

Abstract
Osteoarthritis is a common, complex disease with no curative therapy. In this review, we summarize current knowledge on disease aetiopathogenesis and outline genetics and genomics approaches that are helping catalyse a much-needed improved understanding of the biological underpinning of disease development and progression.
Introduction
Osteoarthritis (OA) is the most prevalent musculoskeletal disease and a leading cause of disability worldwide (1–3). The impact of OA across Europe has been described as immense (4,5). OA affects 40% of individuals over the age of 70 (1), is a major cause of pain (6) and is associated with an increased risk of comorbidity and death (7). Ten million people suffer from OA in the UK alone, with a total indirect cost to the economy of £14.8 billion per annum (7). The most common OA site is the knee, affecting 1 in 5 people over the age of 45 (8,9). The health economic burden of OA is rising, commensurate with longevity and obesity rates (8). There is currently no treatment; disease management targets the main symptoms of pain and loss of function and culminates in joint replacement surgery [1.76 million per year in the EU (10)] with variable patient-reported outcomes (11–13). Thus, there is a large unmet need for therapeutic interventions to alter the natural history of the disease (14). This review will outline established and emerging pathways to improving our understanding of disease aetiopathology through genetics and genomics studies.
OA Is a Disease of The Synovial Joint
The synovial joint is a complex structure, comprising articular cartilage, subchondral bone, synovial lining membrane, fibrous joint capsule and supporting ligaments. The articular cartilage, calcified cartilage and subchondral bone form the osteochondral unit, a biocomposite that is uniquely adapted to transferring loads during weight bearing and joint motion. The osteochondral unit provides tensile strength, compressive resilience and a low-friction articulating surface through the collagen network, proteoglycan aggregates and layer of lubricants, respectively. Chondrocytes are the only cell type in articular cartilage, which is avascular and aneural. Under normal physiological conditions the synovial membrane consists of a thin layer of cells with phenotypic features of macrophages and fibroblasts (15), and serves to produce synovial fluid that is responsible for maintaining nutrition and lubrication of the articular cartilage. The subchondral bone adapts its structural and functional architecture in response to its local mechanical environment through remodelling, regulated by osteocytes via interactions with osteoclasts and osteoblasts (16).
OA is a disease characterized by a gradual process of tissue destruction and remodelling that affects all of the structures of the synovial joint (17,18), with degeneration of articular cartilage, remodelling of the underlying bone, and synovitis (18) as its hallmarks (Fig. 1). The initiating signals that trigger the development of OA remain poorly understood, however established clinical risk factors include increasing age (19–21), female sex (19,21–23), obesity (20,23,24), occupational exposure to high levels of joint loading activity (23,24), previous joint injury and deformity (25,26), smoking status and family history of OA (27–30). Histological changes in OA include synovial hypertrophy and hyperplasia, with macrophage and lymphocyte recruitment, angiogenesis, and fibroblast proliferation. Those within the osteochondral unit includes loss of chondrocytes in the superficial zone with proliferation in deeper zones; loss of extracellular matrix; vascularization and neuronal ingrowth across the tidemark between calcified and non-calcified cartilage; and remodelling of subchondral bone, resulting in sclerosis, cysts and osteophyte formation.
Figure 1.
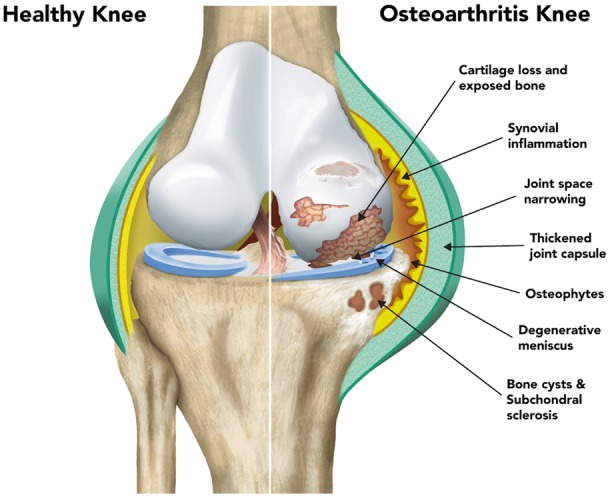
Illustration showing the key pathological features of osteoarthritis. The left side of the image shows the normal knee and the right side shows the diseased joint.
OA Is a Complex Genetic Disease
Both environmental and genetic factors play a role in the aetiology of OA, with genetic factors accounting for half of the variation in OA susceptibility (30). To date, and all within the last 10 years, 21 robustly established OA genetic loci have been reported (Fig. 2). With the exception of GDF5, which was originally identified by a candidate gene-based approach, the remaining loci were established by genome-wide association studies (GWAS). Primarily the studies were carried out in populations of European descent, with two studies performed in Asian populations and none in African. The majority of associations are joint-specific with differences in effect between end-stage and radiographic OA, as well as between males and females. Most of the variants are common (minor allele frequency >4%) with small to moderate effect sizes [largest odds ratio (OR) 1.79 for GDF5]. All of these characteristics typify the polygenic and complex genetic architecture underpinning OA.
Figure 2.

Established OA loci. Each locus is identified by the nearest gene(s) and coloured black to represent association in both sexes, blue for males only and red for females only. The discovery study population is of European descent unless indicated by an asterisk in which case the population is Asian (BTNL2 was identified in a combined Asian and European analysis).
