

Abstract
The photoreceptor outer segment (OS) is a unique modification of the primary cilium, specialized for light perception. Being homologous organelles, the primary cilium and the OS share common building blocks and molecular machinery to construct and maintain them. The OS, however, has several unique structural features that are not seen in primary cilia. Although these unique features of the OS have been well documented, their implications in protein localization have been under-appreciated. In this review, we compare the structural properties of the primary cilium and the OS, and propose a hypothesis that the OS can act as a sink for membrane proteins. We further discuss the implications of this hypothesis in polarized protein localization in photoreceptors and mechanisms of photoreceptor degeneration in retinal ciliopathies.
Introduction
The photoreceptor outer segment (OS) is a primary cilium-derived subcellular organelle. This structure is filled with proteins necessary for the phototransduction cascade. As expected from its ciliary origin, the OS shares its basic structural elements with primary cilia, and common mechanisms are used to build and maintain these two organelles (see review articles (1,2) for more comprehensive discussion on this topic). The parallel between the OS and the primary cilium culminates in the co-existence of pathologic conditions in both photoreceptors and other cells with primary cilia in human genetic diseases associated with ciliary defects. Furthermore, cilia-related molecular defects are one of the major causes of inherited retinal degenerations (3–5). For more comprehensive reviews on ciliopathies, we recommend recent publications (6,7).
Although the OS shares many of its features with primary cilia, there are several features that are unique to the OS. These include the size, the high membrane content, and the renewal process of the OS. These unique features have an important impact on the localization of membrane proteins in both normal and diseased photoreceptors. Here, we overview the structural properties of the OS, propose a hypothesis about the role of the OS, and discuss its implications in membrane protein localization and retinal ciliopathies.
Primary Cilia and Outer Segments: Homologous but Distinct Organelles
The ciliary compartment of the photoreceptor is composed of three main sub-compartments: the basal body, the connecting cilium (CC), and the OS. The basal body is at the base of the ciliary compartment and composed of nine triplet microtubules derived from the mother centriole. It functions as a microtubule-organizing center from which the nine axonemal microtubule doublets grow. Basal bodies are anchored to the periciliary membrane, the border between the ciliary membrane and the cellular plasma membrane, by transition fibers. The periciliary membrane and the transition fibers act as docking stations for proteins bound for the ciliary compartment (8–15). In frog photoreceptors, a prominent structure (named the periciliary ridge complex (PRC)) is observed in this area and rhodopsin-containing vesicles fuse to the PRC for rhodopsin trafficking to the OS (16,17). The periciliary membrane is also where endocytosis of ciliary membrane proteins occurs (18–21). In addition to acting as a platform for ciliary protein delivery and internalization, the periciliary membrane and the transition fibers constitute part of the ciliary gate that prevent random passage of macromolecules into and out of the ciliary compartment (15,22–25).
Distal to the basal body is the photoreceptor CC, equivalent to the transition zone (TZ) in primary cilia. Significant progress has been made during the past decade in our understanding of the constituents and architecture of the CC/TZ, and functions of the proteins that build the CC/TZ. For more in-depth discussions on this topic, we recommend recently published review articles (15,25–27). Proteins that localize to the CC/TZ are involved in ciliogenesis and regulating protein trafficking into and out of the ciliary compartment. Since the OS and the ciliary proper lack the protein synthesis machinery (e.g. ribosomes) and the CC/TZ is the only physical conduit between the OS/ciliary proper and the cell body, all ciliary proteins synthesized in the cell body must pass through the CC/TZ to reach their destination. This makes the CC/TZ an ideal place to control the trafficking of proteins in and out of cilia, and many proteins that localize to the CC/TZ are thought to be a part of the ciliary gate. Indeed, mutations disrupting CC/TZ proteins alter the protein composition of the ciliary compartment and cause various ciliopathies with retinal involvement (28–39). While the size of the OS in photoreceptors and the ciliary proper in primary cilia varies considerably depending on the species and the cell types (see below), the diameter of the TZ and the CC is remarkably invariant (250–300 nm) in all cells investigated (40–43). In contrast, the length of the photoreceptor CC is significantly longer than that of the TZ in primary cilia: 1–1.5 μm long in mouse photoreceptors and 0.25–0.3 μm in typical primary cilia (40–42).
Further distal to the CC is the OS, where considerable deviations from the primary cilium are observed. Furthermore, there are several notable differences between the two major photoreceptor cell types, rod and cone photoreceptors. One of the major differences between the OS and the primary cilium is the size. Typical primary cilia in mammalian cells are 0.25–0.3 μm in diameter and 1–9 μm (mostly 3–6 μm) long (volume = 0.05–0.6 μm3), which corresponds to <0.05% of the total cell volume (44,45). In contrast, human and mouse rod OSs are 1.5–2 μm in diameter and 25–30 μm long (volume = 45–94 μm3) (Fig. 1), corresponding to 30–40% of the total cell volume (41,46,47). Frog rod OSs are even larger: ∼5 μm wide and 50–60 μm long (i.e. ∼1,000 μm3 in volume) (48,49) (Fig. 1). Although cone OSs are smaller than rod OSs, they are still significantly larger than typical primary cilia. Another major difference between the OS and the primary cilium is the membrane content. The OS is highly membrane-enriched compared to primary cilia. In primary cilia, ciliary plasma membrane is the only membranous structure and other vesicular/membranous structures are normally not observed inside the cilium. In contrast, the OS is filled with membranous structures called discs. The vast majority of discs in rod OSs are essentially large vesicles separate from the OS plasma membrane and called ‘closed’ discs (50–52). Approximately 1% of discs most proximal to the CC are topologically continuous with the OS plasma membrane (therefore, essentially part of the OS plasma membrane) and called ‘open’ discs (50–52). Discs in cone OSs are mostly or entirely ‘open’, depending on the species (53–57). Finally, the OS is continuously and rapidly renewed and, particularly in rod OSs, newly delivered membrane proteins are isolated in closed discs at the base of the OS (58–60). Although continuous delivery of proteins and lipids to the ciliary compartment and shedding at the ciliary tip may not be unique to the OS (61,62), the rate, scale, and mechanisms of the renewal are unique to the OS. In mammalian rods, 8∼10% of discs are shed at the distal end every day and engulfed by retinal pigment epithelial cells (58–60,63). To compensate for this loss and maintain the OS, new OS components are continuously delivered to the base of the OS and form new open discs. Previously formed open discs become closed discs as the newly generated open discs displace them. One notable change that occurs to membrane proteins during this open-to-closed disc transition is their separation from the OS plasma membrane. Although cone OSs are thought to be renewed similarly as in rods, our understanding of the cone OS renewal is limited because of the patency of the cone OS.
