

Abstract
Objective
To analyze and describe atypical presentations of Charcot Marie Tooth disease type 4C (CMT 4C).
Methods
We present clinical and physiologic features of five patients with CMT4C caused by biallelic private mutations of SH3TC2.
Results
All patients manifested scoliosis and nerve conduction studies in the demyelinating range. All exhibited signs of motor impairment within the first years of life. We describe two or more different genetic diseases in the same patient, atypical presentations of CMT and 3 new mutations in CMT4C patients.
Discussion
A new era of unbiased genetic testing has led to this small case series of individuals with CMT4C, and highlights the recognition of different genetic diseases in CMT4C patients for accurate diagnosis, genetic risk identification and therapeutic intervention. The phenotype of CMT4C, in addition, appears to be enriched by a number of features unusual for the broad CMT category.
Keywords: Charcot-Marie Tooth disease, autosomal recessive, hereditary motor and sensory neuropathy, CMT4C, SH3TC2
Introduction
Charcot-Marie-Tooth disease (CMT) is a group of clinically and genetically heterogeneous hereditary motor and sensory neuropathies with an overall prevalence of 1 in 2500. Patients present with predominantly distal weakness, muscle atrophy, and sensory loss. 1 Autosomal recessive childhood onset CMT neuropathies are less common than the autosomal dominant CMT neuropathies. 2 CMT type 4C (CMT4C) appears to be the most prevalent (18%) autosomal recessive CMT subtype. 3,4
CMT4C is caused by recessive mutations of the SH3TC2 gene, most commonly by predicted protein null (frameshift and nonsense) mutations 4 but rarely by protein sequence changing missense mutations. This gene encodes an SH3 domain and multiple tetratricopeptide repeats (SH3TC2). It is expressed in myelinating Schwann cells, and localizes to the perinuclear recycling endosomes (RE) and cell membrane. 5
The clinical spectrum of CMT4C appears to be broad in contrast to the slowly progressive distally symmetric phenotype of most forms of CMT. Common features of CMT4C include childhood onset, thoracic spine scoliosis, moderate to severe neuropathy, and cranial nerve deficits including impairment of oculomotor, facial, auditory and hypoglossal nerves. 2, 6, 7 Small groups of individuals sharing a common genotype manifest phenotype variation, suggesting a role for other genetic or environmental factors in phenotypic expression. 6,7 Here, we report 5 patients with atypical phenotypes associated with SH3TC2 mutations to add weight to the evolving understanding of the CMT4C phenotype as a disorder with both consistent and heterogeneous features uncommon for typical CMT.
Methods
Five individuals diagnosed with SH3TC2 mutations were evaluated at the University of Iowa (#1, 2, 4), Johns Hopkins (#2), Wayne State (#3), and the National Institutes of Health (#5). Skeletal muscle strength was tested using the Medical Research Council (MRC) grading system from 0 (no movement) to 5 (normal strength). Four subjects were also evaluated by a modified CMT Neuropathy Score (CMTNS) that assesses CMT-related signs, symptoms and electrophysiologic features with scores of 0–10 marking mild impairment, 11–20 moderate, and >21 significant impairment. 8 Nerve conduction studies (NCS) were obtained routinely from the left ulnar motor nerve and left radial sensory nerve unless otherwise specified. Ulnar and radial responses are used because peroneal and sural potentials are typically absent even in demyelinating CMT. 8
Mutation Identification: The location of each SH3TC2 mutation is described according to the GenBank coding DNA reference sequence NM_024577.3. Mutations were identified by unbiased testing means in all subjects: a targeted commercial CMT panel in 3 and commercial whole exome in 1. Homozygosity mapping (The Children’s Hospital of Philadelphia, PA) and exome sequencing (NIH Intramural Sequencing Center, Bethesda, MD) were done to identify the mutation in Subject #5. Sanger sequencing, performed in a CLIA-certified laboratory (Athena Diagnostics) confirmed the mutations identified by exome sequencing.
