

Summary
The Facilitates Chromatin Transcription (FACT) complex is involved in chromatin remodeling during transcription, replication, and DNA repair. FACT was previously considered to be ubiquitously expressed and not associated with any disease. However, we discovered that FACT is the target of a novel class of anti-cancer compounds and is not expressed in normal cells of adult mammalian tissues, except for undifferentiated and stem-like cells. Here, we show that FACT expression is strongly associated with poorly differentiated aggressive cancers with poor overall survival. In addition, FACT was found to be upregulated during in vitro transformation and to be necessary, but not fully sufficient, to drive transformation. FACT also promoted survival and growth of established tumor cells. Genome-wide mapping of chromatin-bound FACT indicated that FACT’s role in cancer likely involves selective chromatin remodeling of genes that stimulate proliferation, inhibit cell death and differentiation, and regulate cellular stress responses.
Introduction
The Facilitates Chromatin Transcription (FACT) complex is a heterodimer of two subunits: Structure Specific Recognition Protein 1 (SSRP1) and Suppressor of Ty (SPT16). FACT plays a role in chromatin remodeling by modulating nucleosome stability (Reinberg and Sims, 2006; Singer and Johnston, 2004) and has been implicated in multiple processes involving chromatin, including transcription and DNA replication, recombination and repair. (Saunders et al., 2003), (Belotserkovskaya et al., 2003; Birch et al., 2009), (Tan et al., 2006; Tan et al., 2010; Zhou and Wang, 2004), (Kumari et al., 2009), (Heo et al., 2008; Keller et al., 2001) (Ikeda et al., 2011). Our recent discovery that FACT is the molecular target of a new class of anti-cancer compounds, Curaxins, provided the first indication that FACT might play a role in cancer (Gasparian et al., 2011). This possibility is supported by our findings that FACT is expressed at higher levels in tumor cell lines than in normal cells in vitro and that RNAi-mediated knockdown (KD) of FACT expression leads to reduced growth and survival of tumor cells (Gasparian et al., 2011). In addition, FACT expression was found to be elevated during development of mammary carcinomas in transgenic mice expressing the Her2/neu proto-oncogene (Koman et al., 2012). FACT’s pattern of expression in normal (non-tumor) cells is also consistent with a possible role in tumorigenesis. While FACT was previously considered a ubiquitously expressed housekeeping factor (reviewed in (Singer and Johnston, 2004), we did not detect SSRP1 or SPT16 expression in normal organs of adult humans or mice, with the exception of some cell types in hematological and reproductive organs and intestinal crypts (Garcia et al., 2011). Analysis of publically available gene expression data from multiple studies revealed that FACT is expressed at high levels in undifferentiated stem and progenitor cells in different organs and that its expression decreases upon differentiation (Garcia et al., 2011).
Herein, we confirm the association between FACT and cancer by showing that FACT expression increases during in vitro transformation of normal cells and is functionally required for transformation as well as tumor cell survival and growth. We showed that FACT is frequently expressed in multiple different types of tumors and established a statistically significant association between the frequency and level of SSRP1 and tumor aggressiveness. To address the mechanism(s) by which FACT facilitates tumor growth, we assessed genome wide distribution of FACT binding to chromatin in tumor cells. This identified a subset of genes that are likely dependent upon FACT for expression and that have activities associated with malignant and stem-like properties of tumor cells and cellular stress responses.
Results
FACT is elevated during in vitro transformation
To test the hypothesis that FACT plays a role in tumorigenesis, we compared SSRP1 and SPT16 protein levels in cultured cells of mesenchymal or epithelial origin representing different stages of (in vitro) transformation: finite lifespan, immortalized, or transformed. There was essentially no change in FACT levels between normal human fibroblasts and fibroblasts immortalized with human telomerase or between mouse primary fibroblasts from p53 wild-type (finite) or knockout (immortalized) animals (Fig. S1A). However, when we transformed immortalized fibroblasts of either human or mouse origin with activated H-RasV12 oncogene, we observed a dramatic increase in FACT levels (Fig. S1 B and C). Importantly, the fibroblasts (finite lifespan, immortalized, or transformed) did not have significantly different proliferation rates; therefore, FACT upregulation was not a reflection of increased cell proliferation.
To model epithelial cell transformation, we used previously described human mammary epithelial cell (HMEC) strains from reduction mammoplasty specimen (Garbe et al., 2009) and isogenic immortalized and transformed lines derived from these cells via exposure to the chemical carcinogen benzo(a)pyrene (Stampfer and Bartley, 1985) or expression of shRNA against CDKN2A (p16) and/or the cDNA of proto-oncogene c-MYC (Brenner et al., 1998), respectively (Fig. 1). The parental (normal) HMEC strains (184) showed almost no nuclear SSRP1 staining, while transformed derivatives capable of anchorage independent growth (AIG) (184FMY2 and 184AA3) were strongly SSRP1-positive (Fig 1A). Immortalized lines not capable of AIG displayed weak but detectable SSRP1 staining. Increased SSRP1 and SPT16 expression in successive stages of in vitro transformation was confirmed by both Western blotting (Fig. 1B) and qRT-PCR (Fig. 1C). Analysis of PCNA protein expression showed that these differences were not due to differences in proliferation (Fig. 1B).
Figure 1.
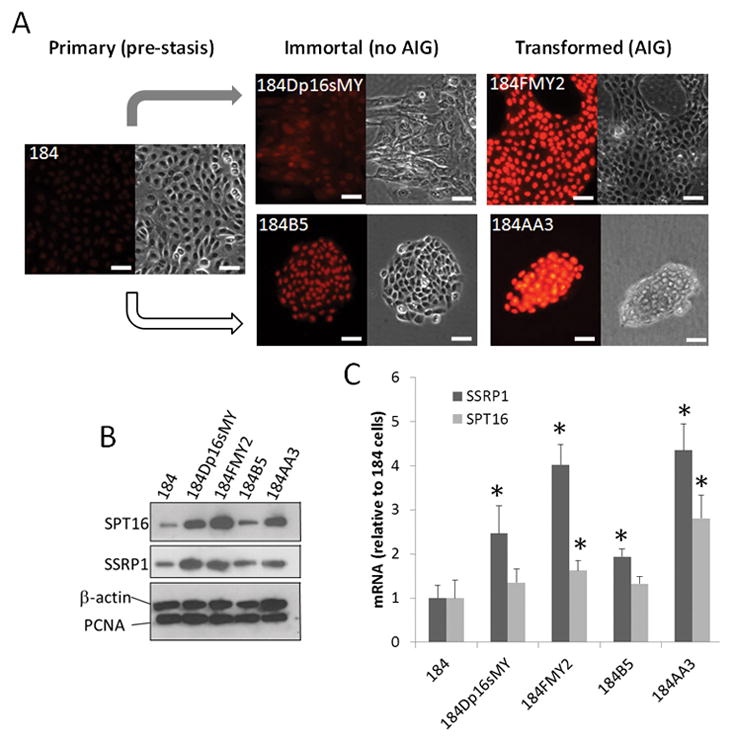
FACT subunits levels are elevated in the process of transformation of HMEC using genetic (grey arrow) or chemical (white arrow) manipulations. Primary (184), immortal (184Dp16sMY, 184B5) and fully transformed (184FMY2, 184AA3) cells were assessed by (A) immunofluorescent staining with antibodies to SSRP1; (B) Western blotting with the indicated antibodies; and (C) qRT-PCR analysis of total RNA with primers specific to SSRP1, or SPT16 or 18S rRNA (loading control). Data in (C)were normalized based on the level of 18S rRNA and are shown relative to the level of the corresponding transcripts in 184 cells (set at 1.0). Bars indicate the mean of three replicates + SDV. * indicates p<0.05 for comparison to 184 cells. See also Fig. S1.
FACT expression is required for transformation and for tumor cell survival and growth
To determine the functional importance of FACT elevation during transformation, we evaluated how changes in FACT levels affected the efficiency of H-RasV12-induced transformation of fibroblasts and epithelial cells. We transduced p53−/− MEF or MCF10A (immortalized non-transformed HMEC) with lentiviral H-RasV12 together with either expression constructs for both FACT subunits or shRNA targeting them. In both cell types, the efficiency of transformation was increased by enforced FACT expression and decreased by FACT knockdown (KD). However, there were some cell type-specific differences. While MEFs proliferated equally well in 2-D culture with or without elevated FACT, growth of epithelial cells was induced by FACT overexpression (Fig. 2A). Moreover, transduction of MCF10A cells with H-Ras V12 led to massive appearance of enlarged flat vacuolated senescent-like cells and a minor population of small, growing, transformed-looking cells which became the majority after replating (Fig. 2A, panel “H-RasV12+empty vectors”). Overexpression of FACT together with H-Ras V12 significantly increased the proportion of actively growing transformed-like cells, which quickly became predominant even without passaging (Fig. 2A, panel “H-RasV12+SSRP1/SPT16”). Transduction of H-RasV12 into fibroblast and epithelial cells leads to appearance of cells able to grow in semisolid medium and in vivo in animals. FACT overexpression significantly increased the proportion of these cells (Fig. S1D, E and 2B), while FACT KD almost completely eliminated them (Fig. 2C, D). Importantly, overexpression of FACT alone (without H-Ras V12) was not sufficient to induce MEF or MCF10A cells to grow in semisolid media (Fig. S1D, E and 2B). These data suggest that FACT promotes, but it not sufficient on its own to drive, cellular transformation.