Figure 1.

Schematic representation of mouse and frog photoreceptors drawn roughly to scale. (From Pearring et al., 2013 (1); adapted from Elsevier Ltd. with permission).
Outer Segment as a Sink for Membrane Proteins
The large size, the high membrane content, and the unique renewal process of the OS predicts that the OS would have different properties than primary cilia with respect to the distribution of membrane proteins in a cell. In typical cells with primary cilia, membrane proteins with no specific targeting signals localize mostly to the plasma membrane (Fig. 2A). In a hypothetical situation in which the ciliary gate is absent or defective, a fraction of such membrane proteins would be found within the cilium, assuming even distribution along the entire plasma membrane. However, the quantity in the ciliary membrane would be negligible compared to that of the rest of the plasma membrane because of the small volume of the primary cilium. Similarly, if the ciliary gate were permissive (not defective) to certain membrane proteins (again with no ciliary targeting or retention signals), a small fraction of such proteins would be found within the cilium. In photoreceptors, however, the large volume and the high membrane content of the OS provides an ample space to accommodate membrane proteins. Therefore, when the ciliary gate is absent or defective, a large fraction of membrane proteins with no targeting signals are expected to localize to the OS (Fig. 2B). If the ciliary gate is permissive to certain membrane proteins, a considerable amount of such proteins are expected to be found within the OS. Besides, the conversion of open discs to closed discs during the renewal process limits further diffusion of membrane proteins out of the OS by trapping them into topologically isolated closed discs. The continuous renewal of the OS (including the removal of older components at the distal tip) allows continuous absorption of membrane proteins from the cell body. Based on this inference, we propose that the OS can act as a sink for membrane proteins, which absorbs and disposes membrane proteins that have access to the OS.
Figure 2.
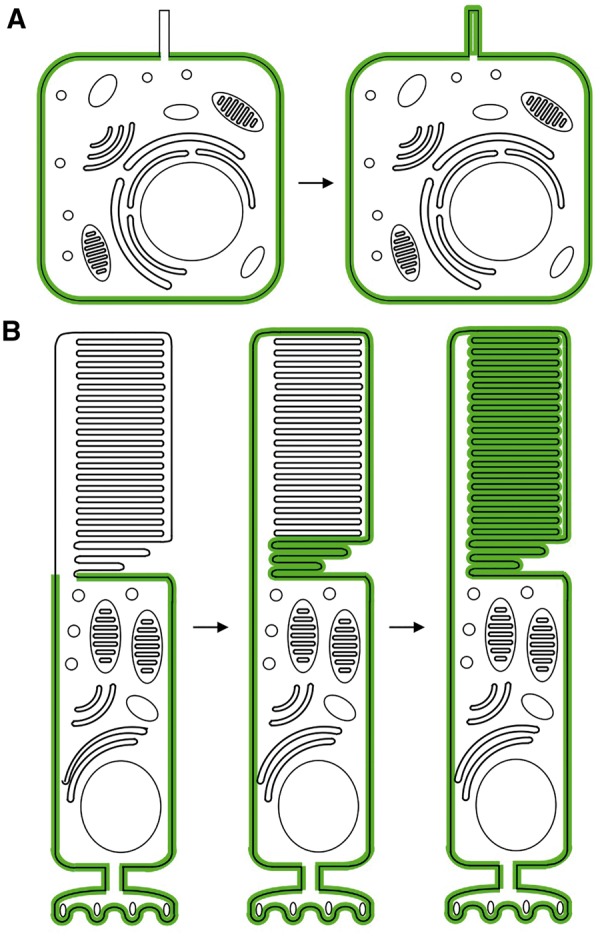
Photoreceptor outer segments as a membrane protein sink. (A) Localization of a hypothetical membrane protein (green) with no targeting signals in typical cells with primary cilia. Left; when the ciliary gate is intact, right; when the ciliary gate is defective or absent. (B) Localization of a hypothetical membrane protein (green) with no targeting signals in rod photoreceptors. Left; when the ciliary gate is intact and the protein is actively excluded from the OS, middle; soon after the ciliary gate is disrupted, right; after the entire outer segment is renewed. Schematic cells are not drawn to scale.
The membrane protein sink hypothesis for the OS can explain the propensity of membrane protein localization to the OS in photoreceptors. The OS was previously shown to be a default destination for membrane proteins with no targeting signals in frog photoreceptors (64,65). In addition, certain inner segment (IS) membrane proteins require active exclusion from the OS for their IS localization (66). Although the propensity is less prominent (presumably because of the relatively smaller size of the mouse OS), similar phenomena were also observed in mouse photoreceptors (67). The OS being a default destination for membrane proteins is consistent with the membrane protein sink hypothesis. Interestingly, the ciliary gate in photoreceptors appears to be permissive to the recombinant proteins used in the aforementioned studies despite their diverse origins, suggesting that it may not be highly selective. In this regard, it is noteworthy that the access of soluble proteins to the ciliary compartment is largely dependent on the size of the protein and diffusion (24,68–72).
