

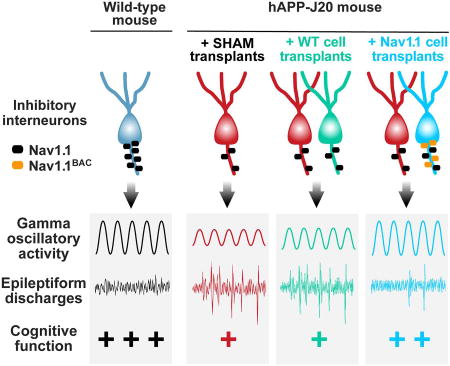
SUMMARY
Inhibitory interneurons regulate oscillatory rhythms and network synchrony that are required for cognitive functions and disrupted in Alzheimer’s disease (AD). Network dysrhythmias in AD and multiple neuropsychiatric disorders are associated with hypofunction of Nav1.1, a voltage-gated sodium channel subunit predominantly expressed in interneurons. We show that Nav1.1-overexpressing, but not wild-type, interneuron transplants derived from the embryonic medial ganglionic eminence (MGE) enhance behavior-dependent gamma oscillatory activity, reduce network hypersynchrony, and improve cognitive functions in human amyloid precursor protein (hAPP) transgenic mice, which simulate key aspects of AD. Increased Nav1.1 levels accelerated action potential kinetics of transplanted fast-spiking and non-fast-spiking interneurons. Nav1.1-deficient interneuron transplants were sufficient to cause behavioral abnormalities in wild-type mice. We conclude that the efficacy of interneuron transplantation and the function of transplanted cells in an AD-relevant context depend on their Nav1.1 levels. Disease-specific molecular optimization of cell transplants may be required to ensure therapeutic benefits in different conditions.
ETOC BLURB
Inhibitory interneurons regulate brain rhythms and cognitive function that are disrupted in Alzheimer’s disease (AD). Martinez-Losa et al. show that the levels of voltage-gated sodium channel Nav1.1 subunit in interneuron transplants regulate their therapeutic efficacy in an AD mouse model.
INTRODUCTION
In Alzheimer’s disease (AD) causes abnormal patterns of neuronal network activity and cognitive decline through mechanisms that remain to be fully elucidated (Palop and Mucke, 2010, 2016). In preclinical and early stages of AD, functional imaging during memory-encoding tasks shows hippocampal hyperactivation and deactivation deficits in regions of the default mode network (Bakker et al., 2012; Dickerson et al., 2005; Sperling et al., 2009). The deactivation deficits correlate with early amyloid deposition and accumulation in these regions (Buckner et al., 2005; Sperling et al., 2009), suggesting an early pathological link between aberrant network activity and amyloid in AD. Aberrant network activity and pathologically elevated amyloid-β (Aβ) levels are also tightly linked in familial AD (FAD)-mutant human amyloid precursor protein (hAPP) transgenic mice, which develop altered fast- and slow-wave oscillatory activity (Busche et al., 2015; Cramer et al., 2012; Rubio et al., 2012; Verret et al., 2012), neuronal hyperactivity (Busche et al., 2008), activity-dependent amyloid deposition (Bero et al., 2011; Yamamoto et al., 2015), and network hypersynchrony resulting in epileptiform activity that is more often non-convulsive and behaviorally silent than convulsive and behaviorally evident (Palop et al., 2007). Humans with AD, particularly with FAD, also have an increased incidence of seizures and epileptiform activity (for review, Palop and Mucke, 2016), and recent studies suggest that the prevalence of these abnormalities among sporadic AD patients may be vastly underestimated (Lam et al., 2017; Vossel et al., 2013; Vossel et al., 2016). Impaired inhibitory function could be a key mechanism of these functional abnormalities in mice and humans (Bakker et al., 2012; Busche et al., 2008; Busche et al., 2015; Palop and Mucke, 2016; Sperling et al., 2009; Verret et al., 2012).
While network epileptiform activity can interfere with cognitive functions (Kleen et al., 2013), physiological neuronal network oscillations (brain rhythms) and network synchrony are required for cognitive functions and are also regulated by inhibitory interneurons (Kepecs and Fishell, 2014; Lapray et al., 2012; Sohal et al., 2009). In humans and mice, increases in gamma oscillatory activity (30–80 Hz) during memory encoding predict effective memory formation (Matsumoto et al., 2013; Sederberg et al., 2007; Yamamoto et al., 2014). Gamma oscillatory activity can be enhanced by optogenetic stimulation of parvalbumin (PV)-positive inhibitory interneurons (PV cells) but not other cell types (Cardin et al., 2009; Sohal et al., 2009), indicating a causal link between PV cell function and gamma oscillations. In humans with epilepsy and in hAPP mice, behaviorally induced gamma oscillatory activity potently suppresses network hypersynchronization (Matsumoto et al., 2013; Verret et al., 2012). Many lines of hAPP mice, including hAPP-J20, TgCRND8, Tg2576, and APP/PS1 mice, have network hypersynchrony and altered gamma oscillatory activity (Cramer et al., 2012; Gurevicius et al., 2013; Hamm et al., 2017; Palop et al., 2007; Palop and Mucke, 2016; Rubio et al., 2012; Verret et al., 2012).
In humans and mice, Nav1.1 is predominantly expressed in interneurons, including PV and somatostatin (SOM) cells (Ogiwara et al., 2007; Rubinstein et al., 2015; Tai et al., 2014; Verret et al., 2012; Wang et al., 2011). Selective Nav1.1 deletion in PV and SOM cells impairs their excitability and action potential (AP) generation (Rubinstein et al., 2015; Tai et al., 2014). Hypofunction of Nav1.1 causes multiple epilepsy syndromes and autism (D'Gama et al., 2015; Yu et al., 2006). Nav1.1 levels are reduced in humans with AD and in multiple mouse models of AD, including hAPP-J20, TgCRND8, and APP/PS1 mice (Hamm et al., 2017; Hu et al., 2017; Verret et al., 2012). Nav1.1 levels can be restored by γ-secretase inhibitors in TgCRND8 mice (Hamm et al., 2017). BACE1-dependent hypofunction of Nav1.1 has also been reported in Tg2576 and BACE1 transgenic mice (Corbett et al., 2013; Kim et al., 2007), which may involve enhanced cleavage of the β2-subunit of voltage-gated sodium channels and impaired trafficking of the α-subunits, including Nav1.1, to the cell membrane. Importantly, restoring Nav1.1 levels by expressing a bacterial artificial chromosome (BAC) enhanced gamma oscillatory activity, reduced network hypersynchrony, and improved cognitive performance in hAPP-J20 mice (Verret et al., 2012), suggesting an important functional role of Nav1.1-dependent interneuron hypofunction in the pathogenesis of AD-related deficits.
Interneurons and neuronal network oscillations are altered in multiple neurological and psychiatric disorders, including AD, epilepsy, schizophrenia, and autism (Fazzari et al., 2010; Kepecs and Fishell, 2014; Sohal et al., 2009; Verret et al., 2012). Transplantation of interneuron progenitors is a potential therapeutic strategy for these conditions (Southwell et al., 2014). Interneuron progenitor cells can be obtained from the embryonic medial ganglionic eminence (MGE), the source of cortical PV and SOM interneurons (Kepecs and Fishell, 2014; Southwell et al., 2014). When transplanted into neonatal or adult brains, MGE-derived progenitor cells (MGE cells) migrate, integrate into neuronal circuits, and mature into functional inhibitory interneurons (Alvarez-Dolado et al., 2006; Henderson et al., 2014; Howard et al., 2014; Hunt et al., 2013). Hippocampal transplants of wild-type MGE cells improve behavioral functions in mouse models with extensive loss of hippocampal hilar interneurons, including pilocarpine-treated mice (Casalia et al., 2017; Hunt et al., 2013) and apolipoprotein E4 knock-in (apoE4-KI) mice (Tong et al., 2014), most likely by restoring interneuron cell numbers. However, it is unknown whether transplanted MGE cells can reduce hAPP/Aβ-dependent behavioral and network dysfunctions in brains with chronically elevated levels of Aβ in the absence of extensive interneuron loss. Here we demonstrate that brain transplants of wild-type, Nav1.1-overexpressing, or Nav1.1-deficient interneuron progenitors have very different effects on network and cognitive functions in hAPP-J20 and wild-type mice and suggest that molecular modification to counteract pathogenic contexts will likely be required to optimize the therapeutic efficacy of cell transplants for neurological disorders.
RESULTS
Transplanted MGE Cells Differentiate into Interneurons and Survive in hAPP-J20 Mice
We chose hAPP-J20 (J20) mice as recipients for interneuron transplants because they have robust cognitive deficits in the Morris water maze and fear conditioning tests, network hypersynchrony, reduced gamma oscillatory power, and multiple other features that resemble AD in humans (Palop et al., 2007; Palop et al., 2003; Palop and Mucke, 2016; Rubio et al., 2012; Verret et al., 2012). To identify cortical and hippocampal inhibitory interneurons derived from the MGE, we used BAC transgenic mice expressing green fluorescent protein (GFP) directed by Lhx6 regulatory sequences (Lhx6GFP mice). Lhx6GFP mice express GFP in embryonic MGE progenitors (Cobos et al., 2006) and in mature inhibitory interneurons, mainly PV and SOM cells (Figure 1A). Consistent with the predominant expression of Nav1.1 in inhibitory interneurons (Ogiwara et al., 2007; Rubinstein et al., 2015; Tai et al., 2014; Verret et al., 2012; Yu et al., 2006), 97% of cells with high Nav1.1 levels in Lhx6GFP mice were GFP positive (Figures 1B and 1C). Thus, Lhx6GFP-postive MGE-derived interneurons express Nav1.1 and mature into PV and SOM interneurons.
Figure 1. Transplanted MGE-derived precursors generate Nav1.1-expressing inhibitory interneurons in host NTG and J20 brains.

(A) Double-labeling for GFP (green) and PV (red, top) or SOM (red, bottom). GFP is expressed by PV (top) and SOM (bottom) inhibitory interneurons in Lhx6GFP mice. Arrowheads indicate double-labeled cells.
(B) Double-labeling for Nav1.1 (red) and GFP (green). Nav1.1 is predominantly expressed by Lhx6GFP inhibitory interneurons. Arrowheads indicate double-labeled cells.
(C) 97% of Nav1.1-positive cells were Lhx6GFP interneurons (n = 4 mice, 104 cells). (D) GFP-positive interneurons that were derived from Lhx6GFP MGENav1.1 precursor cells transplanted into 3–5-day-old J20 mice 7–8 months before analysis had mature morphology and extensive axonal and dendritic arborizations. At 7–8 months of age, J20 mice had Aβ-positive plaques (red).