Figure 2.
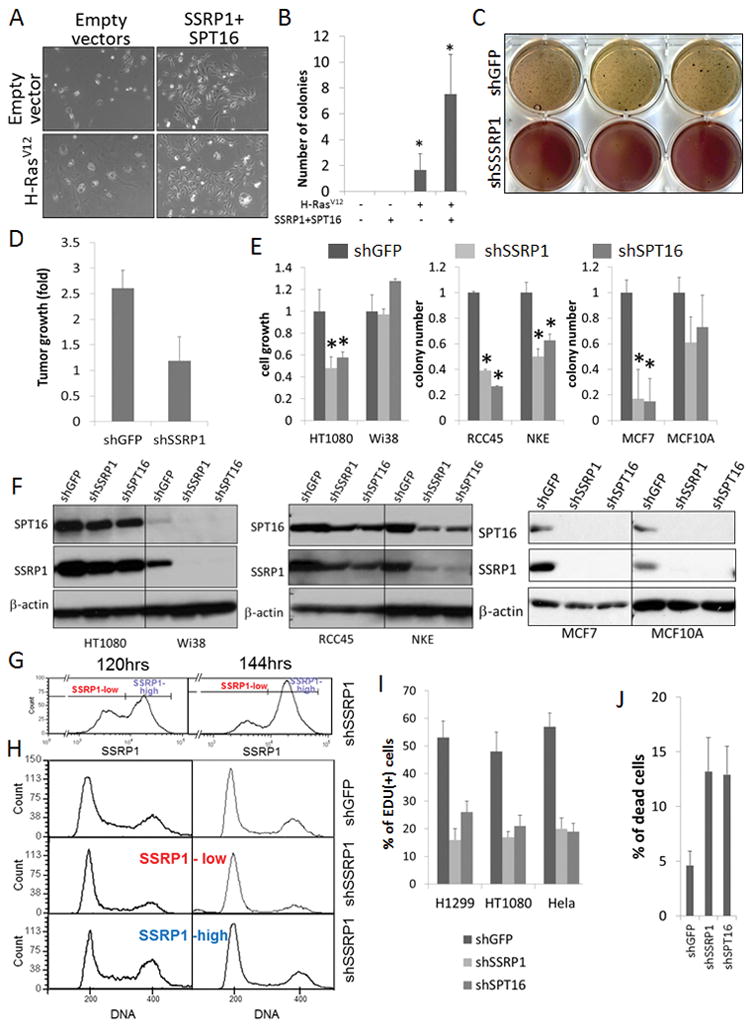
Transformation and tumor, but not normal, cell growth require FACT expression. A–B. Overexpression of FACT increases the efficiency of transformation of MCF10A cells by H-RasV12. A. Microphotographs of 2D colonies 6 days after transduction of cells with the indicated constructs. B. Number of colonies in semi-solid medium for cells transduced with the indicated constructs or empty vectors (-), the mean of triplicates + SDV; * indicates p<0.05 for comparison to cells transfected with both empty vectors. C–D. KD of SSRP1 suppresses H-RasV12induced transformation of MCF10A cells. C. MTT-stained colonies in semi-solid medium in triplicate wells grown for 37 days after transduction with shRNAs. The darker color of shSSRP1 wells is due to unreduced MTT. D. Growth of tumors (n=10) in SCID mice 30 days after inoculation of mice with MCF10A cells transduced with the indicated shRNAs (tumor volume at day 1 after inoculation =1). E. Growth of tumor (HT1080, RCC45, MCF7) and non-transformed (WI38, NKE, MCF10A) cells after shRNA transduction/puromycin selection. Bars show the means of triplicates of methylene blue staining (HT1080/Wi38) or colony number (RCC45/NKE, MCF7/MCF10A) +/− SDV, normalized to shGFP data in the same cell type. * indicates p<0.05. F. Western blot detection of FACT subunits in the cells described in E after puromycin selection. G. FACS analysis of SSRP1 staining in HT1080 cells 120 and 144 hrs after transduction with shSSRP1. H. Cell cycle distribution (FACS with DAPI staining) of HT1080 cells 120 (left column) and 144 hrs (right) after transduction with shGFP or shSSRP1, with the latter population separated based on SSRP1 staining as shown in G. I. EDU incorporation indicative of DNA replication 3 days after transduction of cells with the indicated shRNAs. * indicates p<0.05 for comparison to data with shGFP transduction in the same cells. J. Proportion of dead cells detected using Annexin V and propidium iodide staining (double positive) among HT1080 cells 5 days after transduction with the indicated shRNAs. See also Fig. S2.
To test if FACT is also essential for established transformed cells, we compared the effects of FACT KD on growth of pairs of tumor and non-transformed “normal” cells of the same tissue (fibroblasts, kidney and mammary epithelia, Fig. 2E). It should be noted that unlike primary normal cells in vitro or in vivo, all of tested established cell lines (transformed and non-transformed) express both FACT subunits (Fig. 2F). Since a parallel study demonstrated co-regulation of SSRP1 and SPT16 levels, shRNA against either FACT subunit effectively eliminated both SSRP1 and SPT16 (Safina et al., 2013). We found that FACT KD suppressed growth of all tumor cells, but had a smaller or no effect on growth of non-transformed cells (Fig. 2E). For two out of three cell pairs (kidney and fibroblasts cells), non-transformed cells surviving shRNA transduction showed effective FACT KD, while corresponding tumor cells did not (Fig. 2F). These data suggested that unlike non-transformed cells, tumor cells cannot grow in the absence of FACT. This was subsequently confirmed in the MCF7 (tumor) / MCF10A (non-tumor) cell pair through comparison of cell growth and FACT expression at different times after transduction of shSSRP1 or shSPT16 (Fig. S2).
Further illustrating that FACT is required for tumor cell growth, immunofluorescent staining of shSSRP1-transduced cell cultures revealed that the proportion of cells with low SSRP1 levels decreases with time (Fig. 2G). Moreover, tumor cells with low FACT levels had reduced replication rates (Fig. 2H, I) accumulated in G1 (Fig. 2H), and some died (Fig. 2G red arrow and Fig. 2J). While these data support a role for FACT in DNA replication, the absence of S-phase arrest (which would be expected if FACT is needed only for replication) suggests that signaling leading to G1 arrest and/or other FACT-dependent processes (e.g., transcription) may also be vital for tumor cells.
Chromatin-embedded FACT is enriched at genes associated with cancer and cell pluripotency
The known activities of FACT suggest that it may promote tumor growth by altering chromatin in a way that facilitates transcription of genes important for transformation. FACT does not affect general transcription (Fig. S3A–C), but has been shown to be required for transcription driven by particular transcription factors (TF) such as NF-κB (Gasparian et al., 2011), the activity of which is critical for many types of tumor cells (Gudkov et al., 2011), To identify other FACT-dependent transcriptional programs or genes, we used chromatin immunoprecipitation (ChIP) to examine the distribution of chromatin-bound FACT in HT1080 tumor cells, the growth and survival of which requires FACT (Fig. 2E–J). Three independent ChIP experiments were performed on unsynchronized, growing HT1080 with anti-SSRP1 antibodies shown to be highly specific (LC/MS of iimunoprecipitated complex) and not interfere with either SSRP1/SPT16 association or binding of FACT to chromatin (Fig. S4 and ref. (Gasparian et al., 2011)). As a specificity control for anti-SSRP1 ChIP, we used cells treated with the small molecule Curaxin (CX-137), which causes depletion of FACT from sites of active transcription (Gasparian et al., 2011).
NGS sequencing of DNA fragments that co-precipitated with SSRP1 revealed a non-random genomic distribution of SSRP1 in HT1080 cells (Fig. 3 and S5). 47% of SSRP1 peaks occurred near protein-coding genes, a distribution that is significant relative to a random target list (p<0.0001). FACT distribution in relation to genome features is shown in Fig. 3A and to TSS in Fig. S5B. Gene-associated SSRP1 peaks were much more similar to broad RNAPII peaks than to sharp peaks of sequence-specific TF (Fig. S5C). CX treatment substantially reduced association of FACT with genes (Fig. 3A), confirming our previous findings that CX treatment depletes FACT from areas of gene transcription (Gasparian et al., 2011). As expected, SSRP1 bound NF-κB-dependent genes, and this binding was reduced after CX treatment ((Gasparian et al., 2011) and Fig. S6). In total, we identified 2085 genes in HT1080 cells with significant enrichment of SSRP1 over background (Table S1). For 93% of these genes, SSRP1 binding was reduced (≥2 fold) after CX treatment. To strengthen our gene enrichment analysis, we selected 267 genes with SSRP1 binding >10-fold over background (200kB around the TSS) that was significantly CX-sensitive (Table S1).