Functional studies: To determine whether or not the new missense mutation in Subject 5 affected SH3TC2 localization or recycling endosome (RE) morphology, HA-tagged WT and Tyr680Cys SH3TC2 proteins were co-expressed with Rab11-eGFP in lipofected rat Schwann cells obtained from P3 Sprague-Dawley sciatic nerves, as described previously. 9 The cells were seeded in 4 well dishes and lipofected with FuGene HD (Promega, Madison, WI) following manufacture’s description. After 48 hours, cultures were fixed for 15 min in 4% PFA, permeabilized for 5 min in PBS 0.2% triton X-100, and blocked for one hour with PBS containing 5% normal goat serum and 0.2% triton X-100. The cells were incubated overnight at 4°C with HA (Cell Signaling, Beverly, MA) and GFP (Invitrogen, Carlsbad, CA) antibodies diluted in blocking solution. Detection of primary antibodies was done using anti-mouse-Alexa 594 and, anti-rabbit-Alexa 488 (Invitrogen, Carlsbad, CA). Confocal images were obtained using a Leica SP5. Fifty cells for WT SH3TC2 and sixty for Tyr680Cys mutant were analyzed. Using stacks of confocal images (0.13 μm intervals), the Rab11eGFP positive surface was calculated using Imaris software and compared to the total Schwann cell body surface, in order to estimate the percentage of the surface area of the RE. All analyses were done by blinded investigators.
Standard protocol approvals and patient consents: Research studies were approved by the relevant Institutional Review Boards. Informed written assent and consent were obtained from each subject and parent or guardian before participation in the study.
Results
Subject 1
History
A 12 year old girl born of asymptomatic parents of Chinese descent had delayed motor developmental milestones. She crawled by 14 months and she preferred to cruise (i.e. attempting to ambulate while holding onto furniture) until age 3 years. By age 7 years, she had developed hand weakness, scoliosis, calf cramps, and left hip dysplasia. By age 8 years, she had lost the previously acquired ability to run.
She was diagnosed with short chain acyl coenzyme A dehydrogenase deficiency (SCADD), a rare inborn error of mitochondrial fatty acid disorder during infancy. She did not manifest failure to thrive, low blood sugar or lethargy. She was treated with carnitine, which she continued over the years.
Clinical Exam
Upon examination at age 12, she had horizontal nystagmus in the left eye, mild dysarthria, distal greater than proximal muscle weakness and atrophy, and absent vibratory sensation up to the knees. She could not toe, heel, or tandem walk and had a bilateral steppage gait. She had thoracic scoliosis (Supplementary Table 1, available on-line).
Investigations
The NCS were consistent with a demyelinating motor and sensory neuropathy (Table 1). We identified a novel homozygous SH3TC2 c. 3230C>G, p. Ser1077Ter mutation predicted to be disease causing as it truncates the C-terminus and most likely interferes with the translation of the functional SH3TC2 protein. 10 This mutation is not reported in Broad ExAc database.
Table 1.
Nerve Conduction Studies
| Patient | 1 | 2 | 3 | 4 | 5 | Normal range |
|---|---|---|---|---|---|---|
| Radial Sensory Nerve Latency | 2.8 ms | NR | NR | NR | 3.9 ms (ulnar) | <3.1 ms |
| Radial Sensory Nerve Amplitude | 2.6 uV | NR | NR | NR | 14 uV (ulnar) | >7 μV (radial) >18 μV (ulnar) |
| Radial Sensory Nerve Velocity | 36 m/s | NR | NR | NR | 40 m/s (ulnar) | >46 m/s |
| Ulnar Motor Nerve Latency | 6.4 ms | 8.5 ms | 7.8 ms | 6.4 ms | 4.2 ms | <3.4 ms |
| Ulnar Motor Nerve Amplitude | 1.4 mV | 4.9 mV | 2.79 mV | 4.6 mV | 7.9 mV | >2.8 Mv |
| Ulnar Motor Nerve Velocity | 11 m/s | 15 m/s | 37 m/s | 27 m/s | 35 m/s | >49/s |
Subject 2
History
An 18-year-old girl was born to asymptomatic parents of Native American and African American descent. Her family history was notable for frequent first trimester miscarriages in her mother and maternal aunt. In the first year of life she manifested failure to thrive with multiple sinus and ear infections associated with low IgE levels. Around age 10, she developed episodes of muscle weakness, frequent falls, ataxia, difficulty walking, and numbness lasting two to three days, sometimes associated with urinary retention. Examination during these episodes confirmed worsening of baseline length-dependent weakness and distal lower extremity large and small fiber sensory loss. The presence of patchy demyelinating features on NCS led to prolonged trials of IVIg and corticosteroid therapy without clear effect on her functional abilities or the frequency or severity of her episodes. Because of easy bruising, hematologic evaluation at age 7 years identified absence of measurable plasminogen activator inhibitor-1 (PAI-1), and von Willebrand type 2M. She had a spinal fusion at age 12 years for scoliosis, which was first noted at age 2 years. She developed unilateral sensorineural deafness at age 14 years. She had progressive worsening of a sensory ataxia over the years and lost the ability to ambulate independently by age 15.
