

Abstract
Aminoacyl-tRNA synthetases (ARSs) are responsible for charging amino acids to cognate tRNA molecules, which is the essential first step of protein translation. Interestingly, mutations in genes encoding ARS enzymes have been implicated in a broad spectrum of human inherited diseases. Bi-allelic mutations in ARSs typically cause severe, early-onset, recessive diseases that affect a wide range of tissues. The vast majority of these mutations show loss-of-function effects and impair protein translation. However, it is not clear how a subset cause tissue-specific phenotypes. In contrast, dominant ARS-mediated diseases specifically affect the peripheral nervous system—most commonly causing axonal peripheral neuropathy—and usually manifest later in life. These neuropathies are linked to heterozygosity for missense mutations in five ARS genes, which points to a shared mechanism of disease. However, it is not clear if a loss-of-function mechanism or a toxic gain-of-function mechanism is responsible for ARS-mediated neuropathy, or if a combination of these mechanisms operate on a mutation-specific basis. Here, we review our current understanding of recessive and dominant ARS-mediated disease. We also propose future directions for defining the molecular mechanisms of ARS mutations toward designing therapies for affected patient populations.
An Introduction to Aminoacyl-tRNA Synthetases
The conjugation of tRNA to cognate amino acids is an essential prerequisite for the translation of the genetic code into proteins. This conjugation is performed by a group of enzymes, aminoacyl-tRNA synthetases (ARSs), which are ubiquitously expressed and highly evolutionarily conserved. Each amino acid has a designated ARS enzyme to catalyze a bond with a cognate tRNA. There are 37 members of the ARS gene family—17 encode an enzyme that functions in the cytoplasm, 17 encode an enzyme that functions in the mitochondria, and three encode bi-functional proteins that charge tRNA in both cellular locations (1). The nomenclature for ARS genes and proteins is the single-letter code of the amino acid that the enzyme recognizes followed by ‘ARS’, with mitochondrial enzymes followed by a ‘2’ (e.g. HARS for cytoplasmic histidyl-tRNA synthetase and HARS2 for mitochondrial histidyl-tRNA synthetase) (1).
Despite the essential canonical function and ubiquitous expression of ARS enzymes, mutations in ARS genes have been implicated in a variety of human diseases with both recessive and dominant inheritance patterns. Interestingly, these phenotypes range from later onset peripheral neuropathy to severe, multi-system developmental syndromes. Defining the mechanisms underlying the heterogeneity of ARS-related disease phenotypes is an important first step towards developing treatments for these disorders. Here, we review our current understanding of the molecular pathologies of ARS-related disease in the context of both recessive and dominant phenotypes.
The Role of ARS Mutations in Recessive Disease Phenotypes
Mutations in 31 ARS genes have been implicated in recessive disorders that display a wide range of clinical manifestations (Table 1 and 104). Not surprisingly, pathogenic variants in ARS genes encoding a mitochondrial enzyme tend to cause phenotypes in tissues with a high metabolic demand, particularly in the central nervous system. Leukoencephalopathies (9–12,19–30,70), myopathies (14,93,98–102), and liver disease (33,36) are all common features of mitochondrial ARS disease phenotypes. Additionally, epilepsy (33–37,72,85,86,91), developmental delay, intellectual disability (69,72,73,87), ovarian failure (10–12,51,61), and sensorineural hearing loss (51,54,55,61,62,69,74) are frequently observed in patients with mitochondrial ARS mutations.
Table 1.
ARS loci implicated in dominant and recessive human disease phenotypes
| Gene | Locus | Location of Protein Function | Mode of Inheritance | Disease Phenotype(s) | Unique variants | References |
|---|---|---|---|---|---|---|
| AARS | 16q22 | Cytoplasm | Autosomal Recessive | Early-onset epileptic encephalopathy with myelination defect | 2 | (2) |
| Microcephaly with hypomyelination, epileptic encephalopathy, and spasticity | 2 | (3) | ||||
| Autosomal Dominant | Charcot-Marie-Tooth disease type 2N | 3 | (4,5–7) | |||
| Distal hereditary motor neuropathy | 1 | (8) | ||||
| AARS2 | 6p21.1 | Mitochondria | Autosomal Recessive | Leukoencephalopathy with ovarian failure | 19 | (9–13) |
| Cardiomyopathy | 2 | (14) | ||||
| Multiple respiratory chain complex defects | 4 | (15) | ||||
| CARS2 | 13q34 | Mitochondria | Autosomal Recessive | Epileptic encephalopathy | 2 | (16) |
| Progressive myoclonic epilepsy | 2 | (17) | ||||
| DARS | 2q21.3 | Cytoplasm | Autosomal Recessive | Hypomyelination with brain stem and spinal cord involvement and leg spasticity | 2 | (18) |
| DARS2 | 1q25.1 | Mitochondria | Autosomal Recessive | Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation | 11 | (19–24) |
| EARS2 | 16p12.2 | Mitochondria | Autosomal Recessive | Leukoencephalopathy with thalamus and brainstem involvement and high lactate | 20 | (25–30) |
| Neonatal lactic acidosis, recurrent hypoglycemia, agenesis of corpus callosum | 2 | (31) | ||||
| Multiple respiratory chain complex defects | 3 | (15) | ||||
| FARS2 | 6p25.1 | Mitochondria | Autosomal Recessive | Hereditary spastic paraplegia | 1 | (32) |
| Alpers syndrome | 5 | (33,34) | ||||
| Early onset epilepsy | 5 | (35–37) | ||||
| Global delay, dysarthria and tremor | 2 | (38) | ||||
| GARS | 7p15 | Mitochondria and Cytoplasm | Autosomal Recessive | Systemic mitochondrial disease | 2 | (39) |
| Cardiomyopathy | 1 | (15) | ||||
| Autosomal Dominant | Charcot-Marie-Tooth disease type 2D | 4 | (40–43) | |||
| Distal hereditary motor neuropathy | 10 | (40,41,43–47) | ||||
| HARS | 5q31.3 | Cytoplasm | Autosomal Recessive | Usher syndrome | 1 | (48) |
| Autosomal Dominant | Charcot-Marie-Tooth disease type 2W | 5 | (49,50) | |||
| Distal hereditary motor neuropathy | 2 | (50) | ||||
| HARS2 | 5q31.3 | Mitochondria | Autosomal Recessive | Perrault syndrome | 2 | (51) |
| IARS | 9q22.31 | Cytoplasm | Autosomal Recessive | Prenatal growth retardation, neonatal cholestasis, muscular hypotonia, intellectual disability, infantile hepatopathy | 8 | (52,53) |
| IARS2 | 1q41 | Mitochondria | Autosomal Recessive | Cataracts, growth hormone deficiency, sensory neuropathy, sensorineural hearing loss, skeletal dysplasia syndrome; Leigh syndrome | 4 | (54,55) |
| KARS | 16q23.1 | Mitochondria and Cytoplasm | Autosomal Recessive | Nonsyndromic hearing loss (DFNB89) | 2 | (56) |
| Recessive intermediate Charcot-Marie-Tooth disease type B, dysmorphic features, developmental delay, self-abusive behavior, vestibular Schwannoma | 2 | (57) | ||||
| Visual impairment, microcephaly, developmental delay, seizures | 2 | (58) | ||||
| LARS | 5q32 | Cytoplasm | Autosomal Recessive | Infantile hepatopathy | 4 | (59,60) |
| LARS2 | 3p21.31 | Mitochondria | Autosomal Recessive | Perrault syndrome | 5 | (61,62) |
| Hydrops, lactic acidosis, sideroblastic anemia, multisystem failure | 2 | (63) | ||||
| MARS | 12q13.3 | Cytoplasm | Autosomal Recessive | Interstitial lung disease and liver disease | 7 | (64–66) |
| Autosomal Dominant | Charcot-Marie-Tooth disease type 2U | 2 | (67,68) | |||
| MARS2 | 2q33.1 | Cytoplasm | Autosomal Recessive | Developmental delay, sensorineural hearing loss | 2 | (69) |
| Autosomal recessive spastic ataxia with leukoencephalopathy | 2 | (70) | ||||
| NARS2 | 11q14.1 | Mitochondria | Autosomal Recessive | Alpers syndrome | 1 | (71) |
| Developmental delay, intellectual disability, epilepsy, myopathy | 6 | (72,73) | ||||
| Nonsyndromic deafness | 1 | (74) | ||||
| Leigh syndrome | 2 | (74) | ||||
| PARS2 | 3p21.31 | Mitochondria | Autosomal Recessive | Alpers syndrome | 2 | (71) |
| Infantile-onset developmental delay and epilepsy | 2 | (72) | ||||
| QARS | 3p21.31 | Cytoplasm and Mitochondria | Autosomal Recessive | Progressive microcephaly, cerebral-cerebellar atrophy, hypomyelination, intractable seizures, developmental delay | 6 | (75,76) |
| RARS | 5q34 | Cytoplasm | Autosomal Recessive | Hypomyelination | 5 | (77) |
| RARS2 | 6q16.1 | Mitochondria | Autosomal Recessive | Pontocerebellar hypoplasia | 14 | (78–84) |
| Early onset epileptic encephalopathy | 2 | (85) | ||||
| Lactic acidosis with or without neurological symptoms (microcephaly, seizures, developmental delay) | 1 | (86) | ||||
| Intellectual disability | 1 | (87) | ||||
| SARS | 1p13.3 | Cytoplasm | Autosomal Recessive | Intellectual disability, ataxia, microcephaly, speech impairment, aggressive behavior | 1 | (88) |
| SARS2 | 19q13.2 | Mitochondria | Autosomal Recessive | Hyperuricemia, pulmonary hypertension, renal failure, and alkalosis | 2 | (89,90) |
| TARS2 | 1q21.2 | Mitochondria | Autosomal Recessive | Axial hypotonia and severe psychomotor delay | 2 | (91) |
| VARS | 6p21.33 | Cytoplasm | Autosomal Recessive | Severe developmental delay, microcephaly, seizures | 2 | (92) |
| VARS2 | 6p21.33 | Mitochondria | Autosomal Recessive | Microcephaly and epilepsy | 1 | (91) |
| Encephalocardiomyopathy | 2 | (93) | ||||
| Multiple respiratory chain complex defects | 2 | (15) | ||||
| WARS | 14q32.2 | Cytoplasm | Autosomal Dominant | Distal hereditary motor neuropathy | 1 | (94) |
| WARS2 | 1p12 | Mitochondria | Autosomal Recessive | Intellectual disability, ataxia, microcephaly, speech impairment, aggressive behavior | 2 | (88) |
| YARS | 1p35.1 | Cytoplasm | Autosomal Recessive | Multisystem disease, developmental delay, failure to thrive | 2 | (95) |
| Autosomal Dominant | Dominant intermediate Charcot-Marie-Tooth disease type C | 5 | (96,97) | |||
| YARS2 | 12p11.21 | Mitochondria | Autosomal Recessive | Myopathy, lactic acidosis, sideroblastic anemia, cardiomyopathy, respiratory insufficiency | 7 | (98–103) |
| Multiple respiratory chain complex defects | 1 | (15) |
Mutations in ARS genes encoding cytoplasmic enzymes also cause a spectrum of recessive disorders, which often affect a wider array of tissues but that also typically include a neurological component. The recessive neurological phenotypes associated with cytoplasmic ARSs include hypomyelination (77), microcephaly (3,39,75,92), seizures (2,3,75,76,92), sensorineural hearing loss (48,56), and developmental delay (52,57,64,76,92,95). Some multisystem, cytoplasmic ARS-linked disorders also include liver dysfunction (53,64,65,105) and lung disease (64–66,105).