One prediction from the membrane protein sink hypothesis is that a considerable amount of OS targeted membrane proteins would still localize to the OS (but not exclusively), even when mechanisms for the OS trafficking are impaired. This in fact has been observed in multiple animal models, in which protein trafficking to the OS is disrupted. For example, although the rate of rhodopsin trafficking to the OS decreases in KIF3A-deficient photoreceptors and a fraction of rhodopsin mislocalizes to the IS in various intraflagellar transport (IFT) mutants, a significant subset of rhodopsin still localizes to the OS, obscuring the requirement for IFT proteins in rhodopsin trafficking (73–86). Similar partial mislocalization of presumed cargos (e.g. rhodopsin, GC1, GRK1, and PDE6) was observed in animal models deficient for ARL3 (or overexpressing a constitutively active form of ARL3), PDE6D, RAB8A, TULP1, and UNC119A, as long as the OS forms (87–94). Finally, rhodopsin mutants that misfold or lack the OS targeting signals show partial but not complete mislocalization (see (95) for a recent review on this topic). Although the presence of alternative transport mechanisms and targeting signals cannot be ruled out, these observations are consistent with the membrane protein sink hypothesis and suggest that lateral diffusion along the plasma membrane may partly contribute to the OS localization of the OS-resident membrane proteins.
The membrane protein sink hypothesis further predicts that, to achieve and maintain the polarized protein distribution between the IS and the OS, photoreceptors would need efficient mechanisms to exclude non-OS proteins from the OS. This also suggests that photoreceptors would be prone to the accumulation of membrane proteins in the OS when these mechanisms fail. As mentioned earlier, certain IS proteins need to be actively excluded from the OS for their IS localization (64,66,67). In these studies, removal of short peptides which encompass potential IS targeting signals was sufficient to induce OS accumulation of the protein. In a more recent study, a plethora of non-OS proteins were found to accumulate in the OS when Bardet-Biedl syndrome (BBS) proteins were lost (96). The vast majority of the abnormally accumulated proteins were membrane-associated proteins. These findings support the membrane protein sink hypothesis and suggest that BBS proteins are part of the mechanisms to prevent the encroachment of IS proteins on the OS. Finally, these findings suggest that aberrant accumulation of IS proteins in the OS may be causally involved in the pathogenesis of photoreceptor degeneration in certain retinal ciliopathies.
Relevance to Retinal Ciliopathies and Prospects
One can envisage two main mechanisms to exclude proteins from the OS: 1) preventing OS entry at the ciliary gate and 2) exporting from the OS. As discussed earlier, proteins that localize to the CC/TZ and the transition fibers constitute the ciliary gate. Proteins that belong to this group include (but are not limited to) AHI1, CC2D2A, CEP164, CEP290, FAM161A, MKS1, NPHP5, RPGRIP1, RPGRIP1L, SDCCAG8, SPATA7, TCTN1, and TMEM67 ((15) and references therein). Studies in animal models and human patients with mutations in the genes encoding these proteins showed that they are essential for the OS development and/or homeostasis, and eventually for the photoreceptor survival (97–111) (also see (4,5) for more comprehensive review of retinal ciliopathies). Although most of these proteins are required for the OS development and the trafficking of specific OS-resident proteins, their roles in preventing OS accumulation of IS proteins have not been investigated. Some of them may be necessary mainly or additionally to prevent unauthorized entry of IS proteins into the OS. Future studies should address this question.
With respect to the export of non-OS proteins, two groups of proteins may be involved: i) IFT proteins and ii) BBS proteins. IFT is a mechanism that moves proteins along the ciliary axoneme in both anterograde (from the basal body to the ciliary tip) and retrograde (from the ciliary tip to the basal body) directions. IFT proteins are essential for the OS development at least partly due to their roles in protein trafficking to the OS (73–86,112). Although the precise molecular functions of the BBS proteins in photoreceptors remain to be determined, they associate with IFT particles and are implicated in ciliary trafficking, particularly in the retrograde direction (113–119). These two groups of proteins are thus strong candidates for the export machinery, and retinal ciliopathies caused by mutations in relevant genes may be associated with aberrant accumulation of IS proteins in the OS. However, it should be noted that active export of IS proteins from the OS has not been demonstrated for any proteins. Therefore, the presence of active export mechanisms awaits experimental validation.
Consideration of the OS as a membrane protein sink provides novel insights into the mechanisms by which photoreceptors establish and maintain polarized protein distribution between the IS and the OS, as well as the mechanisms of photoreceptor degeneration in retinal ciliopathies. Mis-trafficking of OS proteins is often suspected as a primary or early defect in retinal ciliopathies. This is probably true in many diseases, considering the high demand for protein trafficking to the OS. However, the membrane protein sink hypothesis based on the structural properties of the OS predicts that aberrant accumulation of non-OS proteins in the OS may be common and causally involved in certain retinal ciliopathies. Future studies should address the prevalence and the significance of this pathologic condition in retinal ciliopathies.
Acknowledgements
This work was supported by a grant from the National Institutes of Health [EY022616 to S.S.]. We thank Brandon Hendrickson for critical reading.
Conflict of Interest statement. None declared.