Clinical Exam
At age four months, she was noted to have pendular nystagmus. After her toddler years, she achieved the fifth percentile in height and weight. At age 5 years she had restricted upgaze.. Neurological exam at age 18 demonstrated pes planus and distal atrophy in her hands and feet. She had grade 4 strength in foot dorsiflexion and 4-strength in hand intrinsic muscles. She had sensory loss to pinprick in her toes, and loss to vibration up to her knees and elbows bilaterally. She was unable to heel, toe, or tandem walk; she had an ataxic gait. (Supplementary Table 1, available on-line).
Investigations
CSF protein was elevated at 416 mg/dL at the onset of an episode at age 14, which decreased to 172 mg/dL after 3 days of IVIg; cell count in CSF was normal. A sural nerve biopsy showed moderate to severe demyelinating polyneuropathy, no inflammation, and small onion bulbs. The NCS initially demonstrated a demyelinating polyneuropathy with side to side and nerve-to-nerve variation and temporal dispersion suggesting patchy involvement; repeat nerve conduction studies however showed a uniformly demyelinating process with no temporal dispersion or conduction block (Table 1). MRI of brain and spinal cord at age 16 demonstrated large caliber facial nerves and femoral nerve with normal caliber ventral and dorsal roots identified in the pelvic portion of the lumbosacral MRI. Spinal myelography (performed because of obscuring artifact from the spinal hardware) did not demonstrate CSF block. A paraneoplastic antibody panel was negative for ANNA-1, ANNA-2, ANNA-3, AGNA-1, PCA-1, PCA-2, PCA-Tr, Amphiphysin Ab, and CRMP-5. Friedreich’s ataxia, vitamin B-12 deficiency, Addison’s disease, and Lyme disease were excluded by appropriate investigations.
A commercial panel of known hereditary demyelinating disorders showed a heterozygous variant in PRX (c.3802G > C, p.Ala1268pro, a variant of unknown significance), a heterozygous variant in LITAF (c.*143,3′UTR), a deep intronic variant of unknown significance, a synonymous variant in PMP22 (c.396C > T, p.Tyr132Tyr), and normal results for GJB1, MPZ, EGR3, and GDAP1. A subsequent commercial whole exome sequencing identified known pathogenic compound heterozygous SH3TC2 mutations, the paternally inherited c. 2860 C>T, p.Arg954Ter mutation 4, 11, 12 and the maternally inherited c.3511C>T p.Arg1171Cys mutation. 13
Subject 3
History
A 29-year-old woman was born to Turkish parents who were first cousins (Figure 1). She was sick at birth and hospitalized for an extended period of time for uncertain reasons. Early motor milestones were delayed as she walked at 1.5 years of age. She had multiple falls due to balance difficulties. Thoracic spine scoliosis was noted at age 3 years. She was diagnosed with retinitis pigmentosa at age 29 years when funduscopy due to visual difficulties identified bilateral pigment displacement, partial optic atrophy, and narrowed vessels. Her father was also diagnosed with retinitis pigmentosa (see pedigree in Figure 1). The patient did not consent for genetic testing of retinitis pigmentosa.
Figure 1.