Figure 3.
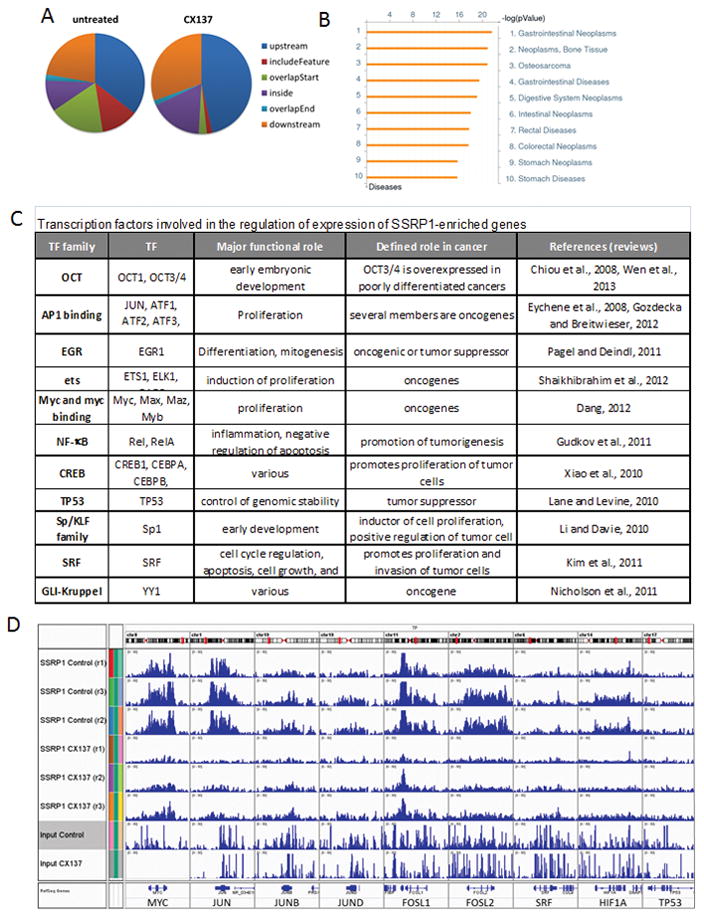
Analysis of genome-wide distribution of SSRP1 in tumor cells. A. MACS statistics of the distribution of SSRP1 tags in relation to genomic features in HT1080 cells untreated or treated with 3μM CX-137 for 1 hr. B. GeneGo analysis of association of SSRP1-enriched genes with diseases; p-values are shown, FDR<0.05. C. Families of TF involved in regulating expression of SSRP1-enriched genes (see details in the text and full lists in Table S1 and S2). D. Enrichment of SSRP1 binding to TF genes. Data are shown as alignments of SSRP1-bound DNA sequencings from three independent ChIP experiments with HT1080 cells left untreated (control, replicates r1-3) or treated with curaxin (CX-137, replicates r1-3) visualized using IGV. See also Fig. S3–S6.
Functional annotation of the list of SSRP1-enriched genes was accomplished by assessing overlap with the Molecular Signature Database (MSigDB, Broad Institute, Harvard University, MIT) curated gene lists. We obtained 52 lists with significant overlap (p<1.0E-5, FDR<0.05), which we divided into several functional categories (Table 1 and S2): (1) MYC–related; (2) stress-induced (3 lists of genes induced by UV, 3 lists of genes induced by hypoxia, and a list of genes induced by TNF or genotoxic drugs); (3) cancer-related (6 lists of genes up- and 4 lists of genes down-regulated in cancer vs normal samples or in high-grade vs low-grade cancers); (4) meiosis and ribosome-related, (5) growth factor-induced, (6) associated with de-differentiation; and (7) miscellaneous (including genes associated with system lupus erythematous (chronic inflammation), genes involved in the cell cycle, genes bound or up-regulated by E2F transcription factors, and several other categories). This set of functional attributes suggests that FACT may be important for regulating expression of genes that stimulate proliferation, inhibit differentiation and/or control stress responses.
Table 1.
Curated gene lists from MSigDB significantly overlapping with the list of SSRP1-enriched genes (only lists with p<0.00E-6 are shown) organized in functional categories. See also Tables S1–S4.
| Functional category | Gene Set Name | p-value |
|---|---|---|
|
| ||
| MYC-related | DANG BOUND BY MYC | 0.00E+00 |
| DANG MYC TARGETS UP | 0.00E+00 | |
| BENPORATH MYC TARGETS WITH EBOX | 5.58E-14 | |
| BENPORATH MYC MAX TARGETS | 1.96E-12 | |
| GGGAGGRR V$MAZ Q6 | 8.47E-09 | |
| LEI MYB TARGETS | 9.08E-09 | |
|
| ||
| Stress-induced | ENK UV RESPONSE KERATINOCYTE UP | 0.00E+00 |
| DAZARD RESPONSE TO UV NHEK UP | 1.11E-16 | |
| KRIEG HYPOXIA NOT VIA KNOCKDOWNM3A | 1.75E-10 | |
| HU GENOTOXIC DAMAGE 24HR | 2.56E-09 | |
| WINTER HYPOXIA METAGENE | 7.04E-09 | |
| HARRIS HYPOXIA | 1.41E-08 | |
| PHONG TNF TARGETS UP | 2.05E-08 | |
| DAZARD UV RESPONSE CLUSTER G2 | 2.25E-08 | |
|
| ||
| Cancer-related | WANG TUMOR INVASIVENESS UP | 0.00E+00 |
| GRADE COLON CANCER UP | 1.11E-16 | |
| OSMAN BLADDER CANCER DN | 3.89E-15 | |
| CHNG MULTIPLE MYELOMA HYPERPLOID UP | 6.55E-15 | |
| LI AMPLIFIED IN LUNG CANCER | 7.78E-11 | |
| ZUCCHI METASTASIS DN | 7.31E-10 | |
| NUTT GBM VS AO GLIOMA DN | 9.06E-10 | |
| SWEET LUNG CANCER KRAS UP | 2.82E-09 | |
| ACEVEDO LIVER CANCER DN | 3.51E-09 | |
| DIAZ CHRONIC MEYLOGENOUS LEUKEMIA DN | 4.35E-08 | |
|
| ||
| Meiosis | REACTOME MEIOSIS | 0.00E+00 |
| REACTOME MEIOTIC RECOMBINATION | 0.00E+00 | |
| REACTOME MEIOTIC SYNAPSIS | 3.15E-14 | |
|
| ||
| Ribosome | KEGG RIBOSOME | 0.00E+00 |
| MIPS RIBOSOME CYTOPLASMIC | 0.00E+00 | |
| MIPS 60S RIBOSOMAL SUBUNIT CYTOPLASMIC | 0.00E+00 | |
| MIPS 40S RIBOSOMAL SUBUNIT CYTOPLASMIC | 2.22E-16 | |
|
| ||
| Stimulated by growth factors | NAGASHIMA EGF SIGNALING UP | 8.58E-13 |
| AMIT EGF RESPONSE 40 HELA | 1.58E-11 | |
| NAGASHIMA NRG1 SIGNALING UP | 6.56E-11 | |
| PEDERSEN METASTASIS BY ERBB2 ISOFORM 1 | 1.12E-09 | |
|
| ||
| Chromatin organization | REACTOME DEPOSITION OF NEW CENPA CONTAINING | 1.11E-16 |
| NUCLEOSOMES AT THE CENTROMERE | 3.33E-12 | |
| REACTOME CHROMOSOME MAINTENANCE | ||
|
| ||
| Differentiation | BENPORATH SOX2 TARGETS | 4.74E-09 |
| ESC V6.5 UP EARLY.V1 DN | 7.69E-07 | |
| ESC J1 UP EARLY.V1 DN | 9.86E-06 | |
As shown previously for NF-κB, FACT may control expression of the SSRP1-associated genes through interactions with particular TF. To identify such TF, we compared our list of SSRP1-enriched genes with (i) a list of genes with promoters containing sequence elements known as TF binding sites using MSigDB (Table S3), and (ii) lists of TF target sequences known from the literature using GenGo (Thomson Reuters) (Table S4). TF identified by both methods are shown in Fig. 3C. Most have well established associations with cancer or embryonic development; importantly, all except one (TP53) promote tumor growth as oncogenes (MYC, JUN, Ets-family, YY1), inducers of cell proliferation (SP1, CREB, SRF), suppressors of apoptosis (NF-κB), or inhibitors of cell differentiation (OCT1, OCT3/4). Moreover, analysis of associations of SSRP1-enriched genes with disease states using GeneGo showed that most significant associations were with different types of neoplasms (Fig. 3B).
In addition, we found that genes for several TF including MYC, JUN, JUNB, JUND, FOSL1 and FOSL2 (but not TP53) were themselves significantly “SSRP1-enriched” (Fig. 3D). Thus, FACT may affect expression of some TF themselves in addition to their targets.