Although mutations in different ARS genes can cause overlapping recessive phenotypes, some tissues appear to be uniquely sensitive to mutations in a specific ARS. For example, retinitis pigmentosa (a component of Usher syndrome) has only been described in patients with bi-allelic HARS mutations (48). Defining the mechanisms that underlie tissue-specific effects of ARS mutations is a significant challenge moving forward (see below).
Although recessive ARS-mediated disorders involve a wide range of phenotypes, they are all, by definition, caused by bi-allelic mutations. Typically, patients with recessive ARS-related disease are homozygous for missense mutations, compound heterozygous for missense mutations, or compound heterozygous for one missense mutation and one null allele. Homozygosity or compound heterozygosity for ARS null alleles would likely be lethal due to the essential nature of the encoded enzymes; this effect has been demonstrated in animal models (106). The genotypes identified in patients with recessive ARS-related disease strongly suggest a loss-of-function mechanism for disease pathogenesis. In support of this, functional studies on disease-causing mutations show a reduction of ARS protein levels (3,16,27,55,63,73,74,78,91,93,100,107,108), and/or severe decreases in mutant enzyme activity (33,36,48,51,57,63,64,75,99). The effect of ARS mutations on enzyme kinetics is often measured directly with in vitro aminoacylation assays. In addition, enzyme function can be inferred by measuring the amount of charged tRNA in patient cells (16,73,79,89,91,108), or by assessing the ability of the mutated gene to support cellular growth in yeast complementation assays (2,12,51,52,57,53,61,66,78,91,98,102). For mutations in mitochondrial ARS enzymes, defects in tRNA charging explain the impaired protein translation of oxidative phosphorylation complexes and respiratory defects observed in patient cells (12,14,16,27,33,34,37,63,69–71,74,79,80,86,91,98–100,108). Furthermore, studies of IARS (53) and QARS in zebrafish (75), and those of HARS2 (51) and LARS2 (61) in C. elegans indicate that knocking down the ARS gene phenocopies key elements of the disorders linked to bi-allelic ARS mutations. This further supports the conclusion that disease-associated mutations lead to a loss of ARS activity, which may ultimately result in impaired protein translation by one of two mechanisms (Fig. 1). First, impaired translation could occur directly due to ribosomal stalling in the environment of reduced aminoacylated tRNA (109–111). Second, reduced translation could occur indirectly due to the recognition of uncharged tRNA molecules by GCN2, and the subsequent phosphorylation of eIF2α and global repression of translation (112).
Figure 1.
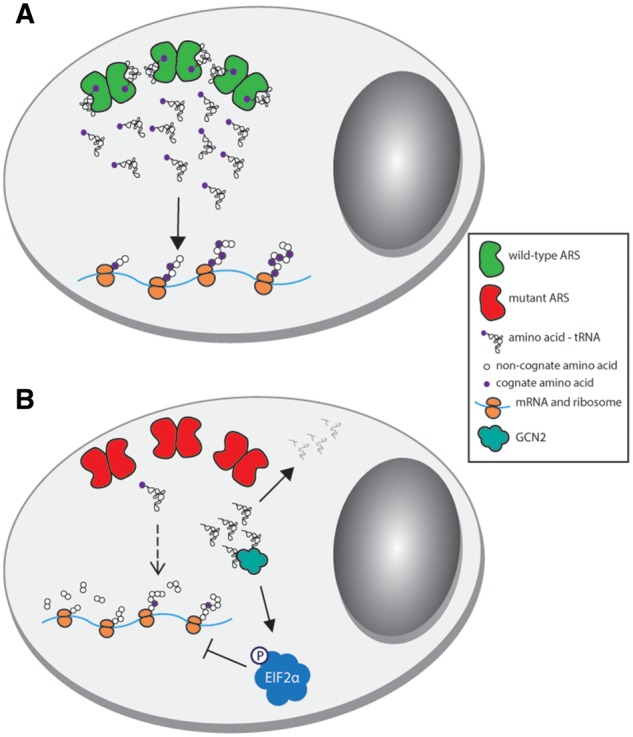
Potential mechanisms of ARS-related recessive disease. (A) Two wild-type ARS alleles supply cells with the requisite charged tRNA for protein translation. (B) Two loss-of-function ARS alleles severely reduce the amount of charged tRNA available for translation, which impairs protein production. Uncharged tRNA is either degraded or binds to GCN2, which phosphorylates eIF2α and inhibits global translation. In both panels, dimeric enzymes functioning in the cytoplasm are shown for simplicity; however, please note that some ARS enzymes act as monomers and that some effects apply to mitochondrial translation.
Moving forward, it will be important to understand the degree to which specific ARS mutations impair translation and how this contributes to phenotypic and tissue-type variability. There is evidence to suggest a link between the severity of the translation defect and the severity of the phenotype. For example, in siblings with RARS2 mutations, the sibling with the greater reduction in mitochondrial OXPHOS protein complex levels presented with lactic acidosis and neurological symptoms, whereas the sibling with a milder OXPHOS reduction had lactic acidosis but no neurological symptoms (86). Further work is needed to elucidate this relationship, particularly with mutations in cytoplasmic and bifunctional ARS enzymes that cause recessive disease, which have not yet been directly linked to protein translation defects. Additionally, there is currently no explanation for the observation that certain ARS mutations cause tissue-specific phenotypes, despite knowledge that ARS enzymes are required for all living cells. One possibility is that mutations in a specific ARS affect the expression of cell type-specific proteins, possibly in a codon-dependent manner. Alternatively, the expression profile of a given tRNA—which can vary between tissues (110)—may modify the cell-specific impact of deficiencies in tRNA charging. Ribosomal profiling to detect stalling at vulnerable codons and tissue-specific tRNA expression studies will be useful approaches for addressing these questions.
The Role of ARS Mutations in Dominant Disease Phenotypes
Although there is a broad spectrum of phenotypes seen in ARS-mediated recessive diseases, dominant ARS-mediated disorders have, to date, a limited phenotypic range. Mutations in five ARS loci have been implicated in dominant Charcot-Marie-Tooth (CMT) disease and related neuropathic phenotypes; these include glycyl-(GARS), tyrosyl-(YARS), alanyl-(AARS), histidyl-(HARS), and tryptophanyl-tRNA synthetase (WARS) (4,40,49,94,96). CMT disease is an inherited peripheral neuropathy characterized by progressive loss of motor and sensory function. The phenotype manifests in an axon-length dependent manner (i.e., symptoms first arise in distal motor and sensory structures innervated by long axons, which can progress proximally over time). Although CMT disease can be caused by a defect in the myelin sheath surrounding the axon (CMT Type 1), ARS-mediated CMT disease is predominantly caused by a defect in peripheral nerve axons (CMT Type 2).
The genetic evidence for the involvement of GARS, YARS, AARS, and HARS mutations in CMT disease is abundant, with numerous pathogenic mutations identified in each gene, many of which were identified in large pedigrees via linkage analysis (4,40,96). In addition, two variants in methionyl-tRNA synthetase (MARS) have been reported in patients with dominant CMT (113,114), but the role of MARS in CMT disease is less clear; although one of the reported MARS variants was shown to cause a loss-of-function effect in yeast complementation assays, it was identified in a small pedigree and was also detected in an unaffected relative (113). This suggests that the MARS variant is either not causal, or that it leads to a phenotype with decreased penetrance; additional research is needed to determine the role of MARS mutations in dominant CMT disease. Recently, a tryptophanyl-tRNA synthetase variant—H257R WARS—was identified in three unrelated families with autosomal dominant distal hereditary motor neuropathy (dHMN) (94). dHMN is phenotypically similar to CMT disease; it is marked by progressive distal limb muscle atrophy, without sensory involvement. This makes WARS the fifth ARS gene to be convincingly implicated in dominant axonal neuropathy. Combined, the genetic analyses implicating five ARS loci in dominant axonal neuropathy strongly suggest that ARS enzymes are particularly important for peripheral nerve axons.
Patients with ARS-mediated axonal neuropathy are typically heterozygous for missense mutations; however, one in-frame deletion has been described (96). A major question moving forward is how these mutations, in the heterozygous state, cause a late-onset, tissue-specific phenotype. It is worth noting that, to date, none of the parents of patients with recessive diseases associated with GARS, YARS, HARS, or AARS mutations have been shown to be affected with a dominant axonal neuropathy. However, interpreting this is complicated by the decreased penetrance and later onset of ARS-related neuropathy (44). This may suggest that some variants only cause recessive disease, while other variants may also cause dominant disease. Further evaluation of the functional differences between these two sets of variants will yield important insights into disease mechanisms.
The Molecular Mechanisms of ARS-Related Human Inherited Disease
Current genetic and functional evidence clearly point to a loss of enzymatic function as the molecular mechanism of ARS-mediated recessive disease. As noted above, causal genotypes correspond to functional data showing reduced gene function. Thus, patient mutations lead to a loss of ARS activity, which in turn results in impaired protein translation and the associated recessive disease phenotype.