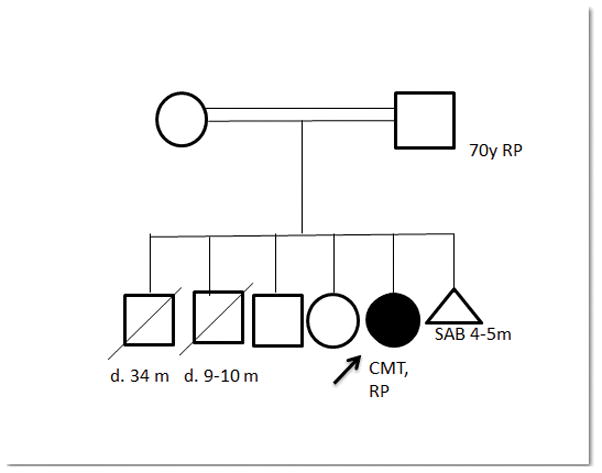
Family history for Subject 3 is significant for one miscarriage between parents and two sons who died in infancy; etiology of deaths unknown. Mother had a brain tumor. There is no history of CMT in the family. Parent ethnicity is Turkish. Her maternal grandfather and paternal grandmother were siblings, making her parents first cousins.
Clinical Exam
At age 29, neurological exam demonstrated limited visual fields with reported tunnel vision on exam, bilateral exotropia, distal greater than proximal muscle weakness and atrophy, reduced vibratory sense to the wrist and knees, and reduced pinprick at the toes. She could walk with mild foot drop, but was unable to tandem or heel walk. (Supplementary Table 1, available on-line).
Investigations
NCS (Table 1) showed a predominantly demyelinating neuropathy. She had a normal electrocardiogram. Nerve biopsy showed onion bulbs with no inflammatory reaction. Genetic testing revealed a known homozygous pathogenic SH3TC2 frameshift c.1894_1897 delinsAAA mutation, p.E632fs. 14
Subject 4
History
A 37 year old man of Irish-Italian decent had delayed motor milestones and walked at age 16 months with balance difficulties. He could ride a bicycle but not roller skate or ice skate. He had burning pain and paresthesias in his feet and ankles and was diagnosed with CMT at age 6 years. He developed scoliosis during childhood. He had a heel cord extension and tendon transfer in his left foot at age 12. Ventricular tachycardia was diagnosed as a child, and cardiac ablation was performed in his teenage years. He was still able to exercise in the gym, and to walk and bike frequently.
Clinical Exam
Examination at 37 years of age was significant for pes cavus, and atrophy in his hands and feet. He had 4+ strength in foot dorsiflexion, plantar flexion, and eversion bilaterally. He had sensory loss to pinprick, vibration, and joint position sense up to his ankles and impaired sensation to pinprick in up to his wrists. He was unable to heel, toe, or tandem walk. (Supplementary Table 1, available on-line)
Investigations
The NCS were consistent with a demyelinating sensorimotor neuropathy (Table 1). A commercial genetic testing showed heterozygous SH3TC2 c.2860 C>T, p. Arg954Ter and c.2128 C>T, p.Gln710Ter mutations inherited on different alleles in trans; the p.Arg954Ter (R954X) mutation is known to be disease-causing. 4, 11, 12 The p.Gln710Ter (Q710X) is a novel variant predicted to be pathogenic through protein truncation or nonsense-mediated mRNA decay. This variant is not detected in the NHLBI exome sequencing project in approximately 6,500 individuals of European and African American ancestry. The Broad ExAc database reports a heterozygous allele count of 1/121402, but no homozygotes were identified. Although this specific variant has not been reported previously there are other pathogenic loss of function variants that have been reported in this gene.15
Subject 5
History
A 40-year-old man was born at full term of non-consanguineous Indian parents. He had normal motor and verbal development. At age 5, he was noted to be a slow runner compared to his peers. He had difficulty arising from the floor and some difficulty in standing and walking. He had thoracic spine scoliosis. He underwent foot surgery to correct an ankle contracture at age 10 years. During his late-teen years, he noticed weakness in the upper extremities causing difficulty lifting his arms above the shoulders and flexing his arms to bring a cup to the mouth. His handwriting at that time deteriorated and he had trouble bending the thumbs and stapling documents. He also developed bilateral foot drop. During his 20s, he had difficulty arising from a chair and required support to climb stairs. He also developed deafness and had trouble with word pronunciation. He had difficulty whistling and blowing out candles. His mouth readily filled with saliva. His leg and arm weakness continued to worsen over the years. In his 30s, he required nightly noninvasive ventilatory support for respiratory insufficiency and carbon dioxide retention. Bilateral sensorineural deafness was noted. He did not report sensory or autonomic symptoms.
