

Abstract
The blood-brain barrier (BBB) plays a vital role in regulating the trafficking of fluid, solutes and cells at the blood-brain interface and maintaining the homeostatic microenvironment of the CNS. Under pathological conditions, such as ischemic stroke, the BBB can be disrupted, followed by the extravasation of blood components into the brain and compromise of normal neuronal function. This article reviews recent advances in our knowledge of the mechanisms underlying BBB dysfunction and recovery after ischemic stroke. CNS cells in the neurovascular unit, as well as blood-borne peripheral cells constantly modulate the BBB and influence its breakdown and repair after ischemic stroke. The involvement of stroke risk factors and comorbid conditions further complicate the pathogenesis of neurovascular injury by predisposing the BBB to anatomical and functional changes that can exacerbate BBB dysfunction. Emphasis is also given to the process of long-term structural and functional restoration of the BBB after ischemic injury. With the development of novel research tools, future research on the BBB is likely to reveal promising potential therapeutic targets for protecting the BBB and improving patient outcome after ischemic stroke.
Keywords: inflammation, neurovascular unit, repair, stroke comorbidities, tight junction
1. Introduction
The blood-brain barrier (BBB) was first identified in the beginning of the 20th century by Lewandowsky and other workers, based on the absence of CNS pharmacological effects of intravenously administered bile acids and ferrocyanide. The concept that this was due to a barrier between blood and brain was fortified by experiments of Goldmann demonstrating the penetration of dyes into brain from cerebrospinal fluid (CSF) but not from blood (Goldmann, 1909, 1913; Zlokovic, 2008). Since those initial studies, our understanding of the BBB has evolved from a physical barrier separating the CNS from periphery, into a dynamic and metabolic interface that bi-directionally regulates the trafficking of fluid, solutes and cells. The concept of neurovascular unit (NVU) has further extended BBB research to include not only endothelial cells (ECs) but also astrocytes, pericytes, neurons and other components. As a site of crosstalk among multiple CNS cell types and blood-borne peripheral cells, the BBB plays a fundamental role in the maintenance of CNS homeostasis and normal neuronal function. BBB dysfunction, referring to its loss of structural integrity and normal functions, is also a prominent pathological feature of many neurological disorders, including stroke (Zlokovic, 2008).
Stroke is the 5th leading cause of death and the leading cause of adult disability in the United States. Ischemia accounts for ~87% of US strokes (Mozaffarian et al., 2016). Intensive basic and clinical research has revealed multiple stroke risk factors and elucidated many mechanisms underlying ischemic brain injury. However, current therapy for acute ischemic stroke remains largely dependent on tissue plasminogen activator (tPA)-mediated thrombolysis in appropriate patients. During and after ischemic stroke, BBB disruption facilitates injury progression and increases the risk of hemorrhage, predicting poor patient outcome and limiting the use of tPA (Keep et al., 2014; Liu et al., 2016b). The existence of stroke comorbid conditions, such as hypertension and hyperglycemia, induces anatomical and functional changes to the CNS vasculature and often exacerbates BBB breakdown after stroke. Having received much less attention than is warranted, BBB research should be better prioritized, with an emphasis on BBB-related mechanisms of neurovascular injury and developing therapeutic strategies to improve BBB integrity after ischemic stroke.
This review describes our current understanding of BBB dysfunction after ischemic stroke, with an emphasis on recent advances elucidating underlying mechanisms. Common and unique mechanisms that contribute to BBB dysfunction in the presence of stroke risk factors and comorbid conditions are summarized, which have been neglected in a large proportion of BBB studies. The concept of BBB restoration is also examined, where approaches enhancing BBB repair may facilitate long-term functional recovery after ischemic stroke and reduce stroke recurrence.
2. Blood-brain barrier: A physical and functional barrier between the CNS and blood
2.1. Structure and functions of the BBB under physiological conditions
A significant structural difference between the cerebral and peripheral vasculatures is the BBB, which strictly regulates the movement of molecules between blood and brain, contributing to CNS homeostasis (Abbott et al., 2010). That regulation includes (a) very limited paracellular diffusion between ECs, (b) low levels of EC transcytosis, (c) an array of endothelial transporters moving substrates from blood to brain or brain to blood, and (d) the presence of cerebrovascular enzymes that metabolize potentially neurotoxic compounds (Fig. 1).
Fig. 1. Regulation of the movement of molecules between blood and brain by microvascular endothelial cells under physiological conditions.
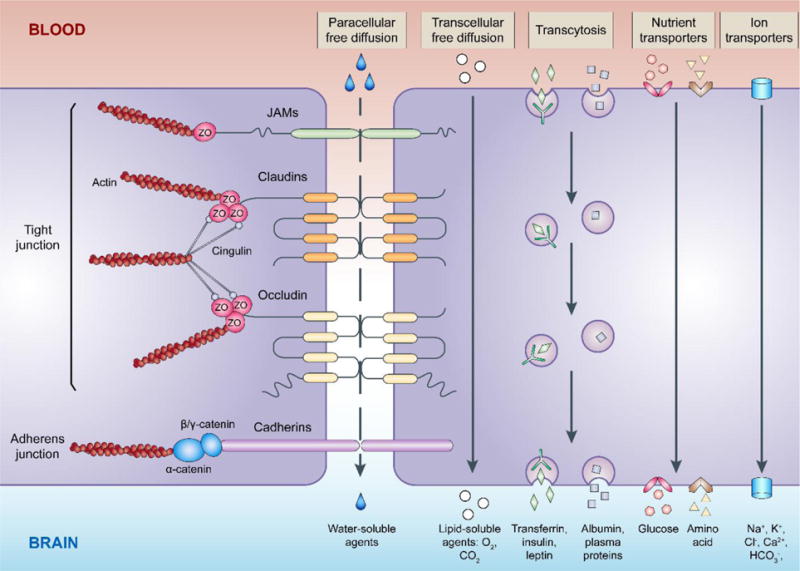
The junctional complexes between brain microvascular endothelial cells include tight junctions and adherens junctions. Major transmembrane proteins of the junctional complexes, such as claudins and occludin (tight junction), cadherins (adherens junction), and junctional adhesion molecules (JAMs), are normally linked to the actin cytoskeleton through plasma proteins zonula occludens (ZO), cingulin, and catenin. Paracellular free diffusion of water-soluble substances along the concentration gradient is highly restricted by tight junctions. Small lipophilic substances effectively diffuse across the endothelium through transcellular pathways. Low levels of transcytosis also occur in endothelial cells, either receptor dependent or receptor independent. An array of endothelial transporters is expressed and responsible for moving nutrients, such as glucose and amino acids. In addition, various ion transporters carrying Na+, K+, Cl−, HCO3−, Ca2+ and other ions are involved in maintaining brain ion homeostasis.
2.1.1. The neurovascular unit
The structural components of the BBB include ECs and their linking tight junctions (TJs), pericytes, astrocytic endfeet and extracellular matrix (ECM) components (Keaney and Campbell, 2015). While ECs form the vessel walls, pericytes are embedded in the vascular basement membrane and astrocytic processes almost completely ensheath brain capillaries (Abbott et al., 2010). Although the ECs and their TJs are the ultimate permeability barrier, pericytes and astrocytes play a major regulatory role. Indeed, the BBB is part of the “neurovascular unit”, a dynamic structure regulated by these and additional cells including neurons, microglia and even peripheral immune cells (Obermeier et al., 2013). Functionally, the concept of the NVU puts more emphasis on cellular interplay in maintaining brain homeostasis and in responding to inflammation and disease.
Pericytes are perivascular mural cells surrounding the ECs. More than supportive cells to ECs, pericytes are vital NVU components involved in many vascular functions including BBB formation and maintenance, vessel maturation, and regulation of blood flow and immune cell trafficking (Armulik et al., 2010; Daneman et al., 2010). During embryogenesis, pericytes are involved in BBB development even earlier than astrocytes. Mouse embryos deficient of pericytes (through null and hypomorphic Pdgfrb mutations) fail to form an intact BBB, display abnormal TJ formation, increased EC vesicular trafficking and immune cell infiltration into CNS (Daneman et al., 2010). In adult mice, pericyte coverage positively correlates with BBB integrity. Pericyte deficiency by ablation of platelet-derived growth factor receptor-beta (PDGFRβ) leads to accumulation of intravenously injected tracers in endothelium and brain parenchyma (Armulik et al., 2010). EC and astrocyte dysfunction may be two important contributing factors to the increased BBB permeability. Endothelial BBB-specific gene and protein expression profiles are altered by pericyte deficiency, partially leading to higher levels of transcytosis. Astrocyte endfeet are also detached from pericyte-deficient vessels (Armulik et al., 2010). In adult pericyte-deficient mice, microcirculation hypoperfusion and increased brain accumulation of vasculotoxic and/or neurotoxic molecules were observed, which would ultimately lead to vascular injury and neuronal degeneration (Bell et al., 2010). Pericytes are multipotent self-renewing cells, and lack of a definitive pan-marker for pericytes is a major limitation in pericyte studies. Two widely used and relatively specific markers for pericytes are PDGFRβ and NG2, the receptor and co-receptor for PDGF, respectively (Hellstrom et al., 1999). Pericytes are able to differentiate into neural and vascular lineage cells under certain stimuli, such as ischemia (Nakagomi et al., 2015).
Astrocytes, the most abundant glial cells in brain, have many housekeeping functions including BBB and cerebral blood flow regulation (Liu and Chopp, 2016; Osborn et al., 2016; Rossi, 2015). Direct EC-astrocyte contact starts from 17 weeks of gestation in human brains (Wilkinson et al., 1990) with eventually perivascular astrocytic endfeet almost completely surrounding the abluminal EC surface (Filous and Silver, 2016; Mathiisen et al., 2010). Gap junctions are present in the astrocyte endfeet enwrapping the blood vessel walls, and mediate intercellular communication and solute movement between astrocytes (Simard et al., 2003). Ablation of gap junction proteins connexin-43 and connexin-30 leads to astrocytic edema and weakens the BBB (Ezan et al., 2012). Besides physical support, astrocytes strengthen the BBB by secreting bioactive substances that lead to TJ modulation (Alvarez et al., 2013; Barreto, 2016; Janzer and Raff, 1987; Neuhaus et al., 1991). Sonic hedgehog (Shh) is the most widely studied molecule released by astrocytes, which acts on EC Hedgehog (Hh) receptors regulating TJ formation and BBB permeability (Alvarez et al., 2011). Other chemical mediators released by astrocytes, such as glial cell-derived neurotrophic factor (GDNF), prostaglandins, nitric oxide (NO), and arachidonic acid, also regulate TJs, blood vessel diameter and blood flow (Iadecola and Nedergaard, 2007; Igarashi et al., 1999).
More than a sturdy barricade, the cerebrovascular endothelium serves as a dynamic regulatory interface linking the blood vessel lumen and smooth muscle, thereby actively modulating cerebral blood flow. Studies suggest a pivotal role of the endothelium in cerebral autoregulation, the processes through which vascular resistance is adjusted to compensate alteration of perfusion pressure and maintain relatively constant cerebral blood flow and microvascular pressure (Lassen, 1964). A variety of vasomodulatory chemical mediators are produced by the endothelium, such as NO, endothelium-derived hyperpolarization factor (EDHF), the eicosanoids, and the endothelins. Furthermore, the endothelium possesses mechanoreceptor properties in response to fluid sheer stress and transmural pressure, which also contribute to cerebral autoregulation (Peterson et al., 2011). ECs are also important participants in the brain’s intrinsic regulatory mechanisms for thrombosis and hemostasis. EC-dependent regulatory pathways of coagulation include the thrombomodulin protein C pathway, the tissue factor pathway inhibitor (TFPI) pathway, and the fibrinolytic pathway (Fisher, 2013). How these pathways are influenced by systemic coagulation factor manipulation are important aspects to consider during stroke pharmacotherapies.