In contrast to the recessive phenotypes, the molecular mechanism of ARS-related dominant axonal neuropathy is less clear. The fact that mutations in five genes encoding an aminoacyl-tRNA synthetase (GARS, YARS, AARS, HARS, and WARS) cause a similar dominant phenotype points to a common disease mechanism. In support of this, over-expression of neuropathy-associated GARS and YARS mutants in a Drosophila model cause a strikingly similar phenotype (115). Although there is a growing body of work defining various non-canonical functions for the above five ARS enzymes (Table 2), none thus far are common to all five nor do they relate to neuron (or axon) function. Thus, it is currently challenging to investigate if a loss of some non-canonical function is responsible for ARS-related neuropathy. Also, given that most neuropathy-associated ARS mutations impair rather than enhance enzyme function (104), it is unlikely that a gain of canonical function is responsible for disease. As a result, there are currently two mechanisms being explored: impaired ARS activity and toxic gain-of-function effects (Fig. 2). It should be emphasized that these pathogenic mechanisms may not be mutually exclusive—for example, impaired tRNA charging may be a prerequisite for a gain-of-function effect, or the two molecular mechanisms may work in concert to modulate phenotypic severity.
Table 2.
Non-canonical functions of ARS enzymes implicated in dominant neuropathy
| ARS | Species | Non-canonical function |
|---|---|---|
| AARS | Homo sapiens | C-terminal splice variant binds DNA (116) |
| GARS | Homo sapiens | Chaperone in neddylation pathway (117) |
| Homo sapiens | Tumorigenesis defense (118) | |
| Saccharomyces cerevisiae | mRNA 3’ end formation (119) | |
| HARS | Homo sapiens | Epitope for autoantibodies in inflammatory myositis (120–122) |
| WARS | Homo sapiens | Activates macrophages in immune response (123) |
| Homo sapiens | Mini-WARS inhibits angiogenesis (124–128) | |
| YARS | Homo sapiens | Locates to the nucleus and protects against DNA damage (129–131) |
| Homo sapiens | Mini-YARS promotes angiogenesis (125,127,128,132,133) |
Figure 2.

Potential mechanisms of ARS-related dominant axonal neuropathy. Neurons are illustrated with the cell body on the left and the axon extending to the right. A wild-type neuron (A) has functional ARS activity (green dimers) facilitating protein translation. There is appropriate NRP1 (orange transmembrane protein) and Trk signaling (blue transmembrane protein). YARS translocates to the nucleus upon oxidative stress and binds TRIM28 (blue), potentially changing the regulation of DNA damage response genes. Proposed mechanisms of ARS-mediated peripheral neuropathy are represented in (B); see text for details. Neuronal function may be compromised by impaired protein translation due to an unknown function of mutant ARS (red subunits) and/or a depletion in available charged tRNA from a significant reduction of aminoacylation activity. For peripheral neuropathy related to GARS mutations, mutant GARS may interfere with NRP1 signaling by preventing VEGFA (magenta) from binding to NRP1. In developing sensory neurons, mutant GARS may also act as a ligand for Trk receptors, aberrantly activating Trk signaling. For peripheral neuropathy related to YARS mutations, increased mutant YARS binding to TRIM28 (blue) may change the expression of DNA damage response genes.
Impaired ARS function in dominant axonal neuropathy
Data from in vitro aminoacylation and yeast complementation assays indicate that neuropathy-associated ARS mutations cause deficits in tRNA charging (104). Additionally, data from three animal models suggest that the mutant proteins are sub-functional. First, in Drosophila projection neurons, the morphological defects caused by neuron-specific homozygosity for a null gars allele were fully rescued by a wild-type human GARS transgene, partially rescued by the neuropathy-associated E71G allele, and not rescued by the neuropathy-associated L129P allele (134). Second, whereas over-expressing wild-type gars rescues the neuromuscular phenotype of zebrafish homozygous for a loss-of-function allele, over-expressing the neuropathy-associated G526R allele showed no rescue (135). Finally, whereas mice heterozygous for P234KY or C201R Gars display a dominant neuropathy, homozygosity for these mutations results in reduced viability (136). In sum, there is an abundance of data showing that neuropathy-associated ARS missense mutations have a deleterious effect on gene function, indicating that this molecular consequence is a component of disease pathogenesis.
In contrast, three lines of evidence argue against a simple loss-of-function effect as the underlying mechanism of ARS-related neuropathy. First, mice heterozygous for a Gars null allele have a wild-type phenotype (106). Second, examination of ARS alleles in human populations reveals null alleles in the heterozygous state. For example, the Genome Aggregation Database (137) annotates many ARS null alleles in the heterozygous state (23 in GARS, 52 in HARS, 97 in AARS, 32 in YARS, and 15 in WARS). Combined, these data rule out haploinsufficiency as the primary mechanism for a penetrant, ARS-related neuropathy. Finally, over-expression of wild-type human GARS in mice heterozygous for either P234KY or C201R Gars did not rescue the phenotype, indicating that the neuropathy caused by these mutations does not arise from a simple loss-of-function effect (136).
One likely explanation for the role of loss-of-function missense mutations in dominant neuropathy is a dominant-negative effect. Prerequisites for a dominant-negative effect include: (1) the mutant protein should be stably expressed; (2) the mutant protein should have reduced or ablated function; and (3) the affected protein should normally dimerize (or oligomerize) and mutant subunits should retain the ability to interact with wild-type subunits. Indeed, AARS, YARS, GARS, HARS, and WARS all charge tRNA as dimers; if an inactive mutant subunit dimerizes with a wild-type subunit, it could result in a dramatic reduction in tRNA charging compared to the happloinsufficient state. This would shift the burden of tRNA charging onto the reduced population (i.e., 25%) of wild-type:wild-type dimers.
Three lines of evidence support a dominant-negative effect for ARS mutations that cause dominant neuropathy. First, yeast cells expressing one wild-type and one mutant copy of tyrosyl-tRNA synthetase showed depleted growth compared to yeast cells expressing only the wild-type enzyme (96). Second, zebrafish homozygous for a loss-of-function gars missense allele show a severe neuromuscular defect, and zebrafish heterozygous for this allele have no phenotype (135). Importantly, the missense mutation (T209K) was shown to ablate dimerization. When T209K was over-expressed in either garsT209K/+ or gars+/+ zebrafish, the fish had no phenotype. However, over-expression of G526R gars, which dimerizes (135) and is non-functional (138), caused enhanced neuromuscular junction defects. Notably, over-expression of T209K in cis with G526R improved the neuromuscular junction phenotype, suggesting that dimerization is required for the toxicity of G526R gars. Finally, the H257R WARS mutation decreases enzyme activity in vitro but does not impact dimerization (94). To measure the potential downstream impact on protein synthesis, cultured cells were co-transfected with a construct to express wild-type or H257R WARS (or an empty vector) and a β-Gal or luciferase construct as a proxy for protein translation. Whereas wild-type WARS increased reporter activity above that of the empty vector, H257R WARS decreased reporter activity below that of the empty vector, indicating that H257R WARS suppressed endogenous levels of protein synthesis. However, a dominant-negative effect may not apply to all neuropathy-associated ARS mutations; as noted above, the effects of P234KY and C201R Gars were not rescued by over-expression of human GARS in affected mice. Thus, more research is needed to explore a dominant-negative mechanism and to determine if it can be alleviated by supplying the wild-type enzyme.
It is important to consider how impaired ARS function (possibly exacerbated by a dominant-negative mechanism) would specifically affect peripheral nerve axons. One possibility is that the long axons of the peripheral nervous system are particularly sensitive to defects in housekeeping functions, such as protein translation. It is quite likely that these functions would have to be maintained throughout the axoplasm of long axons. Indeed, mutations in other ubiquitously expressed genes (e.g. MFN2 and RAB7) have been implicated in axonal neuropathy (139,140).
The role of toxic gain-of-function effects in dominant axonal neuropathy
Another possibility is that neuropathy-associated ARS mutations cause the encoded enzymes to gain a novel, dominantly toxic function that specifically affects the peripheral nervous system. For example, there is evidence that YARS (141) and GARS (142,143) mutations change enzyme conformations and expose amino-acid residues. These structural changes were recently shown to facilitate increased binding of YARS to TRIM28 in the nucleus, which may alter the expression of DNA damage response genes (129); however, the role of this interaction in neuropathy is unclear. Conformational changes have also been described for neuropathy-associated GARS mutations (142,143). A series of pull-down assays showed that some mutant GARS proteins bind to the membrane receptor neuropilin-1 (NRP1), interfering with the binding of VEGF-A165 to an extracellular domain of NRP1 (143). This interaction relies on the secretion of GARS from the cell, which was detected by enriching for exosomes in cell culture medium and performing a western blot for wild-type GARS.
VEGF-NRP1 signaling is important for cardiovascular development, as well as motor neuron cell body migration and axon guidance (144,145). Interestingly, a closer examination of GarsP234KY/+ prenatal mice found a defect in the migration of facial motor neurons similar to that in NRP1 and VEGF-A165 null animals (143). Furthermore, mice double heterozygous for GarsP234KY/+ and Nrp1+/- developed an earlier and more severe neuromuscular phenotype, pointing to a genetic interaction between Gars and Nrp1. When GarsP234KY/+ mice were treated with VEGF-A165, their motor performance improved. Importantly, treating GarsP234KY/+ with other neurotrophic factors or with other isoforms of VEGF-A that do not strongly bind NRP1 did not show the same improvements, pointing to the specificity of the VEGF-A165/NRP1 interaction for improving the neuromuscular phenotype (143).
When considering the timing and location of VEGF-A165/NRP1 signaling, several questions arise. The VEGF-A165/NRP1 interaction is known to be important in cardiovascular development, which is unaffected in the GarsC201R/+ mouse model (146) and in patients with ARS-related neuropathy. Additionally, NRP1 is primarily expressed developmentally, fitting with the migration defects in facial motor neurons of GarsP234KY/+ mice. However, it is unclear how this translates to the human neuropathy phenotype, which is marked by degeneration, not developmental defects. It is also possible that developmental perturbations may play a role in disease later in life—recent work characterizing sensory deficits in GarsC201R/+ mice showed that these mice had fewer large diameter sensory neurons and more small diameter sensory neurons than wild-type mice, and that this imbalance was present at birth (147). This was attributed to mutant GARS binding aberrantly to TrkA, TrkB, and TrkC, receptors that have a role in developmental fate switching in sensory neuron subtypes. In vitro immunoprecipitation experiments revealed that TrkA-C interact with P234KY and C201R GARS, but not wild-type GARS. To show the relevance of this interaction in cells, mutant (LI29P and G240R) and wild-type GARS was added to the media of neuroblastoma cells over-expressing TrkB. Mutant GARS caused a greater increase in the Trk signaling cascade compared to wild-type, suggesting that mutant GARS activates Trk signaling.