(E and F) MGE-derived interneurons predominantly expressed PV or SOM and were distributed in typical subtype-specific fashion across cortical layers. (E) Double-labeling for GFP (green) and PV (red, top) or SOM (red, bottom). Transplanted GFP-positive PV cells were predominantly in intermediate cortical layers (top, layer V), whereas GFP-positive SOM cells were predominantly in superficial cortical layers (bottom, layer I). (F) Proportions of MGEWT- and MGENav1.1-derived interneurons in different cortical layers in NTG and J20 mice expressing PV, SOM, or neuropeptide Y (NPY) (n = 4 mice per group, 1449 GFP-positive cells).
(G) Number of MGEWT- and MGENav1.1-derived interneurons in NTG and J20 mice (n = 6–11 mice per group, 21696 GFP-positive cells).
Values are mean ± SEM. See also Figures S1 and S2.
To generate interneurons with increased Nav1.1 expression (Verret et al., 2012), we bred Lhx6GFP and Scn1a-BAC mice to produce doubly transgenic mice overexpressing wild-type mouse Nav1.1 in GFP-positive interneurons (Lhx6GFPNav1.1-BAC mice). On embryonic day 13.5, we isolated MGE cells with wild-type Nav1.1 levels (MGEWT) from Lhx6GFP mice and MGE cells overexpressing Nav1.1 (MGENav1.1) from Lhx6GFPNav1.1-BAC mice. Nontransgenic (NTG) and J20 littermate mice received transplants of MGEWT or MGENav1.1 cells into cortex and hippocampus on postnatal (P) days 3–5. After 7–8 months, GFP-positive cells in all groups (NTGMGE–WT, J20MGE–WT, NTGMGE–Nav1.1 and J20MGE–Nav1.1) had mature interneuron morphology, predominately expressed the interneuron markers SOM (39%), PV (31%), or neuropeptide Y (7%) and displayed comparable and subtype-appropriate localization across cortical layers (Figures 1D–F). Although transplanted GFP-positive interneurons were in all cortical layers, GFP-positive PV cells were predominantly in intermediate cortical layers (III–V), whereas GFP-positive SOM cells were predominantly in superficial or deeper cortical layers (I–II or VI). Analysis of every tenth serial section throughout the cortex and hippocampus showed that all groups of mice had comparable spatial distribution and numbers of GFP-positive interneurons (Figures 1G and S1B). We estimate that all groups had on average 15,500–17,500 transplanted GFP-positive interneurons per brain. Transplant-derived interneurons did not activate microglia or induce reactive astrocytosis in host brains (Figures S1C and S1D). Thus, transplanted MGE cells differentiate into interneurons and survive in J20 mice.
Low levels of cortical and hippocampal amyloid deposition were present in 100% of 7–8-month-old J20 mice (Figure S2). To determine whether transplant-derived interneurons modulate early amyloid pathology in J20 mice 7–8 months after transplantation, we measured hippocampal Aβ-positive deposits by immunostaining with the anti-Aβ antibody 82E1. Compared to sham-treated mice (J20SHAM), neither MGEWT nor MGENav1.1-derived cell transplants changed the percent area occupied by Aβ deposits or the size or number of deposits in the hippocampus of J20 mice (Figure S2). Hippocampal area per se was also unaffected by MGE transplants (Figure S2).
Nav1.1-Overexpressing, but Not Wild-Type, Interneuron Transplants Prevent and Reverse Behavioral Deficits in J20 Mice
To determine whether Nav1.1 expression levels in MGE cells modulate transplant efficacy in J20 mice, we transplanted MGEWT or MGENav1.1 cells bilaterally into the cortex and hippocampus of J20 and NTG littermate mice at P3–5 and analyzed the mice at 4–7 months of age (Figures 2A–F). Sham-transplanted littermate groups (NTGSHAM and J20SHAM) served as controls. Compared to NTG littermate controls, J20SHAM and J20MGE–WT mice displayed robust hyperactivity in the open field (Figure 2A) and Y-maze (Figure 2B) and excessive freezing during training in the fear-conditioning test (Figures 2C and S3), whereas J20MGE–Nav1.1 mice did not (Figures 2A–C and S3). Compared to J20SHAM mice, J20MGE–Nav1.1 mice displayed less hyperactivity and excessive freezing (Figures 2B and 2C). Thus, MGEWT and MGENav1.1 cells likely differ in their therapeutic efficacy in the presence of elevated levels of hAPP/Aβ.
Figure 2. MGENav1.1 cell transplants reduce behavioral and cognitive deficits in J20 mice.

Mice received bilateral transplants at P3–5 (A–F) or at 2–3 months of age (G–I) and were tested at 4–7 months.
(A–F) P3–5 MGENav1.1, but not MGEWT, transplants reduced behavioral alterations in J20 mice, including hyperactivity in the open field (A) and Y-maze (B) and enhanced freezing in the fear-conditioning test (C). MGENav1.1 cells improved performance in the hidden platform (spatial) component (D and E) and the probe trial (F) of the Morris water maze test. n = 8–10 mice per group. See Figure S3 for the learning curves of the same groups of mice in the fear-conditioning test.
(G) Open-field activity before (trial 1) and after MGE cell transplantation (trials 2–4). J20MGE–Nav1.1 mice habituated faster than J20SHAM and J20MGE–WT mice. n = 12–35 mice per J20 group.
(H) Open-field activity in trial 2.
(I) MGENav1.1, but not MGEWT, cells increased platform location preference in the water maze probe trial.
*P < 0.05, **P < 0.01, ***P < 0.001 vs. NTG littermate controls by ANOVA and Bonferroni post hoc test (A, B, C, and H), or vs. nontarget locations by paired two-tailed t test (F and I). For J20SHAM vs. J20MGE–Nav1.1 comparisons: #P < 0.05, ##P < 0.01, ###P < 0.001 by ANOVA and Bonferroni post hoc test (B, C, F, H, and I) or by repeated-measures ANOVA and Bonferroni test (D and E, days 1–6) and (G, trials 2–4). See Figure S4 for J20WT vs. J20MGE–Nav1 comparisons.
Values are mean ± SEM. Numbers in bars indicate numbers of mice. See also Figures S3 and S4.
To determine whether early MGENav1.1 cell implantation also reduces spatial learning and memory deficits in J20 mice, we first tested NTGSHAM, J20SHAM, NTGMGE–Nav1.1 and J20MGE–Nav1.1 mice in the Morris water maze. Compared to NTGSHAM controls, J20SHAM mice showed markedly impaired learning in the hidden platform (spatial) component, and this deficit was reduced by MGENav1.1 cell transplants (Figures 2D and 2E). In the water maze probe trial, only J20SHAM mice failed to show a significant preference for the original platform location (Figure 2F). J20MGE-Nav1.1 mice had better memory retention than J20SHAM mice (Figure 2F). Thus, Nav1.1-enhanced interneuron transplants reduce hAPP/Aβ-induced behavioral and cognitive deficits.
Next, we determined whether MGEWT and MGENav1.1 cell transplants can reverse behavioral abnormalities in adult, symptomatic J20 mice (Figures 2G–I). To ensure that all J20 groups had similar deficits at baseline, we tested untreated 2–3-month-old J20 and NTG mice in the open field (Figure 2G, trial 1). We then bilaterally transplanted MGEWT or MGENav1.1 cells into equally impaired J20 groups and into NTG controls and re-tested them at 4–7 months of age. Compared to NTG controls, J20SHAM and J20MGE–WT mice had robust and comparable hyperactivity and context-dependent habituation deficits in the open field (Figures 2G and 2H), indicating that MGEWT cells did not reduce these deficits in J20 mice. However, these deficits were reduced in J20MGE-Nav1.1 mice as compared to J20SHAM controls (Figures 2G and 2H). In the water maze probe trial, J20MGE-WT, but not J20MGE-Nav1.1, mice failed to favor the original platform location (Figure 2I). J20MGE-Nav1.1 mice had better memory retention than J20SHAM mice (Figure 2I).
Comparison of J20MGE-WT and J20MGE-Nav1.1 mice by ANOVA and Bonferroni tests (Nieuwenhuis et al., 2011) revealed that Nav1.1 overexpression in MGE cell transplants was required for therapeutic effects to be observed in J20 mice (Figure S4). We confirmed that all adult transplanted groups had similar numbers of MGEWT- and MGENav1.1-derived GFP-positive interneurons (4,797–5,941 GFP-positive cells per brain) (Figure S4A). Thus, transplantation of MGENav1.1, but not MGEWT, cells into the brains of neonatal or adult J20 mice reduced behavioral abnormalities in J20 mice, suggesting that molecular optimization of MGE cells is required for therapeutic benefit in brains with pathologically elevated levels of hAPP/Aβ.
Nav1.1-Deficient Interneuron Transplants Impair Behavioral Functions
Since reduced levels of endogenous Nav1.1 contribute to inhibitory and cognitive dysfunction in J20 mice (Verret et al., 2012), we tested whether reduced Nav1.1 levels in transplanted interneurons alter the behavior of NTG mice. MGE cells with reduced Nav1.1 expression were obtained from heterozygous Nav1.1 knock-in mice in which one Scn1a allele carries a point mutation (R1407X) that introduces a premature stop codon and causes Nav1.1 haploinsufficiency (Ogiwara et al., 2007). MGE cells were unilaterally transplanted into the cortex and hippocampus at P3–5, and mice were analyzed at 6–8 months of age.
MGENav1.1–KI cell transplants impaired learning in the Morris water maze in NTG mice, as shown by longer escape distances and latencies and a lower percentage of trials in which the platform was found (Figure 3A–C). Compared to NTGMGE–Nav1.1 mice, NTGMGE–Nav1.1–KI mice displayed excessive freezing in a fear conditioning test (Figure 3E). Thus, transplantation of Nav1.1-deficient interneurons into otherwise healthy brains caused learning deficits and behavioral alterations, suggesting that loss of Nav1.1 changes the functionality of these cells in ways that actively disrupt normal brain function.
Figure 3. Reduced Nav1.1 levels in interneuron transplants impair behavioral functions.

NTG and J20 mice received MGENav1.1 or MGENav1.1–KI cell transplants unilaterally into the hippocampus and cortex at P3–5 and were tested in the Morris water maze and in a fear-conditioning test at 6–8 months of age.
(A–C) Escape distances (A) and latencies (B) and percentage of trials in which the platform was found (C) in the hidden-platform component of the Morris water maze (n = 5–8 mice per group).
(D) Quadrant location preference during the probe trial. *P < 0.05, **P < 0.01 vs. nontarget quadrants by paired two-tailed t test.
(E and F) Fear-conditioning freezing responses during training and for the 24-h context-test intervals (1–5 min; test) in NTG (E) and J20 (F) mice transplanted with MGENav1.1 or MGENav1.1–KI cells (n = 7–12 mice per group). PSI, post-shock interval.
*P < 0.05, **P < 0.01 by repeated-measures ANOVA and Bonferroni post hoc test (A–C, days 1–5) and (E and F, 1–5 min). Values are mean ± SEM. Numbers in bars indicate numbers of mice.
