

SUMMARY
The spatial localization of hematopoietic stem cells (HSCs) in the bone marrow (BM) remains controversial with studies suggesting that they are maintained in homogenously distributed niches while others have suggested the contributions of distinct niche structures. Subsets of quiescent HSCs have been reported to associate with megakaryocytes (MK) or arterioles in the BM. However, these HSC subsets have not been prospectively defined. Here, we show that platelet and myeloid-biased HSCs, marked by von Willebrand factor (vWF) expression, are highly enriched in MK niches. Depletion of MK selectively expands vWF+ HSCs whereas the depletion of NG2+ arteriolar niche cells selectively depletes vWF− lymphoid-biased HSCs. In addition, MK depletion compromises vWF+ HSC function by reducing their long-term self-renewal capacity, and eliminating their lineage bias after transplantation. These studies demonstrate the existence of two spatially and functionally separate bone marrow niches for HSC subsets with distinct developmental potential.
Keywords: hematopoietic stem cell, lineage bias, von Willebrand factor, hematopoietic stem cell niche, megakaryocyte niche, arteriolar niche
eTOC Blurb
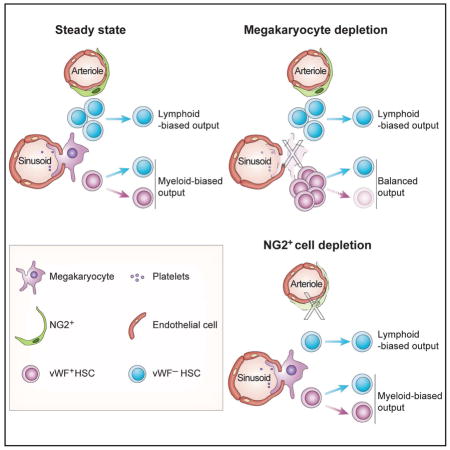
Pinho et al. show that myeloid- and lymphoid-biased HSCs are located in, and regulated by, separate bone marrow niches occupied by megakaryocytes (MK) and arterioles, respectively. MK niches may also regulate HSC fate since MK deletion reprograms myeloid-biased HSCs to balanced-lineage contributions.
Despite intense investigation, there remains controversy about the precise location of hematopoietic stem cells (HSCs) in the bone marrow (BM). Initial studies have suggested a close relationship with osteoblasts in the endosteal region (Zhang et al., 2003). Subsequent studies, with the advent of SLAM markers, have shown that CD150+ CD48− Lineage− CD41− HSCs were distributed broadly in close proximity of endothelial and Nestin-GFP+ perivascular cells (Kiel et al., 2005; Mendez-Ferrer et al., 2010). Further improvement in imaging technologies allowing tridimensional (3D) visualization of endogenous HSCs in the BM have uncovered heterogeneity among Nestin-GFP+ cells with a subset of quiescent HSCs showing a significant association with Nestin-GFP-bright arteriole structures. Upon deletion of periarteriolar NG2+ stromal cells or after inducing HSC proliferation, HSCs were shown to redistribute away from arteriole structures (Kunisaki et al., 2013). Further studies show that another subset of HSCs exhibit a significant association with megakaryocytes (MK) which are broadly distributed in the BM (Bruns et al., 2014). Selective deletion of MK induced HSC proliferation in a manner shown to depend on CXCL4 (also called platelet factor-4, PF4), TGF-β or thrombopoietin production by MK (Bruns et al., 2014; Nakamura-Ishizu et al., 2014; Zhao et al., 2014). These studies thus suggest that both arteriole structures and MK are niche sites promoting HSC quiescence. However, other studies using α-catulin+ c-Kit+ Lineage− to mark HSCs have shown in cleared BM that these HSCs were uniformly distributed near sinusoids in a manner indistinguishable from randomly placed dots (Acar et al., 2015). Other analyses, in support of multiple niche structures, have also suggested the existence of distinct arteriolar and MK niche harboring reactive oxygen species (ROS)-low quiescent HSCs (Itkin et al., 2016), or HSC association with type H endothelial cells bridging arterioles and sinusoids in developing bones (Kusumbe et al., 2016). As sinusoids are evenly distributed throughout the BM (Kunisaki et al., 2013), every hematopoietic cell is expected to be relatively close to them (~15 μm on average), raising questions on the niche specificity. Indeed, the relationship of HSCs with sinusoid vessels was not found to be significantly different from computational modeling random dots (Acar et al., 2015; Kunisaki et al., 2013). However, one study, using a Hoxb5 reporter, found that these HSCs significantly associated with sinusoidal endothelial cells in the BM (Chen et al., 2016). Thus, these studies using different HSC markers have reported differing results about HSC localization in the BM.