Clinical Exam
When evaluated at age 40, he had bilateral facial weakness, scoliosis of thoracic spine, and asymmetric, left greater than right, scapular winging. Muscle bulk was reduced in the lower legs, forearms and around shoulders. There were no visible fasciculations, tremor or myotonia. Muscle strength testing showed bilateral upper and lower extremity weakness proximally and distally (Supplementary Table 1, available on-line) Deep tendon reflexes were absent. Sensory exam was normal except for diminished vibration on the toes. His gait was a broad-based waddle with foot drop.
Investigations
Creatine kinase levels were normal. NCS demonstrated a predominantly motor neuropathy with nerve conduction velocities in the demyelinating range (Table 1). Needle EMG showed chronic denervation with reinnervation in the deltoid, quadriceps, first dorsal interosseous, tibialis anterior, lumbar paraspinal muscles, and trapezius muscle. Exome sequencing identified a homozygous SH3TC2 c.2039A>G p.Tyr680Cys mutation located adjacent to previously reported mutations in a mutation hotspot in exon 11, which encodes a protein interaction domain containing TPR motifs and was confirmed by Sanger sequencing. (See Supplementary Figures 1a, 1b, 1c available on-line). 4 The Tyr680Cys SH3TC2 did not affect intracellular SH3TC2 localization or RE morphology in transfected Schwann cells (Supplementary Figure 1d, available on-line). Both SIFT and PolyPhen-2.2.2. (HumVar) predicted this mutation to be pathogenic. The Tyr680Cys variant is not found in the dbSNP database. The Broad ExAc database includes a heterozygous allele count 2/121374, but the homozygotes were not detected. There were no mutations identified in other candidate neuropathy genes in the exome. We found several regions of extended homozygosity (≥ 3 Mb) including the SH3TC2 locus encompassing at least 197 Mb (6% or 1/16 of the genome) by SNP array-based homozygosity, suggesting that his parents were first cousins. Parental DNA was not available for testing. He was their only child and other family members were not available for segregation analysis. He did not provide consent for a nerve biopsy. The clinically overlapping syndrome of facioscapulohumeral muscular dystrophy types 1 and 2 (MIM #158900 and 158901) was ruled out by genetic testing.5
Discussion
We describe CMT4C patients with two or more independent genetic disease processes, with atypical clinical presentations and new SH3TC2 mutations in our case series. Individual diagnosis of CMT4C patients is challenging due to considerable intra- and interfamilial clinical variability. The diagnostic challenge was further compounded in sporadic CMT4C patients due to the presence of additional extremely rare inherited disorders such as SCADD,16 retinitis pigmentosa,17 and von Willebrand type 2M, 18 and PAI-1 deficiency.19 When considering our patients together with previous reports from the literature, it seems appropriate for CMT 4C to be considered an atypical form of CMT. It is not clear why there is increased phenotypic variability in CMT4C, but this could be secondary to cryptogenic modifiers or to the diversity of SH3TC2 protein function. SH3TC2 has 10 tetratricopeptide repeat domains and multiple regulatory functions each of which could be uniquely targeted by the diverse array of identified mutations. 7
Our patients were from diverse ethnic backgrounds including Native American, African American, Chinese and Indian, in which CMT4C has been rarely or not yet reported. All had the core features of CMT4C including demyelinating neuropathy with early-onset scoliosis. There are reports of CMT 4C patients presenting with side-to-side asymmetry, lack of scoliosis, and phenotypic variability despite identical genotypic mutations. 6,7, 15, 20, 21 Sleep apnea, respiratory failure and cranial nerve involvement such as nystagmus and facial weakness have also been described in some patients.4
Cardiac arrhythmias have not yet been reported in CMT4C. Ventricular tachycardia in the pediatric population is likely a benign condition and unrelated to CMT. Retinitis pigmentosa associated with demyelinating neuropathy of childhood onset is typical of Refsum disease and PHARC (polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataract) syndrome, which are rare autosomal recessive neurodegenerative disorders. 22, 23 In our patient, Refsum disease and PHARC syndrome were ruled out clinically. It is likely that retinitis pigmentosa was an autosomal dominant or X-linked dominant disease because her father was also affected. Whereas proximal muscle weakness and scapular winging have been reported in rare CMT4C patients,17 we report an FSHD phenotype in patient 5. Co-existing rare recessive bleeding disorders, PAI-1 deficiency and von Willebrand type 2M disease, are unusual associations in a patient with CMT4C. Clinically SCADD and CMT4C share neurological features of growth and developmental delay, muscle weakness and scoliosis. It is not yet known whether SCADD affects peripheral nerve function and worsens phenotype of CMT4C.