We next compared the behavioral effects of MGENav1.1 and MGENav1.1–KI cell transplants in J20 mice. J20MGE–Nav1.1 mice performed better than J20MGE–Nav1.1–KI mice in the Morris water maze, as shown by shorter escape distances and latencies and a higher percentage of trials in which the platform was found (Figures 3A–C). In the probe trial, J20MGE–Nav1.1 mice, but not J20MGE–Nav1.1–KI mice, favored the original platform location (Figure 3D). Finally, J20MGE–Nav1.1–KI mice displayed more excessive freezing in a fear-conditioning test than J20MGE–Nav1.1 mice (Figure 3F). Evidently, increased Nav1.1 levels in MGE cells are required for therapeutic efficacy in J20 mice and reduced Nav1.1 levels in these cells cause behavioral deficits in NTG and J20 mice.
Nav1.1 Levels Modulate AP Kinetics and Firing Rates in Transplanted Interneurons
Increasing (MGENav1.1) or reducing (MGENav1.1-KI) Nav1.1 levels in MGE cells strongly modulated their effects upon transplantation into NTG and J20 mice. To explore how Nav1.1 levels modulate intrinsic electrophysiological properties of transplanted MGE cells, we carried out whole-cell patch-clamp recordings of MGEWT-, MGENav1.1- and MGENav1.1–KI-derived interneurons in acute coronal cortical slices from NTG or J20 mice 6–11 months after transplantations. Consistent with our histological data, we identified roughly similar proportions of transplant-derived fast-spiking (FS, n = 62 cells from 23 mice) and non-fast-spiking (N-FS, n = 65 cells from 25 mice) interneurons when combining all recorded cells. The transplant-derived FS and N-FS interneuron subtypes displayed expected differences in electrophysiological properties, including AP firing rate, AP half-width, AP rise and fall times, resting membrane potential, input resistance and membrane time constant (Figures 4 and S5).
Figure 4. Nav1.1 levels regulate AP kinetics in fast-spiking (FS) and non-fast-spiking (N-FS) MGE-derived interneurons transplanted in NTG mice.

NTG mice received MGEWT, MGENav1.1, or MGENav1.1–KI cell transplants into the hippocampus and cortex at P3–5. Whole-cell patch-clamp recordings from interneurons in the cortex of acute coronal brain slices were done at 6–11 months of age. FS: n = 11, 11, and 11 cells from 5 NTGMGE-WT, 4 NTGMGE-Nav1.1, and 3 NTGMGE-Nav1.1-KI mice, respectively. N-FS: n = 11, 26, and 6 from 4 NTGMGE-WT, 8 NTGMGE-Nav1.1, and 3 NTGMGE-Nav1.1-KI mice, respectively.
(A and B) Representative traces of AP firing recorded from cortical GFP-positive MGEWT-, MGENav1.1- or MGENav1.1–KI-derived FS (A) and N-FS (B) interneurons in NTG mice evoked by 1-s current injections of 200 and 400 pA.
(C) Quantification of AP waveform properties of MGEWT-, MGENav1.1-, and MGENav1.1–KI-derived FS and N-FS interneurons in NTG mice. P values by two-way ANOVA assessing cell type (FS and N-FS), MGE genotype (WT, Nav1.1, and Nav1.1-KI), and interaction.
(D) Representative traces of AP waveforms recorded from MGEWT-, MGENav1.1-, and MGENav1.1–KI-derived FS and N-FS interneurons in NTG mice.
(E) Mean number of APs in MGEWT-, MGENav1.1-, and MGENav1.1–KI-derived FS (top) and N-FS (bottom) interneurons evoked by the indicated incremental current pulse intensities. *P < 0.05 and **P < 0.01 by repeated-measures ANOVA and Bonferroni test for 200–500 pA.
(F) Number of APs elicited at the maximum current injection intensity (top) and the AP decrement in the 1-s current pulse (bottom) in MGEWT-, MGENav1.1-, and MGENav1.1–KI-derived interneurons. P values by two-way ANOVA assessing cell type (FS and N-FS), MGE genotype (WT, Nav1.1, and Nav1.1-KI), and interaction.
Values are mean ± SEM. Significant P values are in red. See also Figures S5 and S6.
To determine whether Nav1.1 levels affect AP properties, we compared transplanted MGENav1.1-, MGENav1.1-KI- and MGEWT-derived interneurons in NTG mice in regard to cell type (FS and N-FS), MGE genotype (WT, Nav1.1, and Nav1.1-KI), and the interaction among these variables by two-way ANOVA analyses. Importantly, Nav1.1 expression levels strongly modulated AP kinetics in transplant-derived FS and N-FS interneurons (Figures 4C and 4D). Compared to MGEWT-derived interneurons, MGENav1.1–KI-derived interneurons had a wider AP, slower AP rise and fall times, and a larger AP area, whereas MGENav1.1-derived interneurons had faster AP kinetics. Thus, Nav1.1 levels potently modulated AP kinetics, and MGENav1.1-, MGENav1.1-KI-, and MGEWT-derived interneurons are functionally different, providing potential cellular mechanisms that could explain the benefit of molecular optimization of the transplant-derived interneurons. We confirmed that Nav1.1 levels regulate action potential kinetics using individual cells (Figure 4C) or mice (Figure S6D) as the biological unit to calculate averages. Interestingly, N-FS MGENav1.1-derived cells displayed a distinct input-output relationship consisting of reduced AP firing at lower current injection intensities (0–100 pA; P = 0.020), higher proportional increases of AP firing rates at intermediate current intensities (100–400 pA), and similar AP firing rates at higher current intensities (400–500 pA). Thus, MGENav1.1-derived cells seem to be less responsive at low intensities and to have a better dynamic range to discriminate intermediate current intensities. In addition, FS MGENav1.1-KI-derived cells had slower AP firing rates in response to increased current injections and more pronounced AP amplitude decrement during sustained firing than MGEWT- and MGENav1.1-derived cells (Figures 4E and 4F). Consistent with these results, deletion of Nav1.1 impaired AP kinetics and firing in endogenous PV and SOM cells (Rubinstein et al., 2015; Tai et al., 2014; Yu et al., 2006).
To assess potential effects of the pathological altered host brain, we compared AP properties of transplanted FS and N-FS MGE-derived interneurons in NTG and J20 mice in regard to host brain genotype (NTG and J20), MGE genotype (WT and Nav1.1), and the interaction between them by two-way ANOVA (Figure 5A; and NTG data in Figure 4). Consistent with the above results, the FS and N-FS interneurons in NTG and J20 mice displayed robust Nav1.1-dependent differences in AP kinetics; however, the genotype of the host brain did not affect the AP kinetics of transplanted MGEWT- or MGENav1.1-derived FS and N-FS interneurons (Figures 5B and 5C). We confirmed these results using individual cells (Figure 5B) or mice (Figure S6E) as the biological unit to calculate averages. Comparison of MGEWT and MGENav1.1 cells in J20 mice by two-way ANOVA revealed that Nav1.1 overexpression was required for accelerating AP kinetics in FS and N-FS cells in these mice (Figure S6). In J20 mice, MGENav1.1-derived FS cells also had increased AP firing rates at higher current intensities (200–500 pA) as compared to MGEWT-derived FS cells (Figures 5D and S6). Our results indicate that Nav1.1 overexpression accelerates AP kinetics and increases AP firing rates of MGENav1.1-derived FS interneurons in J20 mice.
Figure 5. Transplanted MGENav1.1-derived interneurons in NTG and J20 mice exhibit faster AP kinetics and their function is not altered by the host genotype.

NTG and J20 mice received MGEWT or MGENav1.1 cell transplants into the hippocampus and cortex at P3–5. Whole-cell patch-clamp recordings from interneurons in the cortex of acute coronal brain slices were done at 6–11 months of age. FS: n = 11, 17, 11, and 12 cells from 5 NTGMGE-WT, 7 J20MGE-WT, 4 NTGMGE-Nav1.1, and 4 J20 MGE-Nav1.1 mice, respectively. N-FS: n = 11, 9, 26, and 13 cells from 4 NTGMGE-WT, 6 J20MGE-WT, 8 NTGMGE-Nav1.1, and 4 J20 MGE-Nav1.1 mice, respectively.
(A) Representative traces of APs recorded from cortical GFP-positive MGEWT- or MGENav1.1-derived FS and N-FS interneurons in NTG and J20 mice evoked by 1-s current injections of 500 pA. Detail of APs (gray area).
(B) Quantification of AP waveform properties of MGEWT- and MGENav1.1-derived FS and N-FS interneurons in NTG (data from Figure 4C) and J20 mice. P values by two-way ANOVA assessing MGE genotype (WT and Nav1.1), host brain genotype (NTG and J20), and interaction.
(C) Representative traces of AP waveforms recorded from MGEWT- and MGENav1.1-derived FS and N-FS interneurons in NTG and J20 mice.
(D) Mean number of APs evoked by the indicated incremental current intensities in MGEWT- and MGENav1.1-derived FS (top) and N-FS (bottom) interneurons transplanted into NTG mice (data from Figure 4E) and J20 mice. J20MGE-WT vs. J20MGE–Nav1.1 comparison, *P < 0.05 by repeated-measures ANOVA and Bonferroni test for 200–500 pA.
(E) Number of APs elicited at the maximum current injection intensity (top) and the AP decrement in the 1-s current pulse (bottom) in MGEWT- and MGENav1.1-derived interneurons transplanted in NTG mice (data from Figure 4F) and J20 mice. P values by two-way ANOVA assessing MGE genotype (WT and Nav1.1), host brain genotype (NTG and J20), and interaction.
Values are mean ± SEM. Numbers in bars indicate numbers of cells. Significant P values are in red. See also Figures S5 and S6.
Nav1.1-Overexpressing, but Not Wild-Type, Interneuron Transplants Counteract Network Hypersynchrony in J20 Mice
Humans with AD, particularly early-onset and familial AD, and hAPP transgenic mice are prone to network hypersynchrony and altered gamma oscillatory activity (Noebels, 2011; Palop and Mucke, 2016; Vossel et al., 2016). To determine whether interneuron transplants suppress hAPP/Aβ-induced network hypersynchrony, we transplanted MGEWT or MGENav1.1 cells bilaterally into the hippocampus and cortex of 2–3-month-old mice and analyzed the mice at 7–8 months of age. We monitored spontaneous epileptiform discharges (spikes/min) by intracranial EEG recordings and locomotor activity (movements; beam breaks/min) for 50 min while mice explored a novel environment. MGENav1.1 cell transplants reduced hyperactivity (Figure 6A) and spike frequency (Figure 6B) in J20 mice.
Figure 6. MGENav1.1, but not MGEWT, cell transplants reduce network hypersynchrony in J20 mice.

Mice were sham-treated or transplanted bilaterally with MGEWT or MGENav1.1 cells at 2–3 months of age. EEGs were recorded at 7–8 months during a 50-min exploration of a novel environment.
(A) Movements (beam breaks/min) during EEG recordings in the open field (n = 9–11 mice per J20 group).