HSCs are known to be functionally heterogeneous (Copley et al., 2012; Muller-Sieburg et al., 2012). For example, transplantation of single HSCs has revealed reproducible bias toward selective differentiation to the myeloid or the lymphoid lineage (Dykstra et al., 2007). Using von Willebrand factor (vWF) promoter to drive GFP in transgenic mice (Vwf-eGFP), GFP was shown to mark a subset of quiescent HSCs that exhibits platelet and myeloid bias upon transplantation whereas the GFP− HSC subset was lymphoid biased (Sanjuan-Pla et al., 2013). This differential expression of vWF may have hierarchical significance since only vWF-eGFP+ HSCs could give rise to vWF-eGFP− HSCs. HSCs with high CD150 expression are enriched in vWF transcripts and are myeloid biased (Beerman et al., 2010; Kent et al., 2009). Expression of CD41 (Gekas and Graf, 2013) or c-Kit (Shin et al., 2014) has also been suggested to mark a megakaryocytic-biased HSC subset. Myeloid-, lymphoid-biased and lymphoid/myeloid balanced HSC subsets prevail in young mice (Dykstra et al., 2007; Muller-Sieburg et al., 2002; Sanjuan-Pla et al., 2013; Sieburg et al., 2006), whereas myeloid-biased HSCs become dominant with age (Cho et al., 2008; Dykstra et al., 2011; Grover et al., 2016) with declined long-term self-renewal functions, possibly contributing to the development of hematologic malignancies. Here, we show using Vwf-eGFP genetic marking that myeloid- and lymphoid-biased HSCs occupy distinct BM niche microenvironments that are differentially regulated.
RESULTS AND DISCUSSION
vWF+ HSCs associate with megakaryocytes, but not arterioles, in the mouse bone marrow
We evaluated the relationships of platelet- and myeloid-biased vWF-eGFP+ and lymphoid-biased vWF-eGFP− HSCs (henceforth designated vWF+ and vWF− HSCs) with MK and arteriolar niches in Vwf-eGFP mice. We prepared whole-mount sternal BM of Vwf-eGFP mice to image by 3D confocal immunofluorescence analyses where HSCs are identified with Lineage− CD48− CD150+ and in which eGFP marks both MK and a subset of HSCs in BM (Asada et al., 2017; Bruns et al., 2014; Kunisaki et al., 2013) (Figure 1A, 1B and S1A). Since 90% of the vWF-eGFP+ HSC subset express CD41 (Sanjuan-Pla et al., 2013), this marker was not included in the analyses and MK progenitors were excluded based on CD48 expression (Oguro et al., 2013), found on > 98% of MK progenitors (Figure S1B). vWF+ HSCs constitute a minor fraction of the Lineage− Sca1+ c-Kit+ (LSK) CD48− CD150+ HSC population (24.1 ± 1.4%, mean ± S.E.M.), as measured by FACS analysis (Figure S1C). However, 77.2 ± 9.9% of vWF+ HSCs were located within 5μm distance from MK (Figure 1C and S1D), with a considerable fraction (70.8 ± 17.2%) laying directly adjacent to MK (Figure 1F). By contrast to vWF− HSCs, vWF+ HSCs did not show any significant association with arterioles (identified by Sca-1High expression, morphology and small diameter) in the mouse BM. vWF+ HSC distribution was statistically different from that of lymphoid-biased vWF− HSCs relative to arterioles (Figure 1D–F and S1E), but statistically indistinguishable from random HSC-like dots (P = 0.9992; Figure 1D and 1E). These data suggest the existence of at least two spatially distinct BM niches for subsets of HSCs with distinct developmental potential.
Figure 1. Platelet- and myeloid-biased vWF+ HSCs are exclusively associated with megakaryocytes in the mouse bone marrow.

(A–B) Representative whole-mount images of Vwf-eGFP mouse sternal bone marrow (BM). The dashed outline in (A) denotes the bone–bone marrow border of a sternum piece. (B) White arrowheads denote a phenotypic Lineage− CD48− CD150+ vWF-eGFP+ HSC. MK are distinguished by their size, morphology and CD150 and vWF expression. Vascular endothelial cells are stained intravenously with antibodies to CD31 and CD144. (C–D) Localization of vWF− (black bars) and vWF+ (green bars) HSCs observed in situ, relative to MK (C) and arterioles (D) in the mouse BM of Vwf-eGFP mice. Yellow bars depict the mean distances between simulated randomly distributed HSCs and MK and arterioles. n = 282 vWF− HSCs and n = 37 vWF+ HSCs. The numbers on the x axis indicate intervals of 10 μm (C; where 5 indicates the interval −5 to 5, 15 indicates 5 to 15, and so on) and 20 μm (D; where 20 indicates the interval 0 to 20, 40 indicates 20 to 40, and so on). (E) P values determined by two-sample KS test (C and D). (F) Percentage of vWF− and vWF+ HSCs located in direct contact (0 μm) with MK or arterioles. Unpaired Student’s t tests. *P<0.05, **P<0.01. Error bars S.E.M. See also Figures S1.
