter mode of HoxB13 regulation contained AREs but not HoxB13 binding motifs, and HoxB13’s interaction with the AR DBD prevented AR from binding and activating gene transcription via AREs. This differential molecular contribution of HoXB13 to AR target gene expression likely underlied observed differential effects of HoxB13 on androgen-responsive CaP cellular processes. Silencing of HoxB13 for instance decreased androgen regulation of cell proliferation but increased aspects of cell differentiation, for instance lipogenesis (Norris et al. 2009a).
The potential relevance of these findings becomes more obvious taking into account the contribution and defining role of HoxB13 in establishing the clinical AR CaP cistrome (Pomerantz et al. 2015). HoxB13 binding sites, along with those for FoxA1, are enriched significantly in AR binding sites isolated in clinical localized CaP compared to those found in benign prostate samples. Co-expression of FoxA1 and HoxB13 in an immortalized prostate epithelial cell line caused the AR cistrome to resemble that of CaP and conferred neoplastic transformation to the cells. ChIP-Seq studies on clinical CaP specimens and cell lines confirmed extensive and selective overlap between CaP-associated but not normal prostate-associated AR binding peaks and HoxB13 peaks. Collectively, these data put HoxB13, and the molecular mechanisms by which it controls select biological processes, forward as key determinants of lineage-specific CaP-relevant AR action.
iii. NANOG
The pluripotent transcription factor NANOG has been shown to be overexpressed gradually during the progression from benign prostate tissue to lethal CaP, and to promote CR progression in CaP models (Jeter, et al. 2009; Jeter, et al. 2011; Jeter, et al. 2016). In CaP cells, NANOG is predominantly derived from a retrogene (NANOG version known as NP8), although it can be expressed also from the NANOG1 locus (version referred to as NANOG1). We will use the term NANOG to refer to both forms as they behave very similarly in the studies outlined below. ChIP-Seq studies were performed to derive the mechanistic underpinnings of the dependency of CR-CaP on NANOG for growth and regeneration (Jeter et al. 2016). ChIP-Seq was done in LNCaP cells that inducibly overexpressed NANOG, as the low levels of endogenously expressed NANOG protein impeded generation of reliable data. The NANOG cistrome in these cells showed little (<10%) overlap with that obtained from embryonic stem cells, indicating the existence of a CaP-specific NANOG cistrome. Motif analyses of NANOG binding peaks indicated significant enrichment for binding sites for FoxA1 and Nkx3.1, both factors that are known to interact with DNA-bound AR (Tan et al. 2012; Wang et al. 2007). Comparison of NANOG’s DNA occupancy with that of AR demonstrated coordinated binding patterns (up to 70% co-occupancy) and revealed that NANOG occupancy under ADT-naïve conditions most closely resembled that of the CR AR cistrome, suggesting that NANOG reprograms AR cistrome to facilitate CR recurrence. Co-localization and functional interaction between NANOG, AR as well as FoxA1 was confirmed using an array of protein-protein interaction assays. RNA-Seq analyses done at different time points in LNCaP cells that were either androgen-stimulated or -deprived identified distinct clusters of genes for which androgen-regulation was activated or repressed, respectively, following overexpressing of NANOG (Jeter et al. 2016). For instance, 2 cluster of genes were repressed by NANOG under both androgen-supplemented and androgen-deprived conditions. These genes correlated with AR-regulated cell differentiation and sensitivity to ADT, and were highly enriched for co-occupancy of AR, FoxA1 and NANOG. These expression data suggested that NANOG suppresses pro-differentiation genes and supports development of CR-CaP. Remarkably, genes belonging to these clusters were able to stratify patients into low versus high risk groups and predicted favorable patient survival. Two other sets of genes were upregulated early in androgen supplemented conditions, and their expression went down at later time points and was downregulated also under androgen-deprived conditions. These expression patterns suggested that these genes are rapidly induced by NANOG to facilitate CR-CaP progression and require ligand binding for their sustained expression. These genes were less enriched for NANOG, AR and FoxA1 co-occupancy, although 40–50% of peaks occupied by NANOG still recruited also AR and/or FoxA1. The molecular basis for the functional interplay between AR and NANOG at select AR target genes that facilitates resistance to ADT and CR-CaP progression has yet to be elucidated. It is tempting to speculate that both competitive and co-operative models of AR-NANOG interaction, similar to those described for HoxB13’s role in AR action, may be at play here and could involve also the actions of FoxA1 and/or NKX31.
The reports discussed here indicate differential co-operation between AR and non-pioneering transcription factors, which translates into diverse composition of the AR cistrome and AR-dependent transcriptome. The examples highlighted here relate to transcription factors with functions in lineage-specificity, pluripotency or cell differentiation. It is likely that AR’s interaction with other factors controls other gene sets that regulate other aspects of androgen-dependent CaP cell biology. Ultimately, these select AR-transcription factor interactions differentially impact on CaP behavior and progression.
Coregulators
AR’s transcriptional output is modulated further by a continuously growing number of coregulators (currently n>274) (DePriest et al. 2016; Heemers and Tindall 2007). Coregulators are recruited by AR to AREs, but do not bind DNA directly. Several AR-associated coregulators are overexpressed in CR-CaP, i.e. in presence of AR that has been “re-activated” under ADT, compared to localized CaP (Table 1) (Agoulnik, et al. 2006; Balasubramaniam, et al. 2013; Comuzzi, et al. 2004; Goodwin, et al. 2013; Gregory, et al. 2001; Groner, et al. 2016; Halkidou, et al. 2003; Karpf, et al. 2009; Logan, et al. 2006; Malik, et al. 2015; Peng, et al. 2008; Puhr, et al. 2014; Schrecengost, et al. 2014; Varambally, et al. 2002). While AR-associated coregulators do regulate AR’s transcriptional output, and some have been claimed to be specific for AR at the time of isolation (Yeh and Chang 1996), it is now clear that their actions are generally not restricted to AR but regulate also other transcription factors. The functional diversity of coregulators reflects the many ways in which coregulators can enhance or repress AR-dependent transcription, which range from facilitating access to chromatin, ensuring proper AR conformation, modulating AR turnover… The ability of these proteins to selectively control the level of androgen regulation of different AR target genes has been recognized for a while but is yet to be fully appreciated. Until recently, the unavailability of genome-wide AR and coregulator-dependent transcriptomes and cistromes has limited most evaluations to a few ARE-driven promoter-reporter genes and a handful of well-characterized AR target genes (e.g. (Heemers, et al. 2009; Ianculescu, et al. 2012; Marshall, et al. 2003). Genome-wide expression profiling and ChIP-based mapping of transcriptional regulators have now become standard assays and allow to more fully capture the intricacies of coregulators’ context-dependent and gene-specific regulation of AR transactivation. A few examples of different mechanisms by which select interaction between AR and its coregulators controls androgen-responsiveness of subsets of AR target genes are discussed in this section. Cases that are selected demonstrate for instance the ability of AR-associated coregulators to act simultaneously as a coactivator for some AR target genes and as a corepressor for others. Other examples document a change in function of the same coregulator from repressor to activator of gene transcription in clinical CaP before and after ADT, or point towards specific coregulator-AR interaction hubs at select AREs that control expression of only a subset of target genes.
Table 1. Representative coregulators that are overexpressed in CR-CaP.