2.1.2. Endothelial cell junctions
The TJs between adjacent ECs are responsible for the extremely low paracellular permeability and high electrical resistance of the BBB. They regulate the movement of polar solutes and macromolecules across the barrier. The junctional complexes between ECs include TJs and adherens junctions (AJs). Claudins (primarily claudin-5) and occludin are major transmembrane TJ proteins. They are phosphoproteins with four transmembrane domains that span the intercellular cleft homotypically binding to proteins on adjacent ECs (Stamatovic et al., 2016). Other transmembrane proteins are the family of junctional adhesion molecules (JAMs) (Martin-Padura et al., 1998). They have a single transmembrane domain and locate at the outside position in TJs. Functionally, they belong to the immunoglobulin superfamily and are involved in both cell-to-cell adhesion and leukocyte transmigration across the BBB (Aurrand-Lions et al., 2001; Sladojevic et al., 2014; Stamatovic et al., 2012). JAMs have been far less studied than claudin-5 and occludin, and more investigation is required to reveal their full function in the normal and injured BBB.
The cytoplasmic terminals of the transmembrane TJ proteins bind to a number of cytoplasmic proteins including zonula occludens (ZO)-1, ZO-2, ZO-3 and cingulin (Stamatovic et al., 2016). These proteins form both a TJ scaffold and regulate TJ function. Such proteins also link TJs to primary cytoskeleton proteins, such as actin, for maintenance of the structural and functional integrity of the endothelium (Ballabh et al., 2004).
Analogous to TJs, AJs have transmembrane proteins, cadherins, that homotypically bind with cadherins on adjacent ECs. The cytoplasmic domains of cadherins bind to cytoplasmic plaque proteins β- or γ-catenin. The cadherin-catenin complex and associated proteins are linked to the actin cytoskeleton. TJ and AJ components, particularly ZO-1 and catenins, are known to interact and influence TJ assembly (Ballabh et al., 2004).
2.2. Endothelial cell transcellular pathways
Small lipophilic substances, such as O2 and CO2, diffuse freely across plasma membranes along their concentration gradient (Grieb et al., 1985) and lipophilicity is a major determinant of BBB permeability for many compounds. Another route across ECs is transcytosis. In normal brain, ECs have few endocytic vesicles that mediate a low rate of transcytosis between blood and brain. Although electron microscopy suggests that brain ECs have 80–84% fewer endocytic vesicles than peripheral capillaries (Claudio et al., 1989), transcytosis still provides the main route to transport large molecular weight solutes, such as proteins, across the BBB.
Transcytosis is receptor dependent or receptor independent (adsorptive transcytosis). In receptor-mediated transcytosis, endocytosis is triggered by a ligand binding to its receptor on the EC luminal membrane. Examples include the transferrin, insulin and leptin receptors (Salameh and Banks, 2014). In contrast, adsorptive transcytosis, a charge interaction between a compound and the EC luminal membrane triggers endocytosis (Salameh and Banks, 2014). In a number of disease states, including stroke, there are increased numbers of EC vesicles, suggesting increased transcytosis (Knowland et al., 2014; Nahirney et al., 2016).
2.3. Tight junction regulation
The TJs connecting adjacent ECs result in high transendothelial electrical resistance (TEER) and low paracellular permeability with size and charge selectivity (Abbott et al., 2006). However, the TJs are regulated physiologically and pathophysiologically by protein modification (e.g. phosphorylation), relocation and degradation (Stamatovic et al., 2016). For example, cytokines, growth factors and hormones can modulate TJs. The Rho/Rho-associated protein kinase (ROCK) (Beckers et al., 2010), protein kinase C (PKC) (Dorfel and Huber, 2012) and mitogen-activated protein kinase (MAPK) (Fujibe et al., 2004) signaling pathways modify TJ protein phosphorylation or expression and are involved in regulating paracellular permeability in the presence of cytokines, noxious agents and other pathological conditions (Krizbai and Deli, 2003).
Claudin-5 is by far the most prevalent claudin at the BBB. However, low levels of claudin-3 are present constitutively and both claudin-1 and claudin-12 expression can be induced (Stamatovic et al., 2016). The impact of such claudins on TJ function is not fully elucidated, but their expression may form another type of TJ regulation. Some claudins (e.g. claudin-2) form water or ion permeable pores (Milatz et al., 2010; Rosenthal et al., 2010; Yu et al., 2010), but these do not appear to be present at the BBB.
2.4. BBB transporters
The cerebral endothelium expresses a wide array of transporters (Fig. 1). Many nutrients, such as glucose and amino acids, cannot permeate the paracellular junctions, and the endothelium contains transporters (e.g. GLUT1) that supply the CNS with such compounds. In addition, there are ion transporters that are involved in brain ion homeostasis and that probably secrete brain extracellular fluid (ECF). The BBB also has efflux transporters involved in preventing the entry of compounds into brain (e.g. P-glycoprotein; P-gp) and clearing waste products from brain. Brain capillary ECs have a mitochondrial density ~2-fold greater than systemic capillaries (Oldendorf et al., 1977). This may reflect the energy requirements of ATP-dependent transport (e.g. Na+/K+-ATPase or transport indirectly dependent on Na+ gradients).
2.4.1. Nutrient transport
D-glucose is the primary energy source for the brain, and a continuous supply is imperative for normal function. Dick et al. first showed the presence of the glucose transporter isoform 1 (GLUT1) in brain ECs (Dick et al., 1984). GLUT1, encoded by the SLC2A1 gene, plays a critical role in glucose brain uptake. The transport is saturable, stereospecific and independent of energy supply, occurring by facilitated diffusion (Bell et al., 1993; Farrell et al., 1992). GLUT1 deficiency causes severe functional deficits and death (De Vivo et al., 1991; Wang et al., 2006).
BBB amino acid transport is either facilitative or Na+-dependent. Two facilitative transporters, primarily L-type amino acid transporter 1 (LAT1, SLC7A5) and LAT2 (SLC7A6), are present on the luminal and abluminal EC membranes. They transport large neutral amino acids providing the brain access to essential amino acids (Dolgodilina et al., 2016). There are also a wide range of Na+-dependent amino acid transporters (Campos-Bedolla et al., 2014). These are predominantly present at the EC abluminal membrane clearing amino acids from the ECF into the cell, from where they can be effluxed into the circulation. This Na+-dependent carrier location underlies the low (~10%) ECF amino acid concentrations compared to plasma (glutamine is an exception) (Hawkins et al., 2006). One group of Na+-dependent transporters is the excitatory amino acid transporter (EAAT) family that transports glutamate and aspartate (Cederberg et al., 2014). The clearance of those amino acids from the ECF may be important in stroke because they may induce excitotoxicity (Cederberg et al., 2014; Heyes et al., 2015).
2.4.2. Ion transporters
Ion transport plays a vital role in the function of all cells (e.g. acid/base and volume regulation, and providing ion gradients that drive secondary active transport of nutrients). However, at the cerebral endothelium, ion transporters are also involved in CNS ion homeostasis and fluid secretion. Regulation of the ionic composition of the brain ECF is vital for CNS function, and the concentrations of certain ions, such as K+ and Ca2+, that regulate neuronal activity, are very tightly controlled (Hladky and Barrand, 2016).
The BBB has an array of ion transporters that carry Na+, K+, Cl−, HCO3−, Ca2+ and other ions. Many of these are asymmetrically distributed between the luminal and abluminal membranes, contributing to vectorial transport across the BBB (Hladky and Barrand, 2016). Thus, for example, there is evidence that a Na+-K+-Cl− cotransporter and a Na+/H+ exchanger present at the EC luminal membrane and Na+/K+-ATPase at the abluminal membrane are involved in the transcellular transport of Na+ (Betz et al., 1980; Lam et al., 2009; O’Donnell et al., 2004). Through functional coupling of luminal and abluminal transporters and channels, the BBB transports Na+, Cl− and other ions and associated water from blood into brain, producing ~30% of brain interstitial fluid in healthy brain (O’Donnell, 2014). Thus, the BBB contributes to the regulation of ECF volume and composition. How such ion and fluid transport is affected under pathological conditions is an important question in brain edema formation. On the one hand, energy-dependent transporters such as Na+/K+-ATPase and Ca2+-ATPase fail to maintain the cellular ion homeostasis in infarct core as a consequence of ATP loss. On the other hand, ischemia stimulates Na+-K+-Cl− cotransport and Na+/H+ exchange, leading to the entry of extracellular Na+. When the Na+/K+-ATPase no longer keeps pace with such transport activities, intracellular Na+ accumulation and endothelial swelling occurs (O’Donnell, 2014). Astrocytes also take up the brain Na+ resulting from transendothelial transport, causing cytotoxic edema (O’Donnell, 2014).
2.4.3. ABC transporters
ATP-binding cassette (ABC) transporters are a protein superfamily containing 48 members grouped into 7 sub-families according to structural homology. At the BBB, the most significant are P-gp (ABCB1), breast cancer resistance protein (ABCG2) and the multidrug resistance-associated proteins (ABCC1, 2, 4, 5 and possibly 3 and 6). They are predominantly localized to the EC luminal membrane, transporting a wide range of substrates from the EC cytoplasm back to blood (Mahringer and Fricker, 2016); i.e. a major role of these transporters is to act as efflux pumps preventing CNS penetration of lipid-soluble compounds. Such compounds include potentially neurotoxic endogenous or xenobiotic molecules. However, while ABC transporters have this neuroprotective function (Dallas et al., 2006), they also limit the penetration of many drugs into brain (Shen and Zhang, 2010), including potential neuroprotectants.
2.5. Metabolic barrier
The BBB also prevents the entry of compounds from blood to brain because of the presence of metabolizing enzymes in the ECs, pericytes or astrocytes. These include monoamine oxidases, endopeptidases, aminopeptidases and cholinesterases (Agundez et al., 2014). These may degrade potentially neuroactive compounds (e.g. circulating catecholamines) before they can have parenchymal actions. This is a relatively understudied area of research in normal brain and in diseases such as stroke.
2.6. Immune cell trafficking
In normal brain, leukocyte trafficking from blood to brain across the cerebrovasculature is restricted compared to other tissues, leading to the concept that the brain is a relatively immune-privileged site (Abbott et al., 2010). This reflects both the tightness of the paracellular route and the paucity of the receptors (e.g. adhesion molecules) involved in leukocyte diapedesis. Both of these parameters change markedly after stroke and in neuroinflammatory states.
3. Mechanisms of blood-brain barrier dysfunction after ischemic stroke
BBB dysfunction, characterized by structural disruption of TJs and increased permeability, is a prominent pathological characteristic of both ischemic and hemorrhagic stroke, and is usually associated with poor prognosis (Keep et al., 2008; Prakash and Carmichael, 2015). With an ischemic stroke, blood-borne cells, chemicals and fluid extravasate into brain parenchyma across the impaired BBB as a result of increased paracellular and transcellular permeability and gross lesioning of the endothelium (Keaney and Campbell, 2015). Water and ion homeostasis of the brain is disrupted, leading to cerebral edema (Rosenberg, 1999). Infiltrating leukocytes further exacerbate inflammatory responses and aggravate brain injury (Huang et al., 2006). While most consequences of BBB dysfunction are detrimental, one potential benefit is that it may enable therapeutic agents to reach brain targets.
3.1. BBB breakdown after ischemic stroke
Ischemic insults can rapidly induce cerebral edema, referring to the excess accumulation of fluid in the intracellular (cytotoxic edema) or extracellular (vasogenic edema) spaces in the brain. Stepwise development of cerebral edema occurs after ischemia, with cytotoxic edema occurring minutes after ischemia onset followed by a relatively late onset of vasogenic edema, the latter in particular related to BBB breakdown (Dharmasaroja, 2016; Stokum et al., 2016). BBB disruption can permit a large inflow of hematogenous fluid into the extravascular space, leading to progressive elevation of brain water content and tissue swelling (Dharmasaroja, 2016; Rosenberg, 1999; Stokum et al., 2016). In patients with acute ischemic stroke, BBB disruption identified by magnetic resonance imaging (MRI) during the first 3 hours after symptom onset is associated with the development of vasogenic edema (Giraud et al., 2015). Consistently, studies based on animal models report cerebral edema formation in the first few hours after ischemia onset.
Ion transporter dysfunction at the BBB is an important mechanism leading to cerebral edema. Soon after ischemia, increased activity of BBB Na+/H+ exchangers, Na+-K+-Cl− cotransporters, or the calcium-activated potassium channel KCa3.1 enhances transcellular transport of Na+ and Cl− from blood into the brain across the BBB which is likely intact (Chen et al., 2015; O’Donnell, 2014; O’Donnell et al., 2013). Subsequent dysregulation of ionic homeostasis, particularly increased brain Na+ uptake, is a major contributor to ischemia-induced edema formation (Chen et al., 2015; O’Donnell, 2014).
