

Abstract
Rationale
Oxidation of 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine generates a group of bioactive oxidized phospholipid products (OxPAPC) with a broad range of biological activities. Barrier-enhancing and anti-inflammatory effects of OxPAPC on pulmonary endothelial cells (EC) are critical for prevention of acute lung injury caused by bacterial pathogens or excessive mechanical ventilation. Anti-inflammatory properties of OxPAPC are associated with its antagonistic effects on toll-like receptors and suppression of RhoA GTPase signaling.
Objective
Because OxPAPC exhibits long lasting anti-inflammatory and lung-protective effects even after single administration in vivo, we tested the hypothesis that these effects may be mediated by additional mechanisms, such as OxPAPC-dependent production of anti-inflammatory and pro-resolving lipid mediator, lipoxin A4 (LXA4).
Methods and Results
Mass spectrometry and ELISA assays detected significant accumulation of LXA4 in the lungs of OxPAPC-treated mice and in conditioned medium of OxPAPC-exposed pulmonary EC. Administration of LXA4 reproduced anti-inflammatory effect of OxPAPC against TNFα in vitro and in the animal model of LPS-induced lung injury. The potent barrier protective and anti-inflammatory effects of OxPAPC against TNFα and LPS challenge were suppressed in human pulmonary EC with siRNA-induced knockdown of LXA4 formyl-peptide receptor-2 (FPR2/ALX) and in mFPR2−/− mice lacking the mouse homolog of human FPR2/ALX.
Conclusions
This is the first demonstration that inflammation- and injury-associated phospholipid oxidation triggers production of anti-inflammatory and pro-resolution molecules such as LXA4. This lipid mediator switch represents a novel mechanism of OxPAPC-assisted recovery of inflamed lung endothelium.
Keywords: Oxidized phospholipids, lipoxin A4, inflammation, endothelial cells, lung injury, vascular biology, pulmonary circulation
Subject Terms: Basic Science Research, Cell signaling/Signal Transduction, Endothelium/Vascular Type/Nitrix Oxide, Inflammation, Pulmonary Biology
INTRODUCTION
The vascular endothelium forms a selective permeable barrier and participates in the regulation of macromolecule transport and blood cell trafficking through the vessel wall. Control of vascular endothelial cell (EC) barrier is achieved via a balance of barrier disruptive and barrier protective signals from circulating bioactive molecules, bacterial and viral pathogens, mechanical microenvironment, and other factors.
Circulating and tissue levels of oxidized phospholipids may become rapidly elevated in a variety of pathological conditions accompanied by oxidative stress including autoimmune diseases, lung injury, and sepsis1. Inflammation-induced generation of oxidants leads to formation of fatty acid hydroperoxides and further accumulation of fragmented and full length oxidized products in cell membranes and lipoproteins. One of the major plasma membrane phospholipids containing polyunsaturated fatty acids (PUFA) is 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (PAPC). Generation of OxPAPC is a result of oxidation of sn-2 unsaturated groups in arachidonic acid contained in the PAPC structure, which leads to formation of full length and fragmented OxPAPC species1, 2.
OxPAPC enriched in full length oxygenated fatty acid residues causes sustained EC barrier-enhancing effect mediated by activation of Rap1 and Rac1 GTPases, remodeling of cortical actin cytoskeleton, and enhancement of EC junctions3–5. OxPAPC also protects against EC barrier dysfunction and vascular leak caused by edemagenic agonists or excessive mechanical stretch via Rap1/Rac1-dependent downregulation of barrier-disruptive RhoA signaling3, 5–7.
In acute settings, OxPAPC also exhibits potent anti-inflammatory effects. In the model of lipopolysaccharide (LPS)-induced acute lung injury OxPAPC decreases inflammatory cell recruitment and cytokine production in the lungs6, 8, 9 and even protects against LPS-mediated lethal shock by blocking the LPS-TLR4 inflammatory cascade10. Additional anti-inflammatory mechanism of OxPAPC is inhibition of RhoA-dependent stimlation of p38 MAPK and NFkB inflammatory pathways11. As a result, OxPAPC-induced inhibition of TLR-NFkB inflammatory cascade and potent barrier enhancing effect preventing vascular leak and lung infiltration of inflammatory cells leads to immediate blockade of inflammatory response in the lung8, 9. A remarkable feature of OxPAPC anti-inflammatory action is its sustained effect in vitro and in vivo, even in the setting of OxPAPC post-treatment11. These observations suggest a possible involvement of other mechanisms contributing to OxPAPC anti-inflammatory properties.
Progression of inflammatory process is associated with a phenomenon called “lipid program switch”, where production of pro-inflammatory lipid molecules (thromboxanes, leukotriens, etc.) in the early phase of inflammation is switched to production of lipid mediators with anti-inflammatory properties (lipoxins and resolvins) which suppress inflammation and promote resolution phase12–14. Lipoxin A4 (LXA4) belongs to a group of lipid mediators generated later in the course of ALI, and contributing to resolution of inflammation12, 15.
Formyl peptide receptors (FPRs) are G protein-coupled receptors known to be important in host defense and inflammation. The three human FPRs (FPR1, FPR2/ALX, and FPR3) share significant sequence homology and are encoded by clustered genes16. These receptors bind a structurally diverse group of N-formyl and nonformyl peptides of bacterial and mitochondrial origin wit N-formylmethionine as the only ligand class common to all three human receptors. High affinity binding of LXA4 to FPR2/ALX17 defines its anti-inflammatory and pro-resolution effects in various models of innate immune response18–20.
The current study tested the hypothesis that pronounced and sustained anti-inflammatory effects of OxPAPC may involve induction of LXA4 production by pulmonary EC. The role of this mechanism was investigated using biochemical, and molecular approaches in cell and animal models including genetic model of lipoxin receptor mFPR2−/− mice.
METHODS
Reagents and cell culture
1-Palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (PAPC) and oxidation-resistant 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) were obtained from Avanti Polar Lipids, Alabaster, AL. PAPC was oxidized by exposure of dry lipid to air as previously described21–23. The extent of oxidation was monitored by positive ion electrospray mass spectrometry (ESI-MS) as described previously23. Lipoxin A4 (LXA4) was obtained from Cayman Chemical (Ann Arbor, MI). Human TNFα was from R&D Systems (Minneapolis, MN). FPR inhibitory peptides BocFLFLF and WRW4 were obtained from Abbiotec (San Diego, CA) and Tocris Bioscience (Minneapolis, MN), respectively. Inhibitors of 15-lipoxygenase PD 146176, and 5-lipoxygenase BW-B 70C were from Tocris Bioscience; sPLA2 inhibitor CAS 393569-31-8 were from Cayman Chemical. All reagents for immunofluorescence were obtained from Molecular Probes (Eugene, OR). Antibodies against di-phospho (Thr18/Ser19) myosin light chains (MLC) and IkBa were from Cell Signaling (Beverly, MA); antibodies to VE-cadherin, ICAM1, VCAM1 were from Santa Cruz Biotechnology (Santa Cruz, CA). Unless specified, biochemical reagents including LPS from Escherichia coli O55:B5 were obtained from Sigma (St. Louis, MO). Human pulmonary artery endothelial cells (HPAEC) and cell culture basal medium with growth supplements were obtained from Lonza Inc (Allendale, NJ), cultured according to the manufacturer’s protocol, and used at passages 5–7.
Measurements of endothelial monolayer permeability
The cellular barrier properties were analyzed by measurements of transendothelial electrical resistance (TER) across confluent human pulmonary artery endothelial monolayers using an electrical cell-substrate impedance sensing system (Applied Biophysics, Troy, NY) as previously described24, 25. Analysis of EC permeability for macromolecules was performed using Vascular Permeability Imaging Assay (XPerT) (Millipore, Bellerica, MA) as described elsewhere26.
Immunofluorescence staining
After agonist treatment endothelial cells grown on glass coverslips were fixed in PBS containing 3.7% formaldehyde, and F-actin was visualized by immunofluorescence staining of cell monolayers with Texas Red conjugated phalloidin. Adherens junction were labeled with antibodies against VE-cadherin, as previously described27, 28.
siRNA transfection of EC cultures
Pulmonary EC were treated with gene-specific siRNA duplexes. Pre-designed human Stealth™ Select siRNA sets of standard purity were ordered from Invitrogen (Carlsbad, CA) and transfection of EC with siRNA was performed as previously described29. The siRNA transfection efficiency according to our protocol exceeded 90%30.
Quantitative reverse-transcription polymerase chain reaction (qRT-PCR)
Analysis of FPR1, FPR2/ALX, FPR3, 15-LO, and ICAM1 mRNA expression by human pulmonary EC was performed by quantitative real-time RT-PCR. Gene expression fold changes were calculated according to the ΔΔCt method31.
Immunoblotting
Western Blot analysis was performed as described elsewhere32. Protein extracts were separated by SDS-PAGE, transferred to polyvinylidene difluoride membranes, and the membranes were incubated with antibodies of interest. Equal protein loading was verified by probing of membranes with antibody to β-tubulin.