A recent study of end-stage hip OA in Icelanders identified two rare variants located in the chondroadherin-like protein (CHADL) and cartilage oligomeric matrix protein (COMP) genes, with substantially larger effect sizes than previously seen (OR 7.7 and 16.7, respectively) (31). The COMP variant c.1141 G > C (allele frequency 0.026%) is a missense variant unique to the Icelandic population. COMP is a functional constituent and abundantly expressed in the extracellular matrix of cartilage (32), and has served as a serum biomarker for cartilage degradation (33,34). The CHADL variant rs532464664 (homozygote frequency 0.15%, recessive model association) is an insertion resulting in a frameshift and has been observed in other, mostly European populations. CHADL is expressed in cartilaginous tissue and is involved in fibrillogenesis and regulation of chondrocyte differentiation (35). Possession of these rare variants in COMP and CHADL significantly decreased the age at which total hip replacement was performed by 13.5 and 4.9 years, respectively.
Among the 21 established OA loci, several have been found to have potentially pleiotropic effects; GDF5 is also associated with height; FTO with body mass index (BMI) (with FTO exerting its effect on OA through BMI); and a recently established OA locus in SMAD3 with bone mineral density (BMD) (36). Furthermore, variants in astrotactin (ASTN2) are associated with total hip replacement in females (37) and also with migraine (38). ASTN2 is highly expressed in the developing brain and is involved with migrating neurones. Shared pathophysiological features between migraine and OA remain unclear, although it is noteworthy that the major symptom of OA is pain. Genome-wide linkage disequilibrium regression analyses can identify genetic correlations between OA and a wide range of complex physiological, molecular and behavioural traits, followed by formal Mendelian randomization approaches to determine the direction of effect.
Several of the established OA loci also have some translational potential. Variants in the carbohydrate (chondroitin 4) sulphotransferase 11 (CHST11) gene are associated with hip OA (37). The protein catalyses the transfer of sulphate groups in chondroitin sulphate, the principal proteoglycan in cartilage. Chondroitin sulphate can be taken as a nutritional supplement for OA and although numerous trials have been performed the clinical benefits and pain relief evidence is inconsistent (39). Variants in the parathyroid hormone-related protein (PTHLH) gene are associated with hip OA (37). PTHLH is involved in the regulation of endochondral bone development and its analogs are prescribed for osteoporosis because of their anabolic actions on bone formation (40). Subchondral bone remodelling is also a consistent feature in OA (17,18), and may provide a novel target for intervention in OA progression.
OA cannot be considered to be a single disease. The requirement for better phenotype definition and homogeneity in much larger sample sizes is a prerequisite for untangling the genetic complexity underlying pathogenesis and progression. Electronic health records provide an excellent opportunity as they provide the ability to study large sample sizes with a wealth of clinical information. Furthermore, longitudinal information can improve phenotype definition and depth, and coupled with large scale, can improve power.
Insights from Rare Musculoskeletal Diseases
Studying severe phenotypes of rare diseases can shed insights into the mechanisms underpinning more common disorders and identify potential therapeutic targets, e.g. the study of rare bone disorders contributed to the development of bisphosphonates (41,42). Similarly, the study of rare cartilage syndromes and developmental skeletal dysplasias can glean insights into important genes and potential targets for OA (43,44).
Pathway to Mechanism of Disease Progression
Unlike in most other common complex diseases, the relevant tissue for OA is readily accessible at joint replacement surgery. OA leads to changes in the joint tissue that can be captured macroscopically (Fig. 3). This provides an opportunity to study the key osteochondral unit components in order to identify signatures of disease progression. There has been great progress in understanding changes in the composition, functional properties and structure of the osteochondral unit during the evolution of OA (16,18), but it is important to also understand at the molecular level how interactions between cartilage and bone cells affect disease development.
Figure 3.

Intact and degraded cartilage-bone interface (osteochondral unit). (A) Cartilage/bone interface showing fibrillation and loss of articular cartilage (blue); (B) magnified image showing clustering of chondrocytes in the deeper cartilage layers; (C) magnified image of the cartilage/bone interface showing blood vessels invading the cartilage layer; (D) loss of proteoglycan indicated by loss of Alcian blue staining.
Recent advances have signalled the development of high-resolution, high-throughput genome-wide technologies for assessing genome function, including spatial transcription (45–47), chromatin accessibility (48) and 3-dimensional conformation (49). The epigenome, transcriptome and proteome are unique for each tissue and cell type. Although systematic cataloguing of molecular maps has been carried out for many tissues (50–52), the landscape of cell types relevant to OA largely remains unknown. The accessibility of relevant tissue at joint replacement surgery enables the deployment of multi-omics to dissect the molecular disease processes in the right cells. Functional genomics is a nascent, but emerging, field in OA (53). Previous work has compared intact and degraded cartilage in modest numbers of patients (median n = 20) (54), investigating genome-wide methylation (55–58), gene (57–60) and/or protein expression (58,61,62), primarily through microarray technologies. Despite overall limited replicability (due to design, technology and analysis differences), pathways such as WNT signalling, angiogenesis, immune response and matrix degradation have been implicated by more than one study (59–61,63–72). The molecular architecture of genome regulation underlying disease mechanisms in OA tissue, for example as elegantly demonstrated through the study of chromatin conformation in T and B-cells in rheumatoid arthritis (73,74), remains unclear.
Integration of Genetics and Genomic Information
Despite progress in identifying genomic regions that harbour OA-associated variants, we know very little about the specific genes involved or the way in which they mediate changes in the joint. Most of the established OA loci discovered to date (75) reside in non-coding sequence, making their biological interpretation challenging. Indeed, even though >80% of all published complex disease loci are found outside of protein-coding exons (76–78), we still have a very limited understanding of the way in which they act on disease pathogenesis (50,79–81). Identification of the causal variants and the genes they affect requires experimental analysis of genome regulation in the right cell type. Evidence is emerging from targeted studies using cell lines and patient samples that OA risk variants are likely to exert their effects on gene expression levels (82–87). Integration of genetic and genomic information is required to define molecular mechanisms through which the non-coding variants underlying association signals exert their effects on OA susceptibility (88).