The infiltration and accumulation of peripheral immune cells and molecules into brain parenchyma following stroke is well-accepted as contributing to BBB dysfunction and injury progression (Gelderblom et al., 2009). Brain resident microglial cells are activated within the first few hours after ischemia and release pro-inflammatory cytokines. Those cytokines, including interleukin (IL)-1 and IL-6, enhance the expression of intercellular adhesion molecule-1 (ICAM-1), P-selectin and E-selectin. These molecules further enable leukocyte adherence, accumulation and transmigration across the endothelium and mediate inflammatory cascades, further exaggerating infarction (McColl et al., 2008; Wang and Doerschuk, 2002). On the other hand, certain leukocyte types, e.g. regulatory T-cells (Tregs) and B-cells, may play disease-limiting protective roles (Li et al., 2013; Liesz et al., 2015; Offner and Hurn, 2012).
BBB dysfunction is also central to the genesis of hemorrhagic transformation and increased mortality after tPA treatment in stroke, especially following delayed tPA treatment (Jickling et al., 2014). tPA-associated hemorrhagic transformation often occurs as a result of the catastrophic breakdown of the BBB, referring to the frank disruption of TJ proteins (Jickling et al., 2014). BBB opening at early stages after cerebral ischemia largely correlates with intracerebral hemorrhage following tPA thrombolysis (Jin et al., 2014). Studies on stroke patients receiving thrombolytic therapy using MRI as a marker for BBB dysfunction indicates early BBB opening as an independent predictor of hemorrhagic transformation (Latour et al., 2004). tPA treatment can elevate brain matrix metalloproteinase (MMP)-9 levels (Jin et al., 2015; Kelly et al., 2006; Sumii and Lo, 2002), but other changes also occur at the endothelial interface upon tPA treatment, such as the phosphorylation of gap junction protein connexin43 (Yang et al., 2016b), which contribute to increased BBB permeability and hemorrhagic transformation.
3.2. Alterations of endothelial junctional proteins after ischemic stroke
TJ disruption is a major reason underlying increased paracellular permeability of the BBB after ischemic stroke (Wolburg and Lippoldt, 2002). Stepwise alterations of TJ proteins take place, such as protein modification, translocation and degradation (Fig. 2). The time course and degree of each process is determined by the severity of ischemic injury, and the processes are linked (e.g. translocation can lead to degradation).
Fig. 2. Stepwise alterations of endothelial junctional proteins after ischemic brain injury.
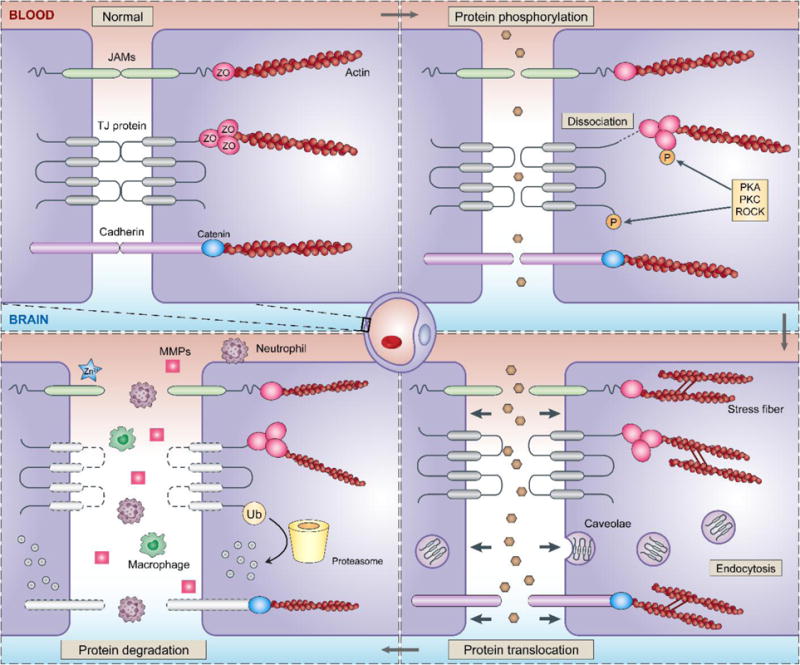
Ischemic insults activate a cascade of signaling in endothelial cells, leading to junctional protein alterations such as phosphorylation, translocation and degradation, each of which could increase BBB permeability and participate in the development of neurovascular injury. The phosphorylation of occludin, claudin-5 and ZO-1 can cause dissociation of the TJ complex and increase BBB permeability. Altered distribution of TJ proteins could result from increased endocytosis, as well as cellular tension transmitted through ischemia-induced actin stress fibers. Such protein translocation weakens the BBB and facilitate the degradation of TJs. TJ degradation could occur extracellularly through activity of MMPs, or intracellularly through proteasomes and lysosomes after ubiquitination. This is accompanied by the infiltration of peripheral immune cells, such as neutrophils and macrophages, which further exacerbates BBB breakdown after ischemia.
3.2.1. Protein modifications
Posttranslational modification of TJ proteins is widely believed to influence BBB permeability, however the consequences of these modifications are heterogeneous, as different kinases act on distinct residues even on the same TJ protein (Cummins, 2012; Gonzalez-Mariscal et al., 2008). The effects of phosphorylation of occludin, claudin-5 and ZO-1 by vascular endothelial growth factor (VEGF), Rho/ROCK, cyclic AMP (cAMP)/PKA are the most comprehensively studied, and usually result in increased barrier permeability (Antonetti et al., 1999; Persidsky et al., 2006; Soma et al., 2004; Yamamoto et al., 2008). Inflammatory mediators released during ischemic brain injury also induce phosphorylation of TJ proteins leading to BBB hyperpermeability. In cultured brain ECs, cytokines or chemokines, such as tumor necrosis factor (TNF)-α, IL-6 and monocyte chemoattractant protein 1 (MCP1)/CCL2, cause significant phosphorylation of ZO-1 at Tyr, Thr and Ser residues (Rochfort and Cummins, 2015; Stamatovic et al., 2006). Co-culture with monocytes activates Rho/ROCK in ECs, which subsequently phosphorylates occludin and claudin-5 at Ser and Tyr residues and facilitates monocyte transmigration across the BBB (Persidsky et al., 2006). In vivo, increased tyrosine phosphorylation of occludin has been reported after cerebral embolism (Kago et al., 2006) and transient middle cerebral artery occlusion (MCAO) (Takenaga et al., 2009).
Modifications of TJ proteins may influence BBB integrity by regulating the expression, interactions and trafficking of TJ proteins. In cultured human brain ECs, inflammation induced by TNF-α and IL-6 downregulates ZO-1 expression and occludin/ZO-1 association, which correlates with ZO-1 phosphorylation at Tyr and Thr sites (Rochfort and Cummins, 2015). In Caco-2 cells, tyrosine phosphorylated occludin fails to bind to ZO-1, -2 and -3, but not F-actin (Kale et al., 2003). That effect remains to be tested in brain ECs. In cultured bovine retinal ECs, VEGF treatment induces TJ fragmentation and occludin trafficking, coincident with the occludin phosphorylation on Ser490 (Murakami et al., 2009). Mutating Ser490 to Ala suppresses VEGF-induced trafficking of TJ proteins and prevents increase in barrier permeability (Murakami et al., 2009). In vivo studies further reveal that VEGF induces PKCβ activation, which phosphorylates occludin at Ser490 and results in vascular impairment by TJ trafficking (Murakami et al., 2012).
Attenuating TJ protein modification may preserve BBB integrity in stroke. PP2, an inhibitor of Src-family tyrosine kinases, blocks occludin phosphorylation as well as BBB leakage after rat MCAO (Takenaga et al., 2009). In rats subjected to hypoxia and post-hypoxic reoxygenation, the PKC inhibitor chelerythrine chloride attenuates hippocampal vascular hyperpermeability and claudin-5 phosphorylation (Willis et al., 2010).
Currently, studies examining TJ protein modifications after stroke have focused on phosphorylation. It should be noted, however, that TJ proteins can also undergo methylation, glycosylation and palmitoylation resulting in altered barrier function (Stamatovic et al., 2016). How those processes are affected by stroke is unknown. In all, there are only a limited number of studies on TJ protein modification in stroke. As regulating TJ modifications may be an appropriate strategy to prevent BBB disruption, further investigations are warranted.
3.2.2. Protein translocation
Altered distribution of TJ proteins is frequently observed in post-ischemic brain microvessels and is usually accompanied by compromised BBB integrity. Such protein translocation is largely mediated by endocytosis. Both in vitro and in vivo experiments have identified the dissociation of claudin-5 from the cytoskeleton after oxygen glucose deprivation (OGD) of EC cultures and post-ischemic brain microvessels (Liu et al., 2012; Song et al., 2007). Co-immunoprecipitation indicated that claudin-5 redistribution is mediated by caveolin-1, a process that may involve endocytosis and vesicular trafficking (Liu et al., 2012). With CCL2-exposure in cultured brain ECs, occludin and claudin-5 become internalized via caveolae, with a concomitant reduction in TEER and can be recycled to the cell surface on CCL2 withdrawal (Stamatovic et al., 2009). This recycling may be important for BBB repair after stroke. JAM-A also redistributes from the interendothelial cell cleft during CCL2 treatment. However, that redistribution is by macropinocytosis and after endocytosis JAM-A is relocated to the apical membrane where it acts as an adhesion molecule (Stamatovic et al., 2012).
Another mechanism promoting translocation of junctional proteins after ischemic stroke is change in the EC actin cytoskeleton, which normally anchors junctional proteins (Burridge and Wittchen, 2013; Shi et al., 2016). Ischemia/reperfusion rapidly enhances actin polymerization in brain ECs through a signaling cascade, leading to robust formation of F-actin-enriched stress fibers and increasing cellular tension (Shi et al., 2017; Shi et al., 2016). These cytoskeletal alterations induce redistribution of junctional proteins from extracellular cell-cell contacts to the cytosol loosening the paracellular pathway. Junctional protein redistribution occurs well before and may actually render TJ proteins more vulnerable to degradation. Preventing or reversing (e.g. by inhibiting actin polymerization) cytoskeletal changes may offer protection against BBB breakdown after ischemic injury (Eira et al., 2016; Shi et al., 2017; Shi et al., 2016).
3.2.3. Protein degradation
The most intensively studied mechanism that mediates TJ protein degradation after ischemic stroke is cleavage by MMPs. MMPs are a family of zinc-containing enzymes that degrade protein substrates based on a conserved mechanism involving Zn2+-mediated activation of a site-bound water molecule (Rempe et al., 2016). Zinc accumulation in microvessels after ischemic stroke activates MMP-9 and MMP-2, leading to the loss of occludin and claudin-5 (Qi et al., 2016). MMP-9 and MMP-2 are also upregulated within hours to days after stroke contributing to TJ protein degradation (Asahi et al., 2001; Liu et al., 2012; Yang et al., 2007) and severe BBB breakdown (Heo et al., 1999; Justicia et al., 2003; Reuter et al., 2015; Romanic et al., 1998). Blocking MMP-2 and MMP-9 using inhibitors (e.g. SB-3CT, GM6001) (Cui et al., 2012; Liu et al., 2012; Yang et al., 2013) or genetic ablation (Asahi et al., 2001; Turner and Sharp, 2016) attenuates TJ protein loss and preserves BBB integrity after stroke.
TJ protein degradation also occurs via intracellular proteasomes and lysosomes after ubiquitination. Immunoprecipitation experiments suggest that occludin ubiquitination occurs in brain lysates of rats after permanent MCAO (Zhang et al., 2013a). Inhibiting the E3 ubiquitin ligase Itch by the γ-secretase blocker DAPT significantly attenuates occludin degradation and alleviates BBB breakdown (Zhang et al., 2013a).