ELISA analysis
Concentrations of tumor necrosis factor-α (TNFα) and soluble ICAM1 (sICAM1) in control and treated cell conditioned medium samples were measured using ELISA kit available from R&D Systems (Minneapolis, MN) according to manufacturer’s instructions. LXA4 detection in EC conditioned medium and lung tissue samples was performed after SPE extraction using ELISA kit from Oxford Biomedical Research (Oxford, MI, Cat# EA45). Absorption was measured using a 2030 Multylabel Reader Victor X5 (Perkin Elmer).
Animal studies
All experimental protocols involving the use of animals were approved by the University of Chicago Institutional Animal Care & Use Committee for the humane treatment of experimental animals. mFPR2−/− mice have been previously described35. C57Bl6 mice used in this study were anesthetized with an intraperitoneal injection of ketamine (75 mg/kg) and acepromazine (1.5 mg/kg). Bacterial lipopolysaccharide (LPS, 0.7 mg/kg body weight; Escherichia coli O55:B5) was injected intratracheally in a small volume (20–30 μl). OxPAPC (1.5 mg/kg) or sterile saline solutions were administrated 5 hrs after LPS instillation by intravenous injection in the external jugular vein. Animals were sacrificed by exsanguination under anesthesia 24 hrs after LPS challenge and used for evaluation of lung injury parameters.
Evaluation of lung injury parameters
Collection of bronchoalveolar lavage (BAL) fluid was performed using 1 ml of sterile Hanks Balanced Saline Buffer. The BAL protein concentration was determined by BCATM Protein Assay kit (Thermo Scientific, Pittsburg, PA). BAL inflammatory cell counting was performed using a standard hemacytometer technique36. As an additional parameter reflecting increased lung vascular leakiness, Evans blue accumulation in the lung tissue was evaluated as described previously36. At the end of the experiment, thoracotomy was performed, and the lungs were perfused in-situ via the left atrium with PBS containing 5 mM EDTA to flush the blood off the lungs. Left lung and right lungs were excised and imaged by a Kodak digital camera.
Statistical analysis
Results are expressed as means ± SD. Experimental samples were compared to controls by unpaired Student’s t-test. For multiple-group comparisons, a one-way variance analysis (ANOVA) and post hoc multiple comparison tests were used. P<0.05 was considered statistically significant.
RESULTS
Lipoxin receptors are expressed in pulmonary EC, but do not affect OxPAPC-induced barrier enhancing effects on endothelial barrier
The levels of mRNA expression of FPR family members FPR1, FPR2/ALX, FPR3 were evaluated by qRT-PCR and showed comparable expression of all three receptors (Figure 1A). Involvement of FPR receptors in barrier-enhancing effects of OxPAPC was tested by measurements of transendothelial electrical resistance (TER) reflecting barrier property of EC monolayer. Selective knockdown of each FPR receptor was performed using gene-specific siRNA. Experiments with selective knockdown of FPR1, FPR2/ALX and FPR3 receptors showed no effect on OxPAPC-induced TER increase (Figure 1B). Knockdown efficiency of FPRs was verified by qRT-PCR. In additional studies, we used two FPR peptide inhibitors: WRW4 (2.5 and 5 μmol/L, Figure 1C) and Boc-FLFLF (10, 20, and 40 μmol/L, Figure 1D). None of these treatments affected OxPAPC-induced sustained increase in TER. These data suggest that FPR receptors do not mediate barrier-enhancing effects of OxPAPC. Immunofluorescence staining of OxPAPC-stimulated EC monolayers (Online Figure I) showed increased VE-cadherin positive areas at the cell-cell junctions, as well as dissolution of central actin stress fibers and pronounced peripheral localization of F-actin. These morphological changes reflect enhancement of adherens junctions and peripheral cytoskeleton underlying increased barrier properties of OxPAPC-stimulated EC monolayer. Pretreatment with BocFLFLF had no effect on basal adherens junctions and cytoskeletal arrangement, or OxPAPC-induced cytoskeletal remodeling.
Figure 1. Inhibition of FPR1-3 receptors does not affect barrier-enhancing effects of OxPAPC on human pulmonary EC.
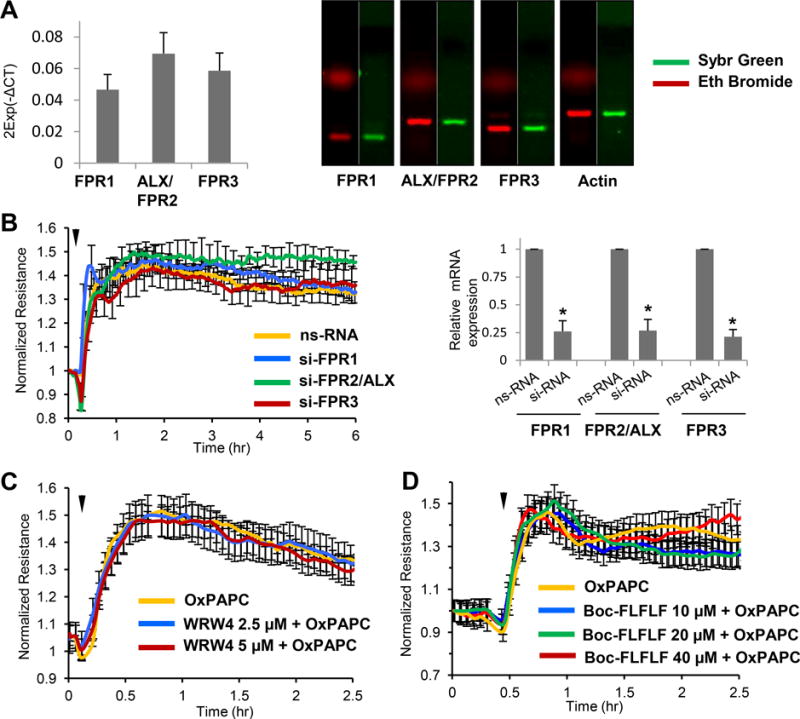
A - Expression of FPR1, FPR2/ALX and FPR3 mRNA in HPAEC was evaluated by qRT-PCR. PCR products corresponding to FPR1, FPR2/ALX and FPR3 were resolved by agarose gel electrophoresis and visualized by gel staining with Ethidium Bromide (red) and SYBR green (green) B - HPAEC plated on microelectrodes were treated with non-specific RNA oligomers (nsRNA) or gene-specific siRNA to FPR1, FPR2/ALX and FPR3, respectively (50 nM, 72 hrs). TER measurements were performed following stimulation with OxPAPC (15 μg/ml, marked by arrow). C and D - TER measurements of HPAEC monolayers preincubated with FPR receptor peptide inhibitors: C - WRW4, 2.5 or 5 μM, or: D - Boc-FLFLF, 10, 20 or 40 μM, 1 hr prior to OxPAPC treatment (marked by arrow). The TER curves represent pooled data from three independent experiments.
FPR receptors are not involved in OxPAPC barrier protective effects against thrombin
Stimulation of pulmonary EC with thrombin induces rapid and reversible permeability increase mediated by RhoA GTPase activation and MLC phosphorylation37. Effects of FPRs knockdown were evaluated in fluorimetry-based permeability assay for macromolecules in 96-well plates described in Methods. Thrombin-induced increase in EC permeability was suppressed by EC co-treatment with OxPAPC. This protective effect of OxPAPC was not affected by siRNA-induced knockdown of FPR1, FPR2/ALX, or FPR3 (Figure 2A).
Figure 2. FPR2/ALX knockdown does not affect barrier protective effect of OxPAPC against thrombin-induced permeability.
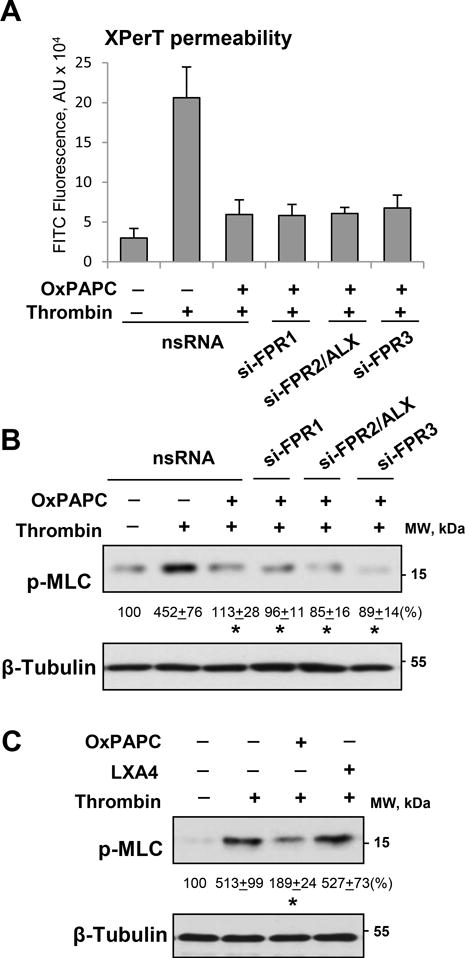
Pulmonary EC transfected with non-specific or FPR1, FPR2/ALX, or FPR3 specific siRNAs were treated with thrombin (0.5 U/ml), with or without OxPAPC pretreatment (15 μg/ml, 30 min). A - Analysis of EC permeability for macromolecules using FITC-labeled avidin as a tracer; n=6, *P < 0.01 vs. thrombin alone. B - MLC di-phosphorylation was evaluated using phospho-MLC specific antibody in control, thrombin, or OxPAPC treated EC. C - Effects of OxPAPC and LXA4 (100 nM) on thrombin-induced MLC phosphorylation were evaluated by Western blotting. Probing for β-tubulin was used as a normalization control. Numerical data depict results of quantitative densitometry; n=4; p<0.05 vs. thrombin alone.