In italics, official gene symbol for coregulators for which the referenced literature report does not use the official gene symbol but an alias. Key reference, representative literature report that describes immunhistochemical overexpression of coregulator in clinical CR-CaP specimens.
| COREGULATOR | KEY REFERENCE |
|---|---|
| BAF57 (SMARCE1) | Balasubramaniam, et al. 2013 |
| CBP (CREBBP) | Comuzzi, et al. 2004 |
| DNA-PK (PRKDC) | Goodwin, et al. 2014 |
| EZH2 | Varambally, et al. 2002 |
| MAGEA11 | Karpf, et al. 2009 |
| MEP50 (WDR77) | Peng, et al. 2008 |
| MLL2 (KMT2) | Malik, et al. 2015 |
| PIAS1 | Puhr, et al. 2014 |
| PIRH2 (RCHY1) | Logan, et al. 2006 |
| SRC1 (NCOA1) | Gregory, et al. 2001 |
| TIF2 (NCOA2) | Agoulnik, et al. 2006 |
| TIP60 (KAT5) | Halkidou, et al. 2003 |
| TRIM24 | Groner, et al. 2016 |
| USP22 | Schrecengost, et al. 2014 |
i. LSD1
Lysine Specific Demethylase 1, or LSD1, can act as a coactivator or a corepressor for different sets of AR target genes, and both these regulatory functions are relevant to CaP progression (Cai, et al. 2011; Cai, et al. 2014; Metzger, et al. 2010). The relevance of its corepressor function was highlighted following the identification of a novel ARE in the second exon of the AR gene (Cai et al. 2011). This ARE, which is distinct from ARE that had been identified before in AR gene 5′UTR (Wang, et al. 2008b), is highly conserved among humans, mice and rats, and mediated active transcriptional repression of AR expression. Strikingly, this repression occurred at higher doses of androgens than those needed to induce expression of other AR target genes such as PSA. DHT treatment induced tight AR binding to this site, to which FoxA1, Oct1 and GATA2 were recruited also. Stimulation of cells with DHT caused a decrease in H3K4me1 and H3K4me2 epigenetic marks specifically at this AR binding site, but not at other AREs in the AR gene or at other representative AR target genes. The histone demethylase LSD1 was found to be recruited to this site after androgen stimulation and association of both AR and LSD1 with this site decreased after androgen deprivation of CaP cell lines and xenografts. Loss of LSD1 did not decrease DHT-stimulated recruitment of AR but did prevent decrease in methylation at this site. LSD1’s corepressor function for androgen regulation of the AR gene was extrapolated to other genes such as the gene encoding AKR1C3, a critical androgen biosynthesis enzyme for which expression increases under ADT in clinical CaP (Adeniji, et al. 2013). These data indicated that lower androgen levels such as those found in CR-CaP are adequate to stimulate AR action on enhancer elements of genes mediated important metabolic functions, but are not sufficient to reduce AR gene expression and cellular proliferation (Cai et al. 2011). These data also implied that the association between AR and LSD1, which had been reported as coactivator for other AR target genes (Metzger et al. 2010; Metzger, et al. 2005), does not determine LSD1’s action as coactivator or corepressor. That distinction may be determined by properties of DNA segment to which LSD1 binds. This hypothesis was supported by the previous literature reports in which LSD1 served as coactivator for AR target genes such as PSA and KLK2. LSD1’s switch from an AR corepressor to an activator has been linked to its differential demethylase activity; i.e. removing mono- and dimethyl marks from H3K4 as a corepressor to removing mono- and dimethyl marks from H3K9 as coactivator, respectively. Phosphorylation of H3T6 by protein kinase C beta I has been identified as the key event that prevents LSD1 from demethylating H3K4 during AR-dependent gene activation (Metzger et al. 2010). Consistent with this model, ChIP-Seq studies showed that 20% of genome-wide LSD1 and AR peaks overlapped in androgen-treated LNCaP cells and co-occupied sites showed significant enrichment for H3T6ph marks (Cai et al. 2014). These observations provide an explanation for the distinct actions of LSD1 at subclasses of differentially epigenetically marked ARE’s to either repress or activate target gene transcription.
ii. EZH2
Enhancer of zeste homolog 2, or EZH2, is of considerable interest in CaP as it is overexpressed in patient specimens, particularly in CR-CaP samples, where cells rely on it to proliferate (Varambally et al. 2002; Xu, et al. 2012). As the catalytic subunit for Polycomb repressive complex 2 (PRC2), EZH2 has been studied intensively for its role in gene repression, e.g. that of the tumor suppressor DAB2IP, via trimethylation of H3K27 (Cao, et al. 2002; Chen, et al. 2005). A comparison of EZH2 function between LNCaP cells and its CR subline abl, uncovered a novel, CR-CaP specific role as an AR-associated coactivator (Xu et al. 2012). EZH2-dependent transcriptome profiling identified a larger gene set of EZH2-stimulated genes in abl and other CR-CaP cells compared to ADT-naïve cells, and these genes were highly expressed also in clinical CR-CaP clinical specimens. Subsequent ChIP-Seq analysis isolated subsets of EZH2 binding sites that lack H3K27 methylation marks, again selectively in CR-CaP cells. These peaks were enriched in epigenetic markers of active transcription, and the associated genes were induced by EZH2 and had prognostic power in CR-CaP. A striking enrichment for AR binding motifs was found in the CR-CaP-specific EZH2 binding sites. The physical interaction between AR and EZH2, found in co-immunoprecipitation and gel filtration assays, was reflected in significant overlap in AR and EZH2 genomic binding peaks. Silencing of EZH2 selectively prevented biding of AR to co-occupied binding sites but not to other peaks. This shift in function of EZH2 from a more general transcriptional repressor to an AR-associated coactivator was caused by a phosphorylation of EZH2 at Ser21, which occurred specifically in CR-CaP cells and may be mediated by Akt action (Xu et al. 2012). This CaP-stage selective role for EZH2 implies that targeting this specific aspect of its function, which is not pursued by available EZH2 inhibitors (Sparmann and van Lohuizen 2006), may lead to a novel CR-specific CaP treatment modality.
iii. SIAH2
Siah2 is an E3 ubiquitin ligase that targets a select pool of chromatin-bound AR for degradation, which leads to preferential control over the growth, survival and tumorigenic capacity of CaP cells, particularly under ADT conditions (Qi, et al. 2013). Initial studies in which Siah2 −/− mice were crossed with TRAMP mice showed a greater reduction in expression of AR target genes such as NKX3.1 and SPINK3 but not other genes like probasin and TMPRSS2 in castrated Siah2-deficient animals. The suggestion that Siah2 regulates the expression of a specific subset of AR target genes was confirmed in human LNCaP and Rv1 CaP cell lines. Gene expression profiling on Rv1 cells after silencing of Siah2 expression showed downregulation of AR target genes with functions in lipid metabolism, steroid metabolism and cholesterol metabolism. Several of these genes were necessary for growth under ADT and were enriched in CR-CaP tissues. Siah2 bound to AR, and degraded AR in an E3 ubiquitin ligase-dependent manner; this Siah2-AR interaction was necessary for AR’s transcriptional activity and control over Siah2-dependent AR target genes. siRNA-mediated silencing of Siah2 did not affect levels of total, nuclear or chromatin bound AR in CaP cells. It did, however, increase the levels of AR and NCoR1, a AR associated corepressor and substrate for Siah2, at AREs of the select genes, but did not affect their binding to others, suggesting that Siah2 selectively degradates NCoR1-bound AR at those target genes. In line with this model, levels of ubiquitinated AR were reduced at those AREs after silencing of Siah2, and knock-down of NCoR1 attenuated Siah2 recruitment to those sites. Co-IP showed stronger association of Siah2 with NCoR1-bound AR than with AR alone (Qi et al. 2013). Siah2 thus uses another molecular route to diversify AR’s transcriptional output, namely degradation of transcriptionally inactive, NCOR1-bound AR on AREs of select AR target genes. The 3 examples of coregulators that are discussed in this section provide a snapshot only of the variety in molecular mechanisms by which coregulators can influence the expression of subsets AR target genes.
Different molecular modes of AR action: opportunities for novel AR-targeting therapies?
Taken together, the evidence discussed above indicates that variability in the sequence of the ARE half-site(s) and the composition of the surrounding DNA regions at different target genes may dictate, or restrict, access of AR to DNA. In addition, differential contribution of pioneer factors has been linked to (re)programming of the AR cistrome. Non-pioneering transcription factors and coregulators use a variety of molecular mechanisms to modulate the output of AR transactivation at a given target genes and, thus induce or repress specific biological processes. These data provide evidence for the diverse composition, and subsequent output, of AR transcriptional complexes across AREs. This possibility fits with the previously proposed model of allosteric effects of DNA sequence on the 3-dimensional structure of DNA-bound GR, a close relative of AR. These allosteric effects have been linked with selective interaction between GR and the coregulators Brm and CARM1 (Meijsing, et al. 2009). The observed specificity recalls also previously described allosteric effects in AR following binding by different ligands, which led to preferentially interacts with different coregulators and coregulatory-derived peptides (Baek, et al. 2006; Norris et al. 2009b). The literature that is reviewed here suggests that allosteric effects with consequences for AR’s interaction with transactivation factors may occur also when AR that has been stimulated by the same ligand is bound to different AREs.