vWF+, but not vWF−, HSC proliferation is regulated by megakaryocytes via CXCL4
To investigate the role of MK in vivo in lineage-biased HSC regulation, we generated Cxcl4-cre;iDTR;Vwf-eGFP mice. Control iDTR;Vwf-eGFP and triple transgenic Cxcl4-cre;iDTR;Vwf-eGFP mice were treated with diphtheria toxin (DT) (Figure 2A) to deplete MK as described (Bruns et al., 2014). Seven days after the initial dose, we evaluated the impact on HSC numbers. Consistent with previous results, MK depletion significantly increased the total number of phenotypic HSCs in BM (Bruns et al., 2014). Strikingly, we found that MK depletion selectively increased (by 11.4-fold, P < 0.0001) vWF+ HSC numbers whereas the number of vWF− HSCs modestly expanded 1.4-fold (Figure 2B–E). In addition, injection of recombinant CXCL4 into Vwf-eGFP mice selectively reduced the number of vWF+ HSCs in the BM while the number of vWF− HSCs was unaltered (Figure 2F and G), which is in line with its role in regulating HSC quiescence (Bruns et al., 2014). To investigate whether MK regulate HSCs directly, we cultured Lineage− cells in the presence or absence of CXCL4. Interestingly, we found that CXCL4 selectively reduced the number (Figure S2A) and proliferation (Figure S2B) of vWF+, but not vWF− HSCs. Evaluation of the bone marrow 3D distribution of vWF− and vWF+ HSCs in steady-state and MK-depleted mice revealed no significant alterations in their relationships with arterioles (Figure S2C and D). These data strongly suggest that MK can selectively regulate a subset of HSCs directly via CXCL4, although the contribution of other MK-derived factors and additional yet unidentified indirect effects emanating from the microenvironment cannot be excluded.
Figure 2. Platelet- and myeloid-biased vWF+ HSC proliferation and reconstitution potential are regulated by megakaryocytes.

(A) Experimental design to test the effect of MK depletion on vWF+ and vWF− HSCs. (B) Representative FACS plots of the frequency of vWF+ and vWF− HSCs in the BM of control (iDTR;Vwf-eGFP) and Cxcl4-cre;iDTR;Vwf-eGFP mice, 7 days after diphtheria toxin (DT) treatment. (C) BM cellularity, (D) absolute number of HSCs per femur and (E) number of HSCs in iDTR;Vwf-eGFP and Cxcl4-cre;iDTR;Vwf-eGFP mice normalized to control iDTR, 7 days after DT treatment. Control group n = 6 and Cxcl4-cre;iDTR;Vwf-eGFP n = 5. (F) Experimental design to test the effect of CXCL4 injection on vWF+ and vWF− HSCs. (G) BM cellularity and number of vWF+ and vWF− HSCs per femur in mice treated with PBS (n = 6) or CXCL4 (n = 4). Bone marrow mononuclear cells (BMNCs). (H–J) Contribution of 100 DAPI− LSK CD48− CD150+ vWF− and vWF+ HSCs from iDTR;Vwf-eGFP and Cxcl4-cre;iDTR;Vwf-eGFP mice after MK depletion, to peripheral blood (H) platelets (16 weeks post-transplantation), (I) myeloid, B and T cells and (J) overall reconstitution at 16 weeks. n = 8 (iDTR groups and Cxcl4-cre;iDTR vWF+ group) and n = 9 (Cxcl4-cre;iDTR vWF− group). *P<0.05, **P<0.01, ***P<0.001; ****P<0.0001. Error bars S.E.M. Unpaired Student’s t tests (C, E, G, H and J). One-way ANOVA analyses followed by Tukey’s multiple comparison tests were used for multiple group comparisons (D). See also Figures S2 and S3.
vWF+ HSC reconstitution potential is regulated by megakaryocytes
We next evaluated the impact of MK depletion on the ability of vWF− and vWF+ HSCs obtained from steady-state and MK-depleted mice to reconstitute hematopoietic lineages after transplantation (Figure S3A). Competitive transplantation of sorted LSK CD48− CD150+ vWF− and vWF+ HSCs revealed that vWF+ HSCs exhibited a bias toward repopulation of platelets (Figures 2H, S3B and S3C) and myeloid cells (Figure 2I and S3C) compared to vWF− HSCs, consistent with prior results (Sanjuan-Pla et al., 2013). The capacity to generate platelets and myeloid cells was preserved after MK depletion (Figure 2H and S3C). In fact, HSCs were more balanced in their lineage generation upon MK depletion (Figure 2I and S3C), suggesting the interesting possibility that MK can regulate HSC fate. While the overall long-term engraftment of vWF− and vWF+ HSCs was not statistically different after MK depletion (Figure 2J), the repopulation capacity of vWF+ HSCs from MK-depleted mice was reduced (Figure 2J, note the y axis values). This is likely due to their proliferative state as the long-term engraftment potential resides predominately in the non-proliferative G0 fraction of HSCs (Glimm et al., 2000; Passegue et al., 2005). In addition, analysis of the BM 16 weeks after transplantation revealed that whereas both HSC subsets from MK-depleted mice were capable to reconstitute the CD45.2+ LSK CD48− CD150+ vWF− HSC compartment (Figure S3D, right plots), the ability of vWF+ HSCs to reconstitute CD45.2+ LSK CD48− CD150+ vWF+ HSCs was compromised (Figure S3D), suggesting a self-renewal defect. Donor vWF− HSCs, harvested from either steady-state or MK-depleted mice, did not regenerate the vWF+ HSC compartment (Figure S3D), indicating that vWF− HSCs are downstream of vWF+ HSCs in the hematopoietic hierarchy (Sanjuan-Pla et al., 2013). These results suggest that MK selectively regulate both the proliferation and fate of the vWF+ HSC subset.