Thrombin-induced MLC phosphorylation associated with increased permeability was significantly attenuated by OxPAPC pretreatment, and this protective effect was not influenced by knockdown of FPR1, FPR2/ALX, or FPR3 (Figure 2B). In line with lack of FPR involvement in EC barrier protection against agonist-induced permeability, pretreatment of thrombin-stimulated pulmonary EC with FPR2/ALX ligand, lipoxin A4 (LXA4), did not affect thrombin-induced MLC phosphorylation (Figure 2C). In turn, OxPAPC suppressed thrombin-induced MLC phosphorylation, as we have described before6, 7.
OxPAPC attenuates TNFα-induced EC inflammatory activation
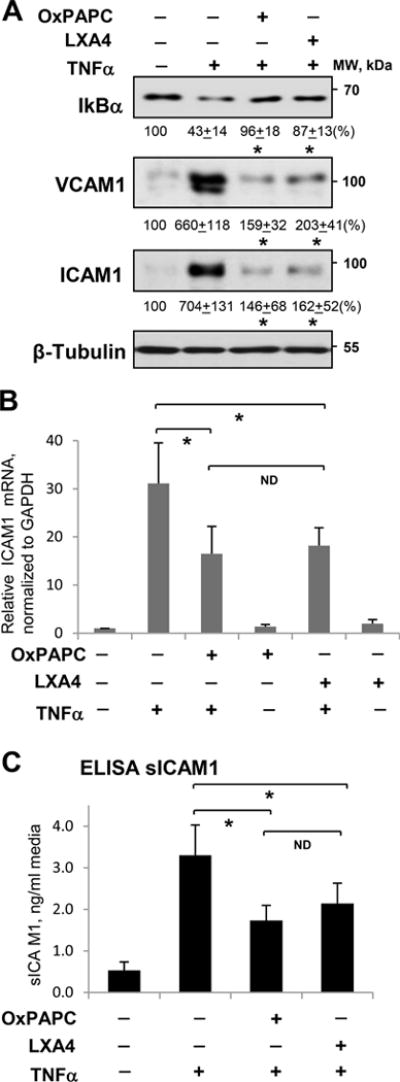
OxPAPC abolishes EC inflammation and barrier dysfunction caused by LPS: a) via induction of LXA4 production (this study); b) inhibition of LPS-induced activation of TLR4 receptor; and c) by direct stimulation of barrier function of inflamed endothelium via Rac1/Rap1-dependent cytoskeletal remodeling7, 10, 29. TNFα is another potent activator of EC inflammation and sustained barrier dysfunction38, 39 acting in a TLR4-independent manner. Therefore, to dissect TLR4 antagonism and TLR4-independent anti-inflammatory mechanisms, we stimulated cultured cells with TNFα. Activation of NFκB-mediated inflammatory signaling cascade by TNFα was reflected by TNFα-induced degradation of NFκB inhibitory subunit, IκBα. This effect was attenuated by pretreatment with OxPAPC. Pretreatment with LXA4 caused similar effect and prevented degradation of NFκB by TNFα (Figure 3A, top panel). LXA4 exhibited dose-dependent anti-inflammatory effects that were saturated between 10 and 100 nM (Online Figure II). These data are consistent with dose dependent increase of LXA4 inhibitory effect on TNFα-induced NFkB activation, neutrophil migration40 and inhibition of neutrophil-endothelial interactions41. Inflammatory activation of vascular endothelium by TNFα also stimulates NFkB-dependent expression of adhesion molecules ICAM1 and VCAM1. These EC receptors are involved in neutrophil adhesion to the vascular EC, leading to neutrophil transmigration to the lung parenchyma and development of lung inflammation. In agreement with inhibition of NFkB signaling, TNFα-induced expression of ICAM1 and VCAM1 was abolished in EC pretreated with OxPAPC or LXA4 (Figure 3A, bottom panels). Complementary qRT-PCR analysis showed strong upregulation of ICAM1 mRNA expression caused by TNFα, which was attenuated by OxPAPC and LXA4 (Figure 3B). OxPAPC and LXA4 also attenuated TNFα-induced secretion of soluble ICAM1 (sICAM1) to the culture medium detected by ELISA assay (Figure 3C).
Figure 3. Effect of OxPAPC and LXA4 on TNFα-induced inflammatory activation of human pulmonary EC.
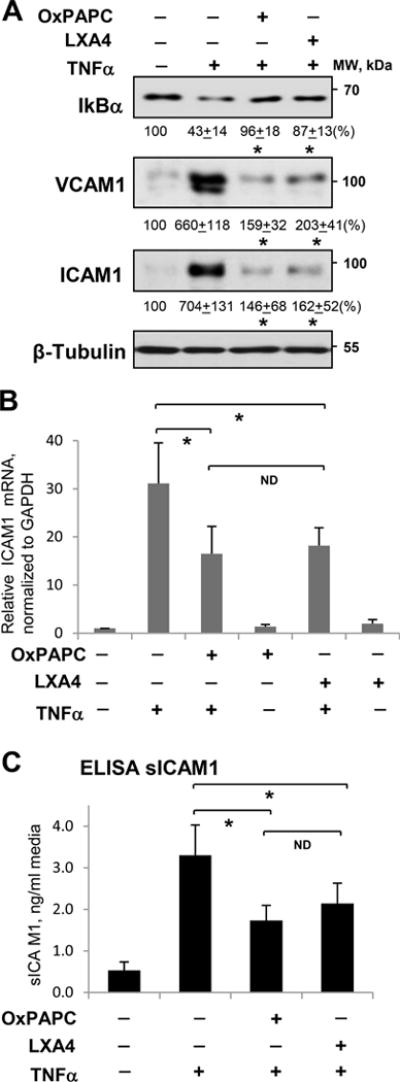
Cells were treated with TNFα (20 ng/ml, 6 hrs) alone or pretreated with OxPAPC (15 μg/ml) or LXA4 (100 nM) for 30 min. A - IκBα degradation, ICAM1 and VCAM1 expression were analyzed by Western blotting. Probing for β-tubulin was used as a normalization control. Numerical data depict results of quantitative densitometry; n=4; p<0.05 vs. TNFα alone. B - Expression of ICAM1 mRNA in control and stimulated HPAEC was evaluated by qRT-PCR; n=3, *P < 0.05 vs. TNFα alone. C - The level of soluble ICAM1 (sICAM1) in EC conditioned medium after stimulations was measured using ELISA assay; n=5, *P < 0.05 vs. TNFα alone.
FPR2/ALX mediates protective effects of OxPAPC against TNFα-induced EC barrier dysfunction and inflammatory activation
In contrast to rapid and reversible EC permeability response to thrombin, EC challenge with TNFα causes sustained EC barrier dysfunction associated with activation of inflammatory signaling pathways39. Analysis of permeability for FITC-avidin in EC monolayers challenged with TNFα showed that EC pretreatment with OxPAPC significantly attenuated TNFα-induced EC permeability increase. Experiments with siRNA-induced knockdown of FPR receptors showed that knockdown of FPR2/ALX, but not FPR1 or FPR3, suppressed OxPAPC-induced barrier protective effect (Figure 4A). Visualization of TNFα-induced EC barrier dysfunction using FITC-avidin as a tracer showed pronounced increase in green fluorescence reflecting increased penetration of FITC-avidin through EC monolayers and immobilization on the substrate underlying EC monolayers. OxPAPC caused pronounced protective effect against TNFα-induced permeability, which was attenuated by pretreatment with FPR2/ALX inhibitor (Figure 4B).
Figure 4. Role of FPR2/ALX signaling in anti-inflammatory and barrier protective effects of OxPAPC on pulmonary EC.
Cells were treated with TNFα (20 ng/ml) alone or pretreated with OxPAPC (15 μg/ml, 30 min). A - Effect of siRNA-induced FPR1, FPR2/ALX or FPR3 knockdown on EC permeability for macromolecules; n=3, *P < 0.05. B - Visualization of FITC-avidin accumulation underneath EC monolayers reflecting TNFα-induced EC barrier dysfunction. Protective effects of OxPAPC were suppressed by FPR2/ALX inhibitor WRW4 (5 μM, 1 hr prior to OxPAPC). Bar = 20 μm. Bar graph depicts quantitative analysis of FITC-avidin fluorescence in control and stimulated EC monolayers; n=6, *P < 0.05. C - TER measurements in HPAEC monolayers preincubated with WRW4 followed by TNFα challenge (marked by second arrow) with or without OxPAPC pretreatment (marked by first arrow). Bar graph depicts TER measurements at the time point of maximal response indicated by dotted line; n=6, *P < 0.05. D - Effect of FPR2/ALX inhibitor on the changes of EC barrier integrity caused by TNFα and OxPAPC. F-actin was visualized by staining with Texas Red phalloidin (red); adherens junctions were visualized by staining for VE-cadherin (green). Paracellular gaps are marked by arrows. Bar = 10 μm. Bar graph represents results of quantitative analysis of paracellular gap formation; n=4, * p<0.05.