(B) Spike rate averaged over the entire exploration period.
(C) Minute-by-minute spike rate. MGENav1.1 cells reduced spike rates in J20 mice.
(D) EEG recordings (top) and minute-by-minute spike rate and movements (bottom) for representative J20SHAM and J20MGE–Nav1.1 mice. Detail of epileptiform spike (gray area).
(E) Spike rates in J20SHAM and J20MGE–Nav1.1 mice (n = 431–493 min of EEG recordings, 9–11 mice per group) by exploratory activity level (left) or movement frequency (right). Treatment (P < 0.0001, F(2, 918) = 22.44), activity level (P < 0.0001, F(2, 918) = 53.67), and interaction (P = 0.572, F(2, 918) = 0.558) effects by two-way ANOVA.
(F) Minute-by-minute spike rate of J20 mice transplanted with MGEWT cells and controls over 50 min. MGEWT cells did not change spike rates.
(G) Average spike frequencies of J20 mice transplanted with MGESHAM, MGEWT and MGENav1.1 cells.
*P < 0.05, ***P < 0.001 by repeated-measures ANOVA and Bonferroni test (A, C, and F) or by one-way ANOVA and Bonferroni test (B, E, G). Values are mean ± SEM. Numbers in or above bars indicate number of mice.
The firing rates of PV-positive interneurons and gamma oscillatory power increase during exploratory activity (Kemere et al., 2013; Lapray et al., 2012). Consistent with this behavior-dependent modulation of network function, spike rates in J20 mice increased as they habituated to the novel environment and their exploratory activity decreased, most markedly in J20SHAM mice (Figure 6C). Movements and spike rates in J20 mice were inversely related (Figure 6D). Spike rates were highest at rest (0–9 beam breaks/min), intermediate during low exploratory activity (10–49 beam breaks/min), and lowest during high exploratory activity (50–150 beam breaks/min) (Figure 6E). Spike rates were lower in J20MGE–Nav1.1 mice than J20SHAM mice (Figure 6E). To determine whether Nav1.1 expression in MGE cells is required for therapeutic effects in J20 mice, we compared J20SHAM, J20MGE-WT, and J20MGE-Nav1.1 mice. Consistent with our behavioral data (Figures 2 and S4), MGENav1.1, but not MGEWT, cells reduced epileptiform activity in J20 mice (Figures 6F and 6G). Thus, Nav1.1 enhancement of MGE cells is required for these transplants to suppress hAPP/Aβ-induced hypersynchrony.
Reduced Behavioral Activity–Associated Increases in Gamma Oscillations Predict Locomotor Hyperactivity in J20 Mice
Gamma oscillatory power in cortical networks increases during sensory or cognitive tasks in humans and mice; the increases predict better memory formation (Matsumoto et al., 2013; Sederberg et al., 2007; Yamamoto et al., 2014) and are associated with reduced rates of epileptiform spikes in humans and mice with network hypersynchrony (Matsumoto et al., 2013; Verret et al., 2012). To determine whether behavior-dependent modulation of gamma oscillations and epileptiform spikes in J20 mice relate to their hyperactivity and habituation deficits, we studied the relationship between gamma oscillatory power, epileptiform spike rate, and locomotor activity at 1-min intervals in J20SHAM and J20MGE–Nav1.1 mice. Consistent with previous findings (Rubio et al., 2012; Verret et al., 2012), J20SHAM mice had reduced gamma power (Figure S7). Gamma power, which is predominantly regulated by PV cells (Cardin et al., 2009; Sohal et al., 2009), was closely related to exploratory activity (Figures 7A–C) (Kemere et al., 2013; Lapray et al., 2012). Linear regression analysis revealed strong positive correlations between exploratory activity and gamma power (Figure 7C). Obtaining data at 1-min intervals allowed us to accurately calculate the slope of the linear regression between exploratory activity and gamma power (relative to resting) for each mouse. Thus, the slope reflects the modulation of gamma power during exploratory activity. J20SHAM and J20MGE–Nav1.1 mice with less behavior-dependent modulation of gamma power (flatter slopes) had higher levels of exploratory activity (Figure 7D, top) and took longer to habituate (Figure 7D, bottom). Total epileptic activity did not correlated with these behavioral alterations in J20 mice (data not shown; P = 0.668, J20SHAM; P = 0.5569, J20MGE-Nav1.1). These results suggest that the extent of behavior-dependent modulation of gamma oscillatory activity predicts behavioral performance and that reduced modulation is closely related to hyperactivity and habituation deficits in hAPP mice.
Figure 7. Reduced behavior-dependent modulation of gamma oscillations predicts behavioral deficits in J20 mice.

Mice were sham-treated or transplanted bilaterally with MGENav1.1 cells at P3–5 or at 2–3 months of age and were tested at 7–8 months (n = 16–18 mice per group).
(A) Minute-by-minute quantifications of gamma power (30–80 Hz; top) and corresponding 30–80 Hz spectrograms (bottom) for representative J20SHAM and J20MGE–Nav1.1 mice.
(B) Exploratory activity and gamma power in representative J20SHAM and J20MGE–Nav1.1 mice.
(C) Linear regression analyses showed strong relationships between gamma power and exploratory activity (mice from A). Data points represent minutes.
(D) Linear regression analyses showed strong relationships between behavior-dependent increases in gamma power (slopes) and total movements in the open field (top) or time to habituate to NTG control level (bottom) for J20SHAM mice (left) and J20MGE–Nav1 mice (right). Data points represent mice. Hyperactivity and habituation deficits were strongly associated with lower behavior-dependent modulation of gamma power. Slopes (m) for J20SHAM and J20MGE–Nav1.1 mice were not significantly different (P = 0.169 and 0.853, respectively) by linear regression slope test.
See also Figure S7.
Nav1.1-Overexpressing Interneuron Transplants Enhance Behavior-Dependent Modulation of Gamma Oscillations
Gamma oscillations aberrantly increase during seizures or hypersynchronized network activity in epilepsy models (Howard et al., 2014). MGE cell transplants reduce these gamma oscillation increases (Howard et al., 2014) and network hypersychrony, including seizures (Baraban et al., 2009; Casalia et al., 2017; Hammad et al., 2015; Henderson et al., 2014; Hunt et al., 2013). However, it is unknown whether MGE cell transplants can enhance normal fluctuations of gamma oscillatory activity during memory encoding (Matsumoto et al., 2013; Sederberg et al., 2007; Yamamoto et al., 2014). To determine whether MGE cell transplants increase behavior-dependent modulation of gamma oscillations, J20 mice were sham-treated or transplanted with MGENav1.1 or MGEWT cells at 2–3 months of age and tested at 4–7 months. Compared to MGESHAM, MGENav1.1 but not MGEWT cell transplants enhanced the increases of gamma power during exploratory activity (Figure 8A), which predict better behavioral performance in J20 mice (Figure 7). J20MGE-Nav1.1 mice had higher increases in gamma power than J20SHAM mice (Figure 8A). Despite the electrophysiological deficits in MGENav1.1–KI-derived FS and N-FS interneurons (Figure 4) and the behavioral abnormalities these cells caused in NTG mice (Figure 3), we were unable to detect epileptiform discharges or altered gamma oscillations in NTGMGE–Nav1.1–KI mice (data not shown).
Figure 8. MGENav1.1, but not MGEWT, cell transplants increase behavior-dependent modulation of gamma oscillations in J20 mice.

Mice were sham-treated or transplanted bilaterally with MGEWT or MGENav1.1 cells at 2–3 months of age and were tested at 7–8 months (n = 20 J20SHAM, 7 J20MGE-WT, and 9 J20MGE-Nav1.1 mice).
(A) Linear regression analyses showed strong relationships between gamma power and exploratory activity for J20 mice transplanted with MGESHAM, MGEWT, or MGENav1.1 cells. Compared to MGESHAM cells, MGENav1.1, but not MGEWT, cells increased behavior-dependent modulation of gamma power (top two panels). J20MGE-Nav1.1 mice had higher increases of gamma power than J20MGE-WT mice (bottom). n = 7–11 mice per group, 431–492 min of EEG recordings per group. ***P < 0.001 by linear regression slope test.
(B) Gamma power as a function of time before and after epileptiform spikes (n = 493 spikes, 11 J20SHAM mice). Gamma oscillatory power decreased steadily 40–50 s before epileptiform discharges and gradually recovered over 50–60 s. Values are mean ± SEM.
(C) MGENav1.1, but not MGEWT, cells reduced the drop in gamma power before the spikes (n = 390–915 spikes per group from 15 J20SHAM, 5 J20MGE–WT, and 8 J20MGE–Nav1.1 mice). ***P < 0.001 by linear regression slope test.
Values are mean ± SEM.
Periods of reduced gamma power tend to be associated with increased rates of epileptiform discharges in humans and mice with network hypersynchrony (Matsumoto et al., 2013; Verret et al., 2012). However, it is unclear whether these phenomena are causally related and, if so, whether epileptiform discharges cause (and therefore precede) reductions in gamma power, or vice versa. To address this issue, we calculated the gamma power before and after each epileptiform discharge in J20 mice. Gamma power decreased abruptly before the discharges, reached the lowest level during the discharges, and rapidly recovered after the discharges (Figure 8B). Thus, decreased gamma power precedes epileptiform discharges, suggesting that the drop in gamma power is causally linked to the emergence of these events. Interestingly, only MGENav1.1 cells prevented the drop in gamma power before epileptiform discharges in J20 mice (Figure 8C), suggesting that the drop is caused by interneuron impairment and can be prevented by Nav1.1-overexpressing interneuron transplants.
DISCUSSION
This study shows that transplantation of Nav1.1-overexpressing MGE-derived interneurons into brains of a mouse model simulating key aspects of AD enhances behavior-dependent modulation of gamma oscillatory activity, reduces network hypersynchrony, and improves cognitive functions. Wild-type interneuron transplants failed to restore brain functions, indicating that the therapeutic effects of the transplanted cells depend on Nav1.1 levels. Moreover, transplantation of Nav1.1-deficient interneurons in NTG mice induced learning deficits and behavioral abnormalities, indicating that Nav1.1 levels in interneurons critically impact cognitive functions. Thus, Nav1.1 deficiency confers an adverse gain of function on interneurons, whereas Nav1.1 overexpression enhances their normal activities beyond a critical threshold or confers a beneficial gain of function. At the cellular level, increased Nav1.1 levels accelerated AP kinetics, whereas decreased Nav1.1 levels slowed AP kinetics in transplant-derived FS and N-FS interneurons in NTG mice. In J20 mice, enhanced Nav1.1 levels in MGENav1.1-derived FS interneurons accelerated AP kinetics and increased AP firing rates. Our findings highlight key differences among MGEWT, MGENav1.1-, and MGENav1.1–KI-derived interneurons, the functional relevance of Nav1.1 levels in transplant-derived interneurons, the potential of Nav1.1 and interneurons as a therapeutic target in AD, and the value of molecularly modifying brain cell transplants to enhance their functions.