References
- 1. Pearring J.N., Salinas R.Y., Baker S.A., Arshavsky V.Y. (2013) Protein sorting, targeting and trafficking in photoreceptor cells. Prog. Retin. Eye Res., 36, 24–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goldberg A.F., Moritz O.L., Williams D.S. (2016) Molecular basis for photoreceptor outer segment architecture. Prog. Retin. Eye Res., 55, 52–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wright A.F., Chakarova C.F., Abd El-Aziz M.M., Bhattacharya S.S. (2010) Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet., 11, 273–284. [DOI] [PubMed] [Google Scholar]
- 4. Estrada-Cuzcano A., Roepman R., Cremers F.P., den Hollander A.I., Mans D.A. (2012) Non-syndromic retinal ciliopathies: translating gene discovery into therapy. Hum. Mol. Genet., 21, R111–R124. [DOI] [PubMed] [Google Scholar]
- 5. Bujakowska K.M., Liu Q., Pierce E.A. (2017) Photoreceptor cilia and retinal ciliopathies. Cold Spring Harb. Perspect. Biol., 9, doi: 10.1101/cshperspect.a028274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hildebrandt F., Benzing T., Katsanis N. (2011) Ciliopathies. N. Engl J. Med., 364, 1533–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brown J.M., Witman G.B. (2014) Cilia and Diseases. Bioscience, 64, 1126–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rosenbaum J.L., Witman G.B. (2002) Intraflagellar transport. Nat. Rev. Mol. Cell Biol., 3, 813–825. [DOI] [PubMed] [Google Scholar]
- 9. Pazour G.J., Bloodgood R.A. (2008) Targeting proteins to the ciliary membrane. Curr. Top. Dev. Biol., 85, 115–149. [DOI] [PubMed] [Google Scholar]
- 10. Schmidt K.N., Kuhns S., Neuner A., Hub B., Zentgraf H., Pereira G. (2012) Cep164 mediates vesicular docking to the mother centriole during early steps of ciliogenesis. J. Cell Biol., 199, 1083–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wei Q., Xu Q., Zhang Y., Li Y., Zhang Q., Hu Z., Harris P.C., Torres V.E., Ling K., Hu J. (2013) Transition fibre protein FBF1 is required for the ciliary entry of assembled intraflagellar transport complexes. Nat. Commun., 4, 2750.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ye X., Zeng H., Ning G., Reiter J.F., Liu A. (2014) C2cd3 is critical for centriolar distal appendage assembly and ciliary vesicle docking in mammals. Proc. Natl Acad. Sci. U S A, 111, 2164–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tanos B.E., Yang H.J., Soni R., Wang W.J., Macaluso F.P., Asara J.M., Tsou M.F. (2013) Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev, 27, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Humbert M.C., Weihbrecht K., Searby C.C., Li Y., Pope R.M., Sheffield V.C., Seo S. (2012) ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc. Natl Acad. Sci. U S A, 109, 19691–19696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garcia-Gonzalo F.R., Reiter J.F. (2017) Open Sesame: How Transition Fibers and the Transition Zone Control Ciliary Composition. Cold Spring. Harb. Perspect. Biol., 9, 10.1101/cshperspect.a028134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peters K.R., Palade G.E., Schneider B.G., Papermaster D.S. (1983) Fine structure of a periciliary ridge complex of frog retinal rod cells revealed by ultrahigh resolution scanning electron microscopy. J. Cell Biol., 96, 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Papermaster D.S., Schneider B.G., Besharse J.C. (1985) Vesicular transport of newly synthesized opsin from the Golgi apparatus toward the rod outer segment. Ultrastructural immunocytochemical and autoradiographic evidence in Xenopus retinas. Invest. Ophthalmol. Vis. Sci., 26, 1386–1404. [PubMed] [Google Scholar]
- 18. Molla-Herman A., Ghossoub R., Blisnick T., Meunier A., Serres C., Silbermann F., Emmerson C., Romeo K., Bourdoncle P., Schmitt A.. et al. (2010) The ciliary pocket: an endocytic membrane domain at the base of primary and motile cilia. J. Cell Sci., 123, 1785–1795. [DOI] [PubMed] [Google Scholar]
- 19. Kaplan O.I., Doroquez D.B., Cevik S., Bowie R.V., Clarke L., Sanders A.A., Kida K., Rappoport J.Z., Sengupta P., Blacque O.E. (2012) Endocytosis genes facilitate protein and membrane transport in C. elegans sensory cilia. Curr. Biol., 22, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clement C.A., Ajbro K.D., Koefoed K., Vestergaard M.L., Veland I.R., Henriques de Jesus M.P., Pedersen L.B., Benmerah A., Andersen C.Y., Larsen L.A.. et al. (2013) TGF-beta signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep., 3, 1806–1814. [DOI] [PubMed] [Google Scholar]
- 21. Langousis G., Shimogawa M.M., Saada E.A., Vashisht A.A., Spreafico R., Nager A.R., Barshop W.D., Nachury M.V., Wohlschlegel J.A., Hill K.L. (2016) Loss of the BBSome perturbs endocytic trafficking and disrupts virulence of Trypanosoma brucei. Proc. Natl Acad. Sci. U S A, 113, 632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vieira O.V., Gaus K., Verkade P., Fullekrug J., Vaz W.L., Simons K. (2006) FAPP2, cilium formation, and compartmentalization of the apical membrane in polarized Madin-Darby canine kidney (MDCK) cells. Proc. Natl Acad. Sci. U S A, 103, 18556–18561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu Q., Milenkovic L., Jin H., Scott M.P., Nachury M.V., Spiliotis E.T., Nelson W.J. (2010) A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science, 329, 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kee H.L., Dishinger J.F., Blasius T.L., Liu C.J., Margolis B., Verhey K.J. (2012) A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat. Cell Biol., 14, 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Verhey K.J., Yang W. (2016) Permeability barriers for generating a unique ciliary protein and lipid composition. Curr. Opin. Cell Biol., 41, 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nachury M.V., Seeley E.S., Jin H. (2010) Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier?. Annu. Rev. Cell Dev. Biol., 26, 59–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takao D., Verhey K.J. (2016) Gated entry into the ciliary compartment. Cell Mol. Life Sci, 73, 119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dryja T.P., Adams S.M., Grimsby J.L., McGee T.L., Hong D.H., Li T., Andreasson S., Berson E.L. (2001) Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am. J. Hum. Genet., 68, 1295–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Otto E.A., Loeys B., Khanna H., Hellemans J., Sudbrak R., Fan S., Muerb U., O'Toole J.F., Helou J., Attanasio M.. et al. (2005) Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat. Genet., 37, 282–288. [DOI] [PubMed] [Google Scholar]