Mechanistic Studies
The molecular mechanisms linking sequence variation to cellular functions remain poorly described. To date, the characterization of OA-associated variants has mainly focused on the effect of the variant on the candidate target gene expression. Only a small number of studies have investigated the cellular and molecular mechanisms played by the identified OA susceptibility genes during OA development.
One of the best functionally characterized OA susceptibility signals is rs143383, which negatively affects the activity of the GDF5 gene promoter, reducing the levels of GDF5 expression (82,83,89). GDF5, a member of the BMP family and TGF-beta superfamily, plays a key role in chondrogenesis during joint development (90) and also in postnatal joint tissue homeostasis. Mice deficient in GDF5 show severe joint damage, decreased subchondral bone density and abnormal arrangement of collagen fibres in the bone (91). Also, recent studies in human primary chondrocytes have shown that GDF5 stimulation reduces the expression of the matrix degrading enzymes MMP13 and ADAMTS4 (92), both implicated in cartilage extracellular matrix degradation in OA (93,94).
Other OA candidate genes studied in cellular or animal models are RUNX2 (rs12206662/rs10948155, associated with hip cartilage thickness and hip OA) (95) and DOT1L (rs1298744, rs11880992, associated with hip cartilage thickness and hip OA) (95–97). For example, knocking down the expression of Dot1l in the chondrogenic cell line ATDC5 results in reduced chondrogenesis differentiation accompanied of up-regulation of matrix metalloproteinase 9, reduced collagen content and less sulphated proteoglycans, suggesting a protective role of DOT1L in OA development (96). Knocking out Runx2 in articular chondrocytes in a mouse model of OA could rescue part of the cartilage degradation and subchondral sclerosis as well as reduce the expression of MMP13 (98).
Despite these recent advances, the mechanism of action of most OA susceptibility loci remains unknown. An important current effort to advance the systematic genetic, molecular and cellular functional study of genetic variation in OA is being carried out by the Origins of Bone and Cartilage Disease (OBCD) consortium, which carries out high-throughput musculoskeletal phenotyping of knockout mice (99). Cellular genetics and phenotyping models can further enhance our understanding of aetiopathogenesis. The combined use of human induced pluripotent stem cells (hiPSC) and genome editing technologies, such as the CRISPR-Cas9 (100) system, allows the targeted modification of the human genome and the generation of isogenic hiPSC lines that differ only at the specific gene or variant of interest (100–103). In the case of OA, the resulting hiPSC lines could then be differentiated towards chondrocytes (104,105), osteoblasts (106) or any other relevant cell type, creating a genetically controlled experimental model of OA. Such a model would allow the systematic study of the individual contribution of OA disease-associated variants to the disease molecular and cellular phenotype (Fig. 4). Furthermore, the possibility to derive hiPSCs from somatic cells from patients and differentiate them into disease-relevant cell types, for example chondrocytes, offers a unique opportunity to recapitulate human development and pathogenic processes.
Figure 4.

Functional characterization of loci associated with osteoarthritis. Schema showing an approach to functionally characterize OA-associated variants and candidate target genes using isogenic hiPSC derived chondrocytes (A) and patient hiPSC-derived chondrocytes carrying the selected variant (B).
Pathway to New Therapies for OA
Osteoarthritis is typically diagnosed on the basis of relevant clinical symptoms and signs, and corroborated with consistent radiographic features. No robust laboratory biomarkers exist for OA. Pharmacological therapies such as paracetamol and non-steroidal anti-inflammatory drugs can be effective in relieving pain but are incapable of reversing cartilage damage (107). In the absence of a curative therapy, management strategies currently focus on interventions early in the OA joint degeneration process and targeting disease progression (18), including emerging regenerative therapies that hold the potential to promote cartilage repair and ultimately restore the original tissue structure and function. A better understanding of the molecular processes underpinning OA in the joint will be crucial to inform and accelerate the success of this new generation of treatments.
Future Perspective
There are currently no approved disease-modifying treatements available for OA and treatment focusses on surgical replacement of the diseased joint. However, the field of OA genetics and genomics is currently witnessing an exciting alignment of opportunities to deploy a multi-pronged attack to solve the current therapeutic impasse. These approaches include massive-scale genetics with linkage to deep clinical phenotypes and patient-reported pain indices through electronic health records, access to primary disease tissue following joint replacement surgery for deep characterization of the local molecular landscape and biomarker discovery, high-throughput genome editing techniques in primary and hiPSC-derived chondrocytes to pin down causal genes, coupling joint imaging to genomics, leveraging lessons from rare musculoskeletal disorders, and emerging regenerative medicine approaches. Taken together, these technologies have the potential to alter the natural history of OA and improve the lives of these patients in the same way that the introduction of biologic treatments has changed the lives of patients with inflammatory arthritis.
Acknowledgements
The authors wish to thank Eleni Zengini and Dániel Süveges for their useful input.
Conflict of Interest statement. None declared.
Funding
Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust.