In a patient with compound heterozygous Arg954Ter and Arg1171Cys mutations, we observed recurrent acute episodes of worsening muscle weakness and numbness with elevated CSF protein and enlarged nerves which may or may not represent superimposed inflammatory neuropathy. The variable nature of the episodes, overall trend to increasing functional debility, and inconsistent response to therapy makes determination of an inflammatory contribution to pathology uncertain. An unclear contribution of inflammatory neuropathy to functional loss has also been reported in a patient with CMT4C due to Arg954Ter homozygous mutation. 24 In our case and the previous reported case, the question of an inflammatory component is raised but not entirely confirmed.
Only one patient in our case series had a pedigree compatible with autosomal recessive inheritance and she had a homozygous known frameshift mutation resulting in premature truncation and loss of functional SH3TC2 protein. In patients without parental consanguinity, compound heterozygosity for two different mutations is the most likely genetic basis for recessive disorders as exemplified by 2 subjects in this series. To be functionally equivalent to homozygosity, it is important to demonstrate that these mutations affect different copies of a gene, which was shown by testing parental DNA in Subject 2 and by an allele-specific PCR in Subject 4.
In Subject 5, a non-consanguineous sporadic patient, a SNP array-based whole genome homozygosity mapping confirmed recessive inheritance and led to the identification of the novel Tyr680Cys mutation in SH3TC2 gene by exome sequencing. Next generation sequencing including candidate gene panels or whole exome sequencing can be the most comprehensive and cost-effective diagnostic testing approach in genetically heterogeneous CMT neuropathies. However, interpretation of the variants of yet unknown significance as described in one of our patients remains a challenging and time-consuming process. Sequencing of a larger number of unaffected individuals is required to differentiate disease-causing mutations from common polymorphisms.25 Family segregation analysis, clinical phenotyping and laboratory-based validation of functional impact of the variants are necessary to establish pathogenicity of these variants. The deep variant databases of over 70,000 individuals available on the Broad Exome Aggregation Consortium browser 26 can provide the information on the frequency and the impact of the identified variants across populations.
We report 3 novel SH3TC2 mutations including homozygous Tyr680Cys and Ser1077Ter mutations and a heterozygous Gln710Ter mutation which co-existed with the common Arg954Ter mutation in this case series. Premature truncating mutations are predicted to result in loss of functional SH3TC2 protein. Studies of missense mutations are important in loss-of-function disorders because these mutations can yield unique insight into the underlying molecular mechanisms in rare diseases. SH3TC2 is specifically expressed in Schwann cells and has been shown to target the recycling endosomes (RE) by associating with the small GTPase Rab11. 5, 27–29 Mis-targeting of SH3TC2 away from RE has been proposed as a disease mechanism in CMT4C. 28 It has been shown that SH3TC2 condenses the RE whereas the CMT4C-associated Asn881Ser mutant impairs RE condensation. 5 In contrast, we found that Tyr680Cys mutation does not affect the RE morphology or the RE-targeting of SH3TC2. Our findings indicate that the Tyr680Cys mutation likely causes hypomyelination by affecting the interaction between SH3TC2 and protein(s) other than Rab11, for example, surface receptors in the SC membrane. 29, 30 Further functional studies of this missense mutation will clarify its pathophysiological consequences in SCs and may more generally provide additional insight into glial cell biology.
In conclusion, phenotypic variability and additional comorbidities have the potential to confound the diagnostic process in CMT4C patients. Whole exome sequencing improves the diagnostic yield among patients with atypical phenotypes and co-existing diseases. It is important to recognize co-existing diseases in patients with CMT4C for genetic risk assessment, prognosis, and potential therapeutic interventions.