NG2+ arteriolar niche cells selectively regulate vWF− lymphoid-biased HSC quiescence and distribution in bone marrow
NG2+ periarteriolar cell depletion leads to an altered localization of the HSC subset associated with arterioles and increases the proportion of proliferative HSCs (Kunisaki et al., 2013). To investigate the impact of arteriolar niche depletion on the vWF+ and vWF− HSC subsets, we generated NG2-creERTM;iDTR;Vwf-eGFP mice. Triple-transgenic NG2-creERTM;iDTR;Vwf-eGFP and control iDTR;Vwf-eGFP mice were treated with tamoxifen and DT to induce NG2+ cell depletion, and analyzed 16 days later by FACS and whole-mount immunofluorescence imaging of the sternal BM (Figure 3A). We found that NG2+ cell depletion led to significant (~50%) reductions in the number of phenotypic LSK CD48− CD150+ lymphoid-biased vWF− HSCs compared to control iDTR mice (Figure 3B–E). However, by contrast to MK depletion, vWF+ HSC numbers were not significantly affected by NG2+ cells depletion and disruption of the arteriolar niche (Figure 3B–E). We next evaluated the spatial relationships of HSCs with MK and arterioles by 3D imaging. Interestingly, while NG2+ cell depletion did not affect the relationship of remaining vWF− (Figure 4A) or vWF+ HSCs (Figure 4B) with MK, it selectively altered vWF− HSC localization relative to arterioles (Figure 4C; P < 0.0001), but not that of vWF+ HSCs (Figure 4D). These results further suggest that MK and arterioles preferentially regulate distinct HSC subsets in the BM.
Figure 3. NG2+ arteriolar niche cells are critical regulators of the vWF− lymphoid-biased HSC quiescence and localization.

(A) Experimental design to test the effect of NG2+ cells depletion on vWF+ and vWF− HSCs. (B) Representative FACS plots of the frequency of vWF+ and vWF− HSCs in the BM of control (iDTR;Vwf-eGFP) and NG2-creERTM;iDTR;Vwf-eGFP mice, 16 days after DT treatment. (C) BM cellularity, (D) absolute number and (E) number of HSCs per femur in iDTR;Vwf-eGFP and NG2-creERTM;iDTR;Vwf-eGFP mice normalized to control iDTR, 16 days after DT treatment. Control group n = 9 and NG2-creERTM;iDTR;Vwf-eGFP n = 6. (F–H) Contribution of 100 DAPI− LSK CD48− CD150+ vWF− and vWF+ HSCs from iDTR and NG2-creERTM;iDTR mice after NG2+ cells depletion, to peripheral blood (F) platelets (16 weeks post-transplantation), (G) overall reconstitution and (H) myeloid, B and T cells at 16 weeks. n = 8 (iDTR groups) and n = 9 (NG2-creERTM;iDTR groups). (I) Summary of BM phenotypes of niche-structure-depleted mice in lineage-biased HSCs. Check marks indicate significant HSC/niche association and cross marks indicate no preferred association. N.D. – not determined due to MK depletion. *P<0.05, **P<0.01, ***P<0.001. Error bars S.E.M. Unpaired Student’s t tests (C, E, F and G). One-way ANOVA analyses followed by Tukey’s multiple comparison tests were used for multiple group comparisons (D). See also Figures S4.
Figure 4. Distribution of lineage-biased HSCs in the mouse BM after NG2+ cells depletion.

Localization of phenotypic Lineage− CD48− CD150+ vWF-eGFP− (A, C) and vWF-eGFP+ (B, D) HSCs in the mouse BM relative to MK (A, B) and arterioles (C, D) in control (iDTR;Vwf-eGFP) and NG2-creERTM;iDTR;Vwf-eGFP mice, 16 days after DT treatment. n = 172 vWF− and n = 32 vWF+ HSCs from control iDTR;Vwf-eGFP; n = 163 vWF− and n = 26 vWF+ HSCs from NG2-creERTM;iDTR;Vwf-eGFP. P values were determined by two-sample KS test. Error bars S.E.M.