Time-resolved analysis of EC permeability using TER measurements showed that pretreatment with OxPAPC caused TER elevation, which decreased following TNFα challenge, but remained at higher levels even 15–20 hrs after TNFα challenge in comparison to EC exposed to TNFα without OxPAPC pretreatment. Pharmacological inhibition of FPR by WRW4 did not affect the initial phase of OxPAPC protective response, but markedly reduced TER levels at later time points (10–20 hrs after TNFα challenge) (Figure 4C).
TNFα-induced sustained EC barrier dysfunction was associated with increased F-actin stress fiber formation and disruption of cell-cell junctions, leading to formation of paracellular gaps observed at later points of TNFα treatment. OxPAPC attenuated TNFα-induced disruption of EC monolayer integrity, but this OxPAPC barrier-protective effect was suppressed by pretreatment with FPR2/ALX inhibitor (Figure 4D).
TNFα triggers EC inflammatory response characterized by activation of NFkB signaling and expression of EC adhesion molecules ICAM1 and VCAM1. This TNFα effect was inhibited by OxPAPC and LXA4 (Figure 5A). Importantly, EC pretreatment with FPR2/ALX inhibitor WRW4 suppressed anti-inflammatory effect of both, OxPAPC and LXA4. (Figure 5A). siRNA-induced FPR2/ALX knockdown used as a complementary approach caused similar effect: inhibition of TNFα-induced VCAM1 expression caused by OxPAPC pretreatment was attenuated in EC with depleted FPR2/ALX (Figure 5B). Inset depicts siRNA-induced FPR2/ALX protein depletion verified by western blot. Pharmacological inhibition of FPR2/ALX also attenuated inhibitory effects of OxPAPC and LXA4 on TNFα-induced secretion of sICAM1 by pulmonary EC which was measured 20 hrs after TNFα addition (Figure 5C).
Figure 5. Role of FPR2/ALX signaling in anti-inflammatory effects of OxPAPC and LXA4.
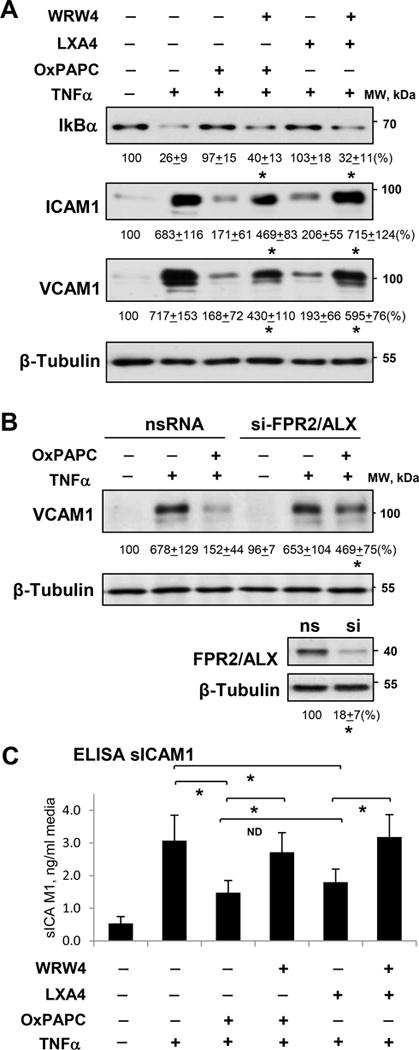
Cells were treated with TNFα (20 ng/ml) alone or pretreated with OxPAPC (15 μg/ml) or LXA4 (100 nM) for 30 min. A - Effect of FPR2/ALX inhibitor WRW4 on IκBα degradation, ICAM1 and VCAM1 expression. Probing for β-tubulin was used as a normalization control. B - Effect of siRNA-induced FPR2/ALX knockdown on VCAM1 expression. Numerical data depict results of quantitative densitometry; n=3; * p<0.05. C - Effect of FPR2/ALX inhibitor on sICAM1 release in culture medium evaluated by ELISA assay; n=5, *P < 0.05.
EC exposure to OxPAPC causes LXA4 accumulation in the conditioned medium
The data described above show involvement of lipoxin receptor FPR2/ALX in protective effects of OxPAPC against TNFα-induced EC inflammation and barrier dysfunction. One potential explanation of this effect is an induction of LXA4 by cell exposure to OxPAPC. This possibility was directly investigated in the following studies. LXA4 levels in the conditioned culture medium from EC exposed to OXPAPC were measured using ELISA. Treatment of pulmonary EC with OxPAPC significantly increased LXA4 levels in a time dependent manner with maximal increase by 24 hrs of OxPAPC exposure (Figure 6A). Importantly, EC incubation with oxidation resistant PAPC analog, DMPC, did not result in LXA4 production. EC preincubation with LPS did not result in considerable elevation of LXA4 production, while stimulation of LPS-challenged cells with OxPAPC administered 1 hr after LPS addition caused a similar increase in LXA4 levels as treatment with OxPAPC alone (Figure 6B). The levels of LXA4 in conditioned medium of OxPAPC-treated EC increased with time of OxPAPC incubation, whereas addition of DMPC did not cause any LXA4 elevation.
Figure 6. Analysis of OxPAPC-induced LXA4 generation.
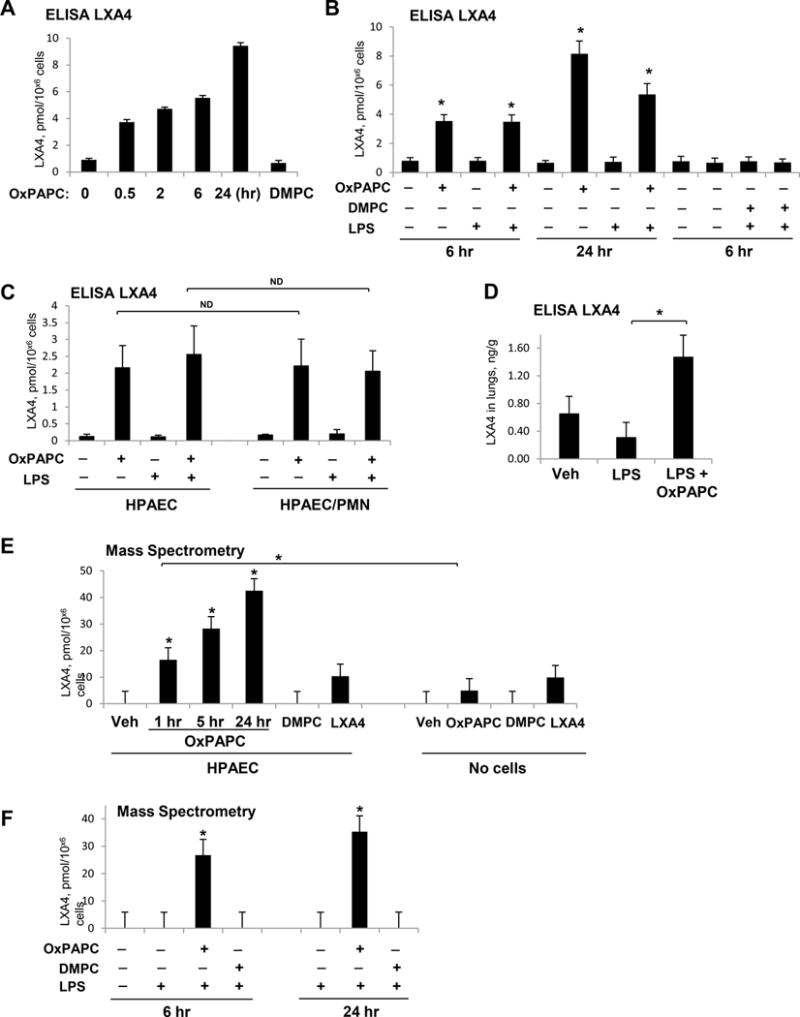
EC were treated with OxPAPC (15 μg/ml), DMPC (15 μg/ml), OxPAPC + LPS (200 ng/ml), or DMPC + LPS. A – ELISA assay: time course of LXA4 generation by EC treated with OxPAPC, DMPC, or vehicle; n=3, *P < 0.05 vs. vehicle. B - LXA4 generation by EC challenged with LPS (1 hr) and post-treated with OxPAPC (6 or 24 hrs), DMPC (6 hrs), or vehicle; n=4, *P < 0.05 vs. vehicle. C - Generation of LXA4 by EC and in EC-PMN co-culture. Similar levels of LXA4 were detected in EC cultured alone and in EC co-cultured with PMN; n=4, ND – no difference. D - Increased LXA4 levels in lung tissue of LPS-challenged mice after 5-hrs post-treatment with OxPAPC; n=3, *P < 0.05. E - Mass spectrometry analysis of LXA4 generation by cultured EC. Cells were treated with OxPAPC (1–24 hrs), DMPC (1 hr) or LXA4 (1 hr) as a positive control. Culture medium with addition of OxPAPC, DMPC, or LXA4 without exposure to the cells was used as an additional control. F - Mass spectrometry analysis of LXA4 in the conditioned medium of EC stimulated with LPS, LPS+OxPAPC, or LPS+DMPC. Cells were incubated with agonists for 6 or 24 hrs. LXA4 was detected in the conditioned medium; n=3, *P < 0.05 vs. LPS alone.