We identified three main cellular and network mechanisms that may underlie the beneficial effects of interneuron transplants, all of which depended on Nav1.1 levels. MGENav1.1, but not MGEWT, cell transplants accelerated AP kinetics, enhanced behavior-dependent increases in gamma oscillatory activity, and reduced network hypersynchrony in hAPP-J20 mice.
Increased Nav1.1 Levels Accelerate AP Kinetics in Transplanted Interneurons
Nav1.1 deficiency in endogenous inhibitory interneurons slows AP kinetics, including AP half-width, and reduces AP firing rates in a gene dose-dependent manner (Rubinstein et al., 2015; Tai et al., 2014; Yu et al., 2006). We observed similar AP deficits in transplanted MGENav1.1-KI-derived interneurons, confirming the functional relevance of Nav1.1 levels in inhibitory cells. Nav1.1 overexpression accelerated AP kinetics (spike waveforms) in transplant-derived FS and N-FS interneurons in NTG and hAPP-J20 mice and increased AP firing rates in MGENav1.1-derived FS interneurons in hAPP-J20 mice. Faster AP kinetics and higher firing rates in MGENav1.1-derived FS interneurons of J20 mice may have several functional benefits, including incomplete sodium channel inactivation during repetitive firing, which enables high-frequency firing in fast-spiking interneurons, resistance to frequency-dependent AP broadening during repetitive firing, and enhanced gamma oscillations (Bean, 2007; Carter and Bean, 2009). Our data suggest that faster AP kinetics and higher AP firing rates are the key cellular mechanisms underlying the therapeutic effects of MGENav1.1-derived interneurons in J20 mice.
MGENav1.1 Cell Transplants Enhance Behavior-Dependent Gamma Oscillations and Cognitive Functions
Since increasing the AP firing rate of PV cells promotes gamma oscillatory activity (Cardin et al., 2009; Sohal et al., 2009), the improvement of gamma oscillations in MGENav1.1-transplanted hAPP-J20 mice likely reflects increased activity or synchrony of transplant-derived PV cells. Indeed, MGENav1.1-derived FS interneurons had higher firing rates than MGEWT-derived FS interneurons in hAPP-J20 mice. It is tempting to speculate that these increased firing rates contributed to the enhanced gamma power in J20MGE-Nav1.1 mice. Importantly, increases in gamma oscillatory activity during cognitive tasks predict effective memory formation in humans and mice (Matsumoto et al., 2013; Sederberg et al., 2007; Yamamoto et al., 2014). Consistent with these findings, reduced behavior-dependent modulation of gamma oscillations predicted habituation deficits and hyperactivity in hAPP-J20 mice. The inability of these mice to enhance gamma oscillatory activity during exploration of a novel environment may contribute to their learning and memory deficits. Indeed, MGENav1.1-derived interneuron transplants increased the behavior-dependent modulation of gamma oscillation and improved cognitive functions.
MGENav1.1 cell transplants also reduced excessive freezing in hAPP-J20 mice during the learning and recall phases of a fear-conditioning test. Excessive freezing was also seen in NTGMGE-Nav1.1-KI mice without detectable epileptic activity, indicating that dysfunction of a small proportion of interneurons is sufficient to cause this behavioral abnormality. Consistent with our results, optogenetic inhibition of PV cells causes excessive fear-like responses in mice without seizures (Courtin et al., 2014).
Interestingly, MGENav1.1 cells transplanted into J20 mice had beneficial effects on behavioral and network functions despite the Nav1.1 hypofunction of endogenous interneurons in these mice. Yet, MGENav1.1-KI cells transplanted into NTG mice had detrimental effects on behavioral functions. These results indicate that relatively small numbers of transplanted MGE cells with modified Nav1.1 function can exert dominant positive as well as dominant negative effects. In principle, these opposing effects could be caused by modulation of the same or distinct cellular and network mechanisms, including complex interactions between transplanted and endogenous neurons.
Network Hypersynchrony and Gamma Oscillations in Mice and Humans
Reduced gamma oscillatory activity seems to be mechanistically linked to network hypersynchrony in hAPP-J20 mice (Verret et al., 2012) and in humans with epilepsy (Matsumoto et al., 2013). Notably, gamma power dropped abruptly seconds before epileptiform discharges, and reached the lowest level during the discharges. Thus, reduced gamma power may be mechanistically linked to the generation of epileptiform discharges, at least in hAPP-J20 mice. In humans with epilepsy, memory encoding is associated with increased gamma oscillations and reduced epileptiform discharges (Matsumoto et al., 2013). Thus, enhancing gamma oscillations by therapeutic interventions may reduce network hypersynchrony. Indeed, MGENav1.1-transplanted hAPP-J20 mice had increased behavior-dependent gamma oscillatory activity and reduced network hypersynchrony, implicating Nav1.1-deficient inhibitory interneurons and impaired gamma oscillations as potential pathogenic drivers.
Interneuron Transplant Efficacy and Disease-Specific Molecular Optimization
MGEWT-derived interneurons did not reduce network and cognitive dysfunctions in hAPP-J20 mice, whereas they did in human apoE4-KI/hAPP-J20 mice (Tong et al., 2014) and pilocarpine-treated nontransgenic mice (Casalia et al., 2017; Hunt et al., 2013). Several mechanisms may account for the greater efficacy of MGEWT cells in these disease models. First, behavioral phenotypes in apoE4-KI/hAPP-J20 mice largely depend on apoE4, as soluble Aβ levels and amyloid deposition are reduced by >75–80% in APP mice that lacked mouse apoE (Bales et al., 1999; Bien-Ly et al., 2012; Kim et al., 2011). Second, pilocarpine-treated (Casalia et al., 2017; Hunt et al., 2013) and apoE4-KI mice with or without hAPP expression (Tong et al., 2014) have severe loss of hippocampal interneurons, particularly of SOM cells in the hilus of the dentate gyrus. Thus, hippocampal MGE transplants may primarily serve to replace the lost interneurons. In hAPP-J20 mice, however, interneurons, particularly PV cells in the parietal cortex, are not depleted but become dysfunctional as a result of Nav1.1 depletion (Verret et al., 2012). Thus, disease-specific molecular optimization of cell transplants might be required to achieve therapeutic benefits in some circumstances but not others.
Translational Opportunities for Brain Cell Transplants
Great caution needs to be applied when trying to extrapolate from animal models to human diseases. Bearing this caveat in mind, we find it promising that transplantation of human MGENav1.1 cells into the brains of symptomatic adult J20 mice reduced cognitive deficits and behavioral alterations, even though it did not change the early amyloid pathology. Because the long-term efficacy and safety of anti-Aβ treatments remains uncertain (Cummings et al., 2017), it is important to explore alternative and complementary strategies to counteract effects that mediate AD-related network dysfunction and cognitive decline. Based on the data presented here and others reviewed elsewhere (Palop and Mucke, 2016), we consider the dysfunction of interneurons and the hypofunction of Nav1.1 worthy targets in this regard.
Notably, interneuron dysfunction and abnormal oscillatory network activities are associated not only with AD, but also with autism, schizophrenia, and other cognitive disorders (Fazzari et al., 2010; Kepecs and Fishell, 2014; Sohal et al., 2009; Verret et al., 2012). Interneuron transplantation strategies might be beneficial in several of these disorders (Southwell et al., 2014). Rapid advances in cell reprogramming approaches may enable the generation of human cells that closely resemble MGE-derived interneuron precursors before long (Cunningham et al., 2014; Liu et al., 2013). Our findings suggest that for these cells to reach their maximal therapeutic potential, they may need to be further engineered to adapt their biological activities to the pathological environment into which they are transplanted.
STAR★ METHODS
• CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be address to and will be fulfilled by the Lead Contact, Jorge J. Palop (jorge.palop@gladstone.ucsf.edu)
• EXPERIMENTAL MODEL AND SUBJECT DETAILS
◦ Mice
We studied heterozygous transgenic mice expressing human amyloid precursor protein (hAPP) with the Swedish and Indiana FAD mutations (line J20) and nontransgenic (NTG) littermate controls (Mucke et al., 2000). F1 offspring from crosses between male J20 C57BL/6J mice and female NTG FVB/N mice were analyzed. To identify inhibitory interneurons derived from the medial ganglionic eminence (MGE), we used BAC transgenic FVB/N mice expressing enhanced green fluorescent protein (eGFP) directed by Lhx6 regulatory sequences (Lhx6GFP mice) (Cobos et al., 2006). To increase Nav1.1 expression levels by ~25% (Verret et al., 2012), we used Scn1a-BAC transgenic FVB/N mice from line 1 overexpressing wildtype murine Nav1.1 (Nav1.1-BAC mice) (Tang et al., 2009). To reduce Nav1.1 expression levels, we used Nav1.1 knock-in mice carrying a premature stop codon point mutation of the mouse Scn1a gene (Nav1.1-R1407X mice) (Ogiwara et al., 2007). Unless indicated otherwise, measurements were done in sex-balanced groups by investigators who were unaware of the genotype and treatment of the mice. Mice had free access to food (Picolab Rodent Diet 20; LabDiet, St Louis, MO) and water and were maintained on a regular light/dark (12 hours on/12 hours off) cycle. For harvesting of brain tissue, mice were deeply anesthetized and flush-perfused transcardially with saline. Brains were drop-fixed in 4% phosphate-buffered paraformaldehyde for 24–48 h. All experiments were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco.
• METHOD DETAILS
◦ MGE Transplants
At embryonic (E) day 13.5, brains from Lhx6GFP, Lhx6GFP/Nav1.1-BAC, or Lhx6GFP/Nav1.1-R1407X FVB/N mice were placed in ice-cold L-15 medium, and the ventricular and subventricular zones of the anterior part of the MGE were dissected as described (Alvarez-Dolado et al., 2006; Zipancic et al., 2010) to isolate MGEWT, MGENav1.1, or MGENav1.1–KI, respectively. To generate MGEWT cells, we crossed homozygous Lhx6GFP+/+ males with wild-type females to produce Lhx6GFP+/− embryos. To generate MGENav1.1 cells, we crossed Lhx6GFP+/+Nav1.1-BAC+/− males with Lhx6GFP+/+Nav1.1-BAC+/− females to produce Lhx6GFP+/+, Lhx6GFP+/+/Nav1.1-BAC+/− and Lhx6GFP+/+/Nav1.1-BAC+/+ embryos. Among 189 offspring from independent breedings between Nav1.1-BAC+/− males and Nav1.1-BAC+/− females 70.4% were Nav1.1-BAC positive, suggesting that such crosses yield both heterozygous and homozygous Nav1.1-BAC transgenic mice. To generate MGENav1.1-KI cells, we crossed Nav1.1-R1407X+/− males with Lhx6GFP+/+ females to produce Lhx6GFP+/−Nav1.1-R1407X+/+ and Lhx6GFP+/−Nav1.1-R1407X+/− embryos. Embryonic MGEs from 2–3 litters (~10–20 embryos) were dissected, pooled, and transplanted into 10–20 mice. Thus, MGENav1.1 and MGENav1.1–KI precursors likely contained ~70% and ~50% cells with increased (Nav1.1-BAC+/− or Nav1.1-BAC+/+) or decreased (Nav1.1-R1407X+/−) Nav1.1 expression, respectively. For the electrophysiological experiments, embryonic MGEs from individual embryos were dissected and transplanted into 1–2 mice. The rest of the brain post MGE dissection was collected from each embryo and genotyped for flag-Nav1.1 or Nav1.1-KI to identify MGE cell genotypes.