References
- 1. Vos T., Flaxman A.D., Naghavi M., Lozano R., Michaud C., Ezzati M., Shibuya K., Salomon J.A., Abdalla S., Aboyans V.. et al. (2012) Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet, 380, 2163–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murray C.J., Vos T., Lozano R., Naghavi M., Flaxman A.D., Michaud C., Ezzati M., Shibuya K., Salomon J.A., Abdalla S.. et al. (2012) Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet, 380, 2197–2223. [DOI] [PubMed] [Google Scholar]
- 3. Cross M., Smith E., Hoy D., Nolte S., Ackerman I., Fransen M., Bridgett L., Williams S., Guillemin F., Hill C.L.. et al. (2014) The global burden of hip and knee osteoarthritis: estimates from the global burden of disease 2010 study. Ann. Rheum. Dis., 73, 1323–1330. [DOI] [PubMed] [Google Scholar]
- 4. Kingsbury S.R., Gross H.J., Isherwood G., Conaghan P.G. (2014) Osteoarthritis in Europe: impact on health status, work productivity and use of pharmacotherapies in five European countries. Rheumatology (Oxford), 53, 937–947. [DOI] [PubMed] [Google Scholar]
- 5. Hilingsmann M., Reginster J.-Y. The economic weight of osteoarthritis in Europe http://www.medicographia.com/2013/10/the-economic-weight-of-osteoarthritis-in-europe/; date last accessed August 08, 2017.
- 6. Dieppe P.A., Lohmander L.S. (2005) Pathogenesis and management of pain in osteoarthritis. Lancet, 365, 965–973. [DOI] [PubMed] [Google Scholar]
- 7. Hiligsmann M., Cooper C., Arden N., Boers M., Branco J.C., Luisa Brandi M., Bruyère O., Guillemin F., Hochberg M.C., Hunter D.J.. et al. (2013) Health economics in the field of osteoarthritis: an expert's consensus paper from the European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis (ESCEO). Semin. Arthritis Rheum., 43, 303–313. [DOI] [PubMed] [Google Scholar]
- 8. Arthritis Research UK. Osteoarthritis in General Practice: Data and Perspectives. 2013.
- 9. Murphy L., Schwartz T.A., Helmick C.G., Renner J.B., Tudor G., Koch G., Dragomir A., Kalsbeek W.D., Luta G., Jordan J.M. (2008) Lifetime risk of symptomatic knee osteoarthritis. Arthritis Rheum., 59, 1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eurostat: Surgical operations and procedures performed in hospitals. http://appsso.eurostat.ec.europa.eu/nui/show.do?dataset=hlth_co_proc2&lang=en; date last accessed August 08, 2017.
- 11. Robertsson O., Dunbar M., Pehrsson T., Knutson K., Lidgren L. (2000) Patient satisfaction after knee arthroplasty: a report on 27, 372 knees operated on between 1981 and 1995 in Sweden. Acta Orthop. Scand., 71, 262–267. [DOI] [PubMed] [Google Scholar]
- 12. Baker P.N., van der Meulen J.H., Lewsey J., Gregg P.J. (2007) National Joint Registry for E, Wales. The role of pain and function in determining patient satisfaction after total knee replacement. Data from the National Joint Registry for England and Wales. J. Bone Joint Surg. Br., 89, 893–900. [DOI] [PubMed] [Google Scholar]
- 13. Baker P.N., Deehan D.J., Lees D., Jameson S., Avery P.J., Gregg P.J., Reed M.R. (2012) The effect of surgical factors on early patient-reported outcome measures (PROMS) following total knee replacement. J. Bone Joint Surg. Br., 94, 1058–1066. [DOI] [PubMed] [Google Scholar]
- 14. Hunter D.J. (2011) Pharmacologic therapy for osteoarthritis–the era of disease modification. Nat. Rev. Rheumatol., 7, 13–22. [DOI] [PubMed] [Google Scholar]
- 15. Scanzello C.R., Goldring S.R. (2012) The role of synovitis in osteoarthritis pathogenesis. Bone, 51, 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goldring S.R., Goldring M.B. (2016) Changes in the osteochondral unit during osteoarthritis: structure, function and cartilage-bone crosstalk. Nat. Rev. Rheumatol., 12, 632–644. [DOI] [PubMed] [Google Scholar]
- 17. Man G.S., Mologhianu G. (2014) Osteoarthritis pathogenesis - a complex process that involves the entire joint. J. Med. Life, 7, 37–41. [PMC free article] [PubMed] [Google Scholar]
- 18. Glyn-Jones S., Palmer A.J., Agricola R., Price A.J., Vincent T.L., Weinans H., Carr A.J.R. (2015) Osteoarthritis. Lancet, 386, 376–387. [DOI] [PubMed] [Google Scholar]
- 19. Felson D.T., Naimark A., Anderson J., Kazis L., Castelli W., Meenan R.F. (1987) The prevalence of knee osteoarthritis in the elderly. The Framingham Osteoarthritis Study. Arthritis. Rheum., 30, 914–918. [DOI] [PubMed] [Google Scholar]
- 20. Dillon C.F., Rasch E.K., Gu Q., Hirsch R. (2006) Prevalence of knee osteoarthritis in the United States: arthritis data from the Third National Health and Nutrition Examination Survey 1991-94. J. Rheumatol., 33, 2271–2279. [PubMed] [Google Scholar]
- 21. van Saase J.L., van Romunde L.K., Cats A., Vandenbroucke J.P., Valkenburg H.A. (1989) Epidemiology of osteoarthritis: Zoetermeer survey. Comparison of radiological osteoarthritis in a Dutch population with that in 10 other populations. Ann. Rheum. Dis., 48, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oliveria S.A., Felson D.T., Reed J.I., Cirillo P.A., Walker A.M. (1995) Incidence of symptomatic hand, hip, and knee osteoarthritis among patients in a health maintenance organization. Arthritis. Rheum., 38, 1134–1141. [DOI] [PubMed] [Google Scholar]