Finally, we examined the reconstitution potential of sorted vWF− and vWF+ HSCs harvested from NG2+ depleted and control animals (Figure S4A). Peripheral blood lineage analyses of reconstituted mice with sorted vWF+ HSCs revealed a similar platelet- and myeloid-biased blood production from both control and NG2+ cell-depleted mice (Figure 3F and S4B). In addition, reconstitution of recipient mice with vWF− HSCs was biased toward the lymphoid lineage in both steady-state and NG2+ cell-depleted mice (Figure 3F–H and S4B). Furthermore, stable platelet-biased blood generation persisted for at least 16 weeks whether vWF+ HSCs were harvested from control or NG2+ cell-depleted animals (Figure 3F and S4B). Interestingly, whereas vWF− HSCs from steady-state mice repopulated, as expected, only the vWF− HSC compartment, instances of vWF+ repopulation (3 out of 9 recipient mice) were observed from vWF− HSCs harvested from NG2+ cell-depleted BM (Figure S4C, third panel).
These results argue for the presence of spatially separate perivascular niches in the mouse BM that regulate functionally distinct subsets of lineage-biased quiescent HSCs. Phenotypic HSCs, like Nestin-GFP+/LepR+/CAR niche cells, are distributed throughout the BM (Acar et al., 2015; Kunisaki et al., 2013; Omatsu et al., 2010). However, the number of niche cells, whether defined by Nestin, LepR or CXCL12 expression, vastly outnumbers that of HSCs (by > 20:1), underscoring the need, if the HSC niche concept is valid, for further stromal fractionation. We have previously suggested that the niche may be formed by the coupling of MSCs and HSCs (Mendez-Ferrer et al., 2010), an idea supported by the segregation of niche factor expression with MSC activity (Pinho et al., 2013). Further analyses fractionating and enriching MSC activity will be helpful to rule in or out this possibility. However, it is becoming clear that multiple cells in the microenvironment contribute to niche activity (Mendelson and Frenette, 2014). Among these are the arteriole-associated stromal cells, which confer HSC quiescence as shown by the depletion of NG2+ cells leading to HSC redistribution and proliferation (Kunisaki et al., 2013). The role of arterioles as a specific microenvironment for HSC maintenance has been questioned by other studies which have used other markers to identify HSC in situ (Acar et al., 2015), but confirmed by others (Itkin et al., 2016). The present studies, using specific disruption of arterial and MK niches, further document their selective contributions in regulating the maintenance of distinct subsets of HSCs.
Vwf-driven GFP marking has allowed for the first time to evaluate the spatial distribution of distinct lineage-biased HSC subsets in situ. Lineage bias in hematopoietic repopulation capacity has been retrospectively identified and defined by single HSC competitive transplantation experiments (Dykstra et al., 2007), but whether such lineage-biased HSCs occupy specific microenvironments or are uniformly distributed in the BM, remained unknown. Our studies show that myeloid-biased vWF+ HSCs are enriched in MK niches whereas lymphoid-biased vWF− HSCs are enriched in arteriolar niches (Figure 3I). This contention is supported by their localization, and perhaps more importantly, by selective disruption of homeostasis using conditional depletion of these niches with genetically engineered models. These results support the notion that HSC niches are not uniform in the BM but rather formed by complex microenvironments with input from several cellular constituents. Serial transplantations of HSCs with defined hues reproduced the same complex pattern in lethally irradiated recipients, suggesting that the lineage contribution array may be intrinsically programmed by epigenetic memory (Yu et al., 2016). Our studies, however, show for the first time that a BM niche constituent (i.e. MK) may also regulate HSC fate since MK deletion appears sufficient to reprogram vWF+ HSCs from myeloid-biased to balanced lineage contributions. As myeloid bias is a hallmark of HSC aging, understanding its molecular underpinnings may provide new armamentarium for the rejuvenation of aged HSCs.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Paul S. Frenette (paul.frenette@einstein.yu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6-Tg(Pf4-iCre)Q3Rsko/J (Cxcl4-cre), B6.Cg-Tg(Cspg4-cre/Esr1*)BAkik/J (NG2-creERTM) and C57BL/6-Gt(ROSA)26Sortm1(HBEGF)Awai/J (iDTR) mice were purchased from the Jackson Laboratory. Vwf-EGFP transgenic mice was generated using a 226 Kb BAC as described (Sanjuan-Pla et al., 2013). The BAC contained near all Vwf gene (110 Kb out of 130 Kb), and an additional 100 Kb upstream sequence. Most of the identified transcriptional regulatory elements of the Vwf gene elements are located within -5kb~5kb from the transcription start site. C57BL/6 (CD45.2) and Bl6-Ly5.1 (CD45.1) mice were purchased from the National Cancer Institute or the Jackson laboratory (B6.SJL-Ptprca Pepcb/BoyJ). Unless indicated otherwise, 8–12-week-old male and female mice were used. No randomization or blinding was used to allocate experimental groups. The Animal Care and Use Committees of Albert Einstein College of Medicine approved all experimental procedures.