The examples discussed here cover only a handful of known AR-affiliated transcription factors and coregulators. As genomic data for additional transcriptional regulators becomes available, it will be important to determine whether similar AR target gene specificity and context-dependency of AR’s transcriptional output also occurs, to define the extent of overlap of such specificity for different AR-associated proteins, and to decipher these patterns in clinical specimens as well as preclinical models. The resulting datasets should increase significantly our knowledge about the modes of protein-protein interaction, and modular hierarchy that may govern such interactions. Modular interaction has been described for steady-state coregulatory complexes in HeLA cells (Malovannaya et al. 2011), but is yet to be examined with respect to ligand-activated transcription.
None-the-less, the evidence derived from the examples discussed here and from others that are not highlighted in this review (e.g. (Toropainen, et al. 2015; Xu, et al. 2009)), does support the presence of different hubs of AR-dependent transcription that control different aspects of androgen-dependent CaP cell biology. The existence of multiple transcriptional codes that result from specific AR-protein and AR-DNA interactions provides the possibility of targets other than ligand-activation domain of AR to inhibit specific portion(s) of AR action that drives CaP progression (Heemers 2014). The opportunity to design those treatment modalities to only the select AR-dependent events that are CaP-specific may also reduce the severe side effects that are associated with systemic inhibition of AR action (Nguyen et al. 2014).
How to go about the daunting task of disruption the output from specific AR transcriptional complexes? Some drug options may be available already. For instance, because a significant portion of them contain enzymatic moieties, coregulators have long been considered as attractive novel targets for CaP treatment (Chmelar, et al. 2007; Heemers and Tindall 2010). For several coregulators, such as SRC family members and LSD1, inhibitors are available that have been tested in preclinical CaP models (Cai et al. 2011; Cai et al. 2014; Wang, et al. 2011b; Wang, et al. 2014). Other therapeutics may be derived from Chem-Seq approaches that map genome-wide interactions between small molecules and their protein targets on chromatin. Chem-Seq assays have identified e.g. SD70 as a novel inhibition of KDM4C to inhibit AR function and CaP cell growth (Jin, et al. 2014). In the case of EZH2, novel inhibitors that target only its Ser21 phosphorylated activator function may lead to CaP stage-specific application (Xu et al. 2012). A potential drawback of this type of compounds is that they will likely simultaneously inhibit both coactivator and corepressor function of the coregulator that is being targeted. This may not be desirable, and/or may impact also on the action of other transcription factors with which the coregulator collaborates. The latter “promiscuity” may lead to significant off-target effects. Other approaches to target more specifically discrete coregulatory-AR interactions of interest have been tested. The use of peptides, such as BAF57 inhibitory peptide (BIPep), which targets the protein-protein interfaces between AR and BAF57, blocks AR residency on chromatin, the resultant AR target gene activation and CaP cell proliferation (Link, et al. 2005). Along the same lines, peptidomimetics have been developed that are able to disrupt interaction between AR and its coregulator PELP1 in several preclinical CaP models and ex vivo CaP explants (Ravindranathan, et al. 2013). Similarly, multivalent peptoid conjugates that block AR coactivator-peptide interactions and prevent AR intermolecular interactions reduced proliferation of CR-CaP cell lines and reduced tumor growth, and AR and Ki67 expression in CR-CaP xenografts (Levine, et al. 2012; Wang, et al. 2016). While the above mentioned tactics strive to disrupt specific AR-protein interactions, successful attempts have been made also at disruption the interaction between AR and AREs and between AR-associated transcription factors and their consensus binding motifs. DNA-binding polyamides that target the consensus ARE half-sites in gene such as PSA, reduce AR occupancy at AREs and effectively block the resulting gene transcription and exert antitumor effects on CaP xenografts (Hargrove, et al. 2015; Nickols and Dervan 2007). Similar approaches in which the Oct1 binding site of a representative AR target gene was targeted also prevented Oct1-dependent transcription as well as the growth of CaP xenografts (Obinata et al. 2016). Alternatively, a small molecule that selectively binds a surface exposed pocket on AR-DBD effectively blocked transcriptional activity of full length AR and constitutively active AR splice variants (Dalal et al. 2014). Targeting what were previously viewed as undruggable interactions within transcriptional complexes provides proof-of-principle for development of novel compounds that inhibit critical protein-protein and protein-DNA interactions that are most relevant to AR-mediated lethal CaP progression. It is conceivable that similar approaches could be extrapolated to disrupt other relevant AR-coregulator, AR-transcription factor, coregulatory-coregulator etc. interactions (Figure 4). Whether such compounds are best administered as monotherapy or as part of a more ‘holistic’ approach in which multiple interaction are targeted at the same time will need to be determined.
Figure 4. Overview of novel therapeutic strategies to disrupt select AR transcriptional complexes.
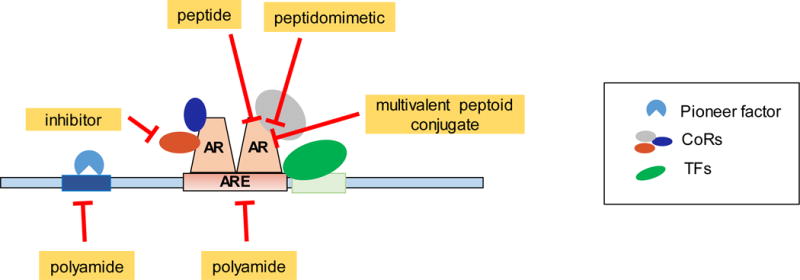
Polyamides inhibit the binding of transcription factors with chromatin. Specific inhibitors target enzymatic moieties of some coregulators. Peptides, peptidomimetics, and multivalent peptoid conjugates disrupt specific AR-coregulatory interactions.
The primary goal of disruption crucial protein-protein and/or protein-DNA interactions is to impair the activity of the corresponding AR transcriptional complexes and to decrease their transcriptional output. Because such interventions may have secondary effects on the stability or the recruitment of different components of the AR complex at the set of AREs that is targeted, it will be important to monitor also their impact on AR, coregulator, transcription factor, RNA pol II … cistromes.
Confounders and facilitators of “selective” forms of androgen deprivation therapy
From a molecular perspective, the output of a specific AR transcriptional complex can be influenced by alterations in the expression of AR, the DNA sequence to which it binds and its protein interactome. In clinical CaP, all of these components have been reported to undergo somatic alterations in a subset of cases. For instance, overexpression of AR and/or (co-)expression of AR variants that lack ligand binding domain impact the level of AR binding throughout the genome and composition of the AR cistrome (Chan et al. 2015; Lu, et al. 2015; Urbanucci, et al. 2012a; Urbanucci, et al. 2012b). Copy number alterations occur in ARE-containing DNA regions and single nucleotide variants affect AREs (Whitington, et al. 2016); both can impact the androgen-dependent expression of the corresponding ARE-containing target gene. Some AR target genes harbor more than 1 ARE, and long distance interactions between AR-bound enhancer with gene promotors mediate transcription of AR target genes (Reid, et al. 2001; Wang, et al. 2005). How the composition of the AR transcriptional complex correlates at different regions of the same gene, and the impact on its activity of cooperativity between multiple AREs within the same gene remains to be determined.
Defining the DNA-binding patterns of components of AR transcriptional complexes, particularly those that do not directly bind DNA, in clinical CaP specimens has been challenging technically because the amounts and the purity of tissue that is required. Nonetheless, efforts to define the AR cistrome in clinical CaP specimens have yielded novel insights in AR action in clinical CaP progression (Pomerantz et al. 2015; Sharma et al. 2013). Elucidation of the recruitment to DNA by AR-associated proteins is likely to be facilitated by the increasing availability of ex vivo explants, organoids, and patient-derived xenografts derived from localized and metastatic CaP that are representative of clinical CaP tissue. Unlike patient specimens, the latter models have the advantage that they allow for androgen supplementation or deprivation, or for silencing or overexpression of genes of interest.