- 23. Felson D.T., Zhang Y., Hannan M.T., Naimark A., Weissman B., Aliabadi P., Levy D. (1997) Risk factors for incident radiographic knee osteoarthritis in the elderly: the Framingham Study. Arthritis Rheum., 40, 728–733. [DOI] [PubMed] [Google Scholar]
- 24. Anderson J.J., Felson D.T. (1988) Factors associated with osteoarthritis of the knee in the first national Health and Nutrition Examination Survey (HANES I). Evidence for an association with overweight, race, and physical demands. Am. J. Epidemiol., 128, 179–189. [DOI] [PubMed] [Google Scholar]
- 25. Cooper C., Snow S., McAlindon T.E., Kellingray S., Stuart B., Coggon D., Dieppe P.A. (2000) Risk factors for the incidence and progression of radiographic knee osteoarthritis. Arthritis. Rheum., 43, 995–1000. [DOI] [PubMed] [Google Scholar]
- 26. Blagojevic M., Jinks C., Jeffery A., Jordan K.P. (2010) Risk factors for onset of osteoarthritis of the knee in older adults: a systematic review and meta-analysis. Osteoarthr. Cartil., 18, 24–33. [DOI] [PubMed] [Google Scholar]
- 27. Spector T.D., Cicuttini F., Baker J., Loughlin J., Hart D. (1996) Genetic influences on osteoarthritis in women: a twin study [see comments]. BMJ, 312, 940–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Spector T.D., MacGregor A.J. (2004) Risk factors for osteoarthritis: genetics. Osteoarthr. Cartil., 12 Suppl A, S39–S44. [DOI] [PubMed] [Google Scholar]
- 29. MacGregor A.J., Li Q., Spector T.D., Williams F.M. (2009) The genetic influence on radiographic osteoarthritis is site specific at the hand, hip and knee. Rheumatology (Oxford), 48, 277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Valdes A.M., Spector T.D. (2011) Genetic epidemiology of hip and knee osteoarthritis. Nat. Rev. Rheumatol., 7, 23–32. [DOI] [PubMed] [Google Scholar]
- 31. Styrkarsdottir U., Helgason H., Sigurdsson A., Norddahl G.L., Agustsdottir A.B., Reynard L.N., Villalvilla A., Halldorsson G.H., Jonasdottir A., Magnusdottir A.. et al. (2017) Whole-genome sequencing identifies rare genotypes in COMP and CHADL associated with high risk of hip osteoarthritis. Nat. Genet., 49, 801–805. [DOI] [PubMed] [Google Scholar]
- 32. Acharya C., Yik J.H., Kishore A., Van Dinh V., Di Cesare P.E., Haudenschild D.R. (2014) Cartilage oligomeric matrix protein and its binding partners in the cartilage extracellular matrix: interaction, regulation and role in chondrogenesis. Matrix Biol., 37, 102–111. [DOI] [PubMed] [Google Scholar]
- 33. Das B.R., Roy A., Khan F.R. (2015) Cartilage oligomeric matrix protein in monitoring and prognostication of osteoarthritis and its utility in drug development. Perspect. Clin. Res., 6, 4–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Verma P., Dalal K. (2013) Serum cartilage oligomeric matrix protein (COMP) in knee osteoarthritis: a novel diagnostic and prognostic biomarker. J. Orthop. Res., 31, 999–1006. [DOI] [PubMed] [Google Scholar]
- 35. Tillgren V., Ho J.C., Onnerfjord P., Kalamajski S. (2015) The novel small leucine-rich protein chondroadherin-like (CHADL) is expressed in cartilage and modulates chondrocyte differentiation. J. Biol. Chem., 290, 918–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hackinger S., Trajanoska K., Styrkarsdottir U., Zengini E., Steinberg S., Ritchie G.R.S., Hatzikotoulas K., Gilly A., Evangelou E., Kemp J.P.. et al. (2017) Evaluation of shared genetic aetiology between osteoarthritis and bone mineral density identifies SMAD3 as a novel osteoarthritis risk locus. Hum. Mol. Genet, 26, 3850–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zeggini E., Panoutsopoulou K., Southam L., Rayner N.W., Day-Williams A.G., Lopes M.C., Boraska V., Esko T., Evangelou E., Hoffman A., arcOGEN Consortium, arcOgen Collaborators. et al. (2012) Identification of new susceptibility loci for osteoarthritis (arcOGEN): a genome-wide association study. Lancet, 380, 815–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Freilinger T., Anttila V., de Vries B., Malik R., Kallela M., Terwindt G.M., Pozo-Rosich P., Winsvold B., Nyholt D.R., van Oosterhout W.P.. et al. (2012) Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat. Genet., 44, 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Uebelhart D. (2008) Clinical review of chondroitin sulfate in osteoarthritis. Osteoarthritis Cartilage, 16 Suppl 3, S19–S21. [DOI] [PubMed] [Google Scholar]
- 40. Esbrit P., Herrera S., Portal-Nunez S., Nogues X., Diez-Perez A. (2016) Parathyroid hormone-related protein analogs as osteoporosis therapies. Calcif. Tissue Int., 98, 359–369. [DOI] [PubMed] [Google Scholar]
- 41. Bonjour J.P., Fleisch H.A. (1973) Diphosphonates and vitamin-D metabolism in Paget's disease. Lancet, 2, 375–376. [DOI] [PubMed] [Google Scholar]
- 42. Fleisch H., Bisaz S. (1962) Mechanism of calcification: inhibitory role of pyrophosphate. Nature, 195, 911. [DOI] [PubMed] [Google Scholar]
- 43. Thomas J.T., Lin K., Nandedkar M., Camargo M., Cervenka J., Luyten F.P. (1996) A human chondrodysplasia due to a mutation in a TGF-beta superfamily member. Nat. Genet., 12, 315–317. [DOI] [PubMed] [Google Scholar]
- 44. Rouault K., Scotet V., Autret S., Gaucher F., Dubrana F., Tanguy D., El Rassi C.Y., Fenoll B., Férec C. (2010) Evidence of association between GDF5 polymorphisms and congenital dislocation of the hip in a Caucasian population. Osteoarthr. Cartil., 18, 1144–1149. [DOI] [PubMed] [Google Scholar]