3.3. Transcellular mechanisms
Besides alterations to junction proteins and the paracellular pathway, emerging evidence suggests that transcellular pathways are also important in ischemia-induced BBB dysfunction. Brain microvascular ECs have an inherently low transcytosis rate, however this increases after brain injury, contributing to BBB hyperpermeability before the degradation of TJ proteins and the ECM (Nag et al., 2007). Studies using electron microscopy (Nahirney et al., 2016) or morphological assessment of TJs by in vivo time-lapse two-photon microscopy (Knowland et al., 2014) consistently report increased vesicles and transcytosis in ECs at early stages (3–6 hours) after ischemia, when the TJs may be intact. Comorbid conditions, such as diabetes or obesity, can exacerbate BBB dysfunction after stroke and this may be linked to increased transcytosis (Haley and Lawrence, 2017; Reeson et al., 2015).
Transcytosis via caveolin-1-coated vesicles is important for the uptake of a number of macromolecules into brain (Simionescu et al., 2009). Caveolins are key proteins in the invagination of plasma membrane to form caveolae. EC caveolin-1 is essential in regulating the microvascular permeability in health and disease (Scherer et al., 1997; Scherer et al., 1996; Song et al., 1996). The role of caveolin-1-mediated transcytosis in BBB dysfunction after ischemic stroke remains largely elusive. Ablation of caveolin-1 reduces endothelial transcytosis, but does not reduce BBB hyperpermeability identified by tracer leakage in the first 24 h after ischemia (Knowland et al., 2014; Shi et al., 2016). It should be noted that besides regulating transcytosis, caveolin-1 may exert additional functions. Caveolin-1 knockout mice show markedly increased infarct volume and BBB damage at later stages after ischemia, compared to wild-type mice (Choi et al., 2016; Gu et al., 2012; Jasmin et al., 2007). Studies reveal that caveolin-binding motif exists in MMPs, and may therefore mediate the interactions between MMPs and caveolin-1. After focal cerebral ischemia and reperfusion, caveolin-1 knockout mice display higher MMP activities, together with enhanced degradation of TJ proteins, which can be reversed by lentiviral-mediated re-expression of caveolin-1 (Choi et al., 2016; Gu et al., 2012; Jin et al., 2015). These results suggest that the role of caveolin-1 in stroke may not only relate to caveolae formation but also involve other mechanisms regulating BBB integrity.
Stroke-induced transcytosis also has implications for CNS drug delivery. Many drugs are unable to enter brain after systemic administration due to the BBB. Recently, neuroprotection has been found by combining therapeutics with nanoparticles that can be transported through the BBB by transcytosis. Those protective effects were suppressed by inhibiting transcytosis (Wong et al., 2012; Xiao and Gan, 2013; Yemisci et al., 2015). Nanoparticle-mediated delivery may be a promising method for enhancing drug delivery to protect against ischemic injury.
3.4. Post-ischemic immune responses and the BBB
Immune cells and molecules act directly or indirectly on BBB components and influence BBB integrity after ischemia. Among the various CNS and peripheral immune cell types, the most well studied cells responding to ischemic insults are microglia/macrophages, neutrophils and T-lymphocytes. Brain resident microglial cells are among the first responders to ischemia (see Section 4.4); this is accompanied by the infiltration of peripheral macrophages, lymphocytes and dendritic cells that precede neutrophil influx (Gelderblom et al., 2009; Jickling et al., 2015). The expression of chemokines or chemoattractive cytokines plays an important role in dictating leukocyte movement to injury sites (Jaerve and Muller, 2012; Kim, 2004).
Infiltrating neutrophils are major promotors of BBB breakdown and deterioration of stroke outcome. Neutrophil integrins, such as VLA-4 (α4β1), Mac-1 (αMβ2) and LFA-1 (αLβ2), and EC adhesion molecules vascular cell adhesion protein 1 (VCAM-1), ICAM-1, E-selectin and P-selectin are important molecules in inducing neutrophil-EC adhesion (Choi et al., 2009; Schnoor and Parkos, 2008; Wang and Doerschuk, 2002), a prerequisite for neutrophil infiltration. Inhibiting the upregulation of these molecules ameliorates inflammatory responses and BBB dysfunction, and reduces brain injury after ischemia (Huang et al., 2016a). Neutrophils are a major source of MMP-9 in the infarct core after ischemic stroke, which subsequently promotes BBB breakdown secondary to microvascular basal lamina proteolysis, eventually contributing to neuronal injury (Justicia et al., 2003; Nguyen et al., 2007; Rosell et al., 2006). MMP-9 also facilitates additional leukocyte recruitment, and knocking out MMP-9 markedly reduces leukocyte recruitment into post-ischemic brain tissue (Gidday et al., 2005).
How lymphocytes modulate BBB permeability after ischemic stroke remains poorly investigated. Tregs are the most studied, but their role is controversial (Liesz and Kleinschnitz, 2016). Adoptive transferred Tregs are shown to preserve BBB integrity after ischemic stroke, without penetrating into the brain parenchyma or affecting resident microglia (Li et al., 2013). Rather, both in vivo and in vitro studies suggest that Tregs suppress peripheral neutrophil-derived MMP-9, thereby ameliorating proteolytic BBB damage (Li et al., 2013). Direct Treg/neutrophil cell-to-cell contact may be essential for the inhibitory effect of neutrophil MMP-9 production after ischemia, with programmed death ligand-1 being a critical mediator of Treg/neutrophil crosstalk (Li et al., 2014b). Other mechanisms may also underlie the Treg-associated BBB protection. Studies with human umbilical vein ECs demonstrate that direct Treg-EC contact and soluble factors are required for Treg-mediated suppression of EC inflammation (Zhang et al., 2014). Further investigation using brain ECs is warranted.
There is controversy over the role of Tregs after ischemia. Depletion of endogenous Tregs can reduce brain infarct and improve neurological function (Kleinschnitz et al., 2013). Adoptive transfer of Tregs potentiates ischemia-induced microvascular dysfunction through their interactions with the endothelium and platelets, and subsequent thrombus formation (Kleinschnitz et al., 2013). Consistent with those results, expansion of Tregs by anti-CD28 antibody before stroke exacerbates thrombus formation in the cerebrovasculature, enlarging thrombo-inflammatory lesions and infarcts (Schuhmann et al., 2015).
The time course of Treg infiltration into brain and their exact role in ischemia-induced BBB breakdown and injury progression require further investigation. Studies on other T-lymphocytes or B-lymphocytes in modulating BBB permeability after ischemic stroke are largely lacking.
3.5. Consequences of BBB dysfunction after ischemic stroke
BBB dysfunction begins from the onset of ischemia and deteriorates with sustained hypoperfusion. The severity of BBB breakdown, as well as its consequence, is topographically heterogeneous. Vascular walls within the ischemic core develop severe and irreversible injury, whereas the vascular endothelium in the penumbra are mildly impaired and potentially salvageable (Nagaraja et al., 2008). The diffuse, mild BBB damage may be reversible with timely reperfusion in the penumbra; however, the severe vascular injury in the core area may be further potentiated by such reperfusion (Fan et al., 2014; Simpkins et al., 2016). The neurovascular toxicity of tPA, together with other pathogenic factors such as vascular oxidative stress and neuroinflammation, confounds the consequence of BBB breakdown (Fan et al., 2014). Although BBB breakdown is usually associated with worse outcome after ischemic stroke, there has been a long-term debate on whether BBB dysfunction is a cause or a consequence of brain parenchymal injury (e.g. the BBB protective effects of certain therapeutics may be due to reduced infarct size). Recent studies, using EC-specific gene manipulation, began to tackle this fundamental question. In mice subjected to MCAO and reperfusion, BBB leakage was identified at 30 minutes after reperfusion, preceding infarct formation and occurring in areas that evolve into infarcts topographically (Shi et al., 2016). Furthermore, EC-specific gene manipulation that blocks early BBB dysfunction is capable of providing parenchymal protection and improving long-term functional outcome (Shi et al., 2017; Shi et al., 2016). These findings suggest that early BBB damage may cause rather than result from parenchymal cell injury. Early BBB dysfunction may be a promising therapeutic target to reduce the adverse effects of thrombolytic therapy, prolong the therapeutic window, and enhance patient outcome. One potential benefit of the opened BBB, though, is that it is easier to deliver therapeutics to brain (Borlongan and Emerich, 2003; Rapoport, 2000). However, some therapies could directly target the BBB, if its opening is a cause instead of a result of parenchymal injury. This makes it vital to fully understand the mechanisms underlying stroke-induced BBB dysfunction and to develop new therapies to target that dysfunction.
4. Modulation of blood-brain barrier permeability by different cell types and chemical mediators
The NVU, consisting of neurons, astrocytes, pericytes, ECM, ECs, and circulating blood elements, illustrates a framework where cell-cell and cell-matrix interactions dictate the brain response to ischemic injury (Lo et al., 2003). As the interface where these interactions occur, the BBB is constantly regulated by different cell types in the NVU (Fig. 3). Various chemical mediators present within the NVU also influence BBB permeability, both under physiological and ischemic conditions.
Fig. 3. Modulation of blood-brain barrier permeability by different cell types after ischemic stroke.
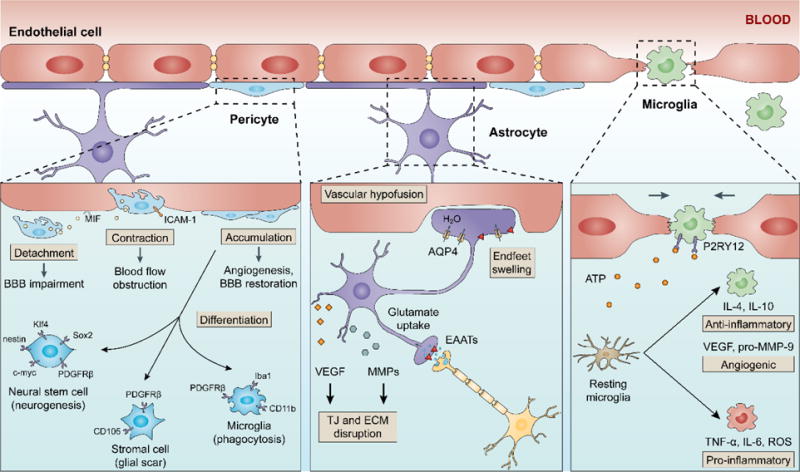
Cell-cell interactions in the neurovascular unit dictate the brain response to ischemic injury and influence BBB damage and repair. Pericytes respond quickly to an ischemic stroke and display characteristics that may be protective or detrimental, such as detachment from the microvessel walls, constriction, migration and accumulation. Furthermore, pericytes are able to differentiate into neural and vascular lineage cells after ischemia. Astrocyte swelling is another early response in the neurovascular unit after ischemia, due to increased uptake of glutamate and water. The swelled astrocytes compress vessels in the ischemic regions and exacerbate vascular hypoperfusion. Post-ischemic astrocytes also produce VEGF and MMPs that degrade TJs and the ECM. Microglia/macrophages undergo phenotypic polarization after ischemia, developing into pro-inflammatory and anti-inflammatory phenotypes. Pro-inflammatory microglia/macrophages may promote BBB disruption, whereas anti-inflammatory microglia/macrophages can facilitate post-stroke angiogenesis and inflammation resolution. Recent studies also suggest a role of microglia/macrophages in vessel repair after injury; two mechanisms proposed being through mechanical stretching and P2RY12 signaling.
4.1. Endothelial cells
As a first-line of defense at the NVU, ECs react to ischemia and hypoxia and the potentially harmful chemicals released from the vascular system. As described above, cytoskeletal rearrangement, increased transcytosis and alterations in TJ proteins occur in ECs after ischemia contributing to stepwise BBB dysfunction. Oxidative stress and inflammation trigger EC injury and irreversible endothelium impairment (Yang et al., 2017; Zhang et al., 2016b). In the subacute stage, autophagosomes are observed in ECs in the ischemic hemisphere (Garbuzova-Davis et al., 2013). Whether enhanced autophagy is pro-survival or pro-death remains unclear, but recent studies support a beneficial role of EC autophagy in ameliorating BBB breakdown and TJ loss after ischemia (Li et al., 2014a).
In addition to infiltrating neutrophils, ECs are a source of MMPs when stimulated by pro-inflammatory cytokines and free radicals after stroke, contributing to TJ and ECM degradation (Reuter et al., 2013). Another molecule synthesized and released by ECs, endothelin-1, decreases local brain blood flow via vasoconstriction and regulates thrombus formation through platelet activation, thereby inducing cerebral infarction (Mamo et al., 2014).