At E13.5, the density of MGE embryonic interneuron progenitors is at its peak, and the MGE is clearly delineated by a sulcus from the lateral ganglionic eminence (LGE). Tissue explants were collected in 1 ml of L-15 medium containing DNase I (10–100 µg/ml) and mechanically dissociated by repeated pipetting through a 200-µl plastic tip. MGE cell number and viability (~80%) were determined by trypan blue exclusion. The cells were pelleted by centrifugation (2 min, 800 × g) and resuspended in L-15 medium at 0.1–0.5×106 cells/µl. MGE cell suspensions were front-loaded into beveled glass micropipettes (~60–150-µm tip diameter) prefilled with pure mineral oil and then with L-15 medium. A microinjector and stereotaxic frame were used to deliver a total of 0.5–1.0×106 MGE cells per mouse distributed bilaterally across 14 (adults) or 8 (neonatal) cortical and hippocampal sites (50–100 nl per site) through four injection sites. Stereotaxic coordinates for adults were: anterior injection sites A −0.7 from the bregma, L ±2.4, D 1.8, 1.4, and 1.0 (from the skull surface); posterior injection sites A −1.7 from the bregma, L±1.4, D 2.0, 1.4, 0.8, and 0.5 (from the skull surface). For neonates, anterior injection sites A −3.0 from the naison at a 30° angle, L ±2.8, D 2.0 and 1.5 (from the skull surface); posterior injection sites A −4.5 from the naison at a 30° angle, L ±3.0, D 3.0 and 1.5 (from the skull surface). For Figure 3, neonatal NTG and J20 mice received unilateral transplants of MGENav1.1 and MGENav1.1–KI cells into the cortex and hippocampus.
For stereotaxic injections, adult mice were anesthetized with avertin (tribromoethanol, 250 mg/kg) and neonates by hypothermia induced by placement on ice until the pedal reflex was abolished. MGEWT or MGENav1.1 cells were transplanted into the hippocampus and cortex of neonatal (P3–5) or 2–3-month-old NTG or J20 mice to generate NTGMGE-WT, J20MGE-WT, NTGMGE-Nav1.1 and J20MGE-Nav1 mice. Sham-transplanted groups (NTGSHAM and J20SHAM) received the same treatment, but the L-15 medium contained no MGE cells.
◦ Immunohistochemistry
Tissue preparation and immunohistochemistry were done as described (Palop et al., 2011). After fixation, brains were cryoprotected in 30% sucrose in phosphate-buffered saline (PBS) for 2 days and sectioned with a sliding microtome (Leica Microsystems) into 30-µm sections. Ten stereological subseries sections from each hemibrain were stored in a solution containing 30% glycerol, 30% ethylene glycol, and 40% PBS at −20°C until staining. Primary antibodies used included rabbit anti-GFP (1:3,000; Life Technologies), chicken anti-GFP (1:500; Aves Labs), rabbit anti-Nav1.1 (1:500; Alomone), mouse anti-parvalbumin (1:3,000; Swant), rabbit anti-parvalbumin (1:3,000; Swant), rat anti-somatostatin (1:50; Novus), rabbit anti-neuropeptide Y (1:2000; Immunostar), rabbit anti-Iba-1 (1:1000; Wako), rabbit anti-GFAP (1:3000; Sigma), and biotinylated 82E1 (1:1000; Immuno-Biological Laboratories). Primary antibodies were detected with biotinylated donkey anti-rabbit (1:500; Jackson ImmunoResearch), Alexa Fluor 488 donkey anti-rabbit (1:300; Life Technologies), Alexa Fluor 488 donkey anti-chicken (1:300; Life Technologies), Alexa Fluor 594 donkey anti-rat (1:300; Life Technologies), Alexa Fluor 594 donkey anti-mouse (1:300; Life Technologies), or Alexa Fluor 594 donkey anti-rabbit (1:300; Life Technologies).
◦ Histological Quantifications
In Lhx6GFP BAC transgenic mice, GFP expression is directed by the interneuron-specific Lhx6 regulatory sequences, and GFP is specifically expressed in embryonic MGE progenitors and in mature inhibitory interneurons derived from them (Cobos et al., 2006). All GFP-positive cells were counted in every tenth serial coronal 30-µm section (300µm apart) throughout the rostrocaudal extent of the cortex and hippocampus of one hemibrain per mouse. On average, we analyzed 14.92 ± 0.22 sections per mouse. A total of 31,529 GFP-positive cells were counted in 6–15 mice per group; from these counts, we estimated the total number of GFP-positive cells per brain. To identify MGE-derived interneuron subtypes, sections were double-labeled by fluorescence immunohistochemistry for GFP (green) and for PV, SOM, or NPY (red). Images were captured with an Axiocam HRc camera and Zen Lite 2012 software (Carl Zeiss) mounted on a fluorescence microscope (Olympus Bx41) at 40× magnification. A total of 1,449 GFP-positive cells (n = 4 mice per group) were analyzed, including PV cells (n = 186/568 GFP-positive cells), SOM (n = 186/487 GFP-positive cells), and NPY (n = 26/394 GFP-positive cells). For quantification of glia activation, brain sections were double-labeled by fluorescence immunohistochemistry for transplanted GFP-positive MGE-derived interneurons (green) and for Iba-1-positive microglia or GFAP-positive astrocytes (red). All images were captured at 20× magnification using the same image acquisition settings with the Keyence BioRevo BZ-9000 microscope and BZ-II Viewer and Analyzer software. To determine glia activation, transplanted GFP-positive MGE-derived interneurons were identified and the fluorescence intensity of Iba-1 or GFAP staining surrounding the transplanted cells or baseline intensity away from the cells were quantified using ImageJ. A total of 80 square blocks of area surrounding transplanted cells and 80 blocks of baseline fluorescence were quantified for microglia and astrocytes respectively (n = 3-4 mice per group).
◦ Whole-Cell Recordings
Mice (aged 8–9 months) were anesthetized with avertin (tribromoethanol, 250 mg/kg) and perfused with ice-cold dissection solution containing (in mM): 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 ascorbic acid, 2 thiourea, 3 sodium pyruvate, 12 N-acetyl-L-cysteine, 10 MgSO4, and 0.5 CaCl2, pH 7.3. Brains were removed and maintained in ice-cold dissection solution for acute slice preparation. Coronal 300-µm-thick slices were prepared using a vibratome and transferred to a recovery chamber for 10 min containing dissection solution warmed to 35°C. Slices were then incubated for 1 h at room temperature in a modified artificial cerebral spinal fluid (ACSF) containing (in mM): 92 NaCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 ascorbic acid, 2 thiourea, 3 sodium pyruvate, 12 N-acetyl-L-cysteine, 2 MgSO4, and 2 CaCl2, pH 7.3. Until recordings were performed, the slices were maintained in oxygenated standard ACSF containing (in mM): 119 NaCl, 2.5 KCl, 1 NaH2PO4, 26.2 NaHCO3, 11 glucose, 1.3 MgSO4, and 2.5 CaCl2.
Slices were transferred to a recording chamber and continuously perfused with standard ACSF at 32°C. Transplanted GFP-expressing interneurons were identified by epifluorescence and infrared differential interference contrast on an upright microscope (BX51WIF, Olympus). Whole-cell patch-clamp recordings were done with internal solution containing (in mM): 135 K-gluconate, 10 HEPES, 0.1 EGTA, 5 KCl, 2 MgCl2, 4 Mg-ATP, 0.3 Na-GTP, pH 7.3. Patch pipette resistance was 3–5 MΩ. Picrotoxin (100 µM), CNQX (10 µM), and D-AP5 (100 µM) were added to the ACSF to block synaptic inputs. Recordings were obtained with a Multiclamp 700B amplifier (Molecular Devices), digitized at 10 kHz, and acquired with WinLTP software (version 1.11b, University of Bristol). Access resistance was continuously monitored throughout the recordings and any neuron with >20 MΩ or >20% change in access resistance was excluded from the analysis. The liquid junction potential was not corrected. Resting membrane potential was determined upon initiating whole-cell configuration. In current clamp mode, neurons were held at −60 mV, and current was injected in incremental steps of 1-sec duration. Current steps ranged from −60 to 500 pA (−60, −40, −20, 20, 40, 60, 80, 100, 200, 300, 400 and 500 pA). The AP waveform kinetics were analyzed by the AP width at half maximum height, the 10–90% rise time, and the 90–10% fall time from the first AP in response to a 1-s current injection at 500 pA. The AP area (ms*mV) was the integral of the of the first AP waveform in response to a 1-s current injection at 500 pA. Maximum AP #/1s at 20–500 pA was the maximum number of APs in response to a 1-s current injection at any current intensity (20–500 pA). AP threshold was defined as the voltage of the action potential where dV/dt first exceeded 15 mV/ms. The initial AP rate at 500 pA was calculated from the intervals between the initial 10 APs in response to a 1-s current injection at 500 pA. Membrane time constant was estimated from the best fit of a single exponential to the initial hyperpolarization during a −60 pA current step. Spike amplitude was calculated as the voltage difference between the resting membrane potential and the peak of the first evoked AP. Recordings were analyzed with pClamp 10.4 (Axon Instruments). Experiments and analysis were done blind to mouse and MGE cell genotype. FS and N-FS cells were defined by their intrinsic properties, mainly the maximum number of APs in response to a 1-s current injection, the AP half-width and the resting membrane potential (Figure S5A–C). From the 130 cells recorded, we excluded 3 cells that we could not properly classify since they had mixed N-FS and FS properties.
◦ Behavioral Tests
Morris water maze
The water maze pool (122-cm diameter) contained opaque white water with a 12×12-cm platform submerged 1.5 cm below the surface. For spatial training sessions, mice were trained to locate the hidden platform during 5 or 6 consecutive days. Each day, mice received two training sessions 4 h apart consisting of two 60-s trials 10 min apart. The hidden platform location remained constant and the entry points were changed semirandomly between trials. Four hours after the last training session, the platform was removed and a 60-s probe trial was done (Verret et al., 2012). Performance was monitored with an EthoVision video tracking system (Noldus Information Technology). Distance swum or time to reach the hidden platform was used to measure performance in the training phase. For the probe trial, we measured the number of times mice crossed the original platform location plus 6 cm around it (target location) or the equivalent positions in the other three quadrants (other locations).