METHOD DETAILS
In vivo treatments
For DT-mediated MK depletion, 250 ng of DT (Sigma) was injected intraperitoneally (i.p.) every 24 hours over 7 days. Mice were subjected to HSC analyses on the day of the last injection. For induction of NG2-creERTM-mediated recombination mice were injected i.p. with 2 mg tamoxifen (Sigma) dissolved in corn oil (Sigma) once a day during 5 consecutive days. For DT-mediated NG2+ cells depletion, 2 days after the last tamoxifen injection, mice were injected i.p. with 250 ng of DT (Sigma) once a day during 2 days. All treatments were similarly applied to control and cre;iDTR groups. CXCL4 (1 μg; PeproTech) or PBS vehicle were injected i.p. every 24 hours over 4 days.
Whole-mount immunofluorescence imaging
Whole-mount tissue preparation, HSC immunofluorescence staining and imaging analysis of the sternum were performed as described previously (Bruns et al., 2014; Kunisaki et al., 2013) with minor modifications. Briefly, bone marrow endothelial cells were stained by i.v. injection of anti-CD31/anti-CD144 or anti-Sca-1 antibodies and after 10 min mice were euthanized. Sternum bones were carefully harvested, cleaned and transected with a surgical blade into individual pieces. Each piece was bisected sagittally in order to expose the marrow cavity. Fragments were fixed with 4% PFA; blocked/permeabilized in PBS with 20% normal goat serum and 0.5% Triton X-100 and stained with primary antibodies (biotin anti-Lineage panel cocktail, biotin anti-CD48 and PE anti-CD150) for 3 days. The tissues were then incubated with streptavidin eFluor 450 for 2 hours. Images were acquired using a ZEISS AXIO examiner D1 microscope (Zeiss) with a confocal scanner unit, CSUX1CU (Yokogawa), and reconstructed in 3D with SlideBook software (Intelligent Imaging Innovations). For each vWF− or vWF+ HSC the Euclidean distance to the closest MK and arteriole was measured in order to generate distribution maps.
Computational modeling of random HSC localization
Computer simulations were performed as described previously (Bruns et al., 2014; Kunisaki et al., 2013) with Matlab software (MathWorks) and minor modifications. 2D z-stack images of whole mount sternal segments were used to generate a representative 3D spatial map of arterioles and MK onto which 20 spots representing HSCs were randomly placed. Thresholds were applied to generate binary maps, followed by median filtering to remove nonspecific background staining to accurately represent arteriole and MK localization within the marrow space. Boundaries of the bone-to-marrow interface were demarcated on the binary maps of MK and arterioles such that random HSC-like spots were only placed in the unoccupied marrow space. HSC simulation coordinates were randomly selected from the region within the boundary, and the shortest Euclidean distance was calculated for each spot to the nearest MK and arteriole. Each simulated run of 20 randomly placed HSC-like spots was repeated 1,000 times on images of arterioles and MK. 3D distance distributions of 2 randomly placed spots (in the same range of frequency of the rarer vWF+ subset) repeated 1,000 times were also generated. Since these random distributions were similar to the random 20-spot distribution, the data from 20 random spots was used in all distribution comparisons to simplify the presentation of results.
Antibodies
The following antibodies were used in this study: anti-Ly6A/E (Sca1) (D7), anti-CD48 (HM48-1), anti-CD41 (MWReg30), anti-CD45.2 (104), anti-CD45.1 (A20), anti-CD4 (GK1.5), anti-CD8a (53-6.7), anti-B220 (RA3-6B2), anti-Ki-67 (SolA15), anti-FcRII/III (93) and anti-Ter119 (TER-119) were all from eBioscience. Anti-CD150 (TC15-12F12.2), anti-CD117 (2B8), anti-CD11b (M1/70), anti-CD31 (MEC13.3), anti-CD144 (BV13) and anti-CD34 (MEC14.7) were from Biolegend. Anti-Lineage panel cocktail (TER-119, RB6-8C5, RA3-6B2, M1/70, 145-2C11 at 1:50 dilution) was from BD Biosciences. Streptavidin eFluor 450 and APC eFluor 780 from eBioscience and streptavidin BV 570 from Biolegend were used for biotinylated antibodies. Unless otherwise specified, all antibodies and streptavidin cocktails were used at a 1:100 dilution.
Flow cytometry and cell sorting
Flow cytometric analyses were carried out in single-cell suspensions of nucleated cells enriched from the peripheral blood or flushed bone marrow using an LSRII flow cytometer (BD Biosciences). Cell sorting experiments were performed using a FACSAria Cell Sorter (BD Biosciences). Dead cells and debris were excluded by FSC, SSC and DAPI (4′,6-diamino-2-phenylindole) staining profiles. Data were analysed with FlowJo (Tree Star) or FACS Diva 6.1 software (BD Biosciences).
Cell cycle analyses
For cell cycle analyses, cells were stained with HSC surface markers, fixed and permeabilized with the Foxp3/Transcription factor staining buffer set from eBioscience according to manufacturer’s instructions, and stained with anti-Ki-67 antibody and Hoechst 33342 (Sigma) at 20 μg ml−1 for 30 min. After washing, cells were immediately analyzed in a LSRII Flow Cytometer (BD Biosciences).