Supplementary Material
Acknowledgments
We thank Ms. Elizabeth Hartnett (NINDS) for help with arranging the Subject 5 visit and evaluations and Dr. Enric Domènech-Estévez for his help with the cell culture experiment. The expression construct for the HA-tagged human SH3TC2 cDNA was kindly provided by Dr. Vincenzo Lupo (Valencia, Spain). This work was supported by intramural funding from the National Institute of Neurological Disorders and Stroke (AM); a grant from the National Institute of Neurological Disorders and Stroke (MES) and Office of Rare Diseases (MES, U54NS065712), Muscular Dystrophy Association (MES), Charcot-Marie-Tooth Association (MES), Swedish Strategic Research Area Neuroscience (StratNeuro) program (HB and RC) and MDA Clinical Research Training grant (NUJ).
Abbreviations
- MNCV
motor nerve conduction velocity
- CMAP
compound muscle action potential
- CMT
Charcot-Marie-Tooth disease
- CMT4
Charcot-Marie-Tooth disease type 4
- SH3TC2
SH3 domain and Tetratricopeptide Repeats 2
Footnotes
Ethical Guidelines: The authors confirm that they have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Conflict of Interest: On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical standard
All human and animal studies have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Informed consent
All persons gave their informed consent prior to their inclusion in the study.
References
- 1.Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clinical genetics. 1974;6:98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x. [DOI] [PubMed] [Google Scholar]
- 2.Cornett KM, Menezes MP, Bray P, et al. Phenotypic Variability of Childhood Charcot-Marie-Tooth Disease. JAMA neurology. 2016 doi: 10.1001/jamaneurol.2016.0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piscosquito G, Saveri P, Magri S, et al. Screening for SH3TC2 gene mutations in a series of demyelinating recessive Charcot-Marie-Tooth disease (CMT4) Journal of the peripheral nervous system: JPNS. 2016;21:142–149. doi: 10.1111/jns.12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Senderek J, Bergmann C, Stendel C, et al. Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy. American journal of human genetics. 2003;73:1106–1119. doi: 10.1086/379525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stendel C, Roos A, Kleine H, et al. SH3TC2, a protein mutant in Charcot-Marie-Tooth neuropathy, links peripheral nerve myelination to endosomal recycling. Brain: a journal of neurology. 2010;133:2462–2474. doi: 10.1093/brain/awq168. [DOI] [PubMed] [Google Scholar]
- 6.Colomer J, Gooding R, Angelicheva D, et al. Clinical spectrum of CMT4C disease in patients homozygous for the p. Arg1109X mutation in SH3TC2. Neuromuscular disorders: NMD. 2006;16:449–453. doi: 10.1016/j.nmd.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Varley TL, Bourque PR, Baker SK. Phenotypic variability of CMT4C in a French-Canadian kindred. Muscle & nerve. 2015;52:444–449. doi: 10.1002/mus.24640. [DOI] [PubMed] [Google Scholar]
- 8.Shy ME, Blake J, Krajewski K, et al. Reliability and validity of the CMT neuropathy score as a measure of disability. Neurology. 2005;64:1209–1214. doi: 10.1212/01.WNL.0000156517.00615.A3. [DOI] [PubMed] [Google Scholar]
- 9.Einheber S, Zanazzi G, Ching W, et al. The axonal membrane protein Caspr, a homologue of neurexin IV, is a component of the septate-like paranodal junctions that assemble during myelination. The Journal of cell biology. 1997;139:1495–1506. doi: 10.1083/jcb.139.6.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gooding R, Colomer J, King R, et al. A novel Gypsy founder mutation, p. Arg1109X in the CMT4C gene, causes variable peripheral neuropathy phenotypes. Journal of medical genetics. 2005;42:e69. doi: 10.1136/jmg.2005.034132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lupski JR, Reid JG, Gonzaga-Jauregui C, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. The New England journal of medicine. 2010;362:1181–1191. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoyer H, Braathen GJ, Busk OL, et al. Genetic diagnosis of Charcot-Marie-Tooth disease in a population by next-generation sequencing. BioMed research international. 2014;2014:210401. doi: 10.1155/2014/210401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayashi M, Abe A, Murakami T, et al. Molecular analysis of the genes causing recessive demyelinating Charcot-Marie-Tooth disease in Japan. Journal of human genetics. 2013;58:273–278. doi: 10.1038/jhg.2013.15. [DOI] [PubMed] [Google Scholar]