Open field behavior
Exploratory locomotor activity was measured in four identical clear plastic open-field chambers (40 × 40 × 30 cm) with two 16 × 16-photobeam arrays to detect horizontal and vertical movements (automated Flex-Field/Open Field Photobeam Activity System). Mice were placed in one of the chambers for 5 min per session. Total horizontal movements (beam breaks), both fine and ambulatory, were recorded. For the habituation studies, each mouse was always retested in the same chamber. The apparatus was cleaned with 70% ethanol between trials.
Y-maze
The Y-maze consisted of three 70-cm arms. The mouse was placed in one of the arms and was allowed to move freely for 6 min. The total distance travelled was measured. Performance was monitored with the EthoVision video-tracking system.
Contextual fear conditioning test
Fear conditioning was performed in four identical computer-controlled chambers (28 × 21 × 22 cm) (Med Associates). The floor of each chamber had 18 stainless steel rods (4-mm diameter, 1.5 cm apart) connected to a shock scrambler and generator, which, together with internal ventilation fans, supplied background noise of 70 dB. For the training session (day 1), after 5-min exposure to the chamber (to record baseline freezing), mice were given four 2-sec shocks (0.45 mA) 2 min apart. For the 24-h context testing, mice were placed in the same chamber for 5 min. Freezing behavior, defined as the absence of any visible movement except that required for respiration, was automatically recorded by a video-tracking system (motion threshold = 18, detection method = linear; Med Associates). The chambers were cleaned with 70% ethanol after each mouse. Mice were singly housed three days before and during the water maze testing but not for the other tests.
◦ Recordings of EEG and Locomotor Activity Levels
Mice were implanted for video EEG monitoring after anesthesia with intraperitoneal avertin (tribromoethanol, 250 mg/kg) as described (Verret et al., 2012). Teflon-coated silver wire electrodes (0.125-mm diameter) soldered to a multichannel electrical connector were implanted into the subdural space over the left frontal cortex (1 mm lateral and anterior to the bregma) and the left and right parietal cortex (2 mm lateral and posterior to the bregma). The left frontal cortex electrode was used as a reference. All EEG recordings were done out at least 10 days after surgery on freely moving mice in an open-field chamber (40 × 40 × 30 cm) during the middle part of the daylight cycle. Exploratory locomotor activity (beam breaks/min) was measured with an automated Flex-Field/Open Field Photobeam Activity System (San Diego Instruments). Total movements included fine and ambulatory horizontal movements and vertical movements. Movement frequencies were defined as follow: resting (0–9 beam breaks/min or ~0–5 cm/min), low (10–49 beam breaks/min or ~5–35 cm/min), and high (50–150 beam breaks/min or ~35–110 cm/min). EEG activity was recorded with Harmonie software at 200 Hz sampling rate (version 5.0b, Stellate) for 50 min.
To determine the time to habituate to NTG control level in the open field (Figure 7C), we calculated the y-intercept at 40 beam breaks/min (habituated basal activity) for each mouse by linear regression analyses, using the 50-min data points of movement frequency (x; beam breaks/min) and time (y; min). The number of sharp-wave discharges (spikes) was automatically detected with a Gotman spike detector (threshold 6; Harmonie, Stellate) or with a custom-made algorithm and LabChart 7 Pro software (AD Instruments). Spikes recorded from left and right hemibrains were added and shown as epileptiform spikes/min.
For spectral analyses, 50-min segments of unfiltered EEG recordings (sampling rate 200 sec−1) from freely moving mice were imported and analyzed using LabChart 7 Pro software (AD Instruments). The 30–90-Hz band was used to calculate gamma power, and a 58–62-Hz notch filter was applied to remove potential electrical noise. To obtain spectral power, we subjected the recordings to a fast Fourier transform (FFT) with a Hann cosine-bell window with 50% overlap between windows of 500-ms epochs. FFT was done with a 512 point FFT size to obtain a resolution of 0.39 Hz. The total signal amplitude or power in V2 in each 500-ms epoch was taken to represent the magnitude of gamma oscillatory power. The gamma power values for each 500-ms epoch were further averaged over 60 sec of EEG recording, using the median values, to remove potential noise. Finally, signals from both hemibrains were averaged to obtain the gamma power values for each mouse.
To calculate relative gamma power, the y-intercept at 0 movements/min was calculated for each mouse by linear regression analyses using the 50-min data points of movement frequency (x; beam breaks/min) and the absolute gamma power (y; V2) (Figures 7 and 8). The y-intercept of the absolute gamma power value was used to normalize the 50 min of EEG data points of gamma power within each mouse, therefore, it was defined as 1.0 (gamma power relative to resting). For calculation of gamma power before spikes (Figure 8), the gamma power for each 100-ms epoch was calculated for the 50 min of EEG recordings for each mouse as described above. Spikes for each 100-ms epoch and epochs to the next spike were calculated. Power spectrum data (Figure 6) was extracted using Spike2 software version 8 with FFT size 1024 (resolution 0.1953) and Hanning window function. NTG and J20 mice were matched based on locomotor activity to avoid locomotor activity-dependent effects on power.
• AVOIDANCE OF BIAS AND STATISTICAL ANALYSIS
◦ Blindcoding and Randomization
Investigators who carried out measurements were blinded with respect to the genotype of mice and the nature of the transplants they had received. Mice of the same genotype were randomly assigned to receive different types of transplants.
◦ Statistical Analysis
Statistical analyses were done with STATA 11.2 or Prism 6.0. Graphs were generated with Prism 6.0. As indicated in the figure legends, differences among multiple means were assessed by one-way or two-way ANOVA and Bonferroni post hoc tests. The P values of the Bonferroni tests are indicated in the figure panels with the following significance levels, *P < 0.05, **P < 0.01, and ***P < 0.001. Two-way ANOVA P values for the factors and interactions are indicated in the figure legends. Number (n) of mice or cells for all statistical analyses is indicated in each graph or in the legend. Learning curves in the Morris water maze, fear-conditioning, and novel-environment, open-field tests were analyzed by repeated-measures ANOVA and Bonferroni post hoc tests. Linear regression analyses were used to calculate slopes or y-intercept and to determine significant differences among groups. Null hypotheses were rejected at the 0.05 level.
Supplementary Material
HIGHLIGHTS.
Nav1.1-enhanced interneuron transplants reduce deficits in Alzheimer model
Nav1.1-deficient interneuron transplants cause behavioral deficits in wild-type mice
Nav1.1 elevation accelerates action potential kinetics in transplanted interneurons
Molecular optimization of cell transplants is required for therapeutic benefits
Acknowledgments
We thank Andrew Escayg for the Nav1.1-BAC mice, John Rubenstein for the Lhx6GFP mice, and Kazuhiro Yamakawa for the Nav1.1-R1407X mice; Gui-Qui Yu, Xin Wang, Daniel Kim and Iris Lo for technical support; the Gladstone Institutes’ Behavioral Core for assistance with the behavioral testing; and G. Howard and Stephen Ordway for editorial review; The study was supported by US National Institutes of Health (NIH) grants AG047313 (J.J.P.), NS041787 (L.M.) and NS065780 (L.M.), AG036884 (L.G.), AG30207 (L.G), training grant AG043301 (T.E.T.), National Center for Research Resource Grant RR18928, Alzheimer’s Association grant IIRG-13-284779 (J.J.P.), S.D. Bechtel, Jr. Young Investigator Award (J.J.P.), BrightFocus Foundation (T.E.T.), Spanish Government grants PRX12/00544 (M.A.D.) and SAF2012-36853 (M.A.D), Sara Borrell postdoctoral fellowship CD08/00201 from the Institute of Health Carlos III (M.M.L.), and a gift from the S.D. Bechtel, Jr. Foundation (L.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
M.M.L., M.A.D., K.M., I.C., and J.J.P performed the transplants; M.M.L, A.C., and J.J.P performed the behavioral testing; A.S.K., M.M.L., K.M., L.V., I.C., K.H., and J.J.P performed the histology; L.V., A.S.K., K.M., M.A.D., and J.J.P performed the electroencephalography; T.E.T. and L.G. performed the slice electrophysiology; J.J.P., M.A.D., K.M., T.E.T., and L.M. wrote the manuscript. All authors critically reviewed and commented on the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Alvarez-Dolado M, Calcagnotto ME, Karkar KM, Southwell DG, Jones-Davis DM, Estrada RC, Rubenstein JL, Alvarez-Buylla A, Baraban SC. Cortical inhibition modified by embryonic neural precursors grafted into the postnatal brain. J Neurosci. 2006;26:7380–7389. doi: 10.1523/JNEUROSCI.1540-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, Gallagher M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74:467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, et al. Apolipoprotein E is essential for amyloid deposition in the APPV717F transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 1999;96:15233–15238. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraban SC, Southwell DG, Estrada RC, Jones DL, Sebe JY, Alfaro-Cervello C, Garcia-Verdugo JM, Rubenstein JL, Alvarez-Buylla A. Reduction of seizures by transplantation of cortical GABAergic interneuron precursors into Kv1.1 mutant mice. Proc Natl Acad Sci USA. 2009;106:15472–15477. doi: 10.1073/pnas.0900141106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8:451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat Neurosci. 2011;14:750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien-Ly N, Gillespie AK, Walker D, Yoon SY, Huang Y. Reducing human apolipoprotein E levels attenuates age-dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. J Neurosci. 2012;32:4803–4811. doi: 10.1523/JNEUROSCI.0033-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, et al. Molecular, structural, and functional characterization of Alzheimer’s disease: Evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- Busche MA, Kekus M, Adelsberger H, Noda T, Forstl H, Nelken I, Konnerth A. Rescue of long-range circuit dysfunction in Alzheimer's disease models. Nat Neurosci. 2015;18:1623–1630. doi: 10.1038/nn.4137. [DOI] [PubMed] [Google Scholar]
- Cardin JA, Carlen M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BC, Bean BP. Sodium entry during action potentials of mammalian neurons: Incomplete inactivation and reduced metabolic efficiency in fast-spiking neurons. Neuron. 2009;64:898–909. doi: 10.1016/j.neuron.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalia ML, Howard MA, Baraban SC. Persistent seizure control in epileptic mice transplanted with gamma-aminobutyric acid progenitors. Ann Neurol. 2017;82:530–542. doi: 10.1002/ana.25021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos I, Long JE, Thwin MT, Rubenstein JL. Cellular patterns of transcription factor expression in developing cortical interneurons. Cereb Cortex. 2006;16(Suppl 1):i82–i88. doi: 10.1093/cercor/bhk003. [DOI] [PubMed] [Google Scholar]