4.2. Pericytes
Pericytes respond quickly to an ischemic stroke and display characteristics that may be protective or detrimental, such as constriction, migration, detachment from the microvessel walls and even cell death (Fernandez-Klett and Priller, 2015; Hall et al., 2014). In rat MCAO, pericytes separate and migrate from the basement membrane as early as 1 hour after occlusion. The space between pericytes and basement membrane was even larger at 3 hours after injury (Duz et al., 2007). These morphological changes and detachment from the microvessel wall may be an initial step in BBB dysfunction. Contracted pericytes are also found on microvessels after MCAO, which leads to capillary constriction and obstructed blood flow. These pericyte morphological changes persist and cannot be reversed by reperfusion (Dalkara et al., 2011; Yemisci et al., 2009).
Inflammation and oxidative stress induced by ischemia may further exacerbate pericyte-mediated BBB destruction. In pericyte-fibroblast co-cultures, pro-inflammatory stimuli induce widespread changes in gene expression of interleukins, chemokines, and cellular adhesion molecules in pericytes (Jansson et al., 2014; Persidsky et al., 2016). In vivo studies suggest that NG2+ pericytes control the pattern and efficacy of leukocyte infiltration into CNS by upregulating ICAM-1 and producing the chemoattractant macrophage migration inhibitory factor (MIF) (Stark et al., 2013). In addition, oxidative stress induces pericyte dysfunction and BBB damage (Amano et al., 2005; Okada et al., 2015b). Inhibition of oxidative-nitrative stress relieves pericyte contraction in mice after MCAO (Yemisci et al., 2009).
On the other hand, pericyte accumulation in the peri-infarct area after ischemic stroke may protect the BBB and improve vascular repair. Rapid accumulation of pericytes is observed in the peri-infarct area (Jeynes, 1985), with concomitant upregulation of PDGFRβ and PDGFβ (Nakamura et al., 2016; Renner et al., 2003). These cells, largely deriving from bone marrow pericyte progenitor cells and adjacent immature pericytes, are involved in angiogenesis and BBB repair after stroke (Kokovay et al., 2006; Lamagna and Bergers, 2006; Renner et al., 2003). Furthermore, as a type of multipotent vascular stem cell, pericytes have additional roles in BBB regulation and neurovascular injury after ischemic stroke. Pericytes are able to differentiate into neural and vascular lineage cells after ischemia (Nakagomi et al., 2015). In human stroke brain tissue, activated pericytes are also found to express microglia markers (Ozen et al., 2014). PDGFRβ+ vascular pericytes isolated from ischemic areas can express stem cell markers (e.g., nestin, c-myc, Klf4, Sox2) and microglial markers (e.g. Iba1, CD11b), suggesting that pericytes may serve as microglia-generating stem cells and have potential phagocytic capacity (Attwell et al., 2016; Sakuma et al., 2016). In brain ischemia and spinal cord injury, PDGFRβ+ cells co-express stromal cell markers and produce scar tissue distinct from glial scar (Fernandez-Klett et al., 2013; Goritz et al., 2011). The mechanisms underlying such transformation and its implication for post-injury repair are unclear, but may be a novel target for regenerative stroke therapies.
4.3. Astrocytes
The water channel aquaporin 4 (AQP4) is highly expressed on astrocyte endfeet and critically regulates water flux between blood and brain (Nagelhus and Ottersen, 2013). AQP4-deficient mice demonstrate reduced cytotoxic brain edema following ischemic stroke (Manley et al., 2000). Interestingly, astrocyte AQP4 is upregulated at delayed stages after ischemia, and this may be involved in BBB repair (Tourdias et al., 2011). Over 80% of glutamate transporters, especially EAAT2, is located on astrocytes, making astrocytes the main site of glutamate uptake at the NVU (Dallerac and Rouach, 2016; Petr et al., 2015). Following ischemia, astrocyte swelling is one of the earliest responses due to increased uptake of glutamate and lactate (Kimelberg, 2005; Landis, 1994; Raiteri and Raiteri, 2015; Verkhratsky et al., 2016). Astrocyte swelling may compress vessels in the ischemic regions exacerbating vascular hypoperfusion (Sykova, 2001).
Astrocytes can facilitate BBB breakdown after ischemic stroke. In EC-astrocyte co-cultures, increased astrocyte apoptosis stimulated by EC-derived microvesicles after OGD is accompanied by increased BBB permeability and downregulation of TJ proteins occludin and claudin-5 (Pan et al., 2016). Furthermore, post-ischemic neurons can stimulate astrocyte production of VEGF, which is responsible for occludin and claudin-5 loss and increased BBB permeability (Li et al., 2014c). Astrocytes are also a sources of MMPs that degrade TJs and the ECM after ischemia (Mun-Bryce and Rosenberg, 1998).
4.4. Microglia
Microglia are resident CNS macrophages that originate from the mesoderm/mesenchyme. After migrating into brain, microglia acquire a specific ramified morphological phenotype with low phagocytic properties, termed “resting microglia” (Kettenmann et al., 2011). Being an integral part of the NVU, microglia actively communicate with endothelium and regulate the BBB both during development and after injury (da Fonseca et al., 2014). Microglia play a crucial role in the development of the cerebral and retinal vasculatures, participating in sprouting, migration and anastomosis of vessels (Arnold and Betsholtz, 2013). Resident microglia, but not monocyte-derived macrophages, serve as cellular chaperones facilitating the stabilization and fusion of brain ECs during embryonic development (Fantin et al., 2010). Microglia are present at vascular junctions and bridge endothelial tip cells, which, in combination with VEGF-induced vessel sprouting, synergistically promotes the formation of the brain vascular network (Fantin et al., 2010). Studies on aortic ring cultures indicate that microglia can stimulate vessel sprouting without direct EC contact, but rather through secreting soluble factors (Rymo et al., 2011).
Microglia are a first responder to ischemic brain injury, rapidly undergoing morphological and genetic changes upon activation (Kettenmann et al., 2011). Activated microglia exert dual roles at the BBB and on ischemic brain injury. They produce a plethora of cytokines and chemokines that upregulate EC adhesion molecules and promote leukocyte infiltration (da Fonseca et al., 2014). However, activated microglia may also have beneficial actions by phagocytosing cellular debris and suppressing inflammatory responses. In a model of neonatal stroke, microglial depletion adversely affected vascular density, increased vascular permeability and intracerebral hemorrhage, effects related to inhibition of microglial transforming growth factor beta 1 (TGFβ1) signaling (Fernandez-Lopez et al., 2016).
Emerging evidence suggests that the dual roles of microglia on the BBB may be related to their phenotypic polarization after ischemic stroke. Depending on stimulus, microglia/macrophages can develop into a spectrum of different but overlapping functional phenotypes, including classically-activated (pro-inflammatory) and alternatively-activated (anti-inflammatory) phenotypes (Franco and Fernandez-Suarez, 2015; Hu et al., 2015; Jiang et al., 2016; Xiong et al., 2016). Lipopolysaccharide (LPS), a potent pro-inflammatory stimulator, primes microglia to induce hyperpermeability in brain ECs in vitro through mechanisms involving TNF-α (Nishioku et al., 2010) and NADPH oxidase (Sumi et al., 2010). Pro-inflammatory microglia also cause P-gp dysfunction in brain ECs through NADPH oxidase activation (Matsumoto et al., 2012), contributing to the accumulation of neurotoxic proteins in brain. On the other hand, microglia can possess anti-inflammatory properties and be associated with long-term neurovascular remodeling and improved neurological functions during recovery after ischemia (Yang et al., 2015). These microglia can also promote angiogenesis through production of VEGF, IL-8 and pro-MMP-9 (Mallucci et al., 2015; Medina et al., 2011; Willenborg et al., 2012; Zajac et al., 2013).
4.5. Chemical mediators
Many chemicals participate in the regulation of BBB permeability after ischemic stroke. These chemicals belong to different categories and function to exacerbate BBB breakdown or benefit BBB integrity (Table 1). Some commonly studied chemicals that induce BBB hyperpermeability and brain edema include inflammatory substances (e.g. bradykinin, histamine, thrombin, substance P, endothelin-1), pro-inflammatory cytokines (e.g. TNF-α, IL-6, IL-1), neurotransmitters (e.g. glutamate, NO), and free radicals. There are also chemical mediators that are beneficial for maintaining BBB integrity after stroke, such as the classic neuroactive steroids 17β-estradiol and progesterone (Johann and Beyer, 2013). The second messenger cAMP also exerts distinct roles on BBB permeability; the cytosolic accumulation of cAMP causes BBB dysfunction, whereas the cAMP in vacuoles preserves BBB integrity (Sayner et al., 2006).
Table 1.
Selected chemical mediators that influence BBB integrity after ischemic stroke.
| Category | Name | Effects on the BBB | Mechanisms | References |
|---|---|---|---|---|
| Inflammatory substance | Bradykinin | Permeability ↑ TEER ↓ | Action on EC B2- receptors; Ca2+-dependent TJ redistribution | (Chen et al., 2016; Marcos-Contreras et al., 2016; Unterberg et al., 1984) |
| Histamine | Permeability ↑ | AJ protein phosphorylation; elevation of intracellular Ca2+; disruption of EC adhesions | (Abbott, 2000; Andriopoulou et al., 1999; Lindsberg et al., 2010; Winter et al., 2004) | |
| Thrombin | Permeability ↑ TEER ↓ Brain edema ↑ | Cleavage and activation of protease-activated receptors (PARs); activation of Src kinases and MMPs in ECs | (Hawkins et al., 2015; Hun Lee et al., 2015; Keep et al., 2014; Li et al., 2015a; Liu et al., 2010; Nguyen et al., 1999; Poggesi et al., 2016) | |
| Substance P | Permeability ↑ Brain edema ↑ | TJ protein redistribution | (Rodriguez et al., 2014; Turner et al., 2011) | |
| Endothelin-1 | Permeability ↑ Brain edema ↑ | Oxidative stress; elevation of MMPs; TJ disruption | (Hostenbach et al., 2016; Hung et al., 2015; Leung et al., 2009; Zhang et al., 2013b) | |
| Cytokine | TNF-α | Permeability ↑ | Oxidative stress; downregulation of TJ proteins | (Aslam et al., 2012; Basuroy et al., 2006; Forster et al., 2008) |
| IL-6 | Permeability ↑ TEER ↓ | Oxidative stress; PKC- dependent cytoskeletal rearrangement; TJ disruption | (Desai et al., 2002; Maruo et al., 1992; Rochfort and Cummins, 2015) | |
| IL-1 | Permeability ↑ | Upregulation of ICAM-1; activation of MMPs; TJ disruption | (McCarron et al., 1993; Wu et al., 2015; Yang et al., 2016a) | |
| Neurotransmitter | Glutamate | Permeability ↑ | Phosphorylation, redistribution and disruption of TJ proteins | (Andras et al., 2007) |
| NO | Permeability ↑ Brain edema ↑ | Upregulation of MMPs | (Gu et al., 2012; Gursoy-Ozdemir et al., 2004) | |
| Steroid | 17β-estradiol | Permeability ↓ Brain edema ↓ | Downregulation of astrocytic AQP4; modulation of ion transporters; reduction of MMPs | (Liu et al., 2005; O’Donnell et al., 2006; Rutkowsky et al., 2011) |
| Progesterone | Permeability ↓ Brain edema ↓ | Downregulation of AQP4; reduction of MMPs; preservation of TJs | (Arbo et al., 2016; Frechou et al., 2015; Ishrat et al., 2010; Li et al., 2015c; Won et al., 2014) | |
| Other | ATP | Permeability ↑ | Activation of MMPs; TJ disruption | (Yang et al., 2016a) |
| VEGF | Permeability ↑ | Phosphorylation, dislocation and downregulation of TJ proteins; enhancement of endocytosis | (Fischer et al., 2002; Horowitz and Seerapu, 2012) | |
| HIF | Permeability ↑ Brain edema ↑ | Upregulation of VEGF; TJ disruption | (Chen et al., 2008; Zhang et al., 2016c) |
5. Influence of stroke risk factors and comorbidities on the blood-brain barrier
Comorbidities occur in most stroke patients and have major impacts on stroke outcome. Some comorbid conditions and risk factors are modifiable, e.g. hypertension and hyperlipidemia, and represent areas of interest to reduce stroke occurrence or improve the efficacy of stroke therapies. Despite an urgent need to understand BBB dysfunction in stroke patients with certain comorbid conditions, the majority of basic and preclinical stroke studies have hitherto focused on healthy young adult male animals, which might impede the translation of potential treatments from bench to bedside. This section reviews BBB alterations associated with common stroke comorbid conditions: hypertension, hyperglycemia and hyperlipidemia, as well as non-modifiable risk factors age and gender. Specific therapeutic strategies are emphasized where applicable.