FACT subunits are overexpressed in multiple types of tumors
To evaluate the clinical significance of our in vitro findings, we compared SSRP1 and SPT16 mRNA levels in human tumor and normal tissue using publically available high-content microarray data and IST Online software (MediSapiens Ltd) for trans-technology and trans-study normalization. This revealed that SSRP1 mRNA, while showing significant variability among different samples, was elevated in the majority of tumors as compared to tissue from patients with no disease or non-cancer related diseases (Fig. 4A). Cultured cell lines included in the analysis had the highest average level of SSRP1 of any category (Fig. 4A), suggesting that in vitro conditions either induce SSRP1 expression or select cells with elevated SSRP1.
Figure 4.
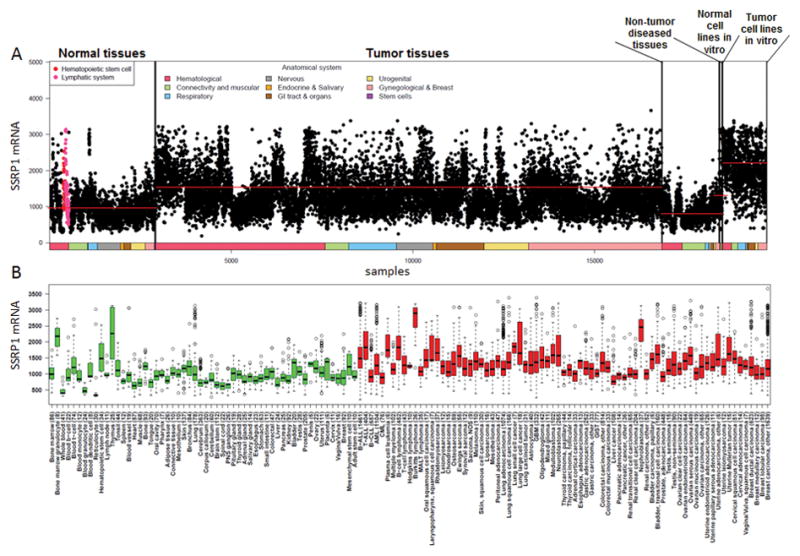
SSRP1 mRNA expression in patient samples and cultured cell lines. A. Dotplot of normalized SSRP1 mRNA levels (y-axis, see details in Experimental Procedures) in all analyzed samples (x-axis, n~20,000) shown in an anatomically and pathologically ordered fashion (x-axis colors corresponds to the legend at top of panel). Colored dots are those with an expression level 1 standard deviation higher than the average expression in all samples of the same type (normal, tumor, non-tumor diseased, etc. as shown above panel), or those in which the 90th percentile of expression was > 2 times the interquartile range plus the 75th percentile of the same type. However, no anatomy or cancer type is colored if there were fewer than ten datapoints per tissue type. Red lines – median for each sample category. B. Tissue box-whisker plot of SSRP1 expression in samples of non-diseased (healthy) and cancer tissues. All results with at least five samples are shown. Green boxes indicate non-diseased samples, while red boxes indicate cancer(s). Boxes span the 25th to 75th percentile of the data with the horizontal line at the median. The whiskers extend to 1.5 times the interquartile range from the edges of the box, and any data points beyond this were considered outliers (hollow circles). See also Fig. S7.
SPT16 mRNA was also elevated in tumors, but to a lesser extent than SSRP1 (Fig. S7). This difference was consistent with our finding that SSRP1 mRNA and protein both increased in the process of HMEC transformation, while for SPT16, only protein (not mRNA) levels increased (Fig. 1B and C). This is most likely due to the demonstrated dependence of SPT16 protein levels on SSRP1 (Safina et al., 2013). Nevertheless, as for SSRP1 mRNA, a significant number of tumors with very high levels of SPT16 mRNA were observed among various types of cancer.
As a more direct evaluation of FACT expression in a clinical setting, we performed IHC staining of SSRP1 on tissue microarrays (TMA) containing primary and metastatic tumors of different types as well as matching normal tissue from 793 patients (see Experimental Procedures). Tumors on the TMAs included invasive breast ductal and lobular carcinoma, non-small cell lung cancer (NSCLC), renal cell carcinoma (RCC), prostatic adenocarcinoma, pancreatic ductal adenocarcinoma (PDA), and colorectal adenocarcinoma. SSRP1 staining was used to assess FACT levels based on the previously established strong correlation between SSRP1 and SPT16 protein levels (Garcia et al., 2011). SSRP1 staining was scored using a semi-quantitative system reflecting both the intensity of staining and the proportion of positive cells (see Experimental Procedures). On the TMAs, all cells in normal tissue samples were SSRP1-negative, with the exception of epithelial cells at the bottom of intestinal crypts (Fig. 5A–C, (Garcia et al., 2011)). Similarly, while tumor samples were frequently SSRP1-positive (see below), stromal cells present in the sample, constituting the tumor microenvironment, were invariably SSRP1-negative (Fig. 5A–C). The highest incidences of SSRP1-positive samples were observed in NSCLC (45–63%), PDA (59%) and colon adenocarcinoma (50%) (Fig. 5D). In contrast, very few cases of prostatic adenocarcinoma and RCC were SSRP1-positive (<10%) (Fig. 5D). Therefore, we deemed the cohort of lung, pancreatic and colon cancers to be “high SSRP1 expressors,” while prostate and kidney cancers appear to be “low SSRP1 expressors.” Notably, all cancers categorized as “high SSRP1 expressors” have a much lower overall survival rate as compared to “low SSRP1 expressors”. In line with this, invasive ductal carcinoma of the breast, which has an intermediate survival rate, was found to have an intermediate incidence of SSRP1-positive/-high samples (18/13%). In contrast to the 100% incidence of SSRP1 expression in human tumor cell lines in vitro, but consistent with our mRNA expression data, a certain proportion of all tumor types were observed to have no SSRP1 staining (Fig. 5D).
Figure 5.
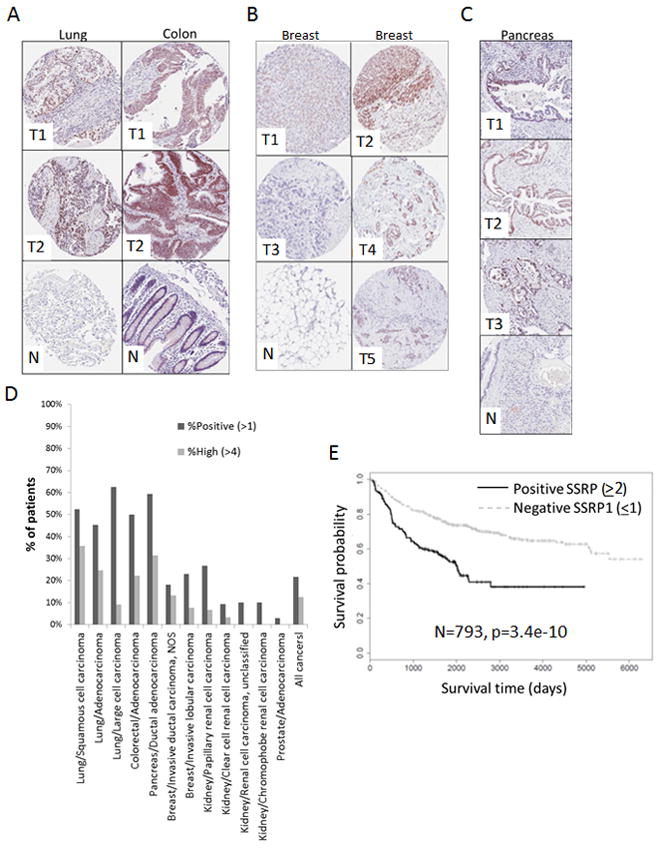
SSRP1 protein expression in human tumors is associated with poor overall patient survival. A–D. Examples of IHC staining with antibodies to SSRP1 containing normal (N) and tumor (T) tissues of lung and colon (A), breast (B) and pancreatic (C) tissues. D. Proportion of patients with SSRP1 expression in their tumors (“positive” – indices >1, “high” indices >4; scoring system is described in Experimental Procedures) out of all analyzed patients with the same type of cancer. E. Patients with SSRP1-negative tumors have better overall survival. Kaplan-Meier survival curves were built using data for all analyzed patients (n=793). The P-value was calculated using Long Rank test. See also Fig.S13 and Tables S5.
Correlation of FACT levels with clinico-pathological features of tumors
Having established that some tumors are SSRP1- and SPT16-positive while others are not, we evaluated whether FACT subunit expression was correlated with any clinico-pathological features of different types of tumors. Analysis of SSRP1 is described below; analysis of SPT16 shown in supplementary materials were generally concordant with SSRP1 (Fig. S7–S12). No correlation between tumor stage and SSRP1 mRNA or protein level was found in any of the analyzed tumor types. This suggests that expression of FACT subunits is an early event in tumorigenesis and does not change with tumor growth (Table S5 and Fig. S8–S12). However, several cancers (breast, lung, colon) showed a correlation between tumor grade and FACT expression, with significantly higher levels of SSRP1 mRNA and protein in high-grade, poorly differentiated tumors (Table S5, Fig. 6C, D, S8C, S9F and S11D,E).
Figure 6.