- 45. Stahl P.L., Salmen F., Vickovic S., Lundmark A., Navarro J.F., Magnusson J., Giacomello S., Asp M., Westholm J.O., Huss M.. et al. (2016) Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science, 353, 78–82. [DOI] [PubMed] [Google Scholar]
- 46. Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods, 5, 621–628. [DOI] [PubMed] [Google Scholar]
- 47. Nagalakshmi U., Wang Z., Waern K., Shou C., Raha D., Gerstein M., Snyder M. (2008) The transcriptional landscape of the yeast genome defined by RNA sequencing. Science, 320, 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods, 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dekker J., Marti-Renom M.A., Mirny L.A. (2013) Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat. Rev. Genet., 14, 390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Encode Project Consortium(2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stunnenberg H.G., International Human Epigenome, C. Hirst M. (2016) The International Human Epigenome Consortium: A Blueprint for Scientific Collaboration and Discovery. Cell, 167, 1145–1149. [DOI] [PubMed] [Google Scholar]
- 52. Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M.J.. et al. (2015) Integrative analysis of 111 reference human epigenomes. Nature, 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Meulenbelt I.M., Bhutani N., den Hollander W., Gay S., Oppermann U., Reynard L.N., Skelton A.J., Young D.A., Beier F., Loughlin J. (2017) The first international workshop on the epigenetics of osteoarthritis. Connect. Tissue Res., 58, 37–48. [DOI] [PubMed] [Google Scholar]
- 54. Steinberg J., Zeggini E. (2016) Functional Genomics in Osteoarthritis: Past, Present, and Future. J. Orthop. Res., 34, 1105–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jeffries M.A., Donica M., Baker L.W., Stevenson M.E., Annan A.C., Humphrey M.B., James J.A., Sawalha A.H. (2014) Genome-wide DNA methylation study identifies significant epigenomic changes in osteoarthritic cartilage. Arthritis Rheumatol., 66, 2804–2815. [DOI] [PubMed] [Google Scholar]
- 56. Moazedi-Fuerst F.C., Hofner M., Gruber G., Weinhaeusel A., Stradner M.H., Angerer H., Peischler D., Lohberger B., Glehr M., Leithner A.. et al. (2014) Epigenetic differences in human cartilage between mild and severe OA. J. Orthop. Res., 32, 1636–1645. [DOI] [PubMed] [Google Scholar]
- 57. den Hollander W., Ramos Y.F., Bos S.D., Bomer N., van der Breggen R., Lakenberg N., de Dijcker W.J., Duijnisveld B.J., Slagboom P.E., Nelissen R.G.. et al. (2014) Knee and hip articular cartilage have distinct epigenomic landscapes: implications for future cartilage regeneration approaches. Ann. Rheum. Dis., 73, 2208–2212. [DOI] [PubMed] [Google Scholar]
- 58. Steinberg J., Ritchie G.R.S., Roumeliotis T.I., Jayasuriya R.L., Brooks R.A., Binch A.L.A., Shah K.M., Coyle R., Pardo M., Le Maitre C.L.. et al. Integrative epigenomics, transcriptomics and proteomics of patient chondrocytes reveal genes and pathways involved in osteoarthritis. bioRxiv2016, doi: https://doi.org/10.1101/038067. [DOI] [PMC free article] [PubMed]
- 59. Karlsson C., Dehne T., Lindahl A., Brittberg M., Pruss A., Sittinger M., Ringe J. (2010) Genome-wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthr. Cartil., 18, 581–592. [DOI] [PubMed] [Google Scholar]
- 60. Tew S.R., McDermott B.T., Fentem R.B., Peffers M.J., Clegg P.D. (2014) Transcriptome-wide analysis of messenger RNA decay in normal and osteoarthritic human articular chondrocytes. Arthritis Rheumatol., 66, 3052–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stenberg J., Rüetschi U., Skiöldebrand E., Kärrholm J., Lindahl A. (2013) Quantitative proteomics reveals regulatory differences in the chondrocyte secretome from human medial and lateral femoral condyles in osteoarthritic patients. Proteome Sci., 11, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ruiz-Romero C., Fernandez-Puente P., Calamia V., Blanco F.J. (2015) Lessons from the proteomic study of osteoarthritis. Expert Rev. Proteomics, 12, 433–443. [DOI] [PubMed] [Google Scholar]
- 63. Chou C.H., Lee M.T., Song I.W., Lu L.S., Shen H.C., Lee C.H., Wu J.Y., Chen Y.T., Kraus V.B., Wu C.C. (2015) Insights into osteoarthritis progression revealed by analyses of both knee tibiofemoral compartments. Osteoarthr. Cartil., 23, 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Delgado-Calle J., Fernández A.F., Sainz J., Zarrabeitia M.T., Sañudo C., García-Renedo R., Pérez-Núñez M.I., García-Ibarbia C., Fraga M.F., Riancho J.A. (2013) Genome-wide profiling of bone reveals differentially methylated regions in osteoporosis and osteoarthritis. Arthritis Rheumatol., 65, 197–205. [DOI] [PubMed] [Google Scholar]
- 65. Fernández-Tajes J., Soto-Hermida A., Vázquez-Mosquera M.E., Cortés-Pereira E., Mosquera A., Fernández-Moreno M., Oreiro N., Fernández-López C., Fernández J.L., Rego-Pérez I., Blanco F.J. (2014) Genome-wide DNA methylation analysis of articular chondrocytes reveals a cluster of osteoarthritic patients. Ann. Rheum. Dis., 73, 668–677. [DOI] [PubMed] [Google Scholar]