5.1. Hypertension
In response to acute or chronic elevations (hypertension) in blood pressure, adaptive vascular remodeling occurs throughout the body to buffer mechanical and pulsatile stresses, leading to multiple end-organ impairment (Scuteri et al., 2011). The brain is an organ particularly affected by high blood pressure. Several crucial cerebrovascular regulatory mechanisms that function to maintain brain energy homeostasis are disrupted by hypertension, which, together with structural alterations, contributes to hypoperfusion and dysfunction of the brain and increases the risk for stroke and dementia. The influence of hypertension on cerebrovascular anatomy and blood flow regulation has been reviewed previously (Faraco and Iadecola, 2013). The present section focuses on BBB changes induced by hypertension.
5.1.1. Anatomical and functional changes at the BBB with hypertension
BBB abnormalities are present from an early stage in patients exhibiting mild symptoms of cognitive impairment during the development of hypertension (Pelisch et al., 2013). Increased BBB permeability has been consistently observed in animal models of hypertension. Spontaneously hypertensive rats (SHRs), the most widely used animal model of genetic and chronic hypertension, share several similarities with human essential hypertension (Folkow, 1982). BBB impairment is observed in cerebral cortex and deep gray matter in SHRs at 5 months and older, when prominent tissue damage has already developed (Fredriksson et al., 1987; Knox et al., 1980). In hippocampus, BBB hyperpermeability occurs in SHRs as young as 3 months, a stage at which neuronal cell loss is not yet developed despite a hypertensive state (Fan et al., 2015b; Ueno et al., 2004). These findings support a causative role of high blood pressure in BBB dysfunction, and also suggest that BBB dysfunction at earlier periods may contribute to the hippocampal neuronal loss observed in 6-month-old SHRs (Ueno et al., 2004). BBB disruption also occurs in acute hypertensive models. Hypertension due to aortic constriction above the renal arteries causes Evans Blue extravasation into brain from 8 days after surgery (Mohammadi and Dehghani, 2014).
The vascular anatomical changes underlying hypertension-induced BBB dysfunction are multifaceted, but alterations in EC junctions likely play a major role. Stroke-prone renal vascular hypertensive rats have progressive morphological changes in BBB TJs, with increasing loss of occludin and ZO-1 from as early as 4 weeks (Fan et al., 2015b). Consistently, chronic administration of Nω-Nitro-L-arginine methyl ester (L-NAME), an inhibitor of nitric oxide synthase (NOS), induces hypertension and loss of occludin and ZO-1 in brain vessels (Kalayci et al., 2009). Short-term hypertension induced by aortic constriction also leads to reduced mRNA levels of claudins (3, 5 and 12) (Mohammadi and Dehghani, 2014). Studies also reveal JAM-A upregulation throughout the body in pre-hypertensive 3-week-old SHRs, which is, therefore, not secondary to elevated blood pressure (Waki et al., 2007). The involvement of elevated JAM-A in BBB dysfunction may be two-fold: JAM-A facilitates leukocyte-EC adhesion promoting leukocyte accumulation in the systemic circulation (Ostermann et al., 2005). This in turn increases hemodynamic resistance and contributes to increased blood pressure in SHRs (Fukuda et al., 2004; Waki et al., 2007). This pro-hypertensive effect triggers other alterations that damage the BBB. Secondly, together with platelet endothelial cell adhesion molecule 1 (PECAM-1), JAM-A promotes leukocyte transmigration across the ECs (Martin-Padura et al., 1998) which also contributes to increased BBB permeability.
Changes also occur in other NVU components that may contribute to the dysregulation of BBB function, such ion and fluid transport. SHRs and stroke-prone spontaneously hypertensive rats (SHRSP) with established hypertension have upregulated astrocytic AQP4 in the frontal cortex, striatum and hippocampus, compared to younger cohorts and age-matched Wistar Kyoto (WKY) rats (Ishida et al., 2006; Tomassoni et al., 2010). Increased AQP4 expression may alter fluid exchange at the BBB interface; however, the exact role of AQP4 in BBB dysfunction with hypertension, e.g. whether it is a cause of or adaptive change to increased BBB permeability, remains elusive.
Multiple studies indicate that inflammation and oxidative stress are major causes of hypertension-induced BBB dysfunction. Pro-inflammatory cytokines such as IL-1β, IL-6 and TNF-α are significantly elevated in brains of 8-month-old SHRs compared to age-matched WKY rats (Liang et al., 2016; Tayebati et al., 2016). 8-hydroxy-2′-deoxyguanosine (8-OHdG), a marker of oxidative stress, is increased in SHRSP but not WKY brains at 16 weeks (Tayebati et al., 2016). Acute hypertension induced by aortic constriction in rats impairs brain antioxidant defense systems and induces brain injury through excessive oxidative stress at 8 days after surgery (Mohammadi and Dehghani, 2014). Enzyme activity of superoxide dismutase (SOD) and catalase, and glutathione content are significantly lower in hypertensive rat brains, whereas malondialdehyde levels are increased (Mohammadi and Dehghani, 2014). Reactive oxygen species (ROS) can alter BBB integrity through cytoskeleton rearrangements and redistribution and degradation of TJ proteins claudin-5 and occludin. Specific signaling pathways, including RhoA and phosphatidylinositide 3-kinases (PI3Ks), mediate these processes, and their specific inhibitors abolish ROS-induced monocyte migration across the BBB (Poulet et al., 2006; Schreibelt et al., 2007). Furthermore, angiotensin II potentiates oxidative stress under hypertension, facilitating leukocyte-EC adhesion and increasing BBB permeability (Zhang et al., 2010).
5.1.2. Hypertension exacerbates BBB breakdown after ischemic stroke
Hypertension worsens the outcome of ischemic stroke, increasing infarct volume, potentiating white matter injury, and augmenting brain edema and cognitive deficits (Choi et al., 2015; Fan et al., 2015a; Hom et al., 2007). Aggravated BBB breakdown in hypertension is one important element contributing to worse outcome. Hypertension exacerbates damage to TJ proteins after ischemic brain injury, such as ZO-1, ZO-2, claudin-5 and occludin (Fan et al., 2015b; Hom et al., 2007). BBB transporters can also be changed; for instance, the Na+/H+ exchanger isoform 1 is markedly increased in hypertensive SHRs after ischemia and may be involved in BBB breakdown and brain edema development (Hom et al., 2007). Additionally, the pro-inflammatory state of the hypertensive brain potentiates BBB breakdown after ischemia. ICAM-1 which mediates leukocyte-leukocyte interaction and leukocyte transmigration, is upregulated in SHRs, enhancing leukocyte infiltration into brain and BBB impairment after stroke (Moller et al., 2015; Nagai et al., 2011).
Controlling blood pressure in patients with chronic hypertension remains one of the most important means of stroke prevention (Hermida et al., 2016). To this end, certain treatments commonly used to lower blood pressure may exert additional effects. For example, BBB AJs and TJs are modulated by calcium. Calcium channel blockers, which are widely used to control blood pressure, may thereby have additional beneficial functions toward improving stroke outcome, through direct action of preserving microvascular integrity in hypertension patients (Brown and Davis, 2002; Farkas et al., 2001).
5.2. Diabetes and hyperglycemia
Diabetes/hyperglycemia is a rapidly increasing risk factor for stroke. It is associated with increased mortality and poor functional recovery (Kruyt et al., 2010; Zhang et al., 2013c). Accumulating evidence indicates that hyperglycemia-induced cerebrovascular complications, especially BBB dysfunction, are major contributors to poor stroke outcome (Baird et al., 2003; Gandhi et al., 2010; Shao and Bayraktutan, 2013; Yu et al., 2016). Hyperglycemia is also a risk factor for intracerebral hemorrhage regardless of tPA treatment, which at least partially results from massive BBB opening (Bruno et al., 1999; Demchuk et al., 1999; Masrur et al., 2015).
5.2.1. Anatomical and functional changes at the BBB with hyperglycemia
In patients with Type 2 diabetes, BBB permeability increases as evidenced by enhanced signal intensity on T1-weighted volumetric images by gadolinium MRI (Iwata et al., 1999; Starr et al., 2003). Consistently, BBB dysfunction is observed in animal or cell culture models of diabetes and hyperglycemia/high glucose (Fujihara et al., 2016; Hawkins et al., 2007; Huber et al., 2006; Mooradian et al., 2005). A major anatomical change accounting for the impaired BBB integrity under hyperglycemia is TJ disruption with increased paracellular permeability. Protein levels of TJ components, including occludin, claudin-5 and ZO-1, are decreased after hyperglycemia (Chao et al., 2016; Xu et al., 2016; Yoo et al., 2016), and therapeutic agents that reverse TJ protein alterations are capable of protecting BBB integrity in diabetic animals (Zanotto et al., 2017). Mechanistically, hyperglycemia-induced activation of upstream signaling molecules, e.g. PKCβ, and subsequent development of oxidative stress in cerebral microvessels play a role in TJ loss (Liao et al., 2005; Shao and Bayraktutan, 2013). ROS are suggested to be a main mediator in BBB breakdown after hyperglycemia, and ROS inhibition preserves TJs and improves BBB integrity (Fukuda et al., 2016; Sun et al., 2015).
Hyperglycemia can also disrupt gap junction communication in other NVU components, such as astrocytes (Gandhi et al., 2010; Prasad et al., 2014). Swollen astrocytic endfeet at the BBB interface are observed in Type 2 diabetic mice KKA(y), and attenuating astrocyte swelling preserves BBB integrity and protects against cognitive decline (Min et al., 2012).
Increased MMP activity is observed in diabetic animal models which may account for TJ protein degradation. After streptozotocin treatment to induce diabetes, mice have elevated expression of both pro- and active-MMP-9, which results in TJ loss and BBB breakdown (Aggarwal et al., 2015; Hawkins et al., 2007). A disrupted BBB further allows infiltration of peripheral monocytes into brain, compromising cognition in obese and diabetic mice (Stranahan et al., 2016). MMP-9 inhibition restores BBB integrity and improves learning and memory in diabetic mice (Aggarwal et al., 2015).
5.2.2. Hyperglycemia exacerbates BBB disruption after stroke
Earlier studies revealed the characteristics of hyperglycemia-induced cerebrovascular changes and EC dysfunction, both during ischemia and during reperfusion (Kawai et al., 1997; Kawai et al., 1998; Kawai et al., 1999; Keep et al., 2005). Both mild and severe hyperglycemia induce marked BBB dysfunction in animals undergoing ischemia and reperfusion (Dietrich et al., 1993; Ennis and Keep, 2007). Diabetic mice exhibit exacerbated BBB breakdown and TJ disruption, increased infarct volume as well as severe neurological deficits after ischemia (Huang et al., 2013; Kamada et al., 2007; Zhang et al., 2016a; Zhang et al., 2016c). Immediate brain swelling and hemorrhagic transformation after ischemia also occur with hyperglycemia (Fan et al., 2014; McBride et al., 2016; Soejima et al., 2012).
Enhanced proteolysis of TJs mediated by the MMPs is an important cause of BBB breakdown after ischemia in hyperglycemia mice (Cipolla et al., 2011; Kamada et al., 2007). Diabetic db/db mice show an increased and more rapid elevation of MMP-9 expression and activity compared to db/+ control mice, resulting in greater degradation of occludin and collagen IV (Kumari et al., 2011). The hypoxia-inducible factor 1α (HIF-1α)/VEGF pathway is also related to TJ protein loss and enhanced BBB paracellular permeability in hyperglycemic mice (Yan et al., 2012). Hyperglycemia induces higher expression of HIF-1α and VEGF in brain microvessels after MCAO/reperfusion. Furthermore, EC-specific knockout of HIF-1α ameliorates BBB leakage and brain infarction in diabetic animals (Zhang et al., 2016c).