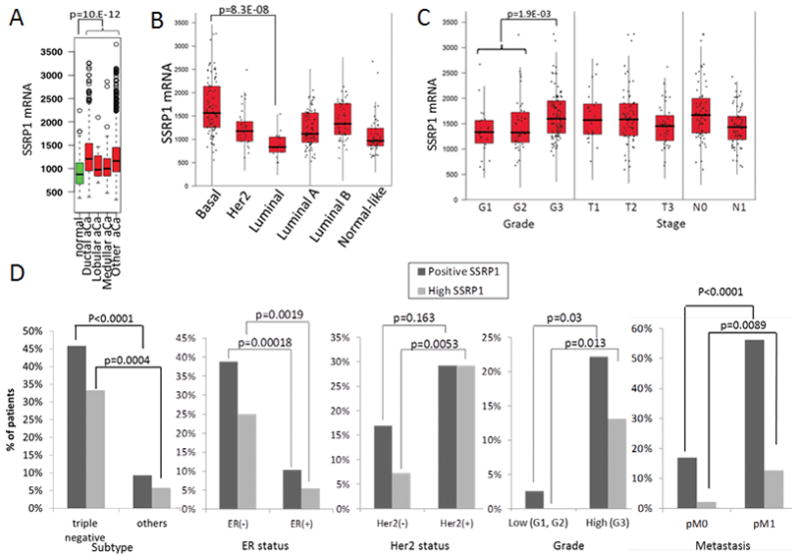
SSRP1 mRNA and protein expression in breast cancer. A–C. Box-whisker plots of SSRP1 mRNA levels in (A) samples of breast tissue (aCa – adenocarcinoma); (B) breast cancer samples categorized by gene expression signature; and (C) breast cancer samples of different grades and stages. P-values from Mann-Whitney-Wilcoxon tests between indicated samples are shown. P-values >0.05 are not shown. D. Comparison of the proportion of SSRP1-positive samples (based on IHC staining) among patients within different categories of breast cancer. P-values from exact Fisher chi-square tests between different categories are shown. E–F. Combination of SSRP1 expression with negative ER (E) and PR (F) status is a significant predictor of poor survival in breast cancer patients. Kaplan-Meier survival curves for each combination of markers. See also Fig. S8–S12.
Among breast cancer patients, SSRP1 mRNA was higher in all tumor types versus normal breast tissue (Fig. 6A), and in basal versus luminal carcinomas (Fig. 6B). SSRP1 protein expression was more frequent in triple negative versus hormone receptor-positive tumors and in ER-negative and Her2-positive versus ER-positive and Her2-negative tumors (Table S5 and Fig. 6D). Similarly, SSRP1 mRNA was higher in NSCLC than in normal lung and the highest level was observed in undifferentiated large cell carcinomas (Fig. S9A, C). The same tendency, although not statistically significant, was observed for SSRP1 protein (Fig. S9E). Notably, among different histological subtypes of breast cancer and NSCLC, “high SSRP1 expressors” were generally tumor subtypes with worse prognoses than “low SSRP1expressors.”
Since most cancer-related deaths are due to metastatic rather than primary disease, we evaluated whether SSRP1 expression is associated with metastatic disease. We found that breast cancer and RCC patients with SSRP1-positive primary tumors had a higher incidence of metastatic disease than patients with SSRP1-negative primary tumors (Fig. 6D, S10D). In addition, SSRP1 mRNA was higher among patients with metastases of lung and prostate cancers than among patients with no metastasis (Fig. S9D and S12A). Overall, there was a strong correlation of SSRP1 status between primary and metastatic lesions in all cancers analyzed by IHC (97%). Therefore, presence of SSRP1 in primary tumors of several types (e.g. breast) may be predictive of metastatic disease.
The data described above suggested that SSRP1 expression might be associated with tumor aggressiveness. To test this, we performed a correlation analysis between SSRP1 protein level and overall survival for all patients as a single cohort irrespective of their tumor classification. To determine whether a particular degree of SSRP1 overexpression had prognostic value, we compared the following groups (defined by semi-quantitative score cut-offs, see Experimental Procedures): (i) “high” SSRP1 vs “low” and negative samples, (ii) positive SSRP1 vs weak/negative samples, and (iii) SSRP1-negative vs all positive samples. For all tumor types, the strongest correlation between survival and SSRP1 level was obtained if SSRP1-positive and -negative samples were compared (Fig. 5E and S13A). For all 793 patients, SSRP1 positivity was significantly associated with worse overall survival (Fig. 5E). The same tendency, although not statistically significant, was observed in lung and colon cancers (Fig. S13). In breast cancer patient’s tumors expression of SSRP1 was significant prognostic markers of poor survival based on univariate analysis (Fig. S13D). The multivariate analysis of SSRP1 and hormone receptors status in breast cancer did not reveal SSRP1 as an independent marker with the number of patients we analyzed, but combination of SSRP1 with estrogen and progesterone receptors significantly improves predictive value of both established markers (Fig. 6E, F).
In summary, analysis of clinical samples indicated that SSRP1 is expressed more frequently and at a higher level in less-differentiated (higher grade) and more aggressive tumors, including (i) types of solid tumors with poor prognosis (lung, pancreatic and colon), (ii) histological subtypes of breast cancer and NSCLC with poor prognosis (triple negative, Her2 positive, large undifferentiated lung carcinoma); (iii) metastatic tumors (breast, lung, renal and prostate cancers); and (iv) tumors from patients with worse overall survival.
Discussion
Although we and others previously noted elevated expression of FACT in tumor cell lines and in ovarian cancer patient samples (Gasparian et al., 2011; Hudson et al., 2007; Koman et al., 2012), this study provides the first comprehensive analysis of FACT’s value as a cancer marker and target. First, we found that both FACT subunits were elevated upon in vitro transformation of fibroblasts and epithelial cells induced by different agents (Fig. 1 and S1). These data, together with the already published observation that FACT is elevated upon Her2/neu-induced transformation of mammary epithelial cells (Koman et al., 2012), suggest that FACT upregulation may be a universal event during in vitro transformation. In epithelial cells, but not fibroblasts, the intermediate step of immortalization was accompanied by modest FACT elevation (Fig. 1A and S1A,); however, the most critical increase in both cell types coincided with transformation and acquisition of malignant properties, such as AIG and/or in vivo tumor growth (Fig. 1A–C and S1B, C). Similarly, ectopic FACT expression induced growth in 2-D cultures for epithelial cells, but not fibroblasts, while increasing the proportion of cells able to grow in semi-solid medium for both cell types (Fig. Fig. 2A–C and S1D, E). Since the same oncogene was used to transform both cell types, these data likely reflect cell type-specific requirements for FACT during transformation.
Overexpression and shRNA-mediated KD experiments demonstrated that FACT was not simply correlated with transformation, but functionally required. However, enforced expression of FACT was not able to substitute for H-RasV12 in driving malignant transformation. This indicates that FACT-mediated chromatin changes are not sufficient to cause transformation, but rather appear to create conditions that promote or accelerate the oncogenic activity of other factors. Therefore FACT cannot be categorized as an oncogene or “driver” of malignant transformation, but at the same time it is also not a “passenger”. We suggest a term “accelerator”, or factor which makes function of “driver” more efficient.
FACT remains important even in established tumors, as illustrated by our finding that all tested tumor cell lines were sensitive to FACT KD (Gasparian et al., 2011) and Fig. 2 and S2). Unlike normal and immortalized non-transformed cells, tumor cell lines with reduced levels of FACT could not be expanded (Fig. 2F and S2). Selective FACT-dependence of tumor, but not normal, cells indicates that targeting of FACT could be a safe and effective anti-cancer strategy.
However, many patient tumor samples are FACT-negative, indicating that FACT is not universally important for tumor transformation in vivo. Most normal tissues in vivo, as well as normal primary cells in culture, are FACT-negative. Passaging of these cells results in elevation of FACT levels (unpublished observation), suggesting that, for normal cells, either in vitro stress induces FACT expression or only cells with elevated FACT (stem or undifferentiated progenitor cells as shown in (Garcia et al., 2011)) can grow in culture. Both of these possibilities are consistent with our observation that many FACT-controlled genes are induced by different types of cellular stress (Table 1), so there may be a feedback mechanism between stress and FACT expression. In line with this hypothesis, all tested cultured tumor cell lines were FACT-positive (Garcia et al., 2011; Gasparian et al., 2011), while many patient tumor samples were FACT-negative. Furthermore, SSRP1 and SPT16 mRNA levels were consistently higher in cultured cell lines as compared to practically all tissues in vivo (Fig. 4 and S7).
Thus, our data shows that normal and tumor cells can be either FACT-positive or -negative in vivo, while both categories are FACT-positive in vitro (although to different extents). The key difference between these cell types is that tumor cells are sensitive to FACT inhibition, while normal cells are not (Fig. 2 and S2). This was also true in vivo, as inhibition of FACT activity by Curaxins had anti-tumor effects in multiple mouse models at non-toxic concentrations (Gasparian et al., 2011).