Cell culture
For in vitro analyses of HSCs, lineage-depleted bone marrow cells were isolated using streptavidin microbeads (Miltenyi Biotech) and cultured for 4 days in StemSpan medium (StemCell Technologies) supplemented with SCF (10 ng ml−1) and fibroblast growth factor 1 (FGF-1) (10 ng ml−1 all PeproTech). Recombinant mouse CXCL4 (100 ng ml−1; ProsPec) or PBS was added to assess CXCL4 effects on lineage-biased HSCs.
Bone marrow transplantation
Competitive repopulation assays were performed using the CD45.1/CD45.2 congenic system. Recipient mice were lethally irradiated (12 Gy, two split doses) in a Cesium Mark 1 irradiator (JL Shepherd & Associates). 100 vWF− or vWF+ CD45.2+ HSCs (defined as LSK CD48− CD150+) were isolated from the bone marrow of Cxcl4-cre;iDTR;Vwf-eGFP or from NG2-creERTM;iDTR;Vwf-eGFP and respective littermate control iDTR;Vwf-eGFP mice. Sorted HSCs were intravenously transplanted together with 0.25 × 106 competitor CD45.1+ total bone marrow mononuclear cells (BMNCs). Before transplantation, dead cells and debris were excluded by FSC, SSC and DAPI (4′,6-diamino-2-phenylindole) staining profiles. CD45.1/CD45.2 chimerism of recipient blood was analysed up to 43months after transplantation using FACS. Blood was harvested by submandibular route and collected in polypropylene tubes containing EDTA. Mice showing more than 1% donor reconstitution in the myeloid (CD11b+), B-cell (B220+) and T-cell (CD4+ CD8+) lineages after 16 weeks were considered engrafted. The percentage of platelet chimerism was calculated as previously described (Sanjuan-Pla et al., 2013) briefly whole peripheral blood was centrifuged for 103min at 100g and platelet-containing supernatant was stained. Plasma-derived platelets were identified based on scatter properties, absence of Ter119 and CD41, CD150 and eGFP expression.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are represented as mean ± S.E.M. Comparisons between two samples were done using the unpaired Student’s t tests. One-way ANOVA analyses followed by Tukey’s multiple comparison tests were used for multiple group comparisons. Two-sample Kolmogorov–Smirnov (KS) tests were used for comparisons of distribution patterns. Statistical analyses were performed with GraphPad Prism. *P3<30.05, **P3<30.01, ***P3<30.001, ****P3<30.0001. Specific statistical parameters (e.g., number of animals used) can be found in the figure legends.
Supplementary Material
HIGHLIGHTS.
Platelet- and myeloid-biased vWF+ HSCs associate with MK, but not arteriolar niches
vWF+, but not lymphoid-biased vWF− HSC proliferation, is regulated by MK
vWF+ HSC reconstitution potential is regulated by MK
NG2+ arteriolar niches selectively regulate vWF− HSC quiescence and localization
Acknowledgments
We would like to thank Colette Prophete and Paul Ciero for technical assistance; Lydia Tesfa and the Einstein Flow Cytometry Core Facility for expert cell sort assistance; Sten Eirik W. Jacobsen for providing the Vwf-eGFP mice; and A. Zahalka and M. Maryanovich for helpful scientific discussions. S.P. was supported by a New York Stem Cell Foundation-Druckenmiller Fellowship; T.M. is supported by the Fondation ARC pour la Recherche sur le Cancer and by the Société Française d’Hématologie. This work was supported by the New York State Department of Health (NYSTEM Program), by the New York Stem Cell Foundation and by R01 grants from the National Institutes of Health (DK056638, HL116340, HL097819 to P.S.F.). The authors have no conflicting financial interests.