One described mechanism of LXA4 production is cooperative interaction between myeloid and non-myeloid cells differently expressing 15-LO, 5-LO, and epoxide hydrolase activities required for LXA4 synthesis42. Published studies suggest that LXA4 production in vivo can be enhanced by EC interaction with leukocytes19, 43. We next tested whether OxPAPC-induced LXA4 generation is further enhanced by EC co-culture with neutrophils (PMN). EC monolayers were treated with vehicle or LPS for 1 hr prior to addition of PMN and OxPAPC. Analysis of LXA4 levels in conditioned medium after 6 hrs of EC-PMN co-culture did not reveal significant difference between EC-PMN co-culture and EC cultured without PMN (Figure 6C).
Analysis of LXA4 levels in the lung tissue extracts from mice with intratracheal administration of LPS, with or without intravenous injection of OxPAPC, showed significant increase of LXA4 levels after 24 hrs of OxPAPC administration, while LPS group did not show LXA4 increase above control levels (Figure 6D).
LXA4 induction by OxPAPC was further verified using mass spectrometry (MS) approach described in Methods. MS analysis of the conditioned medium from the EC stimulated with OxPAPC or oxidation resistant phospholipid, DMPC, confirmed results of ELISA assays. The data showed time-dependent increase in LXA4 generation by EC treated with OxPAPC, but not with DMPC (Figure 6E, left panel). Control studies showed that addition of OxPAPC and DMPC in the absence of cells showed marginal increase in LXA4 signal in the cell-free culture medium (Figure 6E, right panel). Furthermore, in agreement with ELISA results, LXA4 was not detected in the conditioned medium from EC stimulated with LPS alone or LPS + DMPC, but LXA4 levels were increased upon EC incubation with LPS and OxPAPC (Figure 6F). Mass spectrometry profiles (Online Figure III) of the extracts from the medium collected from non-stimulated cells or cells stimulated with DMPC did not show any signal corresponding to LXA4. The employed transition for the detection of LXA4 is highly specific (m/z 351 > 115) and is not characteristic for other naturally occurring products of arachidonic acid oxidation44.
Analysis of OxPAPC-induced LXA4 generation
Our data suggest that production of lipoxins is likely to be involved in the sustained anti-inflammatory and potentially proresolving actions of OxPAPC. Canonical biosynthesis pathway of lipoxin A4 involves 15-lipoxygenase (15-LO). We first tested 15-LO expression levels in the human pulmonary EC. Real time RT-PCR analysis of 15-LO mRNA expression in control and OxPAPC-stimulated EC showed that 15-LO was not expressed in control EC under nonstimulated conditions, in the presence of OxPAPC (15 μg/ml or 30 μg/ml; after 1, 6 or 24 hrs), or in the presence of DMPC (15 μg/ml, 6 hrs). These results are consistent with the lack of basal 15-LOX expression in human EC reported in previous publication45. Furthermore, EC treatment with inhibitors of 15-LO and 5-LO did not affect OxPAPC protective effects against TNFα-induced EC barrier disruption (Figure 7A) and IL-8 production (Figure 7B). EC preincubation with LO-5 and LO-15 inhibitors did not affect OxPAPC-induced elevation of LXA4 signal in EC conditioned medium detected by mass spectrometry (Figure 7C). In contrast, inhibitor of soluble phospholipase type 2 prevented LXA4 accumulation in preconditioned medium of OxPAPC-incubated HPAEC.
Figure 7. Analysis of OxPAPC-induced LXA4 generation.
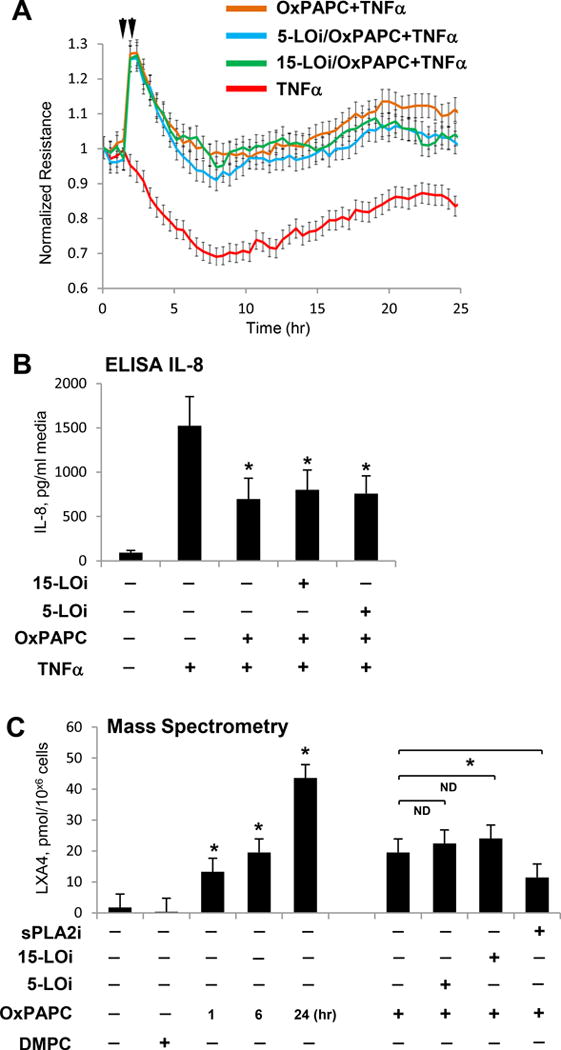
EC were preincubated with 5-LO or 15-LO inhibitor for 1 hr followed by TNFα challenge (20 ng/ml, marked by second arrow) with or without OxPAPC pre-treatment (15 μg/ml, marked by first arrow). A - TER measurements reflecting changes in EC permeability were performed over 25-hr time period. B – IL-8 accumulation in EC conditioned medium after stimulations was measured using ELISA assay. C – EC pretreated with sPLA2, 15-LO or 5-LO inhibitors were incubated with OxPAPC or DMPC (15 μg/ml), and LXA4 accumulation in conditioned medium was measured by mass spectrometry; n=3, *P < 0.05; ND – no difference.
Role of FPR2/ALX in anti-inflammatory and barrier protective effects of OxPAPC in the animal model of acute lung injury
Our findings from cell culture experiments showing involvement of FPR2/ALX in OxPAPC anti-inflammatory effects and the data showing OxPAPC-induced elevation of LXA4 prompted us to test the hypothesis that LXA4 induction may be involved in OxPAPC-induced recovery of ALI caused by Gram-negative pathogens. In the following experiments, C57Bl mice challenged with LPS (intratracheally), were treated with OxPAPC, with or without FPR2/ALX inhibitor WRW4 (2 mg/kg, intravenously, 15 min prior to OxPAPC) and lung injury was evaluated 24 hrs after LPS challenge. LPS caused significant increase in the total cell counts and protein concentration in bronchoalveolar lavage (BAL) samples, which was significantly attenuated by OxPAPC administration (Figure 8A). Treatment with FPR2/ALX inhibitor suppressed protective effects of OxPAPC.
Figure 8. Role of FPR signaling in protective effects of OxPAPC in the model of LPS-induced lung injury.
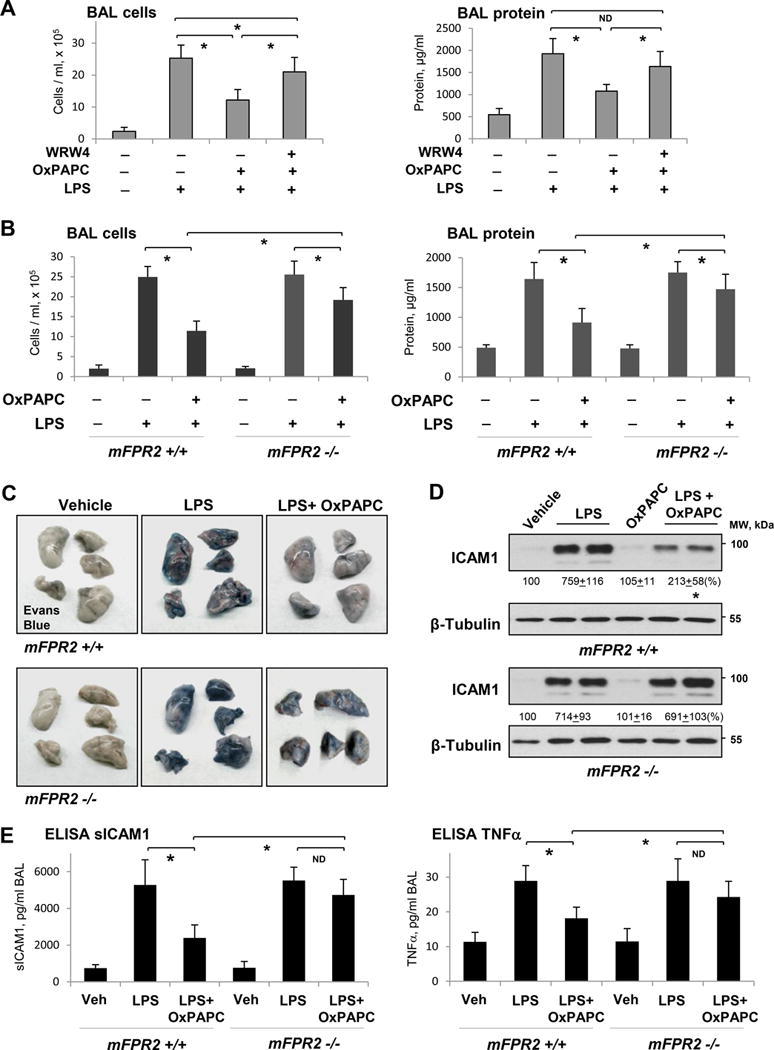
A - Intravenous injection of OxPAPC (1.5 mg/kg) with our without FPR2/ALX inhibitor WRW4 (20 μM, 1 hr prior to OxPAPC) was performed 5 hrs after LPS instillation (0.7 mg/kg, i.t.). BAL cell count and protein content were measured after 24 hrs of LPS challenge; n=5, *P < 0.05. B - BAL cell count and protein content in mFPR−/− mice and matching controls treated with LPS and LPS+OxPAPC; n=4, *P < 0.05. C - Evans Blue accumulation in the lung parenchyma. D - ICAM1 expression in lung tissue samples of control and mFPR-/- mice after LPS or LPS+OxPAPC challenge. E - ELISA assay of TNFα and sICAM1 levels in BAL samples; n=4, *P < 0.05.