To extend the relevance of our findings in cultured cells (see above) and in Curaxin-treated mice towards cancer patients, we investigated FACT expression in a large number of human normal and tumor tissue samples via (i) analysis of publically available microarray-based gene expression data sets, and (ii) IHC staining of TMAs containing an independent set of samples. The bioinformatics approach first suggested that FACT may not be a universal cancer marker, since not all tumor samples displayed elevated FACT levels (Fig. 4 and S7). Trying to clarify the difference between tumors with low and high levels of FACT, we noticed that that the most significant association with clinical features was between FACT-positive and -negative tumors. Thus, whether tumor cells express FACT or not appears to be more important than the level of expression. Notably, multiple specific subtypes of tumors had a high incidence of FACT-positivity and, almost universally, these subtypes behaved more aggressively (overall survival of patients with FACT-positive tumors was significantly worse than that of patients with FACT-negative tumors (Fig. 5E).
In line with FACT expression in normal mouse and human tissues being limited to stem and undifferentiated progenitor cells, FACT expression was positively correlated with grade for several cancer types (Table S5). This suggests that FACT is mostly expressed in poorly differentiated tumors. We did not observe this correlation in pancreatic ductal adenocarcinoma (PDA); however, our PDA sample set did not include any well-differentiated tumors, only moderately- and poorly-differentiated ones (which are aggressive, have poor prognosis and, consistent with our hypothesis, were frequently FACT-positive).
Further supporting the association of FACT with tumor aggressiveness, FACT expression was found to be significantly correlated with metastasis of breast, renal and prostate cancer. Importantly, the FACT status (SSRP1 staining on TMA) of the primary tumor was associated with development of metastatic disease. This, together with the lack of correlation between FACT expression and tumor stage or timing increases the potential value of FACT as a prognostic marker since FACT positivity of a primary tumor could be used even at a very early stage to determine the risk of future metastatic disease.
While all of our data indicate that the SSRP1 and SPT16 subunits of FACT are coordinately expressed in cancer, SSRP1 is the most promising candidate for a diagnostic/prognostic marker since its upregulation is more obvious on both the mRNA and protein levels. This likely derives from the unusual mechanism of SSRP1 and SPT16 co-regulation that we recently described in which the stability of both proteins depends upon formation of the FACT complex and stability of the complex is determined by presence of the SSRP1 mRNA (Safina et al., 2013). In this way, an increase of SSRP1 mRNA is sufficient to drive elevation of both SSRP1 and SPT16 proteins. This was confirmed in the in vitro transformation experiments reported here: while only SSRP1 (not SPT16) mRNA was elevated in all transformed cells, protein levels of both SSRP1 and SPT16 were increased (Fig. 1A–C). This same trend was noted in many types of tumors: SSRP1 mRNA was increased much more universally than SPT16 mRNA (Fig. 4, 6 and S7–S12). At the same time, we did observe tumors with significantly increased SPT16 mRNA (Fig. S7). In the future, it will be interesting to analyze whether these tumors have any selective advantages over those in which only SSRP1 mRNA is elevated.
The mechanism(s) underlying FACT’s role as an accelerator of transformation remain to be fully elucidated. However, our previous demonstration that FACT-assisted NF-κB-dependent transcription is involved suggested that modulation of transcriptional programs driven by other TFs might also contribute to FACT’s pro-cancerous effects. Support for this hypothesis was gained through our ChIP-based exploration of the distribution of chromatin-bound SSRP1. This confirmed significant and selective association of FACT with protein-expressing genes (Fig. 3A,D and S5). SSRP1 peak distribution over the whole body of genes, with additional enrichments at the start and end of the gene territory, indicated involvement of FACT in transcription initiation, elongation, and probably termination, which has not been previously reported. The selectivity of FACT in assisting in transcription of some, but not other, genes was previously demonstrated through comparison of gene expression profiles in yeast (reviewed in (Formosa, 2012), plant, and mammalian cells following inactivation of FACT (Duroux et al., 2004; Li et al., 2005; Lolas et al., 2009).
The list of SSRP1-enriched genes obtained in our study contains many “pro-cancer” genes, including numerous TF targets involved in induction of proliferation, inhibition of cell death and differentiation, maintenance of cell pluripotency, and stress responses (Tables 1, S3, S4, and Fig. 3C, D). The mechanism by which FACT selects these genes is currently under study, although FACT has been found among the direct interactors of several TFs that, based upon SSRP1 binding to their genes, may require FACT for expression (i.e., MYC, SRF and OCT4) (Bunker and Kingston, 1995), (Kihara et al., 2008; Spencer et al., 1999) (Pardo et al., 2010; van den Berg et al., 2010).
We also cannot exclude a non-transcription-related role for FACT in cancer. FACT is known to be involved in replication, and replication is clearly reduced upon FACT KD; however, the availability of cells in vivo and in vitro that can replicate without FACT suggests that this may not be the only FACT-dependent process that is vital for tumor cells. Additional reported functions of FACT that might contribute to its role in cancer are regulation of DNA damage responses and DNA repair. However, genes involved in these processes are usually lost or mutated in cancer (e.g., BRCA, MHS), while neither SPT16 nor SSRP1 display higher-than-background incidences of mutations or deletions in any type of cancer (TCGA, cBio portal). SSRP1, but not SPT16, was reported to be required (in cooperation with many other factors) for proper spindle formation during mitosis (Zeng et al., 2009). We cannot exclude that elevation of SSRP1, and concomitantly, SPT16, levels may be due to the increased rate of mitosis in cancer cells; however, it seems unlikely that an artificial increase in FACT expression would induce transformation by assisting with spindle formation. Understanding the mechanism(s) by which FACT promotes cancer is a major goal of our current work.
In conclusion, the results of this study indicate that FACT is a promising marker and target for subtypes of cancer characterized by high grade and aggressiveness and poor prognosis. This, together with the absence of FACT expression in most normal cells/tissues, suggests that pharmacological inhibition of FACT could be a safe and effective strategy to treat types of cancer for which there are currently few treatment options.
Experimental Procedures
Reagents
Curaxin CX-137 (CBLC137) was provided by Cleveland BioLabs, Inc (Buffalo, NY).
Cells
HT1080, WI-38, MCF7 and MCF10A cells were obtained from ATCC and maintained as suggested.
RCC45 and NKE-hTERT cells have been described (Gurova et al., 2004).
Human mammary epithelial cells were obtained from Martha Stampfer (Lawrence Berkeley National Laboratory, Berkeley, CA) and modified and maintained as described (Garbe et al., 2009). 184B5 and 184AA3 were immortalized following exposure to benzo(a)pyrene (Stampfer and Bartley, 1985; Stampfer et al., 2003); 184Dp16sMY and 240Lp16sMy were immortalized following transduction of c-Myc and shRNAs against p16; 184FMY2 immortalized via expression of c-Myc.
The immortalized BJ fibroblast cell line with tamoxifen-regulated H-Ras V12 was obtained from Reuven Agami (The Netherlands Cancer Institute, Amsterdam, The Netherlands, EU).
Wild-type and p53-knockout Mouse Embryonic Fibroblast (MEF) cells were obtained from 13.5-day pregnant C57/B6 wild-type or p53−/− mice and maintained in DMEM with 10% FBS and antibiotics.
Growth of cells in semisolid media was assessed as described (Yang et al., 2012). Colonies were counted unstained in 10 blindly selected fields of view in each of 3 replicate wells using 10X phase contrast microscopy or stained with 5ug/mL MTT (Sigma-Aldrich) and photographed.
Growth of cells in SCID mice was done according to institution ethical committee approved animal protocol. 5 mln of shRNA transduced MCF10A cells were subcutaneously inoculated into two sites of female 8 weeks old SCID mice (n=5) in 50% matrigel/PBS solution. Tumors were measured once a week using digital caliper and volume was calculated using formula V=length*width2/2. Tumor growth was calculated as fold increase of tumor volume between days 1 and 30 after inoculation.
Plasmids, Transfection and Lentiviral Transduction
pLV-H-RasV12-Bleo or pLV-Bleo lentiviral vectors were kindly provided by Dr. Andrei Gudkov (Roswell Park Cancer Institute, Buffalo, NY). Human SSRP1 cDNA was cloned into the pLV-CMV-Neo lentiviral vector and verified by sequencing. SUPT16H cDNA was synthesized (Invitrogen, GeneArt AG) using a sequence optimized for protein expression by DAPCEL, Inc. (Cleveland, OH) and cloned into the pMLV HygroR lentiviral vector. Mission® shRNAs targeting SSRP1 (TRCN0000019270), Spt16 (TRCN0000001260), and GFP (SHC004) were obtained from Sigma-Aldrich Co., (St. Louis, MO).
siRNAs targeting SSRP1(On-Target plus SMART pool, cat# L-011783-00) or SPT16 (On-Target plus SMART pool, cat# L-009517-00) and siCONTROL non-targeting siRNA (cat# D-001210-01) were from Thermo Scientific Dharmacon (Chicago, IL). Transfection was performed using Lipofectamine 2000 reagent (Life Technologies, Grand Island, NY). Lentivirus packaging and infection was conducted as described (Gurova et al., 2005; Gurova et al., 2004)
Western blotting, Fluorescent-Activated Cell Sorting and Immunofluorescent staining were done as described (Gasparian et al., 2011; Gurova et al., 2005; Gurova et al., 2004). The list of antibodies used can be found in Supplementary Material.