Several AR-interacting proteins undergo somatic mutations that affect their regulatory function during progression to CR-CaP, including MLL2, NCOR1 and FoxA1 (Barbieri, et al. 2012; Grasso et al. 2012). The impact of these alterations on AR-dependent target gene expression and AR cistrome has yet to be fully explored. It is likely, however, that these may have pronounced effects on AR’s function and underlie the heterogeneity that has been observed for AR transcriptional output in patient specimens. As for genomic CaP heterogeneity in general, this variability is more pronounced in localized ADT-naïve CaP than in metastatic CaP that has recurred under ADT (Kumar, et al. 2016; Wei et al. 2016). In addition to alterations that directly affect components and contributors to AR transcription complexes, the expression and mutational status of other genomic markers, can affect AR’s transactivation function. For instance, recurrent point mutation in the E3 ubiquitin ligase SPOP, found in up to 15 percent of CaP specimens (Barbieri et al. 2012), render SPOP incapable of degrading AR, AR variants, and AR coregulators such as SRC3 and TRIM24 (An, et al. 2014; Barbieri et al. 2012; Cancer Genome Atlas Research Network. Electronic address and Cancer Genome Atlas Research 2015; Geng, et al. 2013; Groner et al. 2016). As a consequence, expression of these proteins is elevated in CaPs that express mutant SPOP, which likely impacts the specific AR target gene expression under their control. Alternatively, the TMPRSS2-ERG gene fusion is present in at least half of clinical CaPs (Bostrom et al. 2015). As discussed above, ERG contributes to transcriptional output of a subset of AR target genes in addition to repressing AR expression (Yu et al. 2010). When PTEN is deleted, the impact of TMPRSS2-ERG on aggressive CaP progression is even more pronounced (Chen, et al. 2013; Zong, et al. 2009); it is possible that alterations in AR functions contribute to this behavior. The tumor suppressor Rb negatively regulates AR expression. Consequently, deletion of Rb, a common occurrence in CaP progression, increases significantly AR’s expression and output (Sharma, et al. 2010).
In view of the large number of AR-associated proteins, it is very likely that more than one “selective” AR-dependent transcriptional mechanism is active in a given CaP. Deciding which mechanism(s) to pursue for therapeutic intervention may therefore not be straight-forward. Therapeutic benefit will be most pronounced if the most appropriate mechanism (s) is targeted in a rational, evidence-based manner. The presence of the genomic alterations described above may serve as biomarkers to facilitate the decision to target, or avoid targeting, specific protein-protein or protein-DNA interactions. These genomic markers are measured in routine genomic tests that are performed on clinical CaP biopsies or radical prostatectomy specimens, such as the Foundation One assay (Frampton, et al. 2013). Alternatively, information on the expression level and post-translational modification or mutational status of AR-associated proteins, as well as the specific gene expression programs under their control, may also serve as suitable biomarkers. Moving forward, other potential contributors to AR-mediated transcriptional complexes such as lncRNAs, miRNAs, and pseudogenes, which could impact on the expression levels of the resulting AR mRNA profiles (Tay, et al. 2014), may have to be considered also for the proposed therapeutic strategies. At this time, the knowledge on their respective contribution to specific AR-dependent transcriptional events is too limited for inclusion in the current review.
The proposed therapeutic strategies to target specific AR-dependent transcriptional complexes will require also appropriate clinical endpoints to monitor a patient’s response and CaP progression. Serum levels of PSA, an AR target gene whose expression is evaluated as surrogate for CaP burden during CaP progression, may not be the most suitable measurement. Serum PSA has been recognized to be an imperfect CaP biomarker (Prensner, et al. 2012).
More relevant to this discussion, its expression may not be controlled to the same extent by different AR transcriptional complexes. Determination of the efficacy of the proposed therapies may benefit from disease monitoring as outlined by the Prostate Cancer Clinical Trials Working Group 3 (PCWG3) (Scher, et al. 2016). PCWG3 has taken into consideration evolving CaP treatments and changes in drug development to formulate recommendations that facilitate disease management, clinical decisions and clinical trial design. In addition to biochemical endpoints such as serum levels of PSA, PCWG3 guidelines include evaluation of the number of circulating tumor cells, (radiographic) imaging, and symptomatic progression, as well as taking into account the time to changes in these parameters. Assessment of the response to targeted therapies will benefit also from PCWG3-recommended serial molecular profiling of blood, circulating tumor cells or metastatic tumor sites. Measuring expression of specific target genes or gene signatures in blood, circulating tumor cells or CR-CaP tissue is expected to facilitate the development of predictive biomarkers of response to the proposed therapeutic avenues.
Conclusions
A growing body of evidence indicates that AR-dependent transcription does not occur via a uniform mechanism. Considerable variability in AR-DNA and AR-protein interactions exists that fine-tunes the output of specific AR’s transcriptional complexes. Specific disrupting of those interactions that control aggressive AR-dependent CaP progression may lead to a viable therapeutic strategy in CaP for which ADT that targets androgen biosynthesis has failed.
Acknowledgments
The authors thank the members of the Heemers laboratory for helpful discussions.
Funding
This work was supported the National Cancer Institute (grant number CA166440), the Department of Defense Prostate Cancer Research Program (grant number W81XWH-16-1-0404), and a VeloSano 3 pilot Research Award (to H.V.H.).
Footnotes
Declaration of interest
The authors declare no conflicts of interest.
References
- Adeniji AO, Chen M, Penning TM. AKR1C3 as a target in castrate resistant prostate cancer. J Steroid Biochem Mol Biol. 2013;137:136–149. doi: 10.1016/j.jsbmb.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agoulnik IU, Vaid A, Nakka M, Alvarado M, Bingman WE, 3rd, Erdem H, Frolov A, Smith CL, Ayala GE, Ittmann MM, et al. Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Res. 2006;66:10594–10602. doi: 10.1158/0008-5472.CAN-06-1023. [DOI] [PubMed] [Google Scholar]
- An J, Wang C, Deng Y, Yu L, Huang H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep. 2014;6:657–669. doi: 10.1016/j.celrep.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, Watt K, Tam T, Yang YC, Banuelos CA, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–546. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- Attard G, Cooper CS, de Bono JS. Steroid hormone receptors in prostate cancer: a hard habit to break? Cancer Cell. 2009;16:458–462. doi: 10.1016/j.ccr.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Attard G, Parker C, Eeles RA, Schroder F, Tomlins SA, Tannock I, Drake CG, de Bono JS. Prostate cancer. Lancet. 2016;387:70–82. doi: 10.1016/S0140-6736(14)61947-4. [DOI] [PubMed] [Google Scholar]
- Baek SH, Ohgi KA, Nelson CA, Welsbie D, Chen C, Sawyers CL, Rose DW, Rosenfeld MG. Ligand-specific allosteric regulation of coactivator functions of androgen receptor in prostate cancer cells. Proc Natl Acad Sci U S A. 2006;103:3100–3105. doi: 10.1073/pnas.0510842103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baena E, Shao Z, Linn DE, Glass K, Hamblen MJ, Fujiwara Y, Kim J, Nguyen M, Zhang X, Godinho FJ, et al. ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev. 2013;27:683–698. doi: 10.1101/gad.211011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramaniam S, Comstock CE, Ertel A, Jeong KW, Stallcup MR, Addya S, McCue PA, Ostrander WF, Jr, Augello MA, Knudsen KE. Aberrant BAF57 signaling facilitates prometastatic phenotypes. Clin Cancer Res. 2013;19:2657–2667. doi: 10.1158/1078-0432.CCR-12-3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, Jarosz M, Lipson D, Tagawa ST, Nanus DM, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013;63:920–926. doi: 10.1016/j.eururo.2012.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostrom PJ, Bjartell AS, Catto JW, Eggener SE, Lilja H, Loeb S, Schalken J, Schlomm T, Cooperberg MR. Genomic Predictors of Outcome in Prostate Cancer. Eur Urol. 2015;68:1033–1044. doi: 10.1016/j.eururo.2015.04.008. [DOI] [PubMed] [Google Scholar]