- 30. Craige B., Tsao C.C., Diener D.R., Hou Y., Lechtreck K.F., Rosenbaum J.L., Witman G.B. (2010) CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J. Cell Biol., 190, 927–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Garcia-Gonzalo F.R., Corbit K.C., Sirerol-Piquer M.S., Ramaswami G., Otto E.A., Noriega T.R., Seol A.D., Robinson J.F., Bennett C.L., Josifova D.J.. et al. (2011) A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet., 43, 776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sang L., Miller J.J., Corbit K.C., Giles R.H., Brauer M.J., Otto E.A., Baye L.M., Wen X., Scales S.J., Kwong M.. et al. (2011) Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell, 145, 513–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dowdle W.E., Robinson J.F., Kneist A., Sirerol-Piquer M.S., Frints S.G., Corbit K.C., Zaghloul N.A., van Lijnschoten G., Mulders L., Verver D.E.. et al. (2011) Disruption of a ciliary B9 protein complex causes Meckel syndrome. Am. J. Hum. Genet., 89, 94–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chih B., Liu P., Chinn Y., Chalouni C., Komuves L.G., Hass P.E., Sandoval W., Peterson A.S. (2011) A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat. Cell Biol., 14, 61–72. [DOI] [PubMed] [Google Scholar]
- 35. Huang L., Szymanska K., Jensen V.L., Janecke A.R., Innes A.M., Davis E.E., Frosk P., Li C., Willer J.R., Chodirker B.N.. et al. (2011) TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am. J. Hum. Genet., 89, 713–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Awata J., Takada S., Standley C., Lechtreck K.F., Bellve K.D., Pazour G.J., Fogarty K.E., Witman G.B. (2014) NPHP4 controls ciliary trafficking of membrane proteins and large soluble proteins at the transition zone. J. Cell Sci., 127, 4714–4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roberson E.C., Dowdle W.E., Ozanturk A., Garcia-Gonzalo F.R., Li C., Halbritter J., Elkhartoufi N., Porath J.D., Cope H., Ashley-Koch A.. et al. (2015) TMEM231, mutated in orofaciodigital and Meckel syndromes, organizes the ciliary transition zone. J. Cell Biol., 209, 129–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lambacher N.J., Bruel A.L., van Dam T.J., Szymanska K., Slaats G.G., Kuhns S., McManus G.J., Kennedy J.E., Gaff K., Wu K.M.. et al. (2016) TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nat. Cell Biol., 18, 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shylo N.A., Christopher K.J., Iglesias A., Daluiski A., Weatherbee S.D. (2016) TMEM107 is a critical regulator of ciliary protein composition and is mutated in orofaciodigital syndrome. Hum. Mutat., 37, 155–159. [DOI] [PubMed] [Google Scholar]
- 40. Besharse J.C., Forestner D.M., Defoe D.M. (1985) Membrane assembly in retinal photoreceptors. III. Distinct membrane domains of the connecting cilium of developing rods. J. Neurosci., 5, 1035–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gilliam J.C., Chang J.T., Sandoval I.M., Zhang Y., Li T., Pittler S.J., Chiu W., Wensel T.G. (2012) Three-dimensional architecture of the rod sensory cilium and its disruption in retinal neurodegeneration. Cell, 151, 1029–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang T.T., Su J., Wang W.J., Craige B., Witman G.B., Tsou M.F., Liao J.C. (2015) Superresolution Pattern Recognition Reveals the Architectural Map of the Ciliary Transition Zone. Sci. Rep., 5, 14096.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Battle C., Ott C.M., Burnette D.T., Lippincott-Schwartz J., Schmidt C.F. (2015) Intracellular and extracellular forces drive primary cilia movement. Proc. Natl Acad. Sci. U S A, 112, 1410–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Delling M., Indzhykulian A.A., Liu X., Li Y., Xie T., Corey D.P., Clapham D.E. (2016) Primary cilia are not calcium-responsive mechanosensors. Nature, 531, 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dummer A., Poelma C., DeRuiter M.C., Goumans M.J., Hierck B.P. (2016) Measuring the primary cilium length: improved method for unbiased high-throughput analysis. Cilia, 5, 7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carter-Dawson L.D., LaVail M.M. (1979) Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J. Comp. Neurol., 188, 245–262. [DOI] [PubMed] [Google Scholar]
- 47. Nickell S., Park P.S., Baumeister W., Palczewski K. (2007) Three-dimensional architecture of murine rod outer segments determined by cryoelectron tomography. J. Cell Biol., 177, 917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bownds D., Brodie A.E. (1975) Light-sensitive swelling of isolated frog rod outer segments as an in vitro assay for visual transduction and dark adaptation. J. Gen. Physiol., 66, 407–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kaplan M.W., Iwata R.T., Sears R.C. (1987) Lengths of immunolabeled ciliary microtubules in frog photoreceptor outer segments. Exp. Eye Res., 44, 623–632. [DOI] [PubMed] [Google Scholar]