The role of FPR2/ALX signaling in anti-inflammatory effects of OxPAPC was further investigated using genetic model of mFPR2−/− knockout mice described previously35. We evaluated the magnitude of lung injury in control and FPR−/− mice, which was analyzed 24 hrs after LPS administration. mFPR2−/− mice and matching controls developed similar levels of lung injury after 24 hrs of LPS challenge reflected by increased cell counts and protein content in BAL samples (Figure 8B). Protective effects of OxPAPC against LPS-induced lung injury were significantly attenuated in mFPR2−/− mice.
Effects of FPR2 ablation on protective effects of OxPAPC against the lung vascular leak caused by LPS were further assessed by visualization of Evans blue extravasation into the lung tissue. In agreement with previous reports, OxPAPC intravenous administration significantly reduced LPS-induced Evans blue accumulation in the lung parenchyma. However, this protective effect of OxPAPC was suppressed in mFPR2−/− mice (Figure 8C).
Analysis of other biochemical readouts of lung injury and inflammation showed marked suppression of the LPS-induced ICAM1 expression in the lung tissue and this effect of OxPAPC treatment was attenuated in mFPR2−/− mice (Figure 8D). We also found that OxPAPC suppressed OxPAPC-induced decreases in sICAM1 and TNFα levels in lung BAL samples from mFPR2−/− mice challenged with LPS (Figure 8E).
DISCUSSION
Anti-inflammatory effects of OxPAPC in the models of lung injury caused by cell wall components from Gram-negative and Gram-positive bacteria, CpG DNA, lung injury associated with necrotizing pancreatitis and other conditions associated with activaion of TLR signaling and innate immune response are well recognized8, 9, 11, 46–48. These effects have been previously associated with OxPAPC-directed attenuation of TLR signaling in response to bacterial pathogens and other pathogen associated molecular patterns (PAMPS)1. Importantly, to exclude direct inhibition of TLRs by OxPAPC, in the animal model we performed intravenous injection of OxPAPC 5 hours after intratracheal instillation of LPS. Therefore, OxPAPC protective effects against TNFα- and LPS-induced inflammation and EC barrier disruption observed in this study suggest the presence of other TLR-independent mechanisms. Our data show that OxPAPC also attenuated EC inflammatory and permeability response to TNFα, which is not mediated by the TLR pathway. Interestingly, the OxPAPC protective effects were inhibited by knockdown or pharmacological inhibition of the LXA4 receptor FPR2/ALX. These results strongly suggest that OxPAPC inhibitory effect on TNFα–induced EC inflammation is mediated by TLR-independent mechanism. This study demonstrates for the first time an alternative, TLR-independent antiinflammatory effects of OxPAPC in the models of TNFα and LPS induced inflammation. We describe here the OxPAPC-induced activation of pro-resolution pathway: stimulation of LXA4 production by OxPAPC-exposed pulmonary EC and FPR2/ALX receptor-dependent suppression of lung inflammation and improvement of endothelial dysfunction caused by inflammatory agents.
The canonical pathway of LXA4 production involves conversion of LXA4 precursor, arachidonic acid to LXA4. This process involves sequential combination of 15-LO, 5-LO, and epoxide hydrolase activities42 expressed by different cell types and therefore requiring cell–cell interactions at sites of inflammation. Although LXA4 is present in low abundance during the initiation of acute inflammation, their levels increase substantially during resolution49–51. LXA4, in turn, exhibits multi-facet modalities in suppressing inflammation and promoting lung repair19 including a novel mechanism for control of vascular endothelial inflammation by changes in lung micromechanics recently described by our group18.
Interaction of PMN with resident cells such as EC during inflammation is a previously described model to induce LXA4 production15, 49. While this is the case generally, there are a number of reports documenting single cell types producing lipoxins such as human macrophages and dendritic cells52, primed human peripheral blood neutrophils17 and macrophages53. EC preincubation with LPS for different time periods in this study neither induced LXA4 production by pulmonary EC on its own, nor it affected LXA4 induction caused by EC incubation with OxPAPC (Figure 6). We also did not observe LXA4 upregulation in the LPS-treated lungs 24 hrs after LPS administration, although OxPAPC injection in LPS-treated mice caused the LXA4 increase. This result may be explained by short time period of our experiment. LXA4 induction also depended on phospholipid oxidation, because EC exposure to oxidation resistant phospholipid DMPC did not induce LXA4 production.
The data show that 15-LO and 5-LO did not affect OxPAPC-induced LXA4 production, while inhibitor of soluble PLA2 suppressed LXA4 accumulation in conditioned medium and attenuated OxPAPC anti-inflammatory effect. Interestingly, CYP450 epoxygenase inhibitor also partially blocked OxPAPC-induced LXA4 accumulation detected by MS (data not shown). These data indicate additional involvement of CYP450 in the LXA4 production from OxPAPC. Altogether, our results strongly suggest 15-LOX-independent, sPLA2-dependent mechanism of LXA4 production by pulmonary EC induced by OxPAPC. Further studies are warranted to delineate a precise role of enzymatic and non-enzymatic OxPAPC conversions mediated by EC and resulting in accumulation of LXA4 in culture medium.
From biochemical point of view, PAPC oxidation by air causes formation of epoxy derivatives of PAPC on their arachidonoyl moiety. But upon addition of OxPAPC to cell culture or in the animal experiments, such oxidized (epoxy) lipids are likely to undergo metabolic changes. Then, simultaneous action of cellular epoxide hydrolases and the non-enzymatic formation of intermediate peroxi-intermediates may result in the formation of LXA4-type structures from air oxidized PAPC and explain lipoxygenase-independent LXA4 formation.
Our results show somewhat higher estimation of LXA4 signal by the LC/MS/MS method as compared to ELISA results. First, these differences may be due to variances in experimental conditions such as cell to culture medium ratio and different batches of human pulmonary EC used for ELISA and MS experiments. LC/MS/MS methodology employed in this study allows for highly specific detection of the LXA4 in biological fluids. The extracts of the preconditioned medium from non-stimulated and DMPC-stimulated cells do not show signal corresponding to LXA4. The employed transition for the detection of LXA4 is quite unique (m/z 351 > 115) and is not characteristic for the most known products of arachidonic acid oxidation44 and even for the LXB4. However, we acknowledge one potential limitation of this study. We cannot rule out that oxygenation of PAPC may result in the formation of stereo- and OH-group positional isomers of LXA4 which will be more challenging to separate using our current technique and which could mask and overlay the “true” LXA4. These factors may result in potential LXA4 overestimation by mass spectrometry. Our future work will address such a possibility.
In summary, this study shows a novel anti-inflammatory effect of OxPAPC in cell and animal models of ALI caused by bacterial pathogen and inflammatory cytokine, which is associated with OxPAPC-induced formation of LXA4 and activation of FPR2/ALX receptor. These results demonstrate a new way of lipid program switch triggered by oxidized phospholipids generated as a result of oxidative stress during lung inflammation or injury. We speculate that production of anti-inflammatory lipid mediators by canonical pathway of lipoxin synthesis during lipid mediator program switch49 at advanced stage of inflammation may be accelerated in the presence of OxPAPC. Beneficial role of LXA4-FPR2/ALX signaling has been also shown in other pathologic conditions. FPR2/ALX-LXA4 axis controls vascular inflammatory responses during cerebral ischemia/reperfusion, and induction of LXA4 may be beneficial for patients suffering from stroke54. Induction of LXA4 reduces capillary congestion in malaria model55, and may also influence the clinical management of trauma patients, as higher lipoxin/resolvin scores have been reported in patients with uncomplicated recoveries56. The results of this study strongly suggest that phospholipid oxidation induced by inflammatory insults may represent a built-in mechanism of body recovery after injury. Therefore, the induction of lipoxin production by OxPAPC described in this study may have a broader impact on the outcome of other acute and chronic inflammatory conditions and implication in development of future therapies.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Oxidative stress accompanies tissue injury, infection, and sepsis, and elevates circulating and tissue levels of oxidized phospholipids.
Oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (OxPAPC) exhibits long-lasting anti-inflammatory and vascular protective effects in acute lung injury even after single administration in vivo.
Products of PAPC oxidation attenuate the innate immune response via antagonistic effects on toll-like receptors (TLR) and reduce endothelial permeability by suppressing the RhoA GTPase signaling.
What New Information Does This Article Contribute?
Exposure of endothelial cells to OxPAPC, but not to non-oxidized phospholipids, induces time-dependent generation of the pro-resolving mediator lipoxin A4 (LXA4).
Anti-inflammatory effects of OxPAPC were attenuated in vitro and in vivo by inhibition of the LXA4 receptor, FPR2/ALX
This study demonstrates for the first time alternative protective mechanisms triggered OxPAPC in the models of lung inflammation and endothelial dysfunction caused by inflammatory agents. We show that injury-associated generation of phospholipid oxidation products triggers the production of anti-inflammatory lipid mediators such as LXA4, which may represent a built-in mechanism of vascular and tissue auto recovery after injury.
Acknowledgments
SOURCES OF FUNDING: This work was supported by the grants: HL076259, HL087823, HL107920, HL130431 from the National Heart, Lung, and Blood Institute, and GM114171 from the National Institute of General Medical Sciences.
Nonstandard Abbreviations and Acronyms
- BAL
bronchoalveolar lavage
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine
- EC
endothelial cells
- ECIS
electrical cell-substrate impedance sensing system
- FPR
formyl-peptide receptor
- HPAEC
human pulmonary artery endothelial cells
- LO
lipoxygenase
- LPS
lipopolysaccharide
- LS-ESI-MS
liquid chromatography electrospray ionization tandem mass spectrometry
- LXA4
lipoxin A4
- MLC
myosin light chain
- ns-RNA
non-specific RNA
- OxPAPC
Oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine
- PMN
polymorphonuclear leukocyte
- RDU
relative density units
- TER
transendothelial electrical resistance
- TNFα
tumor necrosis factor-α
- VE-cadherin
vascular endothelial cadherin
- XPerT
express permeability testing assay
Footnotes
DISCLOSURES
None.
References
- 1.Bochkov VN, Oskolkova OV, Birukov KG, Levonen AL, Binder CJ, Stockl J. Generation and biological activities of oxidized phospholipids. Antioxid Redox Signal. 2010;12(8):1009–1059. doi: 10.1089/ars.2009.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Subbanagounder G, Wong JW, Lee H, Faull KF, Miller E, Witztum JL, Berliner JA. Epoxyisoprostane and epoxycyclopentenone phospholipids regulate monocyte chemotactic protein-1 and interleukin-8 synthesis. Formation of these oxidized phospholipids in response to interleukin-1beta. J Biol Chem. 2002;277(9):7271–7281. doi: 10.1074/jbc.M107602200. [DOI] [PubMed] [Google Scholar]
- 3.Zebda N, Tian Y, Tian X, Gawlak G, Higginbotham K, Reynolds AB, Birukova AA, Birukov KG. Interaction of p190RhoGAP with C-terminal domain of p120-catenin modulates endothelial cytoskeleton and permeability. J Biol Chem. 2013;288(25):18290–18299. doi: 10.1074/jbc.M112.432757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birukova AA, Malyukova I, Mikaelyan A, Fu P, Birukov KG. Tiam1 and betaPIX mediate Rac-dependent endothelial barrier protective response to oxidized phospholipids. J Cell Physiol. 2007;211(3):608–617. doi: 10.1002/jcp.20966. [DOI] [PubMed] [Google Scholar]
- 5.Birukova AA, Fu P, Wu T, Dubrovskyi O, Sarich N, Poroyko V, Birukov KG. Afadin controls p120-catenin-ZO-1 interactions leading to endothelial barrier enhancement by oxidized phospholipids. J Cell Physiol. 2012;227(5):1883–1890. doi: 10.1002/jcp.22916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nonas S, Birukova AA, Fu P, Xing J, Chatchavalvanich S, Bochkov VN, Leitinger N, Garcia JG, Birukov KG. Oxidized phospholipids reduce ventilator-induced vascular leak and inflammation in vivo. Crit Care. 2008;12(1):R27. doi: 10.1186/cc6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birukova AA, Zebda N, Fu P, Poroyko V, Cokic I, Birukov KG. Association between adherens junctions and tight junctions via Rap1 promotes barrier protective effects of oxidized phospholipids. J Cell Physiol. 2011;226(8):2052–2062. doi: 10.1002/jcp.22543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma Z, Li J, Yang L, Mu Y, Xie W, Pitt B, Li S. Inhibition of LPS- and CpG DNA-induced TNF-alpha response by oxidized phospholipids. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L808–816. doi: 10.1152/ajplung.00220.2003. [DOI] [PubMed] [Google Scholar]
- 9.Nonas SA, Miller I, Kawkitinarong K, Chatchavalvanich S, Gorshkova I, Bochkov VN, Leitinger N, Natarajan V, Garcia JG, Birukov KG. Oxidized phospholipids reduce vascular leak and inflammation in rat model of acute lung injury. Am J Respir Crit Care Med. 2006;173(10):1130–1138. doi: 10.1164/rccm.200511-1737OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bochkov VN, Kadl A, Huber J, Gruber F, Binder BR, Leitinger N. Protective role of phospholipid oxidation products in endotoxin-induced tissue damage. Nature. 2002;419(6902):77–81. doi: 10.1038/nature01023. [DOI] [PubMed] [Google Scholar]
- 11.Meliton A, Meng F, Tian Y, Sarich N, Mutlu GM, Birukova AA, Birukov KG. Oxidized phospholipids protect against lung injury and endothelial barrier dysfunction caused by heat-inactivated Staphylococcus aureus. Am J Physiol Lung Cell Mol Physiol. 2015 doi: 10.1152/ajplung.00248.2014. ajplung 0024802014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serhan CN, Chiang N. Endogenous pro-resolving and anti-inflammatory lipid mediators: a new pharmacologic genus. Br J Pharmacol. 2008;153(Suppl 1):S200–215. doi: 10.1038/sj.bjp.0707489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohli P, Levy BD. Resolvins and protectins: mediating solutions to inflammation. Br J Pharmacol. 2009;158(4):960–971. doi: 10.1111/j.1476-5381.2009.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ji RR, Xu ZZ, Strichartz G, Serhan CN. Emerging roles of resolvins in the resolution of inflammation and pain. Trends in neurosciences. 2011;34(11):599–609. doi: 10.1016/j.tins.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haworth O, Levy BD. Endogenous lipid mediators in the resolution of airway inflammation. Eur Respir J. 2007;30(5):980–992. doi: 10.1183/09031936.00005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. 2009;61(2):119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiore S, Maddox JF, Perez HD, Serhan CN. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J Exp Med. 1994;180(1):253–260. doi: 10.1084/jem.180.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meng F, Mambetsariev I, Tian Y, Beckham Y, Meliton A, Leff A, Gardel ML, Allen MJ, Birukov KG, Birukova AA. Attenuation of lipopolysaccharide-induced lung vascular stiffening by lipoxin reduces lung inflammation. Am J Respir Cell Mol Biol. 2015;52(2):152–161. doi: 10.1165/rcmb.2013-0468OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonnans C, Levy BD. Lipid mediators as agonists for the resolution of acute lung inflammation and injury. Am J Respir Cell Mol Biol. 2007;36(2):201–205. doi: 10.1165/rcmb.2006-0269TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dufton N, Hannon R, Brancaleone V, Dalli J, Patel HB, Gray M, D’Acquisto F, Buckingham JC, Perretti M, Flower RJ. Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J Immunol. 2010;184(5):2611–2619. doi: 10.4049/jimmunol.0903526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bochkov VN, Mechtcheriakova D, Lucerna M, Huber J, Malli R, Graier WF, Hofer E, Binder BR, Leitinger N. Oxidized phospholipids stimulate tissue factor expression in human endothelial cells via activation of ERK/EGR-1 and Ca(++)/NFAT. Blood. 2002;99(1):199–206. doi: 10.1182/blood.v99.1.199. [DOI] [PubMed] [Google Scholar]
- 22.Leitinger N, Tyner TR, Oslund L, Rizza C, Subbanagounder G, Lee H, Shih PT, Mackman N, Tigyi G, Territo MC, Berliner JA, Vora DK. Structurally similar oxidized phospholipids differentially regulate endothelial binding of monocytes and neutrophils. Proc Natl Acad Sci U S A. 1999;96(21):12010–12015. doi: 10.1073/pnas.96.21.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watson AD, Leitinger N, Navab M, Faull KF, Horkko S, Witztum JL, Palinski W, Schwenke D, Salomon RG, Sha W, Subbanagounder G, Fogelman AM, Berliner JA. Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. J Biol Chem. 1997;272(21):13597–13607. doi: 10.1074/jbc.272.21.13597. [DOI] [PubMed] [Google Scholar]
- 24.Birukova AA, Adyshev D, Gorshkov B, Bokoch GM, Birukov KG, Verin AA. GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2006;290(3):L540–548. doi: 10.1152/ajplung.00259.2005. [DOI] [PubMed] [Google Scholar]