- Boutros PC, Fraser M, Harding NJ, de Borja R, Trudel D, Lalonde E, Meng A, Hennings-Yeomans PH, McPherson A, Sabelnykova VY, et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat Genet. 2015;47:736–745. doi: 10.1038/ng.3315. [DOI] [PubMed] [Google Scholar]
- Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, Nelson PS, Liu XS, Brown M, Balk SP. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011;20:457–471. doi: 10.1016/j.ccr.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C, He HH, Gao S, Chen S, Yu Z, Gao Y, Chen S, Chen MW, Zhang J, Ahmed M, et al. Lysine-specific demethylase 1 has dual functions as a major regulator of androgen receptor transcriptional activity. Cell Rep. 2014;9:1618–1627. doi: 10.1016/j.celrep.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callewaert L, Verrijdt G, Christiaens V, Haelens A, Claessens F. Dual function of an amino-terminal amphipatic helix in androgen receptor-mediated transactivation through specific and nonspecific response elements. J Biol Chem. 2003;278:8212–8218. doi: 10.1074/jbc.M210744200. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Electronic address scmo & Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B, Qi Y, Zhang G, Xu D, Zhan Y, Alvarez X, Guo Z, Fu X, Plymate SR, Sartor O, et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget. 2014;5:1646–1656. doi: 10.18632/oncotarget.1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- Celis L, Claessens F, Peeters B, Heyns W, Verhoeven G, Rombauts W. Proteins interacting with an androgen-responsive unit in the C3(1) gene intron. Mol Cell Endocrinol. 1993;94:165–172. doi: 10.1016/0303-7207(93)90165-g. [DOI] [PubMed] [Google Scholar]
- Chan SC, Selth LA, Li Y, Nyquist MD, Miao L, Bradner JE, Raj GV, Tilley WD, Dehm SM. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic Acids Res. 2015;43:5880–5897. doi: 10.1093/nar/gkv262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Tu SW, Hsieh JT. Down-regulation of human DAB2IP gene expression mediated by polycomb Ezh2 complex and histone deacetylase in prostate cancer. J Biol Chem. 2005;280:22437–22444. doi: 10.1074/jbc.M501379200. [DOI] [PubMed] [Google Scholar]
- Chen Y, Chi P, Rockowitz S, Iaquinta PJ, Shamu T, Shukla S, Gao D, Sirota I, Carver BS, Wongvipat J, et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat Med. 2013;19:1023–1029. doi: 10.1038/nm.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Lan X, Thomas-Ahner JM, Wu D, Liu X, Ye Z, Wang L, Sunkel B, Grenade C, Chen J, et al. Agonist and antagonist switch DNA motifs recognized by human androgen receptor in prostate cancer. EMBO J. 2015;34:502–516. doi: 10.15252/embj.201490306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmelar R, Buchanan G, Need EF, Tilley W, Greenberg NM. Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. Int J Cancer. 2007;120:719–733. doi: 10.1002/ijc.22365. [DOI] [PubMed] [Google Scholar]
- Claessens F, Joniau S, Helsen C. Comparing the rules of engagement of androgen and glucocorticoid receptors. Cell Mol Life Sci. 2017 doi: 10.1007/s00018-017-2467-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comuzzi B, Nemes C, Schmidt S, Jasarevic Z, Lodde M, Pycha A, Bartsch G, Offner F, Culig Z, Hobisch A. The androgen receptor co-activator CBP is up-regulated following androgen withdrawal and is highly expressed in advanced prostate cancer. J Pathol. 2004;204:159–166. doi: 10.1002/path.1609. [DOI] [PubMed] [Google Scholar]
- Culig Z. Androgen receptor cross-talk with cell signalling pathways. Growth Factors. 2004;22:179–184. doi: 10.1080/08977190412331279908. [DOI] [PubMed] [Google Scholar]
- Dai C, Heemers HV, Sharifi N. Androgen signalling in prostate cancer. Cold Spring HArb PErspect MEd. 2017 doi: 10.1101/cshperspect.a030452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalal K, Roshan-Moniri M, Sharma A, Li H, Ban F, Hassona MD, Hsing M, Singh K, LeBlanc E, Dehm S, et al. Selectively targeting the DNA-binding domain of the androgen receptor as a prospective therapy for prostate cancer. J Biol Chem. 2014;289:26417–26429. doi: 10.1074/jbc.M114.553818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Laere B, van Dam PJ, Whitington T, Mayrhofer M, Diaz EH, Van den Eynden G, Vandebroek J, Del-Favero J, Van Laere S, Dirix L, et al. Comprehensive Profiling of the Androgen Receptor in Liquid Biopsies from Castration-resistant Prostate Cancer Reveals Novel Intra-AR Structural Variation and Splice Variant Expression Patterns. Eur Urol. 2017 doi: 10.1016/j.eururo.2017.01.011. [DOI] [PubMed] [Google Scholar]
- Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denmeade SR, Isaacs JT. A history of prostate cancer treatment. Nature reviews Cancer. 2002;2:389–396. doi: 10.1038/nrc801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePriest AD, Fiandalo MV, Schlanger S, Heemers F, Mohler JL, Liu S, Heemers HV. Regulators of Androgen Action Resource: a one-stop shop for the comprehensive study of androgen receptor action. Database (Oxford) 2016;2016 doi: 10.1093/database/bav125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng C, He B, Xu L, Barbieri CE, Eedunuri VK, Chew SA, Zimmermann M, Bond R, Shou J, Li C, et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc Natl Acad Sci U S A. 2013;110:6997–7002. doi: 10.1073/pnas.1304502110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, Ma T, Den RB, Dicker AP, Feng FY, et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013;3:1254–1271. doi: 10.1158/2159-8290.CD-13-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. The androgen receptor gene mutations database: 2012 update. Hum Mutat. 2012;33:887–894. doi: 10.1002/humu.22046. [DOI] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, Wilson EM. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61:4315–4319. [PubMed] [Google Scholar]
- Groner AC, Cato L, de Tribolet-Hardy J, Bernasocchi T, Janouskova H, Melchers D, Houtman R, Cato AC, Tschopp P, Gu L, et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell. 2016;29:846–858. doi: 10.1016/j.ccell.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haelens A, Verrijdt G, Schoenmakers E, Alen P, Peeters B, Rombauts W, Claessens F. The first exon of the human sc gene contains an androgen responsive unit and an interferon regulatory factor element. Mol Cell Endocrinol. 1999;153:91–102. doi: 10.1016/s0303-7207(99)00079-9. [DOI] [PubMed] [Google Scholar]
- Halkidou K, Gnanapragasam VJ, Mehta PB, Logan IR, Brady ME, Cook S, Leung HY, Neal DE, Robson CN. Expression of Tip60, an androgen receptor coactivator, and its role in prostate cancer development. Oncogene. 2003;22:2466–2477. doi: 10.1038/sj.onc.1206342. [DOI] [PubMed] [Google Scholar]
- Hargrove AE, Martinez TF, Hare AA, Kurmis AA, Phillips JW, Sud S, Pienta KJ, Dervan PB. Tumor Repression of VCaP Xenografts by a Pyrrole-Imidazole Polyamide. PLoS One. 2015;10:e0143161. doi: 10.1371/journal.pone.0143161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Lanz RB, Fiskus W, Geng C, Yi P, Hartig SM, Rajapakshe K, Shou J, Wei L, Shah SS, et al. GATA2 facilitates steroid receptor coactivator recruitment to the androgen receptor complex. Proc Natl Acad Sci U S A. 2014;111:18261–18266. doi: 10.1073/pnas.1421415111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, Zhang Y, Xu K, Ni M, Lupien M, et al. Nucleosome dynamics define transcriptional enhancers. Nat Genet. 2010;42:343–347. doi: 10.1038/ng.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemers H, Tindall D. Androgen receptor coregulatory proteins as potential therapeutic targets in the treatment of prostate cancer. Curr Cancer Ther Rev. 2005;1:175–186. [Google Scholar]