- 66. Gobezie R., Kho A., Krastins B., Sarracino D.A., Thornhill T.S., Chase M., Millett P.J., Lee D.M. (2007) High abundance synovial fluid proteome: distinct profiles in health and osteoarthritis. Arthritis Res. Ther., 9, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hopwood B., Tsykin A., Findlay D.M., Fazzalari N.L. (2007) Microarray gene expression profiling of osteoarthritic bone suggests altered bone remodelling, WNT and transforming growth factor-beta/bone morphogenic protein signalling. Arthritis Res. Ther., 9, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lamas J.R., Rodríguez-Rodríguez L., Vigo A.G., Alvarez-Lafuente R., López-Romero P., Marco F., Camafeita E., Dopazo A., Callejas S., Villafuertes E.. et al. (2010) Large-scale gene expression in bone marrow mesenchymal stem cells: a putative role for COL10A1 in osteoarthritis. Ann. Rheum. Dis., 69, 1880–1885. [DOI] [PubMed] [Google Scholar]
- 69. Lambert C., Dubuc J.E., Montell E., Vergés J., Munaut C., Noël A., Henrotin Y. (2014) Gene expression pattern of cells from inflamed and normal areas of osteoarthritis synovial membrane. Arthritis Rheumatol., 66, 960–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ritter S.Y., Subbaiah R., Bebek G., Crish J., Scanzello C.R., Krastins B., Sarracino D., Lopez M.F., Crow M.K., Aigner T.. et al. (2013) Proteomic analysis of synovial fluid from the osteoarthritic knee: comparison with transcriptome analyses of joint tissues. Arthritis Rheumatol., 65, 981–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rushton M.D., Reynard L.N., Barter M.J., Refaie R., Rankin K.S., Young D.A., Loughlin J. (2014) Characterization of the cartilage DNA methylome in knee and hip osteoarthritis. Arthritis Rheumatol., 66, 2450–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xu Y., Barter M.J., Swan D.C., Rankin K.S., Rowan A.D., Santibanez-Koref M., Loughlin J., Young D.A. (2012) Identification of the pathogenic pathways in osteoarthritic hip cartilage: commonality and discord between hip and knee OA. Osteoarthr. Cartil., 20, 1029–1038. [DOI] [PubMed] [Google Scholar]
- 73. Martin P., McGovern A., Orozco G., Duffus K., Yarwood A., Schoenfelder S., Cooper N.J., Barton A., Wallace C., Fraser P.. et al. (2015) Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci. Nat. Commun., 6, 10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. McGovern A., Schoenfelder S., Martin P., Massey J., Duffus K., Plant D., Yarwood A., Pratt A.G., Anderson A.E., Isaacs J.D.. et al. (2016) Capture Hi-C identifies a novel causal gene, IL20RA, in the pan-autoimmune genetic susceptibility region 6q23. Genome Biol., 17, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zengini E., Finan C., Wilkinson J.M. (2016) The genetic epidemiological landscape of hip and knee osteoarthritis: where are we now and where are we going?. J. Rheumatol., 43, 260–266. [DOI] [PubMed] [Google Scholar]
- 76. Pickrell J.K. (2014) Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am. J. Hum. Genet., 94, 559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L.. et al. (2014) The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res., 42, D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U.S.A, 106, 9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. McVicker G., van de Geijn B., Degner J.F., Cain C.E., Banovich N.E., Raj A., Lewellen N., Myrthil M., Gilad Y., Pritchard J.K. (2013) Identification of genetic variants that affect histone modifications in human cells. Science, 342, 747–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M.. et al. (2011) Mapping and analysis of chromatin state dynamics in nine human cell types. Nature, 473, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Schaub M.A., Boyle A.P., Kundaje A., Batzoglou S., Snyder M. (2012) Linking disease associations with regulatory information in the human genome. Genome Res., 22, 1748–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Miyamoto Y., Mabuchi A., Shi D., Kubo T., Takatori Y., Saito S., Fujioka M., Sudo A., Uchida A., Yamamoto S.. et al. (2007) A functional polymorphism in the 5' UTR of GDF5 is associated with susceptibility to osteoarthritis. Nat. Genet., 39, 529–533. [DOI] [PubMed] [Google Scholar]
- 83. Reynard L.N., Bui C., Canty-Laird E.G., Young D.A., Loughlin J. (2011) Expression of the osteoarthritis-associated gene GDF5 is modulated epigenetically by DNA methylation. Hum. Mol. Genet., 20, 3450–3460. [DOI] [PubMed] [Google Scholar]
- 84. Syddall C.M., Reynard L.N., Young D.A., Loughlin J. (2013) The identification of trans-acting factors that regulate the expression of GDF5 via the osteoarthritis susceptibility SNP rs143383. PLoS Genet., 9, e1003557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bos S.D., Bovée J.V., Duijnisveld B.J., Raine E.V., van Dalen W.J., Ramos Y.F., van der Breggen R., Nelissen R.G., Slagboom P.E., Loughlin J.. et al. (2012) Increased type II deiodinase protein in OA-affected cartilage and allelic imbalance of OA risk polymorphism rs225014 at DIO2 in human OA joint tissues. Ann. Rheum. Dis., 71, 1254–1258. [DOI] [PubMed] [Google Scholar]