Although the finding of novel binding interactions with NRP1 and Trk receptors yield new insights into the pathogenesis of some GARS mutations, it would be surprising if this mechanism was shared among neuropathy-associated mutations in different ARS loci. The structures of the five neuropathy-associated ARS enzymes differ significantly, so it is unlikely that they would all have the capacity to bind to the same membrane receptors when mutated. However, if different mutant ARS enzymes aberrantly bind to different proteins that act in a common pathway, it is possible that this may explain the shared pathogenic effect.
Such a common pathway may be related to neuronal signaling, as discussed above, or may be related to protein translation independent of deficits in aminoacylation. Interestingly, the latter possibility could provide an explanation for the translation defects observed in Drosophila models of GARS and YARS mutants. When several neuropathy-associated mutations in human GARS (E71G, G240R, and G526R) or YARS (G41R, 153-156delVKQV, and E196K) are over-expressed in Drosophila motor or sensory neurons, they reduce protein translation rates and cause muscle denervation and morphological defects (115). However, this study concluded that mutant GARS does not impair the endogenous activity of Drosophila gars, and that the reduced translation rate caused by over-expressing G240R human GARS cannot be rescued by over-expressing wild-type Drosophila gars. These findings are particularly interesting since, if the translation defects in flies caused by over-expressing mutant GARS are not a result of mutant GARS suppressing the endogenous protein via a dominant-negative effect, then it is possible that they are caused by aberrant interactions between GARS mutants and the translational machinery.
Future directions
There is currently evidence to support multiple proposed mechanisms of ARS-mediated peripheral neuropathy; however, additional research is needed to determine if either mechanism applies to all neuropathy-associated ARS mutations and loci. For the loss-of-function model, it will be key to determine if dimerization is required for pathogenicity. For example, it will be crucial to determine if monomeric enzymes, such as MARS, are implicated in dominant axonal neuropathy, since a dominant-negative mechanism would not be possible for monomeric enzymes. Identifying MARS variants that segregate with disease in large pedigrees or demonstrating that MARS variants cause neuropathy in animal models would be important steps toward resolving this issue.
For the gain-of-function model, it will be important to show that any novel protein-protein interactions—whether with NRP1, TRIM28, Trk receptors, or other proteins—are specific to mutations associated with neuropathy, and that these interactions do not occur with nonpathogenic protein variants. Additionally, showing that multiple mutant ARS enzymes can participate in the same aberrant interaction, or different aberrant interactions that lead to the same cellular effect, will add weight to this model. Finally, demonstrating that mutations in other components of these pathways also cause peripheral neuropathy would be a strong confirmation of this mechanism.
After refining the loss- and gain-of-function models, the next step will be to determine if there is any interplay between the two mechanisms that may affect phenotypic outcome. For example, some mutations, like G598A GARS (41), are linked to an early-onset, severe spinal muscular atrophy, which may be due to the compound effects of loss-of-function and gain-of-function mechanisms.
Conclusions
ARSs are emerging as a significant cause of rare inherited diseases, particularly recessive mitochondrial disorders, recessive multisystem disorders, and dominant axonal neuropathies. A total of 31 out of the 37 human ARS enzymes have been implicated in a genetic disease phenotype. Identifying and characterizing the remaining ARS loci and alleles associated with disease will provide an important tool for clinicians, and will broaden our understanding of how these enzymes function and tolerate variation. Looking towards potential therapies, it will be necessary to determine the downstream effects of ARS mutations for both dominant and recessive disease-causing alleles, and to determine how certain ARS mutations lead to tissue-specific phenotypes. Finally, we should explore whether improving ARS function itself is a viable mode of therapy for both recessive and dominant phenotypes, and if mutant-allele repression is an effective strategy for alleviating ARS-associated dominant neuropathy.
Acknowledgements
We would like to thank all of the patients and their families for agreeing to participate in the studies reviewed here; and each of our colleagues for contributing to our current knowledge of ARS-associated disease. R.M. is supported by the Michigan Pre-doctoral Training in Genetics Program (GM007544). A.A. is supported by funding from the National Institute of General Medical Sciences (GM118647).
Conflict of Interest statement. None declared.
Funding
Michigan Pre-doctoral Training in Genetics Program (GM007544) and National Institute of General Medical Sciences (GM118647).
References
- 1. Antonellis A., Green E.D. (2008) The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genomics Hum. Genet., 9, 87–107. [DOI] [PubMed] [Google Scholar]
- 2. Simons C., Griffin L.B., Helman G., Golas G., Pizzino A., Bloom M., Murphy J.L.P., Crawford J., Evans S.H., Topper S.. et al. (2015) Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am. J. Hum. Genet., 96, 675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nakayama T., Wu J., Galvin-Parton P., Weiss J., Andriola M.R., Hill R.S., Vaughan D., El-Quessny M., Barry B.J., Partlow J.N.. et al. (2017) Deficient activity of alanyl-tRNA synthetase underlies an autosomal recessive syndrome of progressive microcephaly, hypomyelination, and epileptic encephalopathy. Hum. Mutat., doi: 10.1002/humu.23250, 10.1002/humu.23250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Latour P., Thauvin-Robinet C., Baudelet-Méry C., Soichot P., Cusin V., Faivre L., Locatelli M.-C., Mayençon M., Sarcey A., Broussolle E.. et al. (2010) A major determinant for binding and aminoacylation of tRNAAla in cytoplasmic alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-tooth disease. Am. J. Hum. Genet., 86, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McLaughlin H.M., Sakaguchi R., Giblin W., Wilson T.E., Biesecker L., Lupski J.R., Talbot K., Vance J.M., Züchner S., Lee Y.-C.. et al. (2012) A Recurrent loss‐of‐function alanyl‐tRNA synthetase (AARS ) mutation in patients with charcot‐marie‐tooth disease type 2N (CMT2N). Hum. Mutat., 33, 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin K.-P., Soong B.-W., Yang C.-C., Huang L.-W., Chang M.-H., Lee I.-H., Antonellis A., Antonellis A., Lee Y.-C. (2011) The mutational spectrum in a cohort of Charcot-Marie-Tooth disease type 2 among the Han Chinese in Taiwan. PLoS ONE, 6, e29393.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Motley W.W., Griffin L.B., Mademan I., Baets J., De Vriendt E., De Jonghe P., Antonellis A., Jordanova A., Scherer S.S. (2015) A novel AARS mutation in a family with dominant myeloneuropathy. Neurology, 84, 2040–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao Z., Hashiguchi A., Hu J., Sakiyama Y., Okamoto Y., Tokunaga S., Zhu L., Shen H., Takashima H. (2012) Alanyl-tRNA synthetase mutation in a family with dominant distal hereditary motor neuropathy. Neurology, 78, 1644–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lynch D.S., Zhang W.J., Lakshmanan R., Kinsella J.A., Uzun G.A., Karbay M., Tüfekçioglu Z., Hanagasi H., Burke G., Foulds N.. et al. (2016) Analysis of mutations in AARS2 in a series of CSF1R-negative patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. JAMA Neurol., 73, 1433–1439. [DOI] [PubMed] [Google Scholar]
- 10. Lee J.-M., Yang H.-J., Kwon J.-H., Kim W.-J., Kim S.-Y., Lee E.-M., Park J.-Y., Weon Y.C., Park S.H., Gwon B.-J.. et al. (2017) Two Korean siblings with recently described ovarioleukodystrophy related to AARS2 mutations. Eur. J. Neurol., 24, e21–e22. [DOI] [PubMed] [Google Scholar]
- 11. Hamatani M., Jingami N., Tsurusaki Y., Shimada S., Shimojima K., Asada-Utsugi M., Yoshinaga K., Uemura N., Yamashita H., Uemura K.. et al. (2016) The first Japanese case of leukodystrophy with ovarian failure arising from novel compound heterozygous AARS2 mutations. J Hum. Genet., 61, 899–902. [DOI] [PubMed] [Google Scholar]
- 12. Dallabona C., Diodato D., Kevelam S.H., Haack T.B., Wong L.-J., Salomons G.S., Baruffini E., Melchionda L., Mariotti C., Strom T.M.. et al. (2014) Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology, 82, 2063–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Szpisjak L., Zsindely N., Engelhardt J.I., Vecsei L., Kovacs G.G., Klivenyi P. (2017) Novel AARS2 gene mutation producing leukodystrophy: a case report. J. Hum. Genet., 62, 329–333. [DOI] [PubMed] [Google Scholar]
- 14. Götz A., Tyynismaa H., Euro L., Ellonen P., Hyötyläinen T., Ojala T., Hämäläinen R.H., Tommiska J., Raivio T., Oresic M.. et al. (2011) Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am. J. Hum. Genet., 88, 635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taylor R.W., Pyle A., Griffin H., Blakely E.L., Duff J., He L., Smertenko T., Alston C.L., Neeve V.C., Best A.. et al. (2014) Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. jama, 312, 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coughlin C.R., Scharer G.H., Friederich M.W., Yu H.-C., Geiger E.A., Creadon-Swindell G., Collins A.E., Vanlander A.V., Coster R.V., Powell C.A.. et al. (2015) Mutations in the mitochondrial cysteinyl-tRNA synthase gene, CARS2, lead to a severe epileptic encephalopathy and complex movement disorder. J. Med. Genet., 52, 532–540. [DOI] [PubMed] [Google Scholar]