- Heemers H, Tindall D. Nuclear Receptor Coregulators: Promising Therapeutic Targets for the Treatment of Prostate Cancer. In: Figg WD, Chau CH, Small E, editors. Drug Management of Prostate Cancer. Springer; 2010. pp. 41–52. [Google Scholar]
- Heemers HV. Targeting Androgen Receptor Action for Prostate Cancer Treatment: Does the Post-Receptor Level Provide Novel Opportunities? Int J Biol Sci. 2014;10:576–587. doi: 10.7150/ijbs.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemers HV, Regan KM, Schmidt LJ, Anderson SK, Ballman KV, Tindall DJ. Androgen modulation of coregulator expression in prostate cancer cells. Mol Endocrinol. 2009;23:572–583. doi: 10.1210/me.2008-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28:778–808. doi: 10.1210/er.2007-0019. [DOI] [PubMed] [Google Scholar]
- Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- Henzler C, Li Y, Yang R, McBride T, Ho Y, Sprenger C, Liu G, Coleman I, Lakely B, Li R, et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat Commun. 2016;7:13668. doi: 10.1038/ncomms13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie-Inoue K, Inoue S. Genome-wide integrated analyses of androgen receptor signaling in prostate cancer based on high-throughput technology. Curr Drug Targets. 2013;14:472–480. doi: 10.2174/1389450111314040008. [DOI] [PubMed] [Google Scholar]
- Hu R, Isaacs WB, Luo J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate. 2011;71:1656–1667. doi: 10.1002/pros.21382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Pu Y, Hepps D, Danielpour D, Prins GS. Posterior Hox gene expression and differential androgen regulation in the developing and adult rat prostate lobes. Endocrinology. 2007;148:1235–1245. doi: 10.1210/en.2006-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins C, Hodges CV. Studies on Prostatic Cancer. I. The Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. Cancer Research. 1941;1:293–297. doi: 10.3322/canjclin.22.4.232. [DOI] [PubMed] [Google Scholar]
- Huggins CS. Studies on prostatic cancer: 2. the effects of castration on advanced carcinoma of the prostate gland. Arch Surg. 1941;43:209–212. [Google Scholar]
- Ianculescu I, Wu DY, Siegmund KD, Stallcup MR. Selective roles for cAMP response element-binding protein binding protein and p300 protein as coregulators for androgen-regulated gene expression in advanced prostate cancer cells. J Biol Chem. 2012;287:4000–4013. doi: 10.1074/jbc.M111.300194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeter CR, Badeaux M, Choy G, Chandra D, Patrawala L, Liu C, Calhoun-Davis T, Zaehres H, Daley GQ, Tang DG. Functional evidence that the self-renewal gene NANOG regulates human tumor development. Stem Cells. 2009;27:993–1005. doi: 10.1002/stem.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, Repass J, Zaehres H, Shen JJ, Tang DG. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene. 2011;30:3833–3845. doi: 10.1038/onc.2011.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeter CR, Liu B, Lu Y, Chao HP, Zhang D, Liu X, Chen X, Li Q, Rycaj K, Calhoun-Davis T, et al. NANOG reprograms prostate cancer cells to castration resistance via dynamically repressing and engaging the AR/FOXA1 signaling axis. Cell Discov. 2016;2:16041. doi: 10.1038/celldisc.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Yang L, Xie M, Lin C, Merkurjev D, Yang JC, Tanasa B, Oh S, Zhang J, Ohgi KA, et al. Chem-seq permits identification of genomic targets of drugs against androgen receptor regulation selected by functional phenotypic screens. Proc Natl Acad Sci U S A. 2014;111:9235–9240. doi: 10.1073/pnas.1404303111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin HJ, Kim J, Yu J. Androgen receptor genomic regulation. Transl Androl Urol. 2013;2:157–177. doi: 10.3978/j.issn.2223-4683.2013.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozwik KM, Carroll JS. Pioneer factors in hormone-dependent cancers. Nat Rev Cancer. 2012;12:381–385. doi: 10.1038/nrc3263. [DOI] [PubMed] [Google Scholar]
- Jung C, Kim RS, Zhang HJ, Lee SJ, Jeng MH. HOXB13 induces growth suppression of prostate cancer cells as a repressor of hormone-activated androgen receptor signaling. Cancer Res. 2004;64:9185–9192. doi: 10.1158/0008-5472.CAN-04-1330. [DOI] [PubMed] [Google Scholar]
- Karantanos T, Evans CP, Tombal B, Thompson TC, Montironi R, Isaacs WB. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur Urol. 2015;67:470–479. doi: 10.1016/j.eururo.2014.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpf AR, Bai S, James SR, Mohler JL, Wilson EM. Increased expression of androgen receptor coregulator MAGE-11 in prostate cancer by DNA hypomethylation and cyclic AMP. Mol Cancer Res. 2009;7:523–535. doi: 10.1158/1541-7786.MCR-08-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klezovitch O, Risk M, Coleman I, Lucas JM, Null M, True LD, Nelson PS, Vasioukhin V. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc Natl Acad Sci U S A. 2008;105:2105–2110. doi: 10.1073/pnas.0711711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. 2010;21:315–324. doi: 10.1016/j.tem.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, Etzioni R, Bolouri H, Montgomery B, White T, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016 doi: 10.1038/nm.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine PM, Imberg K, Garabedian MJ, Kirshenbaum K. Multivalent peptidomimetic conjugates: a versatile platform for modulating androgen receptor activity. J Am Chem Soc. 2012;134:6912–6915. doi: 10.1021/ja300170n. [DOI] [PubMed] [Google Scholar]
- Li Z, Alyamani M, Li J, Rogacki K, Abazeed M, Upadhyay SK, Balk SP, Taplin ME, Auchus RJ, Sharifi N. Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy. Nature. 2016;533:547–551. doi: 10.1038/nature17954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Bishop AC, Alyamani M, Garcia JA, Dreicer R, Bunch D, Liu J, Upadhyay SK, Auchus RJ, Sharifi N. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature. 2015;523:347–351. doi: 10.1038/nature14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link KA, Burd CJ, Williams E, Marshall T, Rosson G, Henry E, Weissman B, Knudsen KE. BAF57 governs androgen receptor action and androgen-dependent proliferation through SWI/SNF. Mol Cell Biol. 2005;25:2200–2215. doi: 10.1128/MCB.25.6.2200-2215.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan IR, Gaughan L, McCracken SR, Sapountzi V, Leung HY, Robson CN. Human IRH2 enhances androgen receptor signaling through inhibition of histone deacetylase 1 and is overexpressed in prostate cancer. Mol Cell Biol. 2006;26:6502–6510. doi: 10.1128/MCB.00147-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Van der Steen T, Tindall DJ. Are androgen receptor variants a substitute for the full-length receptor? Nat Rev Urol. 2015;12:137–144. doi: 10.1038/nrurol.2015.13. [DOI] [PubMed] [Google Scholar]
- Magnan S, Zarychanski R, Pilote L, Bernier L, Shemilt M, Vigneault E, Fradet V, Turgeon AF. Intermittent vs Continuous Androgen Deprivation Therapy for Prostate Cancer: A Systematic Review and Meta-analysis. JAMA Oncol. 2015;1:1261–1269. doi: 10.1001/jamaoncol.2015.2895. [DOI] [PubMed] [Google Scholar]
- Malik R, Khan AP, Asangani IA, Cieslik M, Prensner JR, Wang X, Iyer MK, Jiang X, Borkin D, Escara-Wilke J, et al. Targeting the MLL complex in castration-resistant prostate cancer. Nat Med. 2015;21:344–352. doi: 10.1038/nm.3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malovannaya A, Lanz RB, Jung SY, Bulynko Y, Le NT, Chan DW, Ding C, Shi Y, Yucer N, Krenciute G, et al. Analysis of the human endogenous coregulator complexome. Cell. 2011;145:787–799. doi: 10.1016/j.cell.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall TW, Link KA, Petre-Draviam CE, Knudsen KE. Differential requirement of SWI/SNF for androgen receptor activity. J Biol Chem. 2003;278:30605–30613. doi: 10.1074/jbc.M304582200. [DOI] [PubMed] [Google Scholar]