- 14.Fischer C, Trajanoski S, Papic L, et al. SNP array-based whole genome homozygosity mapping as the first step to a molecular diagnosis in patients with Charcot-Marie-Tooth disease. Journal of neurology. 2012;259:515–523. doi: 10.1007/s00415-011-6213-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Human genetics. 2014;133:1–9. doi: 10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shirao K, Okada S, Tajima G, et al. Molecular pathogenesis of a novel mutation, G108D, in short-chain acyl-CoA dehydrogenase identified in subjects with short-chain acyl-CoA dehydrogenase deficiency. Human genetics. 2010;127:619–628. doi: 10.1007/s00439-010-0822-7. [DOI] [PubMed] [Google Scholar]
- 17.Bravo-Gil N, Gonzalez-Del Pozo M, Martin-Sanchez M, et al. Unravelling the genetic basis of simplex Retinitis Pigmentosa cases. Scientific reports. 2017;7:41937. doi: 10.1038/srep41937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hillery CA, Mancuso DJ, Evan Sadler J, et al. Type 2M von Willebrand disease: F606I and I662F mutations in the glycoprotein Ib binding domain selectively impair ristocetin- but not botrocetin-mediated binding of von Willebrand factor to platelets. Blood. 1998;91:1572–1581. [PubMed] [Google Scholar]
- 19.Fay WP, Shapiro AD, Shih JL, Schleef RR, Ginsburg D. Brief report: complete deficiency of plasminogen-activator inhibitor type 1 due to a frame-shift mutation. The New England journal of medicine. 1992;327:1729–1733. doi: 10.1056/NEJM199212103272406. [DOI] [PubMed] [Google Scholar]
- 20.Schreiber O, Schneiderat P, Kress W, et al. Facioscapulohumeral muscular dystrophy and Charcot-Marie-Tooth neuropathy 1A - evidence for “double trouble” overlapping syndromes. BMC medical genetics. 2013;14:92. doi: 10.1186/1471-2350-14-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yger M, Stojkovic T, Tardieu S, et al. Characteristics of clinical and electrophysiological pattern of Charcot-Marie-Tooth 4C. Journal of the peripheral nervous system: JPNS. 2012;17:112–122. doi: 10.1111/j.1529-8027.2012.00382.x. [DOI] [PubMed] [Google Scholar]
- 22.Jansen GA, Ofman R, Ferdinandusse S, et al. Refsum disease is caused by mutations in the phytanoyl-CoA hydroxylase gene. Nature genetics. 1997;17:190–193. doi: 10.1038/ng1097-190. [DOI] [PubMed] [Google Scholar]
- 23.Fiskerstrand T, H’Mida-Ben Brahim D, Johansson S, et al. Mutations in ABHD12 cause the neurodegenerative disease PHARC: An inborn error of endocannabinoid metabolism. American journal of human genetics. 2010;87:410–417. doi: 10.1016/j.ajhg.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houlden H, Laura M, Ginsberg L, et al. The phenotype of Charcot-Marie-Tooth disease type 4C due to SH3TC2 mutations and possible predisposition to an inflammatory neuropathy. Neuromuscular disorders: NMD. 2009;19:264–269. doi: 10.1016/j.nmd.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Bell CJ, Dinwiddie DL, Miller NA, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Science translational medicine. 2011;3:65ra64. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.http://exac.broadinstitute.org
- 27.Arnaud E, Zenker J, de Preux Charles AS, et al. SH3TC2/KIAA1985 protein is required for proper myelination and the integrity of the node of Ranvier in the peripheral nervous system. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:17528–17533. doi: 10.1073/pnas.0905523106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roberts RC, Peden AA, Buss F, et al. Mistargeting of SH3TC2 away from the recycling endosome causes Charcot-Marie-Tooth disease type 4C. Human molecular genetics. 2010;19:1009–1018. doi: 10.1093/hmg/ddp565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vijay S, Chiu M, Dacks JB, Roberts RC. Exclusive expression of the Rab11 effector SH3TC2 in Schwann cells links integrin-alpha and myelin maintenance to Charcot-Marie-Tooth disease type 4C. Biochimica et biophysica acta. 2016;1862:1279–1290. doi: 10.1016/j.bbadis.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gouttenoire EA, Lupo V, Calpena E, et al. Sh3tc2 deficiency affects neuregulin-1/ErbB signaling. Glia. 2013;61:1041–1051. doi: 10.1002/glia.22493. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.