- 17. Hallmann K., Zsurka G., Moskau-Hartmann S., Kirschner J., Korinthenberg R., Ruppert A.-K., Ozdemir O., Weber Y., Becker F., Lerche H.. et al. (2014) A homozygous splice-site mutation in CARS2 is associated with progressive myoclonic epilepsy. Neurology, 83, 2183–2187. [DOI] [PubMed] [Google Scholar]
- 18. Taft R.J., Vanderver A., Leventer R.J., Damiani S.A., Simons C., Grimmond S.M., Miller D., Schmidt J., Lockhart P.J., Pope K.. et al. (2013) Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am. J. Hum. Genet., 92, 774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lan M.-Y., Chang Y.-Y., Yeh T.-H., Lin T.-K., Lu C.-S. (2017) Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL) with a novel DARS2 mutation and isolated progressive spastic paraparesis. J. Neurol. Sci., 372, 229–231. [DOI] [PubMed] [Google Scholar]
- 20. Köhler C., Heyer C., Hoffjan S., Stemmler S., Lücke T., Thiels C., Kohlschütter A., Löbel U., Horvath R., Kleinle S.. et al. (2015) Early-onset leukoencephalopathy due to a homozygous missense mutation in the DARS2 gene. Mol. Cell. Probes., 29, 319–322. [DOI] [PubMed] [Google Scholar]
- 21. Isohanni P., Linnankivi T., Buzkova J., Lönnqvist T., Pihko H., Valanne L., Tienari P.J., Elovaara I., Pirttilä T., Reunanen M.. et al. (2010) DARS2 mutations in mitochondrial leucoencephalopathy and multiple sclerosis. J. Med. Genet., 47, 66–70. [DOI] [PubMed] [Google Scholar]
- 22. Sharma S., Sankhyan N., Kumar A., Scheper G.C., van der Knaap M.S., Gulati S. (2011) Leukoencephalopathy with brain stem and spinal cord involvement and high lactate: a genetically proven case without elevated white matter lactate. J. Child Neurol., 26, 773–776. [DOI] [PubMed] [Google Scholar]
- 23. Galluzzi P., Sacchini M., Bartalini G., Monti L., Cerase A., Lamantea E., Invernizzi F., Zeviani M., Balestri P., Venturi C. (2011) LBSL (leukoencephalopathy with brain stem and spinal cord involvement and high lactate) without sparing of the u-fibers and globi pallidi: A case report. Eur. J. Radiol. Extra, 79, e73–e76. [Google Scholar]
- 24. Lin J., Faria E.C., Da Rocha A.J., Masruha M.R., Vilanova L.C.P., Scheper G.C., van der Knaap M.S. (2010) Leukoencephalopathy with brainstem and spinal cord involvement and normal lactate: a new mutation in the DARS2 gene. J. Child Neurol., 25, 1425–1428. [DOI] [PubMed] [Google Scholar]
- 25. Güngör O, Özkaya A.K., Şahin Y., Güngör G., Dilber C., Aydın K. (2016) A compound heterozygous EARS2 mutation associated with mild leukoencephalopathy with thalamus and brainstem involvement and high lactate (LTBL). Brain Dev., 38, 857–861. [DOI] [PubMed] [Google Scholar]
- 26. Biancheri R., Lamantea E., Severino M., Diodato D., Pedemonte M., Cassandrini D., Ploederl A., Trucco F., Fiorillo C., Minetti C.. et al. (2015) Expanding the clinical and magnetic resonance spectrum of leukoencephalopathy with thalamus and brainstem involvement and high lactate (LTBL) in a patient harboring a novel EARS2 mutation. JIMD Rep. 23, 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oliveira R., Sommerville E.W., Thompson K., Nunes J., Pyle A., Grazina M., Chinnery P.F., Diogo L., Garcia P., Taylor R.W. (2017) Lethal neonatal LTBL associated with biallelic EARS2 variants: case report and review of the reported neuroradiological features. JIMD Rep., 33, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Şahin S., Cansu A., Kalay E., Dinçer T., Kul S., Çakır İM., Kamaşak T., Budak G.Y. (2016) Leukoencephalopathy with thalamus and brainstem involvement and high lactate caused by novel mutations in the EARS2 gene in two siblings. J. Neurol. Sci., 365, 54–58. [DOI] [PubMed] [Google Scholar]
- 29. Steenweg M.E., Ghezzi D., Haack T., Abbink T.E.M., Martinelli D., van Berkel C.G.M., Bley A., Diogo L., Grillo E., Water N., Te J.. et al. (2012) Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain, 135, 1387–1394. [DOI] [PubMed] [Google Scholar]
- 30. Taskin B.D., Karalok Z.S., Gurkas E., Aydin K., Aydogmus U., Ceylaner S., Karaer K., Yilmaz C., Pearl P.L. (2016) Early-onset mild type leukoencephalopathy caused by a homozygous EARS2 mutation. J. Child Neurol., 31, 938–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Danhauser K., Haack T.B., Alhaddad B., Melcher M., Seibt A., Strom T.M., Meitinger T., Klee D., Mayatepek E., Prokisch H.. et al. (2016) EARS2 mutations cause fatal neonatal lactic acidosis, recurrent hypoglycemia and agenesis of corpus callosum. Metabol. Brain Dis., 10.1007/s11011-016-9793-2. [DOI] [PubMed] [Google Scholar]
- 32. Yang Y., Liu W., Fang Z., Shi J., Che F., He C., Yao L., Wang E., Wu Y. (2016) A newly identified missense mutation in FARS2 causes autosomal-recessive spastic paraplegia. Hum. Mutat., 37, 165–169. [DOI] [PubMed] [Google Scholar]
- 33. Elo J.M., Yadavalli S.S., Euro L., Isohanni P., Götz A., Carroll C.J., Valanne L., Alkuraya F.S., Uusimaa J., Paetau A.. et al. (2012) Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum. Mol. Genet, 21, 4521–4529. [DOI] [PubMed] [Google Scholar]
- 34. Raviglione F., Conte G., Ghezzi D., Parazzini C., Righini A., Vergaro R., Legati A., Spaccini L., Gasperini S., Garavaglia B.. et al. (2016) Clinical findings in a patient with FARS2 mutations and early-infantile-encephalopathy with epilepsy. Am. J. Med. Genet., 170, 3004–3007. [DOI] [PubMed] [Google Scholar]
- 35. Cho J.S., Kim S.H., Kim H.Y., Chung T., Kim D., Jang S., Lee S.B., Yoo S.K., Shin J., Kim J.-I.. et al. (2017) FARS2 mutation and epilepsy: Possible link with early-onset epileptic encephalopathy. Epilepsy Res., 129, 118–124. [DOI] [PubMed] [Google Scholar]
- 36. Walker M.A., Mohler K.P., Hopkins K.W., Oakley D.H., Sweetser D.A., Ibba M., Frosch M.P., Thibert R.L. (2016) Novel compound heterozygous mutations expand the recognized phenotypes of FARS2-linked disease. J. Child Neurol., 31, 1127–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Almalki A., Alston C.L., Parker A., Simonic I., Mehta S.G., He L., Reza M., Oliveira J.M.A., Lightowlers R.N., McFarland R.. et al. (2014) Mutation of the human mitochondrial phenylalanine-tRNA synthetase causes infantile-onset epilepsy and cytochrome c oxidase deficiency. Biochim. Biophys. Acta, 1842, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vernon H.J., McClellan R., Batista D.A.S., Naidu S. (2015) Mutations in FARS2 and non-fatal mitochondrial dysfunction in two siblings. Am. J. Med. Genet., 167A, 1147–1151. [DOI] [PubMed] [Google Scholar]
- 39. McMillan H.J., Schwartzentruber J., Smith A., Lee S., Chakraborty P., Bulman D.E., Beaulieu C.L., Majewski J., Boycott K.M., Geraghty M.T. (2014) Compound heterozygous mutations in glycyl-tRNA synthetase are a proposed cause of systemic mitochondrial disease. BMC Med. Genet., 15, 36.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Antonellis A., Ellsworth R.E., Sambuughin N., Puls I., Abel A., Lee-Lin S.-Q., Jordanova A., Kremensky I., Christodoulou K., Middleton L.T.. et al. (2003) Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet., 72, 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. James P.A., Cader M.Z., Muntoni F., Childs A.-M., Crow Y.J., Talbot K. (2006) Severe childhood SMA and axonal CMT due to anticodon binding domain mutations in the GARS gene. Neurology, 67, 1710–1712. [DOI] [PubMed] [Google Scholar]
- 42. Abe A., Hayasaka K. (2009) The GARS gene is rarely mutated in Japanese patients with Charcot-Marie-Tooth neuropathy. J. Hum. Genet., 54, 310–312. [DOI] [PubMed] [Google Scholar]
- 43. Del Bo R., Locatelli F., Corti S., Scarlato M., Ghezzi S., Prelle A., Fagiolari G., Moggio M., Carpo M., Bresolin N.. et al. (2006) Coexistence of CMT-2D and distal SMA-V phenotypes in an Italian family with a GARS gene mutation. Neurology, 66, 752–754. [DOI] [PubMed] [Google Scholar]
- 44. Sivakumar K., Kyriakides T., Puls I., Nicholson G.A., Funalot B., Antonellis A., Sambuughin N., Christodoulou K., Beggs J.L., Zamba-Papanicolaou E.. et al. (2005) Phenotypic spectrum of disorders associated with glycyl-tRNA synthetase mutations. Brain, 128, 2304–2314. [DOI] [PubMed] [Google Scholar]
- 45. Rohkamm B., Reilly M.M., Lochmüller H., Schlotter-Weigel B., Barisic N., Schöls L., Nicholson G., Pareyson D., Laurà M., Janecke A.R.. et al. (2007) Further evidence for genetic heterogeneity of distal HMN type V, CMT2 with predominant hand involvement and Silver syndrome. J. Neurol. Sci., 263, 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee H.J., Park J., Nakhro K., Park J.M., Hur Y.-M., Choi B.-O., Chung K.W. (2012) Two novel mutations of GARS in Korean families with distal hereditary motor neuropathy type V. J. Peripher. Nerv. Syst., 17, 418–421. [DOI] [PubMed] [Google Scholar]