- 50. Ding J.D., Salinas R.Y., Arshavsky V.Y. (2015) Discs of mammalian rod photoreceptors form through the membrane evagination mechanism. J. Cell Biol., 211, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Volland S., Hughes L.C., Kong C., Burgess B.L., Linberg K.A., Luna G., Zhou Z.H., Fisher S.K., Williams D.S. (2015) Three-dimensional organization of nascent rod outer segment disk membranes. Proc. Natl Acad. Sci. U S A, 112, 14870–14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Burgoyne T., Meschede I.P., Burden J.J., Bailly M., Seabra M.C., Futter C.E. (2015) Rod disc renewal occurs by evagination of the ciliary plasma membrane that makes cadherin-based contacts with the inner segment. Proc. Natl Acad. Sci. U S A, 112, 15922–15927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cohen A.I. (1961) The fine structure of the extrafoveal receptors of the Rhesus monkey. Exp. Eye Res., 1, 128–136. [DOI] [PubMed] [Google Scholar]
- 54. Nilsson S.E. (1965) The ultrastructure of the receptor outer segments in the retina of the leopard frog (Rana Pipiens). J. Ultrastruct. Res., 12, 207–231. [DOI] [PubMed] [Google Scholar]
- 55. Cohen A.I. (1968) New evidence supporting the linkage to extracellular space of outer segment saccules of frog cones but not rods. J. Cell Biol., 37, 424–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cohen A.I. (1970) Further studies on the question of the patency of saccules in outer segments of vertebrate photoreceptors. Vision Res., 10, 445–453. [DOI] [PubMed] [Google Scholar]
- 57. Anderson D.H., Fisher S.K. (1976) The photoreceptors of diurnal squirrels: outer segment structure, disc shedding, and protein renewal. J. Ultrastruct. Res., 55, 119–141. [DOI] [PubMed] [Google Scholar]
- 58. Young R.W. (1967) The renewal of photoreceptor cell outer segments. J. Cell Biol., 33, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Young R.W. (1971) The renewal of rod and cone outer segments in the rhesus monkey. J. Cell Biol., 49, 303–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Young R.W., Droz B. (1968) The renewal of protein in retinal rods and cones. J. Cell Biol., 39, 169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nager A.R., Goldstein J.S., Herranz-Perez V., Portran D., Ye F., Garcia-Verdugo J.M., Nachury M.V. (2017) An Actin Network Dispatches Ciliary GPCRs into Extracellular Vesicles to Modulate Signaling. Cell, 168, 252–263 e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Phua S.C., Chiba S., Suzuki M., Su E., Roberson E.C., Pusapati G.V., Setou M., Rohatgi R., Reiter J.F., Ikegami K.. et al. (2017) Dynamic Remodeling of Membrane Composition Drives Cell Cycle through Primary Cilia Excision. Cell, 168, 264–279 e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Young R.W., Bok D. (1969) Participation of the retinal pigment epithelium in the rod outer segment renewal process. J. Cell Biol., 42, 392–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Baker S.A., Haeri M., Yoo P., Gospe S.M. 3rd, Skiba N.P., Knox B.E., Arshavsky V.Y. (2008) The outer segment serves as a default destination for the trafficking of membrane proteins in photoreceptors. J. Cell Biol., 183, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Salinas R.Y., Baker S.A., Gospe S.M. 3rd, Arshavsky V.Y. (2013) A single valine residue plays an essential role in peripherin/rds targeting to photoreceptor outer segments. PLoS One, 8, e54292.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gospe S.M. 3rd, Baker S.A., Arshavsky V.Y. (2010) Facilitative glucose transporter Glut1 is actively excluded from rod outer segments. J. Cell Sci., 123, 3639–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pearring J.N., Lieu E.C., Winter J.R., Baker S.A., Arshavsky V.Y. (2014) R9AP targeting to rod outer segments is independent of rhodopsin and is guided by the SNARE homology domain. Mol. Biol. Cell, 25, 2644–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Calvert P.D., Strissel K.J., Schiesser W.E., Pugh E.N. Jr., Arshavsky V.Y. (2006) Light-driven translocation of signaling proteins in vertebrate photoreceptors. Trends Cell Biol., 16, 560–568. [DOI] [PubMed] [Google Scholar]
- 69. Calvert P.D., Schiesser W.E., Pugh E.N. Jr. (2010) Diffusion of a soluble protein, photoactivatable GFP, through a sensory cilium. J. Gen. Physiol., 135, 173–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Najafi M., Maza N.A., Calvert P.D. (2012) Steric volume exclusion sets soluble protein concentrations in photoreceptor sensory cilia. Proc. Natl Acad. Sci. U S A, 109, 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Breslow D.K., Koslover E.F., Seydel F., Spakowitz A.J., Nachury M.V. (2013) An in vitro assay for entry into cilia reveals unique properties of the soluble diffusion barrier. J. Cell Biol., 203, 129–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lin Y.C., Niewiadomski P., Lin B., Nakamura H., Phua S.C., Jiao J., Levchenko A., Inoue T., Rohatgi R., Inoue T. (2013) Chemically inducible diffusion trap at cilia reveals molecular sieve-like barrier. Nat. Chem. Biol., 9, 437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Trivedi D., Colin E., Louie C.M., Williams D.S. (2012) Live-cell imaging evidence for the ciliary transport of rod photoreceptor opsin by heterotrimeric kinesin-2. J. Neurosci., 32, 10587–10593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pazour G.J., Baker S.A., Deane J.A., Cole D.G., Dickert B.L., Rosenbaum J.L., Witman G.B., Besharse J.C. (2002) The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J. Cell Biol., 157, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tsujikawa M., Malicki J. (2004) Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron, 42, 703–716. [DOI] [PubMed] [Google Scholar]