- Massie CE, Adryan B, Barbosa-Morais NL, Lynch AG, Tran MG, Neal DE, Mills IG. New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep. 2007;8:871–878. doi: 10.1038/sj.embor.7401046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNair C, Urbanucci A, Comstock CE, Augello MA, Goodwin JF, Launchbury R, Zhao SG, Schiewer MJ, Ertel A, Karnes J, et al. Cell cycle-coupled expansion of AR activity promotes cancer progression. Oncogene. 2017;36:1655–1668. doi: 10.1038/onc.2016.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger E, Imhof A, Patel D, Kahl P, Hoffmeyer K, Friedrichs N, Muller JM, Greschik H, Kirfel J, Ji S, et al. Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature. 2010;464:792–796. doi: 10.1038/nature08839. [DOI] [PubMed] [Google Scholar]
- Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH, Yang YC, Tavakoli I, Haile S, Watt K, et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013;123:2948–2960. doi: 10.1172/JCI66398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PL, Alibhai SM, Basaria S, D’Amico AV, Kantoff PW, Keating NL, Penson DF, Rosario DJ, Tombal B, Smith MR. Adverse Effects of Androgen Deprivation Therapy and Strategies to Mitigate Them. Eur Urol. 2014 doi: 10.1016/j.eururo.2014.07.010. [DOI] [PubMed] [Google Scholar]
- Nickols NG, Dervan PB. Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc Natl Acad Sci U S A. 2007;104:10418–10423. doi: 10.1073/pnas.0704217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris JD, Chang CY, Wittmann BM, Kunder RS, Cui H, Fan D, Joseph JD, McDonnell DP. The homeodomain protein HOXB13 regulates the cellular response to androgens. Molecular cell. 2009a;36:405–416. doi: 10.1016/j.molcel.2009.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris JD, Joseph JD, Sherk AB, Juzumiene D, Turnbull PS, Rafferty SW, Cui H, Anderson E, Fan D, Dye DA, et al. Differential presentation of protein interaction surfaces on the androgen receptor defines the pharmacological actions of bound ligands. Chem Biol. 2009b;16:452–460. doi: 10.1016/j.chembiol.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obinata D, Takayama K, Fujiwara K, Suzuki T, Tsutsumi S, Fukuda N, Nagase H, Fujimura T, Urano T, Homma Y, et al. Targeting Oct1 genomic function inhibits androgen receptor signaling and castration-resistant prostate cancer growth. Oncogene. 2016;35:6350–6358. doi: 10.1038/onc.2016.171. [DOI] [PubMed] [Google Scholar]
- Peng Y, Chen F, Melamed J, Chiriboga L, Wei J, Kong X, McLeod M, Li Y, Li CX, Feng A, et al. Distinct nuclear and cytoplasmic functions of androgen receptor cofactor p44 and association with androgen-independent prostate cancer. Proc Natl Acad Sci U S A. 2008;105:5236–5241. doi: 10.1073/pnas.0712262105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz MM, Li F, Takeda DY, Lenci R, Chonkar A, Chabot M, Cejas P, Vazquez F, Cook J, Shivdasani RA, et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat Genet. 2015;47:1346–1351. doi: 10.1038/ng.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prensner JR, Rubin MA, Wei JT, Chinnaiyan AM. Beyond PSA: the next generation of prostate cancer biomarkers. Sci Transl Med. 2012;4:127rv123. doi: 10.1126/scitranslmed.3003180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhr M, Hoefer J, Neuwirt H, Eder IE, Kern J, Schafer G, Geley S, Heidegger I, Klocker H, Culig Z. IAS1 is a crucial factor for prostate cancer cell survival and a valid target in docetaxel resistant cells. Oncotarget. 2014;5:12043–12056. doi: 10.18632/oncotarget.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Tripathi M, Mishra R, Sahgal N, Fazli L, Ettinger S, Placzek WJ, Claps G, Chung LW, Bowtell D, et al. The E3 ubiquitin ligase Siah2 contributes to castration-resistant prostate cancer by regulation of androgen receptor transcriptional activity. Cancer Cell. 2013;23:332–346. doi: 10.1016/j.ccr.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindranathan P, Lee TK, Yang L, Centenera MM, Butler L, Tilley WD, Hsieh JT, Ahn JM, Raj GV. Peptidomimetic targeting of critical androgen receptor-coregulator interactions in prostate cancer. Nat Commun. 2013;4:1923. doi: 10.1038/ncomms2912. [DOI] [PubMed] [Google Scholar]
- Reid KJ, Hendy SC, Saito J, Sorensen P, Nelson CC. Two classes of androgen receptor elements mediate cooperativity through allosteric interactions. J Biol Chem. 2001;276:2943–2952. doi: 10.1074/jbc.M009170200. [DOI] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu B, Laakso M, Ovaska K, Mirtti T, Lundin J, Rannikko A, Sankila A, Turunen JP, Lundin M, Konsti J, et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 2011;30:3962–3976. doi: 10.1038/emboj.2011.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu B, Laakso M, Pihlajamaa P, Ovaska K, Sinielnikov I, Hautaniemi S, Janne OA. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 2012 doi: 10.1158/0008-5472.CAN-12-2350. [DOI] [PubMed] [Google Scholar]
- Sahu B, Pihlajamaa P, Dubois V, Kerkhofs S, Claessens F, Janne OA. Androgen receptor uses relaxed response element stringency for selective chromatin binding and transcriptional regulation in vivo. Nucleic Acids Res. 2014;42:4230–4240. doi: 10.1093/nar/gkt1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarlett CO, Robins DM. In vivo footprinting of an androgen-dependent enhancer reveals an accessory element integral to hormonal response. Mol Endocrinol. 1995;9:413–423. doi: 10.1210/mend.9.4.7659085. [DOI] [PubMed] [Google Scholar]
- Scher HI, Morris MJ, Stadler WM, Higano C, Basch E, Fizazi K, Antonarakis ES, Beer TM, Carducci MA, Chi KN, et al. Trial Design and Objectives for Castration-Resistant Prostate Cancer: Updated Recommendations From the Prostate Cancer Clinical Trials Working Group 3. J Clin Oncol. 2016;34:1402–1418. doi: 10.1200/JCO.2015.64.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt LJ, Tindall DJ. Androgen receptor: past, present and future. Curr Drug Targets. 2013;14:401–407. doi: 10.2174/1389450111314040002. [DOI] [PubMed] [Google Scholar]
- Schrecengost RS, Dean JL, Goodwin JF, Schiewer MJ, Urban MW, Stanek TJ, Sussman RT, Hicks JL, Birbe RC, Draganova-Tacheva RA, et al. USP22 regulates oncogenic signaling pathways to drive lethal cancer progression. Cancer Res. 2014;74:272–286. doi: 10.1158/0008-5472.CAN-13-1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer PL, Jivan A, Dollins DE, Claessens F, Gewirth DT. Structural basis of androgen receptor binding to selective androgen response elements. Proc Natl Acad Sci U S A. 2004;101:4758–4763. doi: 10.1073/pnas.0401123101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Yeow WS, Ertel A, Coleman I, Clegg N, Thangavel C, Morrissey C, Zhang X, Comstock CE, Witkiewicz AK, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest. 2010;120:4478–4492. doi: 10.1172/JCI44239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma NL, Massie CE, Butter F, Mann M, Bon H, Ramos-Montoya A, Menon S, Stark R, Lamb AD, Scott HE, et al. The ETS family member GABPalpha modulates androgen receptor signalling and mediates an aggressive phenotype in prostate cancer. Nucleic Acids Res. 2014;42:6256–6269. doi: 10.1093/nar/gku281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, MacArthur S, Stark R, Warren AY, Mills IG, et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013;23:35–47. doi: 10.1016/j.ccr.2012.11.010. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- Steurer S, Mayer PS, Adam M, Krohn A, Koop C, Ospina-Klinck D, Tehrani AA, Simon R, Tennstedt P, Graefen M, et al. TMPRSS2-ERG fusions are strongly linked to young patient age in low-grade prostate cancer. Eur Urol. 2014;66:978–981. doi: 10.1016/j.eururo.2014.06.027. [DOI] [PubMed] [Google Scholar]
- Svensson MA, LaFargue CJ, MacDonald TY, Pflueger D, Kitabayashi N, Santa-Cruz AM, Garsha KE, Sathyanarayana UG, Riley JP, Yun CS, et al. Testing mutual exclusivity of ETS rearranged prostate cancer. Lab Invest. 2011;91:404–412. doi: 10.1038/labinvest.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadokoro-Cuccaro R, Davies J, Mongan NP, Bunch T, Brown RS, Audi L, Watt K, McEwan IJ, Hughes IA. Promoter-dependent activity on androgen receptor N-terminal domain mutations in androgen insensitivity syndrome. Sex Dev. 2014;8:339–349. doi: 10.1159/000369266. [DOI] [PubMed] [Google Scholar]