- 47. Eskuri J.M., Stanley C.M., Moore S.A., Mathews K.D. (2012) Infantile onset CMT2D/dSMA V in monozygotic twins due to a mutation in the anticodon-binding domain of GARS. J. Peripher. Nerv. Syst., 17, 132–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Puffenberger E.G., Jinks R.N., Sougnez C., Cibulskis K., Willert R.A., Achilly N.P., Cassidy R.P., Fiorentini C.J., Heiken K.F., Lawrence J.J.. et al. (2012) Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS ONE, 7, e28936.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vester A., Velez Ruiz G., McLaughlin H.M., Lupski J.R., Talbot K., Vance J.M., Züchner S., Roda R.H., Fischbeck K.H., Biesecker L.G.. et al. (2013) A loss‐of‐function variant in the human histidyl‐tRNA synthetase (HARS) gene is neurotoxic in vivo. Hum. Mutat., 34, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Safka Brozkova D., Deconinck T., Griffin L.B., Ferbert A., Haberlova J., Mazanec R., Lassuthova P., Roth C., Pilunthanakul T., Rautenstrauss B.. et al. (2015) Loss of function mutations in HARS cause a spectrum of inherited peripheral neuropathies. Brain, 138, 2161–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pierce S.B., Chisholm K.M., Lynch E.D., Lee M.K., Walsh T., Opitz J.M., Li W., Klevit R.E., King M.-C. (2011) Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. U.S.A, 108, 6543–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Orenstein N., Weiss K., Oprescu S.N., Shapira R., Kidron D., Vanagaite Basel L., Antonellis A., Muenke M. (2017) Bi‐allelic IARS mutations in a child with intra‐uterine growth retardation, neonatal cholestasis, and mild developmental delay. Clin. Genet., 91, 913–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kopajtich R., Murayama K., Janecke A.R., Haack T.B., Breuer M., Knisely A.S., Harting I., Ohashi T., Okazaki Y., Watanabe D.. et al. (2016) Biallelic IARS mutations cause growth retardation with prenatal onset, intellectual disability, muscular hypotonia, and infantile hepatopathy. Am. J. Hum. Genet., 99, 414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moosa S., Haagerup A., Gregersen P.A., Petersen K.K., Altmüller J., Thiele H., Nürnberg P., Cho T.-J., Kim O.-H., Nishimura G.. et al. (2017) Confirmation of CAGSSS syndrome as a distinct entity in a Danish patient with a novel homozygous mutation in IARS2. Am. J. Med. Genet., 173, 1102–1108. [DOI] [PubMed] [Google Scholar]
- 55. Schwartzentruber J., Buhas D., Majewski J., Sasarman F., Papillon Cavanagh S., Thiffault I., Sheldon K.M., Massicotte C., Patry L., Simon M.. et al. (2015) Mutation in The Nuclear‐Encoded Mitochondrial Isoleucyl–tRNA Synthetase IARS2 in Patients with Cataracts, Growth Hormone Deficiency with Short Stature, Partial Sensorineural Deafness, and Peripheral Neuropathy or with Leigh Syndrome. Hum. Mutat, 36, 281–281. [DOI] [PubMed] [Google Scholar]
- 56. Santos-Cortez R.L.P., Lee K., Azeem Z., Antonellis P.J., Pollock L.M., Khan S., Irfanullah, Andrade-Elizondo P.B., Chiu I., Adams M.D.. et al. (2013) Mutations in KARS, encoding lysyl-tRNA synthetase, cause autosomal-recessive nonsyndromic hearing impairment DFNB89. Am. J. Hum. Genet., 93, 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McLaughlin H.M., Sakaguchi R., Liu C., Igarashi T., Pehlivan D., Chu K., Iyer R., Cruz P., Cherukuri P.F., Hansen N.F.. et al. (2010) Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am. J. Hum. Genet., 87, 560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McMillan H.J., Humphreys P., Smith A., Schwartzentruber J., Chakraborty P., Bulman D.E., Beaulieu C.L. FORGE Canada Consortium Majewski J., Boycott K.M.. et al. (2015) Congenital visual impairment and progressive microcephaly due to lysyl-transfer ribonucleic Acid (RNA) synthetase (KARS) mutations: the expanding phenotype of aminoacyl-transfer RNA synthetase mutations in human disease. J. Child Neurol., 30, 1037–1043. [DOI] [PubMed] [Google Scholar]
- 59. Casey J.P., McGettigan P., Lynam-Lennon N., McDermott M., Regan R., Conroy J., Bourke B., O'Sullivan J., Crushell E., Lynch S.. et al. (2012) Identification of a mutation in LARS as a novel cause of infantile hepatopathy. Mol. Genet. Metab., 106, 351–358. [DOI] [PubMed] [Google Scholar]
- 60. Casey J.P., Slattery S., Cotter M., Monavari A.A., Knerr I., Hughes J., Treacy E.P., Devaney D., McDermott M., Laffan E.. et al. (2015) Clinical and genetic characterisation of infantile liver failure syndrome type 1, due to recessive mutations in LARS. J. Inherit. Metab. Dis., 38, 1085–1092. [DOI] [PubMed] [Google Scholar]
- 61. Pierce S.B., Gersak K., Michaelson-Cohen R., Walsh T., Lee M.K., Malach D., Klevit R.E., King M.-C., Levy-Lahad E. (2013) Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am. J. Hum. Genet., 92, 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Soldà G., Caccia S., Robusto M., Chiereghin C., Castorina P., Ambrosetti U., Duga S., Asselta R. (2016) First independent replication of the involvement of LARS2 in Perrault syndrome by whole-exome sequencing of an Italian family. J. Hum. Genet., 61, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Riley L.G., Rudinger-Thirion J., Schmitz-Abe K., Thorburn D.R., Davis R.L., Teo J., Arbuckle S., Cooper S.T., Campagna D.R., Frugier M.. et al. (2016) LARS2 variants associated with hydrops, lactic acidosis, sideroblastic anemia, and multisystem failure. JIMD Rep., 28, 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. van Meel E., Wegner D.J., Cliften P., Willing M.C., White F.V., Kornfeld S., Cole F.S. (2013) Rare recessive loss-of-function methionyl-tRNA synthetase mutations presenting as a multi-organ phenotype. BMC Med. Genet, 14, 106.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sun Y., Hu G., Luo J., Fang D., Yu Y., Wang X., Chen J., Qiu W. (2017) Mutations in methionyl-tRNA synthetase gene in a Chinese family with interstitial lung and liver disease, postnatal growth failure and anemia. J. Hum. Genet., 48, 337–651. [DOI] [PubMed] [Google Scholar]
- 66. Hadchouel A., Wieland T., Griese M., Baruffini E., Lorenz-Depiereux B., Enaud L., Graf E., Dubus J.C., Halioui-Louhaichi S., Coulomb A.. et al. (2015) Biallelic mutations of methionyl-tRNA synthetase cause a specific type of pulmonary alveolar proteinosis prevalent on Réunion Island. Am. J. Hum. Genet., 96, 826–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gonzalez M., McLaughlin H., Houlden H., Guo M., Yo-Tsen L., Hadjivassilious M., Speziani F., Yang X.-L., Antonellis A., Reilly M.M.. et al. (2013) Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J. Neurol. Neurosurg. Psychiatr., 84, 1247–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hyun Y.S., Park H.J., Heo S.-H., Yoon B.R., Nam S.H., Kim S.-B., Park C.I., Choi B.-O., Chung K.W. (2014) Rare variants in methionyl- and tyrosyl-tRNA synthetase genes in late-onset autosomal dominant Charcot-Marie-Tooth neuropathy. Clin. Genet., 86, 592–594. [DOI] [PubMed] [Google Scholar]
- 69. Webb B.D., Wheeler P.G., Hagen J.J., Cohen N., Linderman M.D., Diaz G.A., Naidich T.P., Rodenburg R.J., Houten S.M., Schadt E.E. (2015) Novel, compound heterozygous, single-nucleotide variants in MARS2 associated with developmental delay, poor growth, and sensorineural hearing loss. Hum. Mutat., 36, 587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bayat V., Thiffault I., Jaiswal M., Tétreault M., Donti T., Sasarman F., Bernard G., Demers-Lamarche J., Dicaire M.-J., Mathieu J.. et al. (2012) Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol., 10, e1001288.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sofou K., Kollberg G., Holmström M., Dávila M., Darin N., Gustafsson C.M., Holme E., Oldfors A., Tulinius M., Asin-Cayuela J. (2015) Whole exome sequencing reveals mutations in NARS2 and PARS2, encoding the mitochondrial asparaginyl-tRNA synthetase and prolyl-tRNA synthetase, in patients with Alpers syndrome. Mol. Genet. Genomic Med., 3, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mizuguchi T., Nakashima M., Kato M., Yamada K., Okanishi T., Ekhilevitch N., Mandel H., Eran A., Toyono M., Sawaishi Y.. et al. (2017) PARS2 and NARS2 mutations in infantile-onset neurodegenerative disorder. J. Hum. Genet., 10.1038/jhg.2016.163. [DOI] [PubMed] [Google Scholar]
- 73. Vanlander A.V., Menten B., Smet J., De Meirleir L., Sante T., De Paepe B., Seneca S., Pearce S.F., Powell C.A., Vergult S.. et al. (2015) Two siblings with homozygous pathogenic splice-site variant in mitochondrial asparaginyl-tRNA synthetase (NARS2). Hum. Mutat., 36, 222–231. [DOI] [PubMed] [Google Scholar]
- 74. Simon M., Richard E.M., Wang X., Shahzad M., Huang V.H., Qaiser T.A., Potluri P., Mahl S.E., Davila A., Nazli S.. et al. (2015) Mutations of human NARS2, encoding the mitochondrial asparaginyl-tRNA synthetase, cause nonsyndromic deafness and Leigh syndrome. PLoS Genet., 11, e1005097.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang X., Ling J., Barcia G., Jing L., Wu J., Barry B.J., Mochida G.H., Hill R.S., Weimer J.M., Stein Q.. et al. (2014) Mutations in QARS, encoding glutaminyl-tRNA synthetase, cause progressive microcephaly, cerebral-cerebellar atrophy, and intractable seizures. Am. J. Hum. Genet., 94, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Salvarinova R., Ye C.X., Rossi A., Biancheri R., Roland E.H., Pavlidis P., Ross C.J., Tarailo-Graovac M., Wasserman W.W., van Karnebeek C.D.M. (2015) Expansion of the QARS deficiency phenotype with report of a family with isolated supratentorial brain abnormalities. Neurogenetics, 16, 145–149. [DOI] [PubMed] [Google Scholar]
- 77. Wolf N.I., Salomons G.S., Rodenburg R.J., Pouwels P.J.W., Schieving J.H., Derks T.G.J., Fock J.M., Rump P., van Beek D.M., van der Knaap M.S.. et al. (2014) Mutations in RARS cause hypomyelination. Ann. Neurol., 76, 134–139. [DOI] [PubMed] [Google Scholar]
- 78. Cassandrini D., Cilio M.R., Bianchi M., Doimo M., Balestri M., Tessa A., Rizza T., Sartori G., Meschini M.C., Nesti C.. et al. (2013) Pontocerebellar hypoplasia type 6 caused by mutations in RARS2: definition of the clinical spectrum and molecular findings in five patients. J. Inherit. Metab. Dis., 36, 43–53. [DOI] [PubMed] [Google Scholar]