Replication and transcription rates in cells were measured using Click-iT EDU Alexa Fluor 594 HCS Assay and Click-iT RNA Alexa Fluor 488 HCS Assay kits (Invitrogen, Eugene, OR).
Quantitative RT-PCR was done as described (Safina et al., 2013)
Patient samples
Patients included in this study (n=793) were diagnosed with cancer between March 1992 and January 2010 at Roswell Park Cancer Institute. The RPCI Institutional Review Board gave approval for this study. We selected all patients from this time period with adequate material in the RPCI archive for immunohistochemistry and with follow-up information in the RPCI Tumor Registry or various RPCI Research Program Databases. The 793 patients included 143 invasive ductal carcinoma of the breast, 13 invasive lobular carcinoma of the breast, 54 colorectal adenocarcinoma, 10 chromophobe renal cell carcinoma, 235 clear cell renal cell carcinoma, 15 papillary renal cell carcinoma, 10 renal cell carcinoma unclassified, 73 lung adenocarcinoma, 11 lung large cell carcinoma, 42 lung squamous cell carcinoma, 54 ductal adenocarcinoma of the pancreas, and 133 prostatic adenocarcinoma.
Demographic details of patients are provided in Supplementary Materials.
Tissue Microarrays
SSRP1 protein expression in the clinical cohort was assessed using 16 TMAs comprising 6 cancer types collected from patients described above. All RPCI TMAs are built in a standardized fashion with three 1-millimeter tissue cores from formalin-fixed paraffin embedded donor blocks precisely arrayed into a new recipient paraffin block, including tumor specimens as well as controls. For most TMAs, 3 cores of matching normal tissue were also evaluated. Additional controls within each TMA consisted of multiple cores of normal tissue from 10 different organs including heart, colon, kidney, adrenal, ovary, myometrium, brain, thyroid, lung, and prostate representing slightly more than 20% of all cores per TMA.
TMA scoring
TMAs were stained as described (Garcia et al., 2011). Results were recorded according to the American Society of Clinical Oncology Guideline Recommendations. The neoplastic cells for any given core were scored as a semi-quantitative/ordered categorical manner for intensity (range of scores 0–3) and percentage of cells 0 (0%), 1 (1–9%), 2 (10–49%), 3 (50–100%) staining. The results for all cores for one patient in a TMA format were averaged for a final score. An immunohistochemistry (IHC) index (range of scores 0–9) was defined as the product of the intensity and percentage of cells staining. Cores were excluded from evaluation due to an absence of tumor cells, or core drop-off. Cases were also excluded if there was insufficient clinical or outcome data.
Statistical analysis of TMA staining
The SSRP1 IHC data was dichotomized as described in the TMA scoring section. Fisher’s exact tests were performed to test the association between the dichotomized SSRP1 expression indices (0 vs all others; <2 vs ≥2; ≤4 vs >4) and other dichotomized categorical variables, such as age (≥60/<60), tumor grade (high/low), stage (early/late) and expression of disease specific markers, where available. Chi-square tests were performed to test for association with categorical variables of more than two levels. Kaplan-Meier survival analyses with log-rank tests (Peto and Peto, 1972) were employed to assess the correlation between patient survival and SSRP1 expression index. P-values <0.05 were considered significant. Multivariate survival analysis was conducted using Cox regression. All statistical analyses were performed using the R statistical programming language (R Team, 2012).
In silico analysis of SSRP1 and SPT16 expression
Analysis of gene expression data from 251 different studies was performed using the In Silico Transcriptomics Online – Integrated gene expression reference database, IST Online (MediSapiens Ltd). Details of this analysis are provided in Supplementary Materials
Immunoprecipitation of SSRP1/SPT16 complex
HT1080 cells were lysed in NP40 buffer and rotated at 4°C for 15 min. Then, lysates were centrifuged at 14,000g for 15 minutes, and supernatants were precleared by rotating them with protein A/G agarose beads at 4°C for 1 h. Cell lysates at 1mg/ml concentrations were rotated overnight with anti- SSRP1 (cat# 609702, BioLegend, Inc., San Diego, CA) or control mouse IgG2b (cat#400302, Biolegend, Inc., San Diego, CA ) - 5 μg/500 μl lysate at 4°C. To capture immunocomplexes, Dynabeads Protein A (Life Technologies, Grand Island, NY, USA) were added to the lysates according to the manufacturer’s instructions and then rotated for 1 h at 4°C. Beads were collected, washed 3 times with cold PBS, and either used for LC/MS sample preparation or boiled in 60 μl of 2X loading buffer for 5 min, after being centrifuged briefly to separate the supernatants for SDS-PAGE and western blotting.
Sample preparation for LC-MS/MS analysis, LC-MS/MS Analysis, Database Search and Peptide and Protein Identification are described in Supplementary Materials.
ChIP-sequencing and analysis
ChIP was performed (3 independent experiments) using HT1080 cells left untreated or treated with 3uM CX-137 for 1 hour withe mouse monoclonal anti-SSRP1 (cat# 609702, BioLegend, Inc., San Diego, CA) and mouse IgG2b antibody (cat#400302, Biolegend, Inc.). ChIP was performed with a kit from Upstate (EMD Millipore, Billerica, MA) as outlined by the manufacturer except that Dynabeads Protein A (Invitrogen, Eugene, OR) were used instead of Protein A agarose beads. ChIP-isolated DNA was treated using the standard ChIP-seq protocol from Illumina except that after adaptor ligation, the library was separated on a 2% agarose gel, and the 150 to 500 bp region was excised and purified. The resulting ChIP libraries were single-end sequenced on an Illumina HiSeq2000 with 50 bp reads. Each sample was sequenced in a single flow cell lane and generated 89 to 190 million reads. The resulting raw sequencing reads were filtered for quality and aligned to the most recent build of the human genome (hg19) with BowTie. Peaks were identified, averaged and normalized against the background, and input using PeakRanger (Feng et al., 2011). The peak positions in relation to genome features were calculated using MACS (Zhang et al., 2008). For this, the data from replicated samples were concatenated together. The comparison of peaks in untreated and CX-137-treated samples and peak annotation was performed using an Integrated Genomic Viewer (Broad Institute, MIH, Harvard University, Cambridge, MA).
Supplementary Material
Highlights.
FACT is expressed in aggressive undifferentiated cancers with poor overall survival.
FACT increases the efficiency of oncogene-induced transformation.
Neoplastic, but not normal, cell growth depends on FACT activity.
FACT binds genes that are involved in cancer.
Acknowledgments
We would like to thank Dr. Bruce Specht for his constant and enormous support of our research, Dr. Andrei Gudkov for critical discussion of the work and manuscript; Dr. Patricia Stanhope Baker for editing of the manuscript, Monica Murphy for help with organization of the clinical data, Jonathan Bard for help with processing of sequencing files, and Yekaterina Leonova for help with mouse embryonic fibroblasts. We also thank Natalia Tararova and Anton Komar from Dapcel, Inc for the help with generation of SPT16 mammalian expression construct. Biospecimens and research pathology services for this study were provided by the Pathology Resource Network, which is funded by the National Cancer Institute (NCI) and is a RPCI Cancer Center Support Grant (CCSG) shared resource. Clinical Data Delivery and Honest Broker services for this study were provided by the Clinical Data Network, which is funded by the NCI and is a RPCI CCSG shared resource. This study was funded by grants to KVG from Incuron, Inc. and from The Komen Foundation (CCR13264604).