Footnotes
AUTHOR CONTRIBUTIONS
S.P. designed the study, performed the majority of the experiments and analysed data. T.M. helped with the whole-mount imaging experiments and analysed data. E.Y. performed computational modelling. Q.W. performed FACS analyses. C.N. provided the Vwf-eGFP mice and advised on experimental setup. P.S.F. supervised the study. S.P. and P.S.F. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, Jaiyeola C, Zhao Z, Luby-Phelps K, Morrison SJ. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. 2015;526:126–130. doi: 10.1038/nature15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asada N, Kunisaki Y, Pierce H, Wang Z, Fernandez NF, Birbrair A, Ma’ayan A, Frenette PS. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat Cell Biol. 2017;19:214–223. doi: 10.1038/ncb3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, Rossi DJ. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:5465–5470. doi: 10.1073/pnas.1000834107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, Scheiermann C, Schiff L, Poncz M, Bergman A, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nature medicine. 2014;20:1315–1320. doi: 10.1038/nm.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Miyanishi M, Wang SK, Yamazaki S, Sinha R, Kao KS, Seita J, Sahoo D, Nakauchi H, Weissman IL. Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature. 2016;530:223–227. doi: 10.1038/nature16943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RH, Sieburg HB, Muller-Sieburg CE. A new mechanism for the aging of hematopoietic stem cells: aging changes the clonal composition of the stem cell compartment but not individual stem cells. Blood. 2008;111:5553–5561. doi: 10.1182/blood-2007-11-123547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copley MR, Beer PA, Eaves CJ. Hematopoietic stem cell heterogeneity takes center stage. Cell Stem Cell. 2012;10:690–697. doi: 10.1016/j.stem.2012.05.006. [DOI] [PubMed] [Google Scholar]
- Dykstra B, Kent D, Bowie M, McCaffrey L, Hamilton M, Lyons K, Lee SJ, Brinkman R, Eaves C. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell. 2007;1:218–229. doi: 10.1016/j.stem.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. The Journal of experimental medicine. 2011;208:2691–2703. doi: 10.1084/jem.20111490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekas C, Graf T. CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood. 2013;121:4463–4472. doi: 10.1182/blood-2012-09-457929. [DOI] [PubMed] [Google Scholar]
- Glimm H, Oh IH, Eaves CJ. Human hematopoietic stem cells stimulated to proliferate in vitro lose engraftment potential during their S/G(2)/M transit and do not reenter G(0) Blood. 2000;96:4185–4193. [PubMed] [Google Scholar]
- Grover A, Sanjuan-Pla A, Thongjuea S, Carrelha J, Giustacchini A, Gambardella A, Macaulay I, Mancini E, Luis TC, Mead A, et al. Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat Commun. 2016;7:11075. doi: 10.1038/ncomms11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, Ledergor G, Jung Y, Milo I, Poulos MG, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature. 2016;532:323–328. doi: 10.1038/nature17624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent DG, Copley MR, Benz C, Wohrer S, Dykstra BJ, Ma E, Cheyne J, Zhao Y, Bowie MB, Zhao Y, et al. Prospective isolation and molecular characterization of hematopoietic stem cells with durable self-renewal potential. Blood. 2009;113:6342–6350. doi: 10.1182/blood-2008-12-192054. [DOI] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, Mizoguchi T, Wei Q, Lucas D, Ito K, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502:637–643. doi: 10.1038/nature12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumbe AP, Ramasamy SK, Itkin T, Mae MA, Langen UH, Betsholtz C, Lapidot T, Adams RH. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature. 2016;532:380–384. doi: 10.1038/nature17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nature medicine. 2014;20:833–846. doi: 10.1038/nm.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, Frenette PS. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Sieburg CE, Cho RH, Thoman M, Adkins B, Sieburg HB. Deterministic regulation of hematopoietic stem cell self-renewal and differentiation. Blood. 2002;100:1302–1309. [PubMed] [Google Scholar]
- Muller-Sieburg CE, Sieburg HB, Bernitz JM, Cattarossi G. Stem cell heterogeneity: implications for aging and regenerative medicine. Blood. 2012;119:3900–3907. doi: 10.1182/blood-2011-12-376749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura-Ishizu A, Takubo K, Fujioka M, Suda T. Megakaryocytes are essential for HSC quiescence through the production of thrombopoietin. Biochem Biophys Res Commun. 2014;454:353–357. doi: 10.1016/j.bbrc.2014.10.095. [DOI] [PubMed] [Google Scholar]
- Oguro H, Ding L, Morrison SJ. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell. 2013;13:102–116. doi: 10.1016/j.stem.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, Nagasawa T. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010;33:387–399. doi: 10.1016/j.immuni.2010.08.017. [DOI] [PubMed] [Google Scholar]
- Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. The Journal of experimental medicine. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S, Lacombe J, Hanoun M, Mizoguchi T, Bruns I, Kunisaki Y, Frenette PS. PDGFRalpha and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. The Journal of experimental medicine. 2013;210:1351–1367. doi: 10.1084/jem.20122252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan-Pla A, Macaulay IC, Jensen CT, Woll PS, Luis TC, Mead A, Moore S, Carella C, Matsuoka S, Jones TB, et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature. 2013 doi: 10.1038/nature12495. [DOI] [PubMed] [Google Scholar]
- Shin JY, Hu W, Naramura M, Park CY. High c-Kit expression identifies hematopoietic stem cells with impaired self-renewal and megakaryocytic bias. The Journal of experimental medicine. 2014;211:217–231. doi: 10.1084/jem.20131128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieburg HB, Cho RH, Dykstra B, Uchida N, Eaves CJ, Muller-Sieburg CE. The hematopoietic stem compartment consists of a limited number of discrete stem cell subsets. Blood. 2006;107:2311–2316. doi: 10.1182/blood-2005-07-2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu VW, Yusuf RZ, Oki T, Wu J, Saez B, Wang X, Cook C, Baryawno N, Ziller MJ, Lee E, et al. Epigenetic Memory Underlies Cell-Autonomous Heterogeneous Behavior of Hematopoietic Stem Cells. Cell. 2016;167:1310–1322. e1317. doi: 10.1016/j.cell.2016.10.045. [DOI] [PubMed] [Google Scholar]
- Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, Ahamed J, Li L. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nature medicine. 2014;20:1321–1326. doi: 10.1038/nm.3706. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.