- 79. Edvardson S., Shaag A., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O. (2007) Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet., 81, 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Glamuzina E., Brown R., Hogarth K., Saunders D., Russell-Eggitt I., Pitt M., de Sousa C., Rahman S., Brown G., Grunewald S. (2012) Further delineation of pontocerebellar hypoplasia type 6 due to mutations in the gene encoding mitochondrial arginyl-tRNA synthetase, RARS2. J. Inherit. Metab. Dis., 35, 459–467. [DOI] [PubMed] [Google Scholar]
- 81. Li Z., Schonberg R., Guidugli L., Johnson A.K., Arnovitz S., Yang S., Scafidi J., Summar M.L., Vezina G., Das S.. et al. (2015) A novel mutation in the promoter of RARS2 causes pontocerebellar hypoplasia in two siblings. J. Hum. Genet., 60, 363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ngoh A., Bras J., Guerreiro R., Meyer E., McTague A., Dawson E., Mankad K., Gunny R., Clayton P., Mills P.B.. et al. (2016) RARS2 mutations in a sibship with infantile spasms. Epilepsia, 57, e97–e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rankin J., Brown R., Dobyns W.B., Harington J., Patel J., Quinn M., Brown G. (2010) Pontocerebellar hypoplasia type 6: A British case with PEHO-like features. Am. J. Med. Genet, 152A, 2079–2084. [DOI] [PubMed] [Google Scholar]
- 84. Namavar Y., Barth P.G., Kasher P.R., van Ruissen F., Brockmann K., Bernert G., Writzl K., Ventura K., Cheng E.Y., Ferriero D.M.. et al. (2011) Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain, 134, 143–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nishri D., Goldberg-Stern H., Noyman I., Blumkin L., Kivity S., Saitsu H., Nakashima M., Matsumoto N., Leshinsky-Silver E., Lerman-Sagie T.. et al. (2016) RARS2 mutations cause early onset epileptic encephalopathy without ponto-cerebellar hypoplasia. Eur. J. Paediatr. Neurol., 20, 412–417. [DOI] [PubMed] [Google Scholar]
- 86. Lühl S., Bode H., Schlötzer W., Bartsakoulia M., Horvath R., Abicht A., Stenzel M., Kirschner J., Grünert S.C. (2016) Novel homozygous RARS2 mutation in two siblings without pontocerebellar hypoplasia - further expansion of the phenotypic spectrum. Orphanet. J. Rare Dis., 11, 140.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Alkhateeb A.M., Aburahma S.K., Habbab W., Thompson I.R. (2016) Novel mutations in WWOX, RARS2, and C10orf2 genes in consanguineous Arab families with intellectual disability. Metab. Brain Dis., 31, 901–907. [DOI] [PubMed] [Google Scholar]
- 88. Musante L., Püttmann L., Kahrizi K., Garshasbi M., Hu H., Stehr H., Lipkowitz B., Otto S., Jensen L.R., Tzschach A.. et al. (2017) Mutations of the aminoacyl-tRNA-synthetases SARS and WARS2 are implicated in the etiology of autosomal recessive intellectual disability. Hum. Mutat., 38, 621–636. [DOI] [PubMed] [Google Scholar]
- 89. Belostotsky R., Ben-Shalom E., Rinat C., Becker-Cohen R., Feinstein S., Zeligson S., Segel R., Elpeleg O., Nassar S., Frishberg Y. (2011) Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am. J. Hum. Genet., 88, 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rivera H., Martín-Hernández E., Delmiro A., García-Silva M.T., Quijada-Fraile P., Muley R., Arenas J., Martín M.A., Martínez-Azorín F. (2013) A new mutation in the gene encoding mitochondrial seryl-tRNA synthetase as a cause of HUPRA syndrome. BMC Nephrol., 14, 195.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Diodato D., Melchionda L., Haack T.B., Dallabona C., Baruffini E., Donnini C., Granata T., Ragona F., Balestri P., Margollicci M.. et al. (2014) VARS2 and TARS2 mutations in patients with mitochondrial encephalomyopathies. Hum. Mutat., 35, 983–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Karaca E., Harel T., Pehlivan D., Jhangiani S.N., Gambin T., Coban Akdemir Z., Gonzaga-Jauregui C., Erdin S., Bayram Y., Campbell I.M.. et al. (2015) Genes that affect brain structure and function identified by rare variant analyses of Mendelian neurologic disease. Neuron, 88, 499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Baertling F., Alhaddad B., Seibt A., Budaeus S., Meitinger T., Strom T.M., Mayatepek E., Schaper J., Prokisch H., Haack T.B.. et al. (2017) Neonatal encephalocardiomyopathy caused by mutations in VARS2. Metabol. Brain Dis., 32, 267–270. 10.1007/s11011-016-9890-2. [DOI] [PubMed] [Google Scholar]
- 94. Tsai P.-C., Soong B.-W., Mademan I., Huang Y.-H., Liu C.-R., Hsiao C.-T., Wu H.-T., Liu T.-T., Liu Y.-T., Tseng Y.-T.. et al. (2017) A recurrent WARS mutation is a novel cause of autosomal dominant distal hereditary motor neuropathy. Brain, 10.1093/brain/awx058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Nowaczyk M.J.M., Huang L., Tarnopolsky M., Schwartzentruber J., Majewski J., Bulman D.E. FORGE Canada Consortium, Care4Rare Canada Consortium Hartley T., Boycott K.M. (2016) A novel multisystem disease associated with recessive mutations in the tyrosyl-tRNA synthetase (YARS) gene. Am. J. Med. Genet., 173, 126–134. [DOI] [PubMed] [Google Scholar]
- 96. Jordanova A., Irobi J., Thomas F.P., Van Dijck P., Meerschaert K., Dewil M., Dierick I., Jacobs A., De Vriendt E., Guergueltcheva V.. et al. (2006) Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet., 38, 197–202. [DOI] [PubMed] [Google Scholar]
- 97. Gonzaga-Jauregui C., Harel T., Gambin T., Kousi M., Griffin L.B., Francescatto L., Ozes B., Karaca E., Jhangiani S.N., Bainbridge M.N.. et al. (2015) Exome sequence analysis suggests that genetic burden contributes to phenotypic variability and complex neuropathy. CellRep., 12, 1169–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sommerville E.W., Ng Y.S., Alston C.L., Dallabona C., Gilberti M., He L., Knowles C., Chin S.L., Schaefer A.M., Falkous G.. et al. (2017) Clinical features, molecular heterogeneity, and prognostic implications in YARS2-related mitochondrial myopathy. JAMA Neurol., 74, 686–694 10.1001/jamaneurol.2016.4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Riley L.G., Cooper S., Hickey P., Rudinger-Thirion J., McKenzie M., Compton A., Lim S.C., Thorburn D., Ryan M.T., Giegé R.. et al. (2010) Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia–MLASA syndrome. Am. J. Hum. Genet., 87, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sasarman F., Nishimura T., Thiffault I., Shoubridge E.A. (2012) A novel mutation in YARS2 causes myopathy with lactic acidosis and sideroblastic anemia. Hum. Mutat, 33, 1201–1206. [DOI] [PubMed] [Google Scholar]
- 101. Nakajima J., Eminoglu T.F., Vatansever G., Nakashima M., Tsurusaki Y., Saitsu H., Kawashima H., Matsumoto N., Miyake N. (2014) A novel homozygous YARS2 mutation causes severe myopathy, lactic acidosis, and sideroblastic anemia 2. J Hum Genet., 59, 229–232. [DOI] [PubMed] [Google Scholar]
- 102. Ardissone A., Lamantea E., Quartararo J., Dallabona C., Carrara F., Moroni I., Donnini C., Garavaglia B., Zeviani M., Uziel G. (2015) A Novel Homozygous YARS2 Mutation in Two Italian Siblings and a Review of Literature. JIMD Rep, 20, 95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Shahni R., Wedatilake Y., Cleary M.A., Lindley K.J., Sibson K.R., Rahman S. (2013) A distinct mitochondrial myopathy, lactic acidosis and sideroblastic anemia (MLASA) phenotype associates with YARS2 mutations. Am. J. Med. Genet., 161A, 2334–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Oprescu S.N., Griffin L.B., Beg A.A., Antonellis A. (2017) Predicting the pathogenicity of aminoacyl-tRNA synthetase mutations. Methods, 113, 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Nowaczyk M.J.M., Huang L., Tarnopolsky M., Schwartzentruber J., Majewski J., Bulman D.E., Hartley T., Boycott K.M. (2017) A novel multisystem disease associated with recessive mutations in the tyrosyl‐tRNA synthetase (YARS) gene. Am. J. Med. Genet., 173, 126–134. [DOI] [PubMed] [Google Scholar]
- 106. Seburn K.L., Nangle L.A., Cox G.A., Schimmel P., Burgess R.W. (2006) An active dominant mutation of Glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-tooth 2D mouse model. Neuron, 51, 715–726. [DOI] [PubMed] [Google Scholar]
- 107. Danhauser K., Haack T.B., Alhaddad B., Melcher M., Seibt A., Strom T.M., Meitinger T., Klee D., Mayatepek E., Prokisch H.. et al. (2016) EARS2 mutations cause fatal neonatal lactic acidosis, recurrent hypoglycemia and agenesis of corpus callosum. Metabol. Brain Dis., 31, 717–721. [DOI] [PubMed] [Google Scholar]
- 108. Jiang P., Jin X., Peng Y., Wang M., Liu H., Liu X., Zhang Z., Ji Y., Zhang J., Liang M.. et al. (2016) The exome sequencing identified the mutation in YARS2 encoding the mitochondrial tyrosyl-tRNA synthetase as a nuclear modifier for the phenotypic manifestation of Leber's hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum. Mol. Genet., 25, 584–596. [DOI] [PubMed] [Google Scholar]
- 109. Ishimura R., Nagy G., Dotu I., Chuang J.H., Ackerman S.L. (2016) Activation of GCN2 kinase by ribosome stalling links translation elongation with translation initiation. eLife Sciences, 5, e14295.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ishimura R., Nagy G., Dotu I., Zhou H., Yang X.-L., Schimmel P., Senju S., Nishimura Y., Chuang J.H., Ackerman S.L. (2014) RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science, 345, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhang G., Fedyunin I., Miekley O., Valleriani A., Moura A., Ignatova Z. (2010) Global and local depletion of ternary complex limits translational elongation. Nucleic Acids Res., 38, 4778–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Dong J., Qiu H., Garcia-Barrio M., Anderson J., Hinnebusch A.G. (2000) Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell, 6, 269–279. [DOI] [PubMed] [Google Scholar]