Footnotes
Accession numbers
Sequencing data in the form of bed files are available at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE45393
References
- Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003;301:1090–1093. doi: 10.1126/science.1085703. [DOI] [PubMed] [Google Scholar]
- Birch JL, Tan BC, Panov KI, Panova TB, Andersen JS, Owen-Hughes TA, Russell J, Lee SC, Zomerdijk JC. FACT facilitates chromatin transcription by RNA polymerases I and III. EMBO J. 2009;28:854–865. doi: 10.1038/emboj.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner AJ, Stampfer MR, Aldaz CM. Increased p16 expression with first senescence arrest in human mammary epithelial cells and extended growth capacity with p16 inactivation. Oncogene. 1998;17:199–205. doi: 10.1038/sj.onc.1201919. [DOI] [PubMed] [Google Scholar]
- Bunker CA, Kingston RE. Identification of a cDNA for SSRP1, an HMG-box protein, by interaction with the c-Myc oncoprotein in a novel bacterial expression screen. Nucleic Acids Res. 1995;23:269–276. doi: 10.1093/nar/23.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duroux M, Houben A, Ruzicka K, Friml J, Grasser KD. The chromatin remodelling complex FACT associates with actively transcribed regions of the Arabidopsis genome. Plant J. 2004;40:660–671. doi: 10.1111/j.1365-313X.2004.02242.x. [DOI] [PubMed] [Google Scholar]
- Feng X, Grossman R, Stein L. PeakRanger: a cloud-enabled peak caller for ChIP-seq data. BMC Bioinformatics. 2011;12:139. doi: 10.1186/1471-2105-12-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formosa T. The role of FACT in making and breaking nucleosomes. Biochim Biophys Acta. 2012;1819:247–255. doi: 10.1016/j.bbagrm.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbe JC, Bhattacharya S, Merchant B, Bassett E, Swisshelm K, Feiler HS, Wyrobek AJ, Stampfer MR. Molecular distinctions between stasis and telomere attrition senescence barriers shown by long-term culture of normal human mammary epithelial cells. Cancer Res. 2009;69:7557–7568. doi: 10.1158/0008-5472.CAN-09-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia H, Fleyshman D, Kolesnikova K, Safina A, Commane M, Paszkiewicz G, Omelian A, Morrison C, Gurova KV. Expression of Facilitates Chromatin Transcription complex in mammalian tissues suggests FACT role in maintaining of undifferentiated state of cells. Oncotarget. 2011;2:783–796. doi: 10.18632/oncotarget.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparian AV, Burkhart CA, Purmal AA, Brodsky L, Pal M, Saranadasa M, Bosykh DA, Commane M, Guryanova OA, Pal S, et al. Curaxins: Anticancer Compounds That Simultaneously Suppress NF-{kappa}B and Activate p53 by Targeting FACT. Sci Transl Med. 2011;3:95ra74. doi: 10.1126/scitranslmed.3002530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudkov AV, Gurova KV, Komarova EA. Inflammation and p53: A Tale of Two Stresses. Genes Cancer. 2011;2:503–516. doi: 10.1177/1947601911409747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurova KV, Hill JE, Guo C, Prokvolit A, Burdelya LG, Samoylova E, Khodyakova AV, Ganapathi R, Ganapathi M, Tararova ND, et al. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-kappaB-dependent mechanism of p53 suppression in tumors. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:17448–17453. doi: 10.1073/pnas.0508888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurova KV, Hill JE, Razorenova OV, Chumakov PM, Gudkov AV. p53 pathway in renal cell carcinoma is repressed by a dominant mechanism. Cancer Res. 2004;64:1951–1958. doi: 10.1158/0008-5472.can-03-1541. [DOI] [PubMed] [Google Scholar]
- Heo K, Kim H, Choi SH, Choi J, Kim K, Gu J, Lieber MR, Yang AS, An W. FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol Cell. 2008;30:86–97. doi: 10.1016/j.molcel.2008.02.029. [DOI] [PubMed] [Google Scholar]
- Hudson ME, Pozdnyakova I, Haines K, Mor G, Snyder M. Identification of differentially expressed proteins in ovarian cancer using high-density protein microarrays. Proc Natl Acad Sci U S A. 2007;104:17494–17499. doi: 10.1073/pnas.0708572104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Kinoshita Y, Susaki D, Iwano M, Takayama S, Higashiyama T, Kakutani T, Kinoshita T. HMG domain containing SSRP1 is required for DNA demethylation and genomic imprinting in Arabidopsis. Dev Cell. 2011;21:589–596. doi: 10.1016/j.devcel.2011.08.013. [DOI] [PubMed] [Google Scholar]
- Keller DM, Zeng X, Wang Y, Zhang QH, Kapoor M, Shu H, Goodman R, Lozano G, Zhao Y, Lu H. A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Mol Cell. 2001;7:283–292. doi: 10.1016/s1097-2765(01)00176-9. [DOI] [PubMed] [Google Scholar]
- Kihara T, Kano F, Murata M. Modulation of SRF-dependent gene expression by association of SPT16 with MKL1. Exp Cell Res. 2008;314:629–637. doi: 10.1016/j.yexcr.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Koman IE, Commane M, Paszkiewicz G, Hoonjan B, Pal S, Safina A, Toshkov I, Purmal AA, Wang D, Liu S, et al. Targeting FACT Complex Suppresses Mammary Tumorigenesis in Her2/neu Transgenic Mice. Cancer Prev Res (Phila) 2012;5:1025–1035. doi: 10.1158/1940-6207.CAPR-11-0529. [DOI] [PubMed] [Google Scholar]
- Kumari A, Mazina OM, Shinde U, Mazin AV, Lu H. A role for SSRP1 in recombination-mediated DNA damage response. J Cell Biochem. 2009;108:508–518. doi: 10.1002/jcb.22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Keller DM, Scott JD, Lu H. CK2 phosphorylates SSRP1 and inhibits its DNA-binding activity. J Biol Chem. 2005;280:11869–11875. doi: 10.1074/jbc.M413944200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolas IB, Himanen K, Gronlund JT, Lynggaard C, Houben A, Melzer M, Van Lijsebettens M, Grasser KD. The transcript elongation factor FACT affects Arabidopsis vegetative and reproductive development and genetically interacts with HUB1/2. Plant J. 2009;61:686–697. doi: 10.1111/j.1365-313X.2009.04096.x. [DOI] [PubMed] [Google Scholar]
- Pardo M, Lang B, Yu L, Prosser H, Bradley A, Babu MM, Choudhary J. An expanded Oct4 interaction network: implications for stem cell biology, development, and disease. Cell stem cell. 2010;6:382–395. doi: 10.1016/j.stem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peto R, Peto J. Asymptotically Efficient Rank Invariant Test Procedures. Journal of the Royal Statistical Society Series A (General) Blackwell Publishing. 1972;135:185–207. [Google Scholar]
- Reinberg D, Sims RJ., 3rd de FACTo nucleosome dynamics. J Biol Chem. 2006;281:23297–23301. doi: 10.1074/jbc.R600007200. [DOI] [PubMed] [Google Scholar]
- Safina A, Garcia H, Commane M, Guryanova O, Degan S, Kolesnikova K, Gurova KV. Complex mutual regulation of Facilitates Chromatin Transcription (FACT) subunits on mRNA and protein levels in human cells. J Biol Chem. 2013 doi: 10.4161/cc.25452. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Werner J, Andrulis ED, Nakayama T, Hirose S, Reinberg D, Lis JT. Tracking FACT and the RNA polymerase II elongation complex through chromatin in vivo. Science. 2003;301:1094–1096. doi: 10.1126/science.1085712. [DOI] [PubMed] [Google Scholar]
- Singer RA, Johnston GC. The FACT chromatin modulator: genetic and structure/function relationships. Biochem Cell Biol. 2004;82:419–427. doi: 10.1139/o04-050. [DOI] [PubMed] [Google Scholar]
- Spencer JA, Baron MH, Olson EN. Cooperative transcriptional activation by serum response factor and the high mobility group protein SSRP1. J Biol Chem. 1999;274:15686–15693. doi: 10.1074/jbc.274.22.15686. [DOI] [PubMed] [Google Scholar]
- Stampfer MR, Bartley JC. Induction of transformation and continuous cell lines from normal human mammary epithelial cells after exposure to benzo[a]pyrene. Proc Natl Acad Sci U S A. 1985;82:2394–2398. doi: 10.1073/pnas.82.8.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampfer MR, Garbe J, Nijjar T, Wigington D, Swisshelm K, Yaswen P. Loss of p53 function accelerates acquisition of telomerase activity in indefinite lifespan human mammary epithelial cell lines. Oncogene. 2003;22:5238–5251. doi: 10.1038/sj.onc.1206667. [DOI] [PubMed] [Google Scholar]
- Tan BC, Chien CT, Hirose S, Lee SC. Functional cooperation between FACT and MCM helicase facilitates initiation of chromatin DNA replication. EMBO J. 2006;25:3975–3985. doi: 10.1038/sj.emboj.7601271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan BC, Liu H, Lin CL, Lee SC. Functional cooperation between FACT and MCM is coordinated with cell cycle and differential complex formation. J Biomed Sci. 2010;17:11. doi: 10.1186/1423-0127-17-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg DL, Snoek T, Mullin NP, Yates A, Bezstarosti K, Demmers J, Chambers I, Poot RA. An Oct4-centered protein interaction network in embryonic stem cells. Cell stem cell. 2010;6:369–381. doi: 10.1016/j.stem.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Morrison CD, Liu P, Miecznikowski J, Bshara W, Han S, Zhu Q, Omilian AR, Li X, Zhang J. TAZ induces growth factor-independent proliferation through activation of EGFR ligand amphiregulin. Cell Cycle. 2012;11:2922–2930. doi: 10.4161/cc.21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng SX, Li Y, Jin Y, Zhang Q, Keller DM, McQuaw CM, Barklis E, Stone S, Hoatlin M, Zhao Y, Lu H. Structure-specific recognition protein 1 facilitates microtubule growth and bundling required for mitosis. Mol Cell Biol. 2009;30:935–947. doi: 10.1128/MCB.01379-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome biology. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang TS. A coordinated temporal interplay of nucleosome reorganization factor, sister chromatin cohesion factor, and DNA polymerase alpha facilitates DNA replication. Mol Cell Biol. 2004;24:9568–9579. doi: 10.1128/MCB.24.21.9568-9579.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.