- Corbett BF, Leiser SC, Ling HP, Nagy R, Breysse N, Zhang X, Hazra A, Wood A, Pangalos MN, Reinhart PH, et al. Sodium channel cleavage is associated with aberrant neuronal activity and cognitive deficits in a mouse model of Alzheimer's disease. J Neurosci. 2013;33:7020–7026. doi: 10.1523/JNEUROSCI.2325-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtin J, Chaudun F, Rozeske RR, Karalis N, Gonzalez-Campo C, Wurtz H, Abdi A, Baufreton J, Bienvenu TC, Herry C. Prefrontal parvalbumin interneurons shape neuronal activity to drive fear expression. Nature. 2014;505:92–96. doi: 10.1038/nature12755. [DOI] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings J, Lee G, Mortsdorf T, Ritter A, Zhong K. Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement (NY) 2017;3:367–384. doi: 10.1016/j.trci.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham M, Cho JH, Leung A, Savvidis G, Ahn S, Moon M, Lee PK, Han JJ, Azimi N, Kim KS, et al. hPSC-derived maturing GABAergic interneurons ameliorate seizures and abnormal behavior in epileptic mice. Cell Stem Cell. 2014;15:559–573. doi: 10.1016/j.stem.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Gama AM, Pochareddy S, Li M, Jamuar SS, Reiff RE, Lam AT, Sestan N, Walsh CA. Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron. 2015;88:910–917. doi: 10.1016/j.neuron.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, Bertram L, Mullin K, Tanzi RE, Blacker D, et al. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology. 2005;65:404–411. doi: 10.1212/01.wnl.0000171450.97464.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazzari P, Paternain AV, Valiente M, Pla R, Lujan R, Lloyd K, Lerma J, Marin O, Rico B. Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature. 2010;464:1376–1380. doi: 10.1038/nature08928. [DOI] [PubMed] [Google Scholar]
- Gurevicius K, Lipponen A, Tanila H. Increased cortical and thalamic excitability in freely moving APPswe/PS1dE9 mice modeling epileptic activity associated with Alzheimer's disease. Cereb Cortex. 2013;23:1148–1158. doi: 10.1093/cercor/bhs105. [DOI] [PubMed] [Google Scholar]
- Hamm V, Héraud C, Bott J-B, Herbeaux K, Strittmatter C, Mathis C, Goutagny R. Differential contribution of APP metabolites to early cognitive deficits in a TgCRND8 mouse model of Alzheimer's disease. Science Advances. 2017;3 doi: 10.1126/sciadv.1601068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammad M, Schmidt SL, Zhang X, Bray R, Frohlich F, Ghashghaei HT. Transplantation of GABAergic interneurons into the neonatal primary visual cortex reduces absence seizures in stargazer mice. Cereb Cortex. 2015;25:2970–2979. doi: 10.1093/cercor/bhu094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson KW, Gupta J, Tagliatela S, Litvina E, Zheng X, Van Zandt MA, Woods N, Grund E, Lin D, Royston S, et al. Long-term seizure suppression and optogenetic analyses of synaptic connectivity in epileptic mice with hippocampal grafts of GABAergic interneurons. J Neurosci. 2014;34:13492–13504. doi: 10.1523/JNEUROSCI.0005-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard MA, Rubenstein JL, Baraban SC. Bidirectional homeostatic plasticity induced by interneuron cell death and transplantation in vivo. Proc Natl Acad Sci USA. 2014;111:492–497. doi: 10.1073/pnas.1307784111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T, Xiao Z, Mao R, Chen B, Lu MN, Tong J, Mei R, Li SS, Xiao ZC, Zhang LF, et al. Navβ2 knockdown improves cognition in APP/PS1 mice by partially inhibiting seizures and APP amyloid processing. Oncotarget. 2017;8:99284–99295. doi: 10.18632/oncotarget.21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RF, Girskis KM, Rubenstein JL, Alvarez-Buylla A, Baraban SC. GABA progenitors grafted into the adult epileptic brain control seizures and abnormal behavior. Nat Neurosci. 2013;16:692–697. doi: 10.1038/nn.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemere C, Carr MF, Karlsson MP, Frank LM. Rapid and continuous modulation of hippocampal network state during exploration of new places. PLoS One. 2013;8:e73114. doi: 10.1371/journal.pone.0073114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepecs A, Fishell G. Interneuron cell types are fit to function. Nature. 2014;505:318–326. doi: 10.1038/nature12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DY, Carey BW, Wang H, Ingano LA, Binshtok AM, Wertz MH, Pettingell WH, He P, Lee VM, Woolf CJ, et al. BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat Cell Biol. 2007;9:755–764. doi: 10.1038/ncb1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H, Basak JM, Finn MB, Holtzman DM. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J Neurosci. 2011;31:18007–18012. doi: 10.1523/JNEUROSCI.3773-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleen JK, Scott RC, Holmes GL, Roberts DW, Rundle MM, Testorf M, Lenck-Santini PP, Jobst BC. Hippocampal interictal epileptiform activity disrupts cognition in humans. Neurology. 2013;81:18–24. doi: 10.1212/WNL.0b013e318297ee50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam AD, Deck G, Goldman A, Eskandar EN, Noebels J, Cole AJ. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat Med. 2017;23:678–680. doi: 10.1038/nm.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapray D, Lasztoczi B, Lagler M, Viney TJ, Katona L, Valenti O, Hartwich K, Borhegyi Z, Somogyi P, Klausberger T. Behavior-dependent specialization of identified hippocampal interneurons. Nat Neurosci. 2012;15:1265–1271. doi: 10.1038/nn.3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Weick JP, Liu H, Krencik R, Zhang X, Ma L, Zhou GM, Ayala M, Zhang SC. Medial ganglionic eminence-like cells derived from human embryonic stem cells correct learning and memory deficits. Nat Biotechnol. 2013;31:440–447. doi: 10.1038/nbt.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto JY, Stead M, Kucewicz MT, Matsumoto AJ, Peters PA, Brinkmann BH, Danstrom JC, Goerss SJ, Marsh WR, Meyer FB, et al. Network oscillations modulate interictal epileptiform spike rate during human memory. Brain. 2013;136:2444–2456. doi: 10.1093/brain/awt159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu G-Q, Mallory M, Rockenstein E, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwenhuis S, Forstmann BU, Wagenmakers EJ. Erroneous analyses of interactions in neuroscience: A problem of significance. Nat Neurosci. 2011;14:1105–1107. doi: 10.1038/nn.2886. [DOI] [PubMed] [Google Scholar]
- Noebels J. A perfect storm: Converging paths of epilepsy and Alzheimer's dementia intersect in the hippocampal formation. Epilepsia. 2011;52(Suppl 1):39–46. doi: 10.1111/j.1528-1167.2010.02909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, et al. Na(v)1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: A circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu G-Q, Kreitzer A, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc Natl Acad Sci USA. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-β-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci. 2016;17:777–792. doi: 10.1038/nrn.2016.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L, Roberson ED. Quantifying biomarkers of cognitive dysfunction and neuronal network hyperexcitability in mouse models of Alzheimer’s disease: Depletion of calcium-dependent proteins and inhibitory hippocampal remodeling. Methods Mol Biol. 2011;670:245–262. doi: 10.1007/978-1-60761-744-0_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein M, Han S, Tai C, Westenbroek RE, Hunker A, Scheuer T, Catterall WA. Dissecting the phenotypes of Dravet syndrome by gene deletion. Brain. 2015;138:2219–2233. doi: 10.1093/brain/awv142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio SE, Vega-Flores G, Martinez A, Bosch C, Perez-Mediavilla A, del Rio J, Gruart A, Delgado-Garcia JM, Soriano E, Pascual M. Accelerated aging of the GABAergic septohippocampal pathway and decreased hippocampal rhythms in a mouse model of Alzheimer's disease. FASEB J. 2012;26:4458–4467. doi: 10.1096/fj.12-208413. [DOI] [PubMed] [Google Scholar]
- Sederberg PB, Schulze-Bonhage A, Madsen JR, Bromfield EB, McCarthy DC, Brandt A, Tully MS, Kahana MJ. Hippocampal and neocortical gamma oscillations predict memory formation in humans. Cereb Cortex. 2007;17:1190–1196. doi: 10.1093/cercor/bhl030. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwell DG, Nicholas CR, Basbaum AI, Stryker MP, Kriegstein AR, Rubenstein JL, Alvarez-Buylla A. Interneurons from embryonic development to cell-based therapy. Science. 2014;344:1240622. doi: 10.1126/science.1240622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Laviolette PS, O'Keefe K, O'Brien J, Rentz DM, Pihlajamaki M, Marshall G, Hyman BT, Selkoe DJ, Hedden T, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai C, Abe Y, Westenbroek RE, Scheuer T, Catterall WA. Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA. 2014;111:E3139–E3148. doi: 10.1073/pnas.1411131111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Dutt K, Papale L, Rusconi R, Shankar A, Hunter J, Tufik S, Yu FH, Catterall WA, Mantegazza M, et al. A BAC transgenic mouse model reveals neuron subtype-specific effects of a Generalized Epilepsy with Febrile Seizures Plus (GEFS+) mutation. Neurobiol Dis. 2009;35:91–102. doi: 10.1016/j.nbd.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong LM, Djukic B, Arnold C, Gillespie AK, Yoon SY, Wang MM, Zhang O, Knoferle J, Rubenstein JL, Alvarez-Buylla A, et al. Inhibitory interneuron progenitor transplantation restores normal learning and memory in ApoE4 knock-in mice without or with Abeta accumulation. J Neurosci. 2014;34:9506–9515. doi: 10.1523/JNEUROSCI.0693-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012;149:708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, Cornes SB, Henry ML, Nelson AB, et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013;70:1158–1166. doi: 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Ranasinghe KG, Beagle AJ, Mizuiri D, Honma SM, Dowling AF, Darwish SM, Van Berlo V, Barnes DE, Mantle M, et al. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann Neurol. 2016;80:858–870. doi: 10.1002/ana.24794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WZ, Takashima S, Segawa Y, Itoh M, Shi XY, Hwang SK, Nabeshima K, Takeshita M, Hirose S. The developmental changes of Na(v)1.1 and Na(v)1.2 expression in the human hippocampus and temporal lobe. Brain Res. 2011;1389:61–70. doi: 10.1016/j.brainres.2011.02.083. [DOI] [PubMed] [Google Scholar]
- Yamamoto J, Suh J, Takeuchi D, Tonegawa S. Successful execution of working memory linked to synchronized high-frequency gamma oscillations. Cell. 2014;157:845–857. doi: 10.1016/j.cell.2014.04.009. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Tanei Z, Hashimoto T, Wakabayashi T, Okuno H, Naka Y, Yizhar O, Fenno LE, Fukayama M, Bito H, et al. Chronic optogenetic activation augments abeta pathology in a mouse model of Alzheimer disease. Cell Rep. 2015;11:859–865. doi: 10.1016/j.celrep.2015.04.017. [DOI] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- Zipancic I, Calcagnotto ME, Piquer-Gil M, Mello LE, Alvarez-Dolado M. Transplant of GABAergic precursors restores hippocampal inhibitory function in a mouse model of seizure susceptibility. Cell Transplant. 2010;19:549–564. doi: 10.3727/096368910X491383. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.