- 113. Gonzalez M., McLaughlin H., Houlden H., Guo M., Yo-Tsen L., Hadjivassilious M., Speziani F., Yang X.-L., Antonellis A., Reilly M.M.. et al. (2013) Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J. Neurol. Neurosurg. Psychiatry, 84, 1247–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hirano M., Oka N., Hashiguchi A., Ueno S., Sakamoto H., Takashima H., Higuchi Y., Kusunoki S., Nakamura Y. (2016) Histopathological features of a patient with Charcot-Marie-tooth disease type 2U/AD-CMTax-MARS. J. Peripher. Nerv. Syst., 21, 370–374. [DOI] [PubMed] [Google Scholar]
- 115. Niehues S., Bussmann J., Steffes G., Erdmann I., Köhrer C., Sun L., Wagner M., Schäfer K., Wang G., Koerdt S.N.. et al. (2015) Impaired protein translation in Drosophila models for Charcot–Marie–tooth neuropathy caused by mutant tRNA synthetases. Nat. Commun., 6, 7520–7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sun L., Song Y., Blocquel D., Yang X.-L., Schimmel P. (2016) Two crystal structures reveal design for repurposing the C-Ala domain of human AlaRS. Proc. Natl. Acad. Sci. U.S.A, 113, 14300–14305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Mo Z., Zhang Q., Liu Z., Lauer J., Shi Y., Sun L., Griffin P.R., Yang X.-L. (2016) Neddylation requires glycyl-tRNA synthetase to protect activated E2. Nat. Struct. Mol. Biol., 23, 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Park M.C., Kang T., Jin D., Han J.M., Kim S.B., Park Y.J., Cho K., Park Y.W., Guo M., He W.. et al. (2012) Secreted human glycyl-tRNA synthetase implicated in defense against ERK-activated tumorigenesis. Proc. Natl. Acad. Sci. U.S.A, 109, E640–E647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Johanson K., Hoang T., Sheth M., Hyman L.E. (2003) GRS1, a yeast tRNA synthetase with a role in mRNA 3' end formation. J. Biol. Chem., 278, 35923–35930. [DOI] [PubMed] [Google Scholar]
- 120. Zhou J.J., Wang F., Xu Z., Lo W.-S., Lau C.-F., Chiang K.P., Nangle L.A., Ashlock M.A., Mendlein J.D., Yang X.-L.. et al. (2014) Secreted histidyl-tRNA synthetase splice variants elaborate major epitopes for autoantibodies in inflammatory myositis. J. Biol. Chem., 289, 19269–19275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Raben N., Nichols R., Dohlman J., McPhie P., Sridhar V., Hyde C., Leff R., Plotz P. (1994) A motif in human histidyl-tRNA synthetase which is shared among several aminoacyl-tRNA synthetases is a coiled-coil that is essential for enzymatic activity and contains the major autoantigenic epitope. J. Biol. Chem., 269, 24277–24283. [PubMed] [Google Scholar]
- 122. Howard O.M.Z., Dong H.F., Yang D., Raben N., Nagaraju K., Rosen A., Casciola-Rosen L., Härtlein M., Kron M., Yang D.. et al. (2002) Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J. Exp. Med., 196, 781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Ahn Y.H., Park S., Choi J.J., Park B.-K., Rhee K.H., Kang E., Ahn S., Lee C.-H., Lee J.S., Inn K.-S.. et al. (2016) Secreted tryptophanyl-tRNA synthetase as a primary defence system against infection. Nat. Microbiol., 2, 16191.. [DOI] [PubMed] [Google Scholar]
- 124. Wakasugi K., Slike B.M., Hood J., Otani A., Ewalt K.L., Friedlander M., Cheresh D.A., Schimmel P. (2002) A human aminoacyl-tRNA synthetase as a regulator of angiogenesis. Proc. Natl.Acad. Sci. USA, 99, 173–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zeng R., Wang M., You G.-Y., Yue R.-Z., Chen Y.-C., Zeng Z., Liu R., Qiang O., Zhang L. (2016) Effect of mini-tyrosyl-tRNA synthetase/mini-tryptophanyl-tRNA synthetase on angiogenesis in Rhesus monkeys after acute myocardial infarction. Cardiovasc. Ther., 34, 4–12. [DOI] [PubMed] [Google Scholar]
- 126. Otani A., Slike B.M., Dorrell M.I., Hood J., Kinder K., Ewalt K.L., Cheresh D., Schimmel P., Friedlander M. (2002) A fragment of human TrpRS as a potent antagonist of ocular angiogenesis. Proc. Natl. Acad. Sci. USA, 99, 178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Zeng R., Chen Y.-C., Zeng Z., Liu W.-Q., Jiang X.-F., Liu R., Qiang O., Li X. (2011) Effect of mini-tyrosyl-tRNA synthetase/mini-tryptophanyl-tRNA synthetase on ischemic angiogenesis in rats: proliferation and migration of endothelial cells. Heart Vessels, 26, 69–80. [DOI] [PubMed] [Google Scholar]
- 128. Zeng R., Chen Y.-C., Zeng Z., Liu X.-X., Liu R., Qiang O., Li X. (2012) Inhibition of mini-TyrRS-induced angiogenesis response in endothelial cells by VE-cadherin-dependent mini-TrpRS. Heart Vessels, 27, 193–201. [DOI] [PubMed] [Google Scholar]
- 129. Wei N., Shi Y., Truong L.N., Fisch K.M., Xu T., Gardiner E., Fu G., Hsu Y.-S.O., Kishi S., Su A.I.. et al. (2014) Oxidative stress diverts tRNA synthetase to nucleus for protection against DNA damage. Mol. Cell, 56, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Fu G., Xu T., Shi Y., Wei N., Yang X.-L. (2012) tRNA-controlled nuclear import of a human tRNA synthetase. J. Biol. Chem., 287, 9330–9334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Cao X., Li C., Xiao S., Tang Y., Huang J., Zhao S., Li X., Li J., Zhang R., Yu W. (2017) Acetylation promotes TyrRS nuclear translocation to prevent oxidative damage. Proc. Natl Acad. Sci. USA, 114, 687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Cheng G., Zhang H., Yang X., Tzima E., Ewalt K.L., Schimmel P., Faber J.E. (2008) Effect of mini-tyrosyl-tRNA synthetase on ischemic angiogenesis, leukocyte recruitment, and vascular permeability. Am. J. Physiol. Regul. Integr. Comp. Physiol., 295, R1138–R1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Greenberg Y., King M., Kiosses W.B., Ewalt K., Yang X., Schimmel P., Reader J.S., Tzima E. (2008) The novel fragment of tyrosyl tRNA synthetase, mini-TyrRS, is secreted to induce an angiogenic response in endothelial cells. faseb J., 22, 1597–1605. [DOI] [PubMed] [Google Scholar]
- 134. Chihara T., Luginbuhl D., Luo L. (2007) Cytoplasmic and mitochondrial protein translation in axonal and dendritic terminal arborization. Nat. Neurosci., 10, 828–837. [DOI] [PubMed] [Google Scholar]
- 135. Malissovas N., Griffin L.B., Antonellis A., Beis D. (2016) Dimerization is required for GARS-mediated neurotoxicity in dominant CMT disease. Hum. Mol. Genet., 25, 1528–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Motley W.W., Seburn K.L., Nawaz M.H., Miers K.E., Cheng J., Antonellis A., Green E.D., Talbot K., Yang X.-L., Fischbeck K.H.. et al. (2011) Charcot-Marie-tooth-linked mutant GARS is toxic to peripheral neurons independent of wild-type GARS levels. PLoS Genet., 7, e1002399.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O'Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B.. et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Antonellis A., Lee-Lin S.-Q., Wasterlain A., Leo P., Quezado M., Goldfarb L.G., Myung K., Burgess S., Fischbeck K.H., Green E.D. (2006) Functional analyses of glycyl-tRNA synthetase mutations suggest a key role for tRNA-charging enzymes in peripheral axons. J. Neurosci., 26, 10397–10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Züchner S., Mersiyanova I.V., Muglia M., Bissar-Tadmouri N., Rochelle J., Dadali E.L., Zappia M., Nelis E., Patitucci A., Senderek J.. et al. (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-tooth neuropathy type 2A. Nat. Genet., 36, 449–451. [DOI] [PubMed] [Google Scholar]
- 140. Meggouh F., Bienfait H.M.E., Weterman M.A.J., de Visser M., Baas F. (2006) Charcot-Marie-Tooth disease due to a de novo mutation of the RAB7 gene. Neurology, 67, 1476–1478. [DOI] [PubMed] [Google Scholar]
- 141. Blocquel D., Li S., Wei N., Daub H., Sajish M., Erfurth M.-L., Kooi G., Zhou J., Bai G., Schimmel P.. et al. (2017) Alternative stable conformation capable of protein misinteraction links tRNA synthetase to peripheral neuropathy. Nucleic Acids Res., 10.1093/nar/gkx455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. He W., Zhang H.-M., Chong Y.E., Guo M., Marshall A.G., Yang X.-L. (2011) Dispersed disease-causing neomorphic mutations on a single protein promote the same localized conformational opening. Proc. Natl. Acad. Sci. U.S.A, 108, 12307–12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. He W., Bai G., Zhou H., Wei N., White N.M., Lauer J., Liu H., Shi Y., Dumitru C.D., Lettieri K.. et al. (2015) CMT2D neuropathy is linked to the neomorphic binding activity of glycyl-tRNA synthetase. Nature, 526, 710–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Neufeld G., Cohen T., Shraga N., Lange T., Kessler O., Herzog Y. (2002) The neuropilins: multifunctional semaphorin and VEGF receptors that modulate axon guidance and angiogenesis. Trends Cardiovasc. Med., 12, 13–19. [DOI] [PubMed] [Google Scholar]
- 145. Huettl R.-E., Soellner H., Bianchi E., Novitch B.G., Huber A.B. (2011) Npn-1 contributes to axon-axon interactions that differentially control sensory and motor innervation of the limb. PLoS Biol., 9, e1001020.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Sleigh J.N., Gómez-Martín A., Schiavo G. (2016) Neuropilin 1 sequestration by neuropathogenic mutant glycyl-tRNA synthetase is permissive to vascular development and homeostasis. bioRxiv, 10.1101/081513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Sleigh J.N., Dawes J.M., West S.J., Wei N., Spaulding E.L., Gómez-Martín A., Zhang Q., Burgess R.W., Cader M.Z., Talbot K.. et al. (2017) Trk receptor signaling and sensory neuron fate are perturbed in human neuropathy caused by Gars mutations. Proc. Natl. Acad. Sci. U.S.A, 10.1073/pnas.1614557114. [DOI] [PMC free article] [PubMed] [Google Scholar]