- Tan PY, Chang CW, Chng KR, Wansa KD, Sung WK, Cheung E. Integration of regulatory networks by NKX3-1 promotes androgen-dependent prostate cancer survival. Mol Cell Biol. 2012;32:399–414. doi: 10.1128/MCB.05958-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–352. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari AK, Yardimci GG, Shibata Y, Sheffield NC, Song L, Taylor BS, Georgiev SG, Coetzee GA, Ohler U, Furey TS, et al. Chromatin accessibility reveals insights into androgen receptor activation and transcriptional specificity. Genome Biol. 2012;13:R88. doi: 10.1186/gb-2012-13-10-r88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Longen CG, Oyer HM, Chen N, Maher CM, Salvino JM, Kania B, Anderson KN, Ostrander WF, Knudsen KE, et al. Sigma1 Targeting to Suppress Aberrant Androgen Receptor Signaling in Prostate Cancer. Cancer Res. 2017 doi: 10.1158/0008-5472.CAN-16-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, Helgeson BE, Cao Q, Prensner JR, Rubin MA, Shah RB, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10:177–188. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- Toropainen S, Malinen M, Kaikkonen S, Rytinki M, Jaaskelainen T, Sahu B, Janne OA, Palvimo JJ. SUMO ligase IAS1 functions as a target gene selective androgen receptor coregulator on prostate cancer cell chromatin. Nucleic Acids Res. 2015;43:848–861. doi: 10.1093/nar/gku1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanucci A, Marttila S, Janne OA, Visakorpi T. Androgen receptor overexpression alters binding dynamics of the receptor to chromatin and chromatin structure. Prostate. 2012a;72:1223–1232. doi: 10.1002/pros.22473. [DOI] [PubMed] [Google Scholar]
- Urbanucci A, Sahu B, Seppala J, Larjo A, Latonen LM, Waltering KK, Tammela TL, Vessella RL, Lahdesmaki H, Janne OA, et al. Overexpression of androgen receptor enhances the binding of the receptor to the chromatin in prostate cancer. Oncogene. 2012b;31:2153–2163. doi: 10.1038/onc.2011.401. [DOI] [PubMed] [Google Scholar]
- Valenca LB, Sweeney CJ, Pomerantz MM. Sequencing current therapies in the treatment of metastatic prostate cancer. Cancer Treat Rev. 2015;41:332–340. doi: 10.1016/j.ctrv.2015.02.010. [DOI] [PubMed] [Google Scholar]
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- Verrijdt G, Tanner T, Moehren U, Callewaert L, Haelens A, Claessens F. The androgen receptor DNA-binding domain determines androgen selectivity of transcriptional response. Biochem Soc Trans. 2006;34:1089–1094. doi: 10.1042/BST0341089. [DOI] [PubMed] [Google Scholar]
- Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011a;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Cai Y, Yu W, Ren C, Spencer DM, Ittmann M. Pleiotropic biological activities of alternatively spliced TMPRSS2/ERG fusion gene transcripts. Cancer Res. 2008a;68:8516–8524. doi: 10.1158/0008-5472.CAN-08-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LG, Johnson EM, Kinoshita Y, Babb JS, Buckley MT, Liebes LF, Melamed J, Liu XM, Kurek R, Ossowski L, et al. Androgen receptor overexpression in prostate cancer linked to Pur alpha loss from a novel repressor complex. Cancer Res. 2008b;68:2678–2688. doi: 10.1158/0008-5472.CAN-07-6017. [DOI] [PubMed] [Google Scholar]
- Wang Q, Carroll JS, Brown M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol Cell. 2005;19:631–642. doi: 10.1016/j.molcel.2005.07.018. [DOI] [PubMed] [Google Scholar]
- Wang Q, Li W, Liu XS, Carroll JS, Janne OA, Keeton EK, Chinnaiyan AM, Pienta KJ, Brown M. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. 2007;27:380–392. doi: 10.1016/j.molcel.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Dehigaspitiya DC, Levine PM, Profit AA, Haugbro M, Imberg-Kazdan K, Logan SK, Kirshenbaum K, Garabedian MJ. Multivalent Peptoid Conjugates Which Overcome Enzalutamide Resistance in Prostate Cancer Cells. Cancer Res. 2016;76:5124–5132. doi: 10.1158/0008-5472.CAN-16-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG, O’Malley BW. Small molecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1. Mol Endocrinol. 2011b;25:2041–2053. doi: 10.1210/me.2011-1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG, Wang J, Qi R, Matzuk AJ, Song X, Madoux F, et al. Bufalin is a potent small-molecule inhibitor of the steroid receptor coactivators SRC-3 and SRC-1. Cancer Res. 2014;74:1506–1517. doi: 10.1158/0008-5472.CAN-13-2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Wang J, Lampert E, Schlanger S, DePriest AD, Hu Q, Gomez EC, Murakam M, Glenn ST, Conroy J, et al. Intratumoral and Intertumoral Genomic Heterogeneity of Multifocal Localized Prostate Cancer Impacts Molecular Classifications and Genomic Prognosticators. Eur Urol. 2016 doi: 10.1016/j.eururo.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J, Simon R, Feuerbach L, Schlangen K, Weichenhan D, Minner S, Wuttig D, Warnatz HJ, Stehr H, Rausch T, et al. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell. 2013;23:159–170. doi: 10.1016/j.ccr.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Whitington T, Gao P, Song W, Ross-Adams H, Lamb AD, Yang Y, Svezia I, Klevebring D, Mills IG, Karlsson R, et al. Gene regulatory mechanisms underpinning prostate cancer susceptibility. Nat Genet. 2016;48:387–397. doi: 10.1038/ng.3523. [DOI] [PubMed] [Google Scholar]
- Wilson S, Qi J, Filipp FV. Refinement of the androgen response element based on ChIP-Seq in androgen-insensitive and androgen-responsive prostate cancer cell lines. Sci Rep. 2016;6:32611. doi: 10.1038/srep32611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Sunkel B, Chen Z, Liu X, Ye Z, Li Q, Grenade C, Ke J, Zhang C, Chen H, et al. Three-tiered role of the pioneer factor GATA2 in promoting androgen-dependent gene expression in prostate cancer. Nucleic Acids Res. 2014;42:3607–3622. doi: 10.1093/nar/gkt1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W, Gambhir SS, Lee P, Sartor O, Flemington EK, et al. Androgen Receptor Splice Variants Dimerize to Transactivate Target Genes. Cancer Res. 2015;75:3663–3671. doi: 10.1158/0008-5472.CAN-15-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Shimelis H, Linn DE, Jiang R, Yang X, Sun F, Guo Z, Chen H, Li W, Kong X, et al. Regulation of androgen receptor transcriptional activity and specificity by RNF6-induced ubiquitination. Cancer Cell. 2009;15:270–282. doi: 10.1016/j.ccr.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Lin PJ, Beraldi E, Zhang F, Kawai Y, Leong J, Katsumi H, Fazli L, Fraser R, Cullis PR, et al. siRNA Lipid Nanoparticle Potently Silences Clusterin and Delays Progression When Combined with Androgen Receptor Cotargeting in Enzalutamide-Resistant Prostate Cancer. Clin Cancer Res. 2015;21:4845–4855. doi: 10.1158/1078-0432.CCR-15-0866. [DOI] [PubMed] [Google Scholar]
- Yeh S, Chang C. Cloning and characterization of a specific coactivator, ARA70, for the androgen receptor in human prostate cells. Proc Natl Acad Sci U S A. 1996;93:5517–5521. doi: 10.1073/pnas.93.11.5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X, Wu L, Li J, Hu M, Gong Y, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17:443–454. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeltser L, Desplan C, Heintz N. Hoxb-13: a new Hox gene in a distant region of the HOXB cluster maintains colinearity. Development. 1996;122:2475–2484. doi: 10.1242/dev.122.8.2475. [DOI] [PubMed] [Google Scholar]
- Zhan Y, Zhang G, Wang X, Qi Y, Bai S, Li D, Ma T, Sartor O, Flemington EK, Zhang H, et al. Interplay between Cytoplasmic and Nuclear Androgen Receptor Splice Variants Mediates Castration Resistance. Mol Cancer Res. 2017;15:59–68. doi: 10.1158/1541-7786.MCR-16-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang S, Murtha PE, Zhu W, Hou SS, Young CY. Identification of two novel cis-elements in the promoter of the prostate-specific antigen gene that are required to enhance androgen receptor-mediated transactivation. Nucleic Acids Res. 1997;25:3143–3150. doi: 10.1093/nar/25.15.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JC, Fong KW, Jin HJ, Yang YA, Kim J, Yu J. FOXA1 acts upstream of GATA2 and AR in hormonal regulation of gene expression. Oncogene. 2016;35:4335–4344. doi: 10.1038/onc.2015.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong Y, Xin L, Goldstein AS, Lawson DA, Teitell MA, Witte ON. ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proc Natl Acad Sci U S A. 2009;106:12465–12470. doi: 10.1073/pnas.0905931106. [DOI] [PMC free article] [PubMed] [Google Scholar]