Inflammation and oxidative stress both enhance TJ disruption in diabetic animals after cerebral ischemia (Kamada et al., 2007; Won et al., 2011). Hyperglycemic rats show enhanced formation of superoxide by NADPH oxidase in brain parenchyma and the vasculature during reperfusion, which may contribute to increased BBB permeability (Won et al., 2011). Inhibiting inflammation, e.g. by blockade of the high-mobility group box1 (HMGB1) and NF-κB signaling pathway, alleviates diabetic cerebral ischemia/reperfusion injury and attenuates BBB breakdown (Luan et al., 2013).
5.3. Hyperlipidemia
Hyperlipidemia refers to abnormal elevation of blood lipids or lipoproteins. According to the type lipid excess, hyperlipidemia is classified into hypercholesterolemia, hypertriglyceridemia, or both in combined hyperlipidemia (Nelson, 2013). Specific genetic abnormalities cause primary hyperlipidemia, while most hyperlipidemia results from environmental factors, such as a high fat diet (HFD).
5.3.1. Anatomical and functional changes at the BBB with hyperlipidemia
A commonly used animal model for studying hyperlipidemia is the apolipoprotein E-deficient (ApoE−/−) mouse. ApoE is a class of apolipoprotein essential for lipid and cholesterol metabolism, the ablation of which leads to hyperlipidemia and atherosclerosis (Plump et al., 1992). Young, 6–8 weeks, ApoE−/− mice already have prominent BBB hyperpermeability, manifested by 70% more spontaneous leakage of plasma albumin into the brain compared to controls (Methia et al., 2001). Furthermore, BBB dysfunction associated with aging is exacerbated by ApoE knockout (Hafezi-Moghadam et al., 2007). However, elevated blood lipids may not solely account for the compromised BBB integrity in ApoE−/− mice. It is possible that ApoE directly modulates BBB integrity through mechanisms affecting the cerebrovasculature, e.g. inducing TJ formation or suppressing inflammation in the NVU (Bell et al., 2012; Nishitsuji et al., 2011). In another hyperlipidemia model, 10 weeks of HFD in 6-week-old mice increases blood cholesterol, triglycerides, and low-density lipoprotein (LDL) (Deng et al., 2014). Both cerebral macrovessels and microvessels undergo remodeling during HFD feeding, including increased cerebral vascular tortuosity index and decreased MCA inner diameters (Deng et al., 2014). This remodeling may be mediated at least in part by MMP-9, as HFD induces similar level of obesity, hyperglycemia and hyperlipidemia in MMP-9−/− mice, but there is no cerebrovascular remodeling (Deng et al., 2014). Acute injection of human triglyceride-rich lipoprotein, which releases lipolysis products upon hydrolysis, increases BBB permeability in rats within 20 min (Ng et al., 2016). Exactly how hyperlipidemia leads to BBB dysfunction remains largely unknown, although oxidative stress may play a role (Dias et al., 2014). The changes in the cerebrovasculature render it more vulnerable to ischemic attack, where BBB disruption may be exacerbated.
5.3.2. The influence of hyperlipidemia on BBB integrity after stroke
Hyperlipidemia exacerbates ischemic brain injury (Langdon et al., 2011). In both patients and animal models, hyperlipidemia is associated with EC dysfunction. Chronic hyperlipidemia induces profound vascular remodeling, such as increased tortuosity index (Deng et al., 2014), increased collagen deposition and vessel stiffness (Deutsch et al., 2009), which worsens the perfusion defects and BBB breakdown after ischemic brain injury (Ayata et al., 2013). In animal models with HFD or ApoE deficiency, hyperlipidemia exacerbates BBB breakdown and brain edema after ischemia, as shown in several reports (Deng et al., 2014; ElAli et al., 2011; Lynch et al., 2002; Methia et al., 2001).
Several mechanisms may underlie hyperlipidemia-exacerbated BBB breakdown after ischemic stroke, including MMP activation (Deng et al., 2014; ElAli et al., 2011), inflammation (Cao et al., 2015; Fang et al., 2015), enhanced oxidative stress (Cao et al., 2015), and impaired EC-pericyte interactions (Zechariah et al., 2013). Hyperlipidemia elevates MMP-2 and -9 activity in post-ischemic brain and downregulates microvessel TJ proteins (Deng et al., 2014; ElAli et al., 2011). HFD induces cerebrovascular remodeling and worsens neurological outcome after MCAO, effects abolished in MMP-9 knockout mice despite similar increases in blood lipid levels compared to controls (Deng et al., 2014). MMP activation in turn induces RhoA/ROCK activation, which is an important mechanism regulating BBB integrity (Allen et al., 2010; ElAli et al., 2011; Shin et al., 2014; Sugimoto et al., 2009). A pharmacological inhibitor of ROCK, fasudil, decreased blood pressure and cerebrovascular resistance in hyperlipidemic mice and improved tissue perfusion after MCAO (Shin et al., 2014). HFD-induced hyperlipidemia also enhances the expression of pro-inflammatory factors TNF-α and IL-6, as well as ICAM-1 and VCAM-1 after ischemia/reperfusion injury (Cao et al., 2015). Hyperlipidemia decreases serum superoxide dismutase activity and glutathione peroxide content, and increases lipid peroxidation and LDL oxidation in brain after cerebral ischemia/reperfusion injury (Cao et al., 2015; ElAli et al., 2011).
Hyperlipidemia also influences post-stroke recovery through altering cell-cell interactions at the BBB interface. VEGF-induced capillary formation after ischemia is dose-dependently attenuated by hyperlipidemia, with reduced brain EC pericyte coverage. Increased expression of N-cadherin in ischemic brain microvessels upon VEGF treatment, which mediates EC-pericyte interactions, is blunted by hyperlipidemia. These changes impair cerebral blood flow and reduce the metabolic penumbra increasing infarct size (Zechariah et al., 2013).
5.4. Aging
5.4.1. Anatomical and functional changes at the BBB during aging
Aging is accompanied by complicated and progressive disturbances in the structural integrity and physiological functions of cells and organs (Lopez-Otin et al., 2013). BBB dysfunction during aging leads to disruption of brain homeostasis and plays a crucial role in the pathogenesis of various neurodegenerative diseases. For many years, studies investigating whether increased BBB permeability is associated with healthy aging in humans generated controversial results (Gorle et al., 2016). Nevertheless, a large-scale meta-analysis on 31 BBB permeability studies reports increased BBB permeability with normal aging, as evaluated by the CSF/serum albumin ratio (Farrall and Wardlaw, 2009). A more recent study using advanced MRI to quantify regional BBB integrity further reveals that BBB dysfunction is an early event in aging brain, which starts from the hippocampus and may contribute to cognitive impairment (Montagne et al., 2015). Consistently, in animal models, enhanced IgG extravasation is observed in 24-month-old mice compared to young controls (Elahy et al., 2015).
Age-related BBB changes are well documented by early studies, e.g. altered transport functions (Mooradian, 1988a, b), increased glycosylation of microvessel proteins (Mooradian and Meredith, 1992) and free radical damage (Mooradian and Smith, 1992), all of which may contribute to age-related changes in BBB permeability. Anatomically, there is reduced capillary density and cerebral blood flow in the aged brain, accompanied by ultrastructural abnormalities in microvessels, such as microvascular fibrosis, basement membrane thickening and loss of TJ proteins (Farkas and Luiten, 2001). Aged mice that are 24 months old have significantly less expression of occludin and, to a smaller degree, ZO-1, compared to young adult mice (Elahy et al., 2015). In addition, pericytes degenerate and are lost in aging brains, which may compromise BBB integrity and contribute to mild cognitive impairment during aging (Montagne et al., 2015).
Another important mechanism that may account for increased BBB permeability during aging is inflammation. Aged brains can have a low-grade but progressive inflammatory state, where the normal balance between pro- and anti-inflammatory mediators is shifted toward a pro-inflammatory state (Franceschi and Campisi, 2014). Inflammatory mediators, such as IL-1β, IFNγ and TNF-α, increase in brain during aging, with concomitant activation of microglia (Elahy et al., 2015; Kumagai et al., 2007). Despite the pro-inflammatory state, cell adhesion molecules ICAM-1 and VCAM-1 at the BBB are not upregulated in aged mice (Elahy et al., 2015). Consistently, no leukocyte transmigration is seen in the cortex and hippocampus of aged mice (Elahy et al., 2015). Studies on humans also show similar expression level of ICAM-1 in cortical blood vessels in aged and young brains (Miguel-Hidalgo et al., 2007).
5.4.2. Aging exacerbates ischemia/reperfusion-induced BBB disruption
Age is the most important non-modifiable risk factor for stroke. The AHA reports that the chance of having a stroke approximately doubles for each decade of life after age 55 (Mozaffarian et al., 2016). Profound changes occur in brain with aging which render the BBB more susceptible to ischemia/reperfusion injury, e.g. arterial remodeling, activation of glial cells, and apoptosis (Diaz-Otero et al., 2016; Roussel et al., 2009). In embolic MCAO models involving tPA-mediated reperfusion, BBB breakdown evaluated by plasma albumin extravasation occurs early and is increased nearly two-fold in 18-month-old aged compared to 3-month-old young rats (DiNapoli et al., 2008). This enhanced BBB breakdown precede and is associated with larger infarcts, reduced functional recovery and more severe neuronal injury (DiNapoli et al., 2008). TJ changes, i.e. the phosphorylation and disassembly of both claudin-5 and occludin, may underlie exacerbated BBB disruption in aged mice after ischemia/reperfusion (Kaur et al., 2011). However, a more recent ultrastructural analysis of the BBB using electron microscopy indicates that transcellular pathway by EC caveolae/vacuoles, rather than TJ protein loss, accounts for BBB changes in both young (3–4 months old) and aged (18 months old) mice within the first 72 hours after photothrombotic stroke (Nahirney et al., 2016).
To date, the majority of basic and preclinical studies on BBB dysfunction after stroke are based on young animals. Although certain common mechanisms involved in post-stroke BBB breakdown are shared by young and aged subjects, e.g. endoplasmic reticulum stress, ROS and glutamate excitotoxicity (Curcio et al., 2016; Lucke-Wold et al., 2014), treatment against BBB breakdown in aged brains may not be as efficient as in younger brains due to the confounding effects of aging. A recent study reported differential effects of erythropoietin, where ischemia-induced BBB breakdown and neuronal loss can be rescued by erythropoietin in young animals but not in aged littermates (Theriault et al., 2016). In summary, BBB hyperpermeability after stroke in aged subjects can be much earlier and more severe than in young subjects. Age is an important parameter in stroke pathogenesis and should be taken into consideration in future studies developing therapeutic strategies to protect the BBB after stroke.
5.5. Gender
Gender differences are well known in ischemic stroke and may impact the efficacy of stroke treatments (Ahnstedt et al., 2016). Clinically, women have a lower risk for stroke before menopause compared to men of similar age (Lisabeth and Bushnell, 2012). The risk is substantially elevated after menopause in women with usually poorer outcome than in men, coincident with subsiding circulating estrogen and progesterone levels (Wenger et al., 1993). Recovery of neurological functions in response to tPA therapy after ischemic stroke is also different between men and women (Kent et al., 2005). All these suggest that gender differences should be taken into consideration when investigating ischemic brain injury, including BBB dysfunction.
5.5.1. Gender-related changes of the BBB
Changes in BBB integrity in response to different stimuli vary among males and females as a result of the influence of reproductive hormones. Estrogen declines during aging in female mice with a concomitant increase of gonadotropins, which is associated with increased BBB permeability compared to young adult female mice (Bake and Sohrabji, 2004; Wilson et al., 2008). Upon LPS-induced transient inflammation, BBB integrity is compromised in adult young male mice but not in young females (Maggioli et al., 2016). This BBB protection in young females is likely mediated by estrogen, as similar BBB breakdown is observed in old, reproductively senescent females or ovariectomy-operated young females, and can be rescued by estradiol replacement (Maggioli et al., 2016).