- 86. Egli R.J., Southam L., Wilkins J.M., Lorenzen I., Pombo-Suarez M., Gonzalez A., Carr A., Chapman K., Loughlin J. (2009) Functional analysis of the osteoarthritis susceptibility-associated GDF5 regulatory polymorphism. Arthritis Rheum., 60, 2055–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Styrkarsdottir U., Thorleifsson G., Helgadottir H.T., Bomer N., Metrustry S., Bierma-Zeinstra S., Strijbosch A.M., Evangelou E., Hart D., Beekman M.. et al. (2014) Severe osteoarthritis of the hand associates with common variants within the ALDH1A2 gene and with rare variants at 1p31. Nat. Genet., 46, 498–502. [DOI] [PubMed] [Google Scholar]
- 88. Gaulton K.J., Ferreira T., Lee Y., Raimondo A., Mägi R., Reschen M.E., Mahajan A., Locke A., Rayner N.W., Robertson N.. et al. (2015) Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat. Genet., 47, 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Southam L., Rodriguez-Lopez J., Wilkins J.M., Pombo-Suarez M., Snelling S., Gomez-Reino J.J., Chapman K., Gonzalez A., Loughlin J. (2007) An SNP in the 5'-UTR of GDF5 is associated with osteoarthritis susceptibility in Europeans and with in vivo differences in allelic expression in articular cartilage. Hum. Mol. Genet., 16, 2226–2232. [DOI] [PubMed] [Google Scholar]
- 90. Francis-West P.H., Abdelfattah A., Chen P., Allen C., Parish J., Ladher R., Allen S., MacPherson S., Luyten F.P., Archer C.W. (1999) Mechanisms of GDF-5 action during skeletal development. Development, 126, 1305–1315. [DOI] [PubMed] [Google Scholar]
- 91. Daans M., Luyten F.P., Lories R.J. (2011) GDF5 deficiency in mice is associated with instability-driven joint damage, gait and subchondral bone changes. Ann. Rheum. Dis., 70, 208–213. [DOI] [PubMed] [Google Scholar]
- 92. Enochson L., Stenberg J., Brittberg M., Lindahl A. (2014) GDF5 reduces MMP13 expression in human chondrocytes via DKK1 mediated canonical Wnt signaling inhibition. Osteoarthr. Cartil., 22, 566–577. [DOI] [PubMed] [Google Scholar]
- 93. Song R.H., Tortorella M.D., Malfait A.M., Alston J.T., Yang Z., Arner E.C., Griggs D.W. (2007) Aggrecan degradation in human articular cartilage explants is mediated by both ADAMTS-4 and ADAMTS-5. Arthritis Rheum., 56, 575–585. [DOI] [PubMed] [Google Scholar]
- 94. Tetlow L.C., Adlam D.J., Woolley D.E. (2001) Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum., 44, 585–594. [DOI] [PubMed] [Google Scholar]
- 95. Castaño-Betancourt M.C., Evans D.S., Ramos Y.F., Boer C.G., Metrustry S., Liu Y., den Hollander W., van Rooij J., Kraus V.B., Yau M.S.. et al. (2016) Novel Genetic Variants for Cartilage Thickness and Hip Osteoarthritis. PLoS Genet., 12, e1006260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Castaño Betancourt M.C., Cailotto F., Kerkhof H.J., Cornelis F.M., Doherty S.A., Hart D.J., Hofman A., Luyten F.P., Maciewicz R.A., Mangino M.. et al. (2012) Genome-wide association and functional studies identify the DOT1L gene to be involved in cartilage thickness and hip osteoarthritis. Proc. Natl. Acad. Sci. U.S.A, 109, 8218–8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Evangelou E., Valdes A.M., Castano-Betancourt M.C., Doherty M., Doherty S., Esko T., Ingvarsson T., Ioannidis J.P., Kloppenburg M., Metspalu A.. et al. (2013) The DOT1L rs12982744 polymorphism is associated with osteoarthritis of the hip with genome-wide statistical significance in males. Ann. Rheum. Dis., 72, 1264–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Liao L., Zhang S., Gu J., Takarada T., Yoneda Y., Huang J., Zhao L., Oh C.D., Li J., Wang B.. et al. (2017) Deletion of Runx2 in articular chondrocytes decelerates the progression of DMM-induced osteoarthritis in adult mice. Sci. Rep., 7, 2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Origins of Bone and Cartilage Disease consortium. http://www.boneandcartilage.com/. I; date last accessed August 08, 2017.
- 100. Ding Q., Regan S.N., Xia Y., Oostrom L.A., Cowan C.A., Musunuru K. (2013) Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell, 12, 393–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A.. et al. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science, 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jinek M., East A., Cheng A., Lin S., Ma E., Doudna J. (2013) RNA-programmed genome editing in human cells. Elife, 2, e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. (2013) RNA-guided human genome engineering via Cas9. Science, 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Cheng A., Kapacee Z., Peng J., Lu S., Lucas R.J., Hardingham T.E., Kimber S.J. (2014) Cartilage repair using human embryonic stem cell-derived chondroprogenitors. Stem Cells Transl. Med, 3, 1287–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Craft A.M., Rockel J.S., Nartiss Y., Kandel R.A., Alman B.A., Keller G.M. (2015) Generation of articular chondrocytes from human pluripotent stem cells. Nat. Biotechnol., 33, 638–645. [DOI] [PubMed] [Google Scholar]
- 106. Phillips M.D., Kuznetsov S.A., Cherman N., Park K., Chen K.G., McClendon B.N., Hamilton R.S., McKay R.D., Chenoweth J.G., Mallon B.S.. et al. (2014) Directed differentiation of human induced pluripotent stem cells toward bone and cartilage: in vitro versus in vivo assays. Stem Cells Transl. Med., 3, 867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zhang W., Ouyang H., Dass C.R., Xu J. (2016) Current research on pharmacologic and regenerative therapies for osteoarthritis. Bone Res., 4, 15040. [DOI] [PMC free article] [PubMed] [Google Scholar]