- 76. Jimeno D., Feiner L., Lillo C., Teofilo K., Goldstein L.S., Pierce E.A., Williams D.S. (2006) Analysis of kinesin-2 function in photoreceptor cells using synchronous Cre-loxP knockout of Kif3a with RHO-Cre. Invest. Ophthalmol. Vis. Sci., 47, 5039–5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sukumaran S., Perkins B.D. (2009) Early defects in photoreceptor outer segment morphogenesis in zebrafish ift57, ift88 and ift172 Intraflagellar Transport mutants. Vision Res., 49, 479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hudak L.M., Lunt S., Chang C.H., Winkler E., Flammer H., Lindsey M., Perkins B.D. (2010) The intraflagellar transport protein ift80 is essential for photoreceptor survival in a zebrafish model of jeune asphyxiating thoracic dystrophy. Invest. Ophthalmol. Vis. Sci., 51, 3792–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Krock B.L., Perkins B.D. (2008) The intraflagellar transport protein IFT57 is required for cilia maintenance and regulates IFT-particle-kinesin-II dissociation in vertebrate photoreceptors. J. Cell Sci., 121, 1907–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Keady B.T., Le Y.Z., Pazour G.J. (2011) IFT20 is required for opsin trafficking and photoreceptor outer segment development. Mol. Biol. Cell, 22, 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Boldt K., Mans D.A., Won J., van Reeuwijk J., Vogt A., Kinkl N., Letteboer S.J., Hicks W.L., Hurd R.E., Naggert J.K.. et al. (2011) Disruption of intraflagellar protein transport in photoreceptor cilia causes Leber congenital amaurosis in humans and mice. J. Clin. Invest., 121, 2169–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Crouse J.A., Lopes V.S., Sanagustin J.T., Keady B.T., Williams D.S., Pazour G.J. (2014) Distinct functions for IFT140 and IFT20 in opsin transport. Cytoskeleton (Hoboken), 71, 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Boubakri M., Chaya T., Hirata H., Kajimura N., Kuwahara R., Ueno A., Malicki J., Furukawa T., Omori Y. (2016) Loss of ift122, a Retrograde Intraflagellar Transport (IFT) Complex Component, Leads to Slow, Progressive Photoreceptor Degeneration Due to Inefficient Opsin Transport. J. Biol. Chem., 291, 24465–24474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jiang L., Wei Y., Ronquillo C.C., Marc R.E., Yoder B.K., Frederick J.M., Baehr W. (2015) Heterotrimeric kinesin-2 (KIF3) mediates transition zone and axoneme formation of mouse photoreceptors. J. Biol. Chem., 290, 12765–12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Avasthi P., Watt C.B., Williams D.S., Le Y.Z., Li S., Chen C.K., Marc R.E., Frederick J.M., Baehr W. (2009) Trafficking of membrane proteins to cone but not rod outer segments is dependent on heterotrimeric kinesin-II. J. Neurosci., 29, 14287–14298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bujakowska K.M., Zhang Q., Siemiatkowska A.M., Liu Q., Place E., Falk M.J., Consugar M., Lancelot M.E., Antonio A., Lonjou C.. et al. (2015) Mutations in IFT172 cause isolated retinal degeneration and Bardet-Biedl syndrome. Hum. Mol. Genet., 24, 230–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hanke-Gogokhia C., Wu Z., Gerstner C.D., Frederick J.M., Zhang H., Baehr W. (2016) Arf-like protein 3 (ARL3) regulates protein trafficking and ciliogenesis in mouse photoreceptors. J. Biol. Chem., 291, 7142–7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wright Z.C., Singh R.K., Alpino R., Goldberg A.F., Sokolov M., Ramamurthy V. (2016) ARL3 regulates trafficking of prenylated phototransduction proteins to the rod outer segment. Hum. Mol. Genet., 25, 2031–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhang H., Li S., Doan T., Rieke F., Detwiler P.B., Frederick J.M., Baehr W. (2007) Deletion of PrBP/delta impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc. Natl Acad. Sci. U S A, 104, 8857–8862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ying G., Gerstner C.D., Frederick J.M., Boye S.L., Hauswirth W.W., Baehr W. (2016) Small GTPases Rab8a and Rab11a are dispensable for rhodopsin transport in mouse photoreceptors. PLoS One, 11, e0161236.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Grossman G.H., Watson R.F., Pauer G.J., Bollinger K., Hagstrom S.A. (2011) Immunocytochemical evidence of Tulp1-dependent outer segment protein transport pathways in photoreceptor cells. Exp. Eye Res., 93, 658–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hagstrom S.A., Adamian M., Scimeca M., Pawlyk B.S., Yue G., Li T. (2001) A role for the Tubby-like protein 1 in rhodopsin transport. Invest. Ophthalmol. Vis. Sci., 42, 1955–1962. [PubMed] [Google Scholar]
- 93. Hagstrom S.A., Duyao M., North M.A., Li T. (1999) Retinal degeneration in tulp1-/- mice: vesicular accumulation in the interphotoreceptor matrix. Invest. Ophthalmol. Vis. Sci., 40, 2795–2802. [PubMed] [Google Scholar]
- 94. Zhang H., Constantine R., Vorobiev S., Chen Y., Seetharaman J., Huang Y.J., Xiao R., Montelione G.T., Gerstner C.D., Davis M.W.. et al. (2011) UNC119 is required for G protein trafficking in sensory neurons. Nat. Neurosci., 14, 874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Nemet I., Ropelewski P., Imanishi Y. (2015) Rhodopsin trafficking and mistrafficking: signals, molecular components, and mechanisms. Prog. Mol. Biol. Transl Sci., 132, 39–71. [DOI] [PubMed] [Google Scholar]
- 96. Datta P., Allamargot C., Hudson J.S., Andersen E.K., Bhattarai S., Drack A.V., Sheffield V.C., Seo S. (2015) Accumulation of non-outer segment proteins in the outer segment underlies photoreceptor degeneration in Bardet-Biedl syndrome. Proc. Natl Acad. Sci. U S A, 112, E4400–E4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lessieur E.M., Fogerty J., Gaivin R.J., Song P., Perkins B.D. (2017) The Ciliopathy Gene Ahi1 is required for zebrafish cone photoreceptor outer segment morphogenesis and survival. Invest. Ophthalmol. Vis. Sci., 58, 448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Westfall J.E., Hoyt C., Liu Q., Hsiao Y.C., Pierce E.A., Page-McCaw P.S., Ferland R.J. (2010) Retinal degeneration and failure of photoreceptor outer segment formation in mice with targeted deletion of the Joubert syndrome gene, Ahi1. J. Neurosci., 30, 8759–8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Louie C.M., Caridi G., Lopes V.S., Brancati F., Kispert A., Lancaster M.A., Schlossman A.M., Otto