- 25.Birukova AA, Birukov KG, Smurova K, Adyshev DM, Kaibuchi K, Alieva I, Garcia JG, Verin AD. Novel role of microtubules in thrombin-induced endothelial barrier dysfunction. FASEB J. 2004;18(15):1879–1890. doi: 10.1096/fj.04-2328com. [DOI] [PubMed] [Google Scholar]
- 26.Dubrovskyi O, Birukova AA, Birukov KG. Measurement of local permeability at subcellular level in cell models of agonist- and ventilator-induced lung injury. Lab Invest. 2013;93:254–263. doi: 10.1038/labinvest.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Birukova AA, Chatchavalvanich S, Rios A, Kawkitinarong K, Garcia JG, Birukov KG. Differential regulation of pulmonary endothelial monolayer integrity by varying degrees of cyclic stretch. Am J Pathol. 2006;168(5):1749–1761. doi: 10.2353/ajpath.2006.050431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Birukova AA, Cokic I, Moldobaeva N, Birukov KG. Paxillin is Involved in the Differential Regulation of Endothelial Barrier by HGF and VEGF. Am J Respir Cell Mol Biol. 2008 doi: 10.1165/rcmb.2008-0099OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singleton PA, Chatchavalvanich S, Fu P, Xing J, Birukova AA, Fortune JA, Klibanov AM, Garcia JG, Birukov KG. Akt-mediated transactivation of the S1P1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circ Res. 2009;104(8):978–986. doi: 10.1161/CIRCRESAHA.108.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Birukova AA, Alekseeva E, Mikaelyan A, Birukov KG. HGF attenuates thrombin-induced permeability in the human pulmonary endothelial cells by Tiam1-mediated activation of the Rac pathway and by Tiam1/Rac-dependent inhibition of the Rho pathway. FASEB J. 2007;21(11):2776–2786. doi: 10.1096/fj.06-7660com. [DOI] [PubMed] [Google Scholar]
- 31.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Birukova AA, Malyukova I, Poroyko V, Birukov KG. Paxillin - {beta}-catenin interactions are involved in Rac/Cdc42-mediated endothelial barrier-protective response to oxidized phospholipids. Am J Physiol Lung Cell Mol Physiol. 2007;293(1):L199–211. doi: 10.1152/ajplung.00020.2007. [DOI] [PubMed] [Google Scholar]
- 33.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Canadian journal of biochemistry and physiology. 1959;37(8):911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 34.Meliton AY, Munoz NM, Meliton LN, Binder DC, Osan CM, Zhu X, Dudek SM, Leff AR. Cytosolic group IVa phospholipase A2 mediates IL-8/CXCL8-induced transmigration of human polymorphonuclear leukocytes in vitro. Journal of inflammation (London, England) 2010;7:14. doi: 10.1186/1476-9255-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen K, Le Y, Liu Y, Gong W, Ying G, Huang J, Yoshimura T, Tessarollo L, Wang JM. A critical role for the g protein-coupled receptor mFPR2 in airway inflammation and immune responses. J Immunol. 2010;184(7):3331–3335. doi: 10.4049/jimmunol.0903022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu P, Birukova AA, Xing J, Sammani S, Murley JS, Garcia JG, Grdina DJ, Birukov KG. Amifostine reduces lung vascular permeability via suppression of inflammatory signalling. Eur Respir J. 2009;33(3):612–624. doi: 10.1183/09031936.00014808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Birukova AA, Smurova K, Birukov KG, Kaibuchi K, Garcia JGN, Verin AD. Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc Res. 2004;67(1):64–77. doi: 10.1016/j.mvr.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 38.Petrache I, Birukova A, Ramirez SI, Garcia JG, Verin AD. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am J Respir Cell Mol Biol. 2003;28(5):574–581. doi: 10.1165/rcmb.2002-0075OC. [DOI] [PubMed] [Google Scholar]
- 39.Xing J, Birukova AA. ANP attenuates inflammatory signaling and Rho pathway of lung endothelial permeability induced by LPS and TNFalpha. Microvasc Res. 2010;79(1):26–62. doi: 10.1016/j.mvr.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fierro IM, Colgan SP, Bernasconi G, Petasis NA, Clish CB, Arita M, Serhan CN. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit human neutrophil migration: comparisons between synthetic 15 epimers in chemotaxis and transmigration with microvessel endothelial cells and epithelial cells. J Immunol. 2003;170(5):2688–2694. doi: 10.4049/jimmunol.170.5.2688. [DOI] [PubMed] [Google Scholar]
- 41.Filep JG, Zouki C, Petasis NA, Hachicha M, Serhan CN. Anti-inflammatory actions of lipoxin A(4) stable analogs are demonstrable in human whole blood: modulation of leukocyte adhesion molecules and inhibition of neutrophil-endothelial interactions. Blood. 1999;94(12):4132–4142. [PubMed] [Google Scholar]
- 42.Bannenberg G, Serhan CN. Specialized pro-resolving lipid mediators in the inflammatory response: An update. Biochim Biophys Acta. 2010;1801(12):1260–1273. doi: 10.1016/j.bbalip.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brady HR, Serhan CN. Adhesion promotes transcellular leukotriene biosynthesis during neutrophil-glomerular endothelial cell interactions: inhibition by antibodies against CD18 and L-selectin. Biochem Biophys Res Commun. 1992;186(3):1307–1314. doi: 10.1016/s0006-291x(05)81548-7. [DOI] [PubMed] [Google Scholar]
- 44.Dumlao DS, Buczynski MW, Norris PC, Harkewicz R, Dennis EA. High-throughput lipidomic analysis of fatty acid derived eicosanoids and N-acylethanolamines. Biochim Biophys Acta. 2011;1811(11):724–736. doi: 10.1016/j.bbalip.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee YW, Kuhn H, Kaiser S, Hennig B, Daugherty A, Toborek M. Interleukin 4 induces transcription of the 15-lipoxygenase I gene in human endothelial cells. J Lipid Res. 2001;42(5):783–791. [PubMed] [Google Scholar]
- 46.Walton KA, Cole AL, Yeh M, Subbanagounder G, Krutzik SR, Modlin RL, Lucas RM, Nakai J, Smart EJ, Vora DK, Berliner JA. Specific phospholipid oxidation products inhibit ligand activation of toll-like receptors 4 and 2. Arterioscler Thromb Vasc Biol. 2003;23(7):1197–1203. doi: 10.1161/01.ATV.0000079340.80744.B8. [DOI] [PubMed] [Google Scholar]
- 47.Erridge C, Kennedy S, Spickett CM, Webb DJ. Oxidized phospholipid inhibition of toll-like receptor (TLR) signaling is restricted to TLR2 and TLR4: roles for CD14, LPS-binding protein, and MD2 as targets for specificity of inhibition. J Biol Chem. 2008;283(36):24748–24759. doi: 10.1074/jbc.M800352200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oskolkova OV, Afonyushkin T, Preinerstorfer B, Bicker W, von Schlieffen E, Hainzl E, Demyanets S, Schabbauer G, Lindner W, Tselepis AD, Wojta J, Binder BR, Bochkov VN. Oxidized Phospholipids Are More Potent Antagonists of Lipopolysaccharide than Inducers of Inflammation. J Immunol. 2010;185(12):7706–7712. doi: 10.4049/jimmunol.0903594. [DOI] [PubMed] [Google Scholar]
- 49.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2(7):612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 50.Haworth O, Cernadas M, Yang R, Serhan CN, Levy BD. Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat Immunol. 2008;9(8):873–879. doi: 10.1038/ni.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barnig C, Levy BD. Innate immunity is a key factor for the resolution of inflammation in asthma. Eur Respir Rev. 2015;24(135):141–153. doi: 10.1183/09059180.00012514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. 2012;120(15):e60–72. doi: 10.1182/blood-2012-04-423525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Norris PC, Gosselin D, Reichart D, Glass CK, Dennis EA. Phospholipase A2 regulates eicosanoid class switching during inflammasome activation. Proc Natl Acad Sci U S A. 2014;111(35):12746–12751. doi: 10.1073/pnas.1404372111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith HK, Gil CD, Oliani SM, Gavins FN. Targeting formyl peptide receptor 2 reduces leukocyte-endothelial interactions in a murine model of stroke. Faseb J. 2015;29(5):2161–2171. doi: 10.1096/fj.14-263160. [DOI] [PubMed] [Google Scholar]
- 55.Souza MC, Padua TA, Torres ND, Souza Costa MF, Candea AP, Maramaldo T, Seito LN, Penido C, Estato V, Antunes B, Silva L, Pinheiro AA, Caruso-Neves C, Tibirica E, Carvalho L, Henriques MG. Lipoxin A4 attenuates endothelial dysfunction during experimental cerebral malaria. Int Immunopharmacol. 2015;24(2):400–407. doi: 10.1016/j.intimp.2014.12.033. [DOI] [PubMed] [Google Scholar]
- 56.Orr SK, Butler KL, Hayden D, Tompkins RG, Serhan CN, Irimia D. Gene Expression of Proresolving Lipid Mediator Pathways Is Associated With Clinical Outcomes in Trauma Patients. Crit Care Med. 2015;43(12):2642–2650. doi: 10.1097/CCM.0000000000001312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.