E.A., Leitges M., Grone H.J.. et al. (2010) AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat. Genet., 42, 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Bachmann-Gagescu R., Phelps I.G., Stearns G., Link B.A., Brockerhoff S.E., Moens C.B., Doherty D. (2011) The ciliopathy gene cc2d2a controls zebrafish photoreceptor outer segment development through a role in Rab8-dependent vesicle trafficking. Hum. Mol. Genet., 20, 4041–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chang B., Khanna H., Hawes N., Jimeno D., He S., Lillo C., Parapuram S.K., Cheng H., Scott A., Hurd R.E.. et al. (2006) In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum. Mol. Genet., 15, 1847–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Karlstetter M., Sorusch N., Caramoy A., Dannhausen K., Aslanidis A., Fauser S., Boesl M.R., Nagel-Wolfrum K., Tamm E.R., Jagle H.. et al. (2014) Disruption of the retinitis pigmentosa 28 gene Fam161a in mice affects photoreceptor ciliary structure and leads to progressive retinal degeneration. Hum. Mol. Genet., 23, 5197–5210. [DOI] [PubMed] [Google Scholar]
- 103. Downs L.M., Scott E.M., Cideciyan A.V., Iwabe S., Dufour V., Gardiner K.L., Genini S., Marinho L.F., Sumaroka A., Kosyk M.S.. et al. (2016) Overlap of abnormal photoreceptor development and progressive degeneration in Leber congenital amaurosis caused by NPHP5 mutation. Hum. Mol. Genet., 25, 4211–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ronquillo C.C., Hanke-Gogokhia C., Revelo M.P., Frederick J.M., Jiang L., Baehr W. (2016) Ciliopathy-associated IQCB1/NPHP5 protein is required for mouse photoreceptor outer segment formation. faseb J., 30, 3400–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Won J., Gifford E., Smith R.S., Yi H., Ferreira P.A., Hicks W.L., Li T., Naggert J.K., Nishina P.M. (2009) RPGRIP1 is essential for normal rod photoreceptor outer segment elaboration and morphogenesis. Hum. Mol. Genet., 18, 4329–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lheriteau E., Libeau L., Stieger K., Deschamps J.Y., Mendes-Madeira A., Provost N., Lemoine F., Mellersh C., Ellinwood N.M., Cherel Y.. et al. (2009) The RPGRIP1-deficient dog, a promising canine model for gene therapy. Mol Vis, 15, 349–361. [PMC free article] [PubMed] [Google Scholar]
- 107. Khanna H., Davis E.E., Murga-Zamalloa C.A., Estrada-Cuzcano A., Lopez I., den Hollander A.I., Zonneveld M.N., Othman M.I., Waseem N., Chakarova C.F.. et al. (2009) A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat. Genet., 41, 739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Airik R., Slaats G.G., Guo Z., Weiss A.C., Khan N., Ghosh A., Hurd T.W., Bekker-Jensen S., Schroder J.M., Elledge S.J.. et al. (2014) Renal-retinal ciliopathy gene Sdccag8 regulates DNA damage response signaling. J. Am. Soc. Nephrol., 25, 2573–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Eblimit A., Nguyen T.M., Chen Y., Esteve-Rudd J., Zhong H., Letteboer S., Van Reeuwijk J., Simons D.L., Ding Q., Wu K.M.. et al. (2015) Spata7 is a retinal ciliopathy gene critical for correct RPGRIP1 localization and protein trafficking in the retina. Hum. Mol. Genet., 24, 1584–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Collin G.B., Won J., Hicks W.L., Cook S.A., Nishina P.M., Naggert J.K. (2012) Meckelin is necessary for photoreceptor intraciliary transport and outer segment morphogenesis. Invest. Ophthalmol. Vis. Sci., 53, 967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Tiwari S., Hudson S., Gattone V.H. 2nd, Miller C., Chernoff E.A., Belecky-Adams T.L. (2013) Meckelin 3 is necessary for photoreceptor outer segment development in rat Meckel syndrome. PLoS One, 8, e59306.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Krock B.L., Mills-Henry I., Perkins B.D. (2009) Retrograde intraflagellar transport by cytoplasmic dynein-2 is required for outer segment extension in vertebrate photoreceptors but not arrestin translocation. Invest. Ophthalmol. Vis. Sci., 50, 5463–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Blacque O.E., Reardon M.J., Li C., McCarthy J., Mahjoub M.R., Ansley S.J., Badano J.L., Mah A.K., Beales P.L., Davidson W.S.. et al. (2004) Loss of C. elegans BBS-7 and BBS-8 protein function results in cilia defects and compromised intraflagellar transport. Genes Dev., 18, 1630–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ou G., Blacque O.E., Snow J.J., Leroux M.R., Scholey J.M. (2005) Functional coordination of intraflagellar transport motors. Nature, 436, 583–587. [DOI] [PubMed] [Google Scholar]
- 115. Lechtreck K.F., Johnson E.C., Sakai T., Cochran D., Ballif B.A., Rush J., Pazour G.J., Ikebe M., Witman G.B. (2009) The Chlamydomonas reinhardtii BBSome is an IFT cargo required for export of specific signaling proteins from flagella. J. Cell Biol., 187, 1117–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Lechtreck K.F., Brown J.M., Sampaio J.L., Craft J.M., Shevchenko A., Evans J.E., Witman G.B. (2013) Cycling of the signaling protein phospholipase D through cilia requires the BBSome only for the export phase. J. Cell Biol., 201, 249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Eguether T., San Agustin J.T., Keady B.T., Jonassen J.A., Liang Y., Francis R., Tobita K., Johnson C.A., Abdelhamed Z.A., Lo C.W.. et al. (2014) IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev Cell, 31, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Liew G.M., Ye F., Nager A.R., Murphy J.P., Lee J.S., Aguiar M., Breslow D.K., Gygi S.P., Nachury M.V. (2014) The intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev. Cell, 31, 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Yen H.J., Tayeh M.K., Mullins R.F., Stone E.M., Sheffield V.C., Slusarski D.C. (2006) Bardet-Biedl syndrome genes are important in retrograde intracellular trafficking and Kupffer's vesicle cilia function. Hum. Mol. Genet., 15, 667–677. [DOI] [PubMed] [Google Scholar]