TJ proteins and their regulators are believed to be major sites where estrogen affects BBB permeability. Thus, estradiol treatment increases TEER in cultured brain ECs and upregulates claudin-5 (Burek et al., 2010). Annexin A1, a central modulator of TJ integrity, is diminished in aged females and dramatically upregulated by estradiol, and may underlie the gender difference of BBB integrity after LPS-induced inflammation (Maggioli et al., 2016). Ovariectomy in 3-month-old female mice induces extravasation of Evans Blue into brain (Wilson et al., 2008). The expression and localization of microvascular ZO-1 is not altered by ovariectomy, but there is a redistribution of a gap junction protein connexin-43 at the endothelium (Wilson et al., 2008). Instead of reduced serum estrogen, elevated serum gonadotropins may account for these changes, as they are abolished by a gonadotropin-releasing hormone (GnRH) agonist leuprolide acetate (Wilson et al., 2008).
5.5.2. Gender differences in BBB permeability changes after stroke
BBB permeability differences after ischemic stroke between male and female is largely mediated by estrogen. Estrogen elicits a cascade of protective mechanisms in the NVU after ischemic insults, including cerebrovascular dilation and improved blood flow (Hurn et al., 1995; Mendelsohn and Karas, 1994), suppression of inflammation (Mori et al., 2004; Wen et al., 2004), and upregulation of cellular pro-survival mediators (Alkayed et al., 2001; Vagnerova et al., 2008; Xu et al., 2006), all of which may have beneficial effects on the BBB. In cultured brain ECs after OGD/reperfusion, estrogen improves mitochondrial efficiency, reduces free radical production and enhances cell survival (Guo et al., 2010). In animal models, estrogen treatment ameliorates ischemia-induced BBB disruption and edema formation through multifaceted actions (Liu et al., 2005; O’Donnell et al., 2006). Na+-K+-Cl− cotransporter activity in brain ECs is reduced by estradiol treatment before MCAO, leading to less Na+ and Cl− transport from blood to brain and subsequent edema formation (O’Donnell et al., 2006). Estradiol also inhibits the transcription and activity of MMPs and attenuates associated junctional protein degradation after ischemia (Liu et al., 2005; Na et al., 2015). The protective effects of estrogen are possibly via estrogen receptors (ERs), which include both classical ERs (ERα and ERβ) and non-classical ER (G protein-coupled estrogen receptor 1, GPER-1) (Schreihofer and Ma, 2013). ERα- and ERβ-specific agonists reduce TJ disruption in cultured brain ECs after OGD, but the role of GPER-1 in ischemia-induced BBB disruption remains unclear (Shin et al., 2016). In mouse MCAO, an ERβ-selective agonist reduced the expression of VEGF and its inducer HIF-1α, thereby alleviating VEGF-induced TJ disruption and BBB breakdown (Shin et al., 2016; van Bruggen et al., 1999; Zhang et al., 2000).
6. Blood-brain barrier recovery and repair
6.1. Time course of recovery
Multiple studies have examined the time course of BBB permeability after ischemic stroke in rodents (e.g. (Lin et al., 2008; Moisan et al., 2014; Strbian et al., 2008)). These have shown a peak in permeability in the acute/subacute phase of stroke (~1–7 days) followed by a gradual reduction. However, it should be noted that studies have still found BBB hyperpermeability 3–4 weeks after ischemia (Lin et al., 2008; Moisan et al., 2014; Strbian et al., 2008) indicating there can be long-term derangement in barrier function. Indeed in human stroke patients, there is evidence that there may be low level BBB dysfunction at one month (Liu et al., 2013). Such long-term dysfunction may lead to neuroinflammation which, in turn, may increase the propensity for stroke recurrence.
Longer-term studies on barrier function in vitro have focused on OGD with reoxygenation rather than OGD alone. In endothelial monocultures, such studies have generally shown rapid (hours) recovery of barrier function during the reoxygenation phase (Andjelkovic et al., 2003; Kuntz et al., 2014a). However, that recovery time course is impacted by co-culture with other elements of the NVU. Thus, Kuntz et al. found that endothelial/astrocyte co-cultures had enhanced barrier permeabilities at 24 hours after reoxygenation compared to endothelial cells where astrocytes were absent in the reoxygenation phase (Kuntz et al., 2014b). Dimitrijevic et al. also reported longer term (48 hours) barrier disruption after OGD + reoxygenation in endothelial/astrocyte co-cultures (Dimitrijevic et al., 2006). These results suggest that factors secreted by astrocytes can delay BBB recovery after OGD.
It should also be noted that inflammation plays a role in long-term BBB dysfunction after stroke in vivo (see Section 3.4). Thus, the general absence of microglia and leukocytes in in vitro models may alter (compress) the time course of recovery. In addition, in vivo, a number of co-morbidities (such as diabetes and hypertension) impact BBB dysfunction after stroke (see Section 5). The effects of such co-morbidities are difficult to model in vitro.
6.2. Mechanisms underlying BBB recovery after stroke
Multiple processes may be involved in the restoration of BBB permeability after stroke (Fig. 4). As described above, ischemic brain injury results in the production of many factors (e.g. ROS, cytokines, chemokines and VEGF-A) in the brain which can induce BBB hyperpermeability. For many factors, the production peaks in the acute/subacute phase after ischemia (Fagan et al., 2004) and falling levels during stroke recovery may help restore barrier function. For example, exposure to the chemokine CCL2 induces hyperpermeability in brain endothelial monolayers by causing the internalization of TJ proteins from the cell membrane. Removal or neutralization of CCL2 results in the return of those junction proteins to the cell membrane and normalization of permeability (Stamatovic et al., 2009).
Fig. 4. Restoration of BBB permeability after stroke.
In response to ischemic stroke, there is the release of multiple factors that enhance BBB permeability, e.g. ROS, inflammatory mediators and pro-angiogenic factors such as VEGF. Some of the restoration in barrier tightness with time after a stroke is related to reduced levels of those factors, but there is also an upregulation of mediators of barrier quiescence that enhance barrier properties, such as Ang-1. Those mediators can also serve (directly or indirectly) to diminish the effects of the factors enhancing barrier permeability (red lines). Progenitor cells may be directly (integration) or indirectly (mediator release) involved in barrier repair after stroke.
A crucial control point that regulates barrier breakdown versus barrier recovery after TJ protein internalization is at the level of the early endosomes. There proteins may be sorted towards late endosomes and degradation or towards recycling endosomes and return to the plasma membrane with barrier restoration (Stamatovic et al., 2017). There is still relatively little known about how these processes are controlled at the BBB and such regulation may be particularly important in the response to transient ischemia (long-term barrier disruption or rapid recovery). Studies on a variety of barrier tissues do, however, pinpoint three crucial types of regulation: a) proteins involved in membrane trafficking, particularly the Rab family of small GTPases; b) cell signaling pathways; c) sorting signals within the TJ protein itself (Stamatovic et al., 2017). Understanding this regulation and redirecting TJ proteins to the plasma membrane may be a fruitful line of research for promoting barrier restoration. BBB damage can upregulate angiogenic growth factors in an attempt to salvage and repair the ischemic penumbra (Beck and Plate, 2009). ECs are the main targets in angiogenesis, a process regulated by several signaling pathways. Those include activation of the VEGFR-2 receptor and its downstream effectors Ras/Raf/MEK, the PI3K-AKT/PKB pathway and the p38/MAPK-HSP27 pathway. These pathways promote EC proliferation, survival and migration (Suzuki et al., 2016). Increased production of endothelial nitric oxide synthase (eNOS) accompanies the increase in VEGF expression and benefits angiogenesis (Chen et al., 2005). The increased eNOS activity causes a marked release of NO, inducing vessel dilation and fostering vessel remodeling (Lapi et al., 2013; Veltkamp et al., 2002). Activation of EC integrin matrix receptors, such as αvβ3, also plays an important and therapeutically significant role in angiogenesis after ischemic brain injury (Abumiya et al., 1999; Guell and Bix, 2014).
As well as brain-derived factors that induce barrier hyperpermeability, there are factors that restore/stabilize BBB permeability, such as angiopoietin-1 (Ang-1), sphingosine-1-phosphate and activated protein C (Rodrigues and Granger, 2015; Siddiqui et al., 2015). Ang-1 has a delayed increase in expression after stroke, and this upregulation may contribute to barrier repair (Moisan et al., 2014). Exogenous Ang-1 can reduce the acute BBB disruption linked to tPA-induced reperfusion (Kawamura et al., 2014), but the effects of such a treatment on normal BBB restoration after stroke has not been directly examined. While Ang-1 maintains endothelial cells in a quiescent state, promoting cell-cell and cell-extracellular matrix interactions, another angiopoietin, Ang-2, destabilizes vascular endothelial cells and promotes angiogenesis (Rubio and Adamis, 2016). Ang-1 and -2 have generally antagonistic actions at the Tie-2 receptor (Rubio and Adamis, 2016) and the relative expression of these angiopoietins impacts BBB permeability (Moisan et al., 2014).
Two processes that may also contribute to the restoration of BBB permeability after stroke are degeneration of damaged leaky vessels (Yoshida et al., 1989) and the formation of new vessels (angiogenesis) (Prakash and Carmichael, 2015). Such vascular remodeling lasts for up to three weeks after ischemia and it is associated with improved outcome following stroke (Lapi and Colantuoni, 2015). The effect of angiogenesis long-term is to increase blood flow to the ischemic brain which will aid in reducing brain-derived factors that enhance BBB permeability. Activated Shh signaling in astrocytes can also strengthen the BBB by upregulating TJ proteins, decreasing the expression of pro-inflammatory mediators and reducing leukocyte adhesion and migration (Alvarez et al., 2011; Wang et al., 2014). However, factors that promote angiogenesis, such as VEGF-A and Ang-2, can also cause barrier hyperpermeability (Nag et al., 2011; Rubio and Adamis, 2016).
Another process that may contribute to BBB structural repair after stroke is the integration of progenitor cells into the damaged cerebrovasculature. Such cells can home to sites of injury and, as well as directly integrating into the endothelium and surrounding tissue, they secrete factors that promote angiogenesis and barrier repair (Pu et al., 2016; Tenreiro et al., 2016). Administration of human umbilical cord blood cells to diabetic rats after MCAO decreased BBB hyperpermeability, promoted vascular remodeling and reduced brain injury and functional deficits, effects which were at least in part mediated by Ang-1 (Yan et al., 2014).
Recent studies also highlight the role of microglia/macrophages in vessel repair after injury. Using in vivo time-lapse imaging, Liu et al. examined vascular repair in a zebrafish cerebrovascular rupture model with laser-induced lesions with two endothelial ends (Liu et al., 2016a). Macrophages migrated to the lesion site, extended filopodia/lamellipodia that attached to the endothelium, and pulled and ligated the endothelial ends together, thereby repairing the rupture (Liu et al., 2016a). While the majority of the repairing macrophages were resident microglia, peripheral macrophages also participated in this process. Similarly in a mouse laser model of capillary injury, local BBB opening was repaired by juxtavascular microglia through P2RY12 activity [purinergic receptor P2Y, G-protein coupled, 12] (Lou et al., 2016). Information on the phenotype of these repairing microglia and whether such repair occurs in more widespread injuries (e.g. MCAO) remains lacking and warrants further investigation.
6.3. Therapeutic approaches to enhance recovery
One potential approach to enhance BBB repair after stroke is to reduce the signals (such as inflammatory mediators) that induce hyperpermeability. In this regard, the agents that have been most extensively examined for their potential to tighten the BBB are the glucocorticoids. In brain tumors, dexamethasone is a mainstay for the treatment of edema due to reductions in BBB permeability that are probably linked to effects on inflammation and TJ protein expression (Kotsarini et al., 2010). While there is some preclinical evidence that dexamethasone can reduce BBB permeability and brain edema after stroke (Betz and Coester, 1990), dexamethasone has undergone multiple clinical trials in both ischemic stroke and intracerebral hemorrhage without evidence of benefit on patient outcome (Poungvarin, 2004). This lack of benefit maybe due to the side-effects of dexamethasone but it may also reflect degradation of the glucocorticoid receptor limiting the effects of the glucocorticoid after stroke. Kleinschnitz et al. found that co-administration of a proteasome inhibitor, Bortezomib, with dexamethasone resulted in reduced BBB permeability and brain edema (Kleinschnitz et al., 2011).
The use of steroids is not the only potential method to limit the inflammatory cascade induced by stroke. However, none of the anti-inflammatory approaches that have been examined clinically so far have improved outcome in stroke patients (Petrovic-Djergovic et al., 2016). While there are many potential reasons for this, it should be noted that inflammation can have beneficial as well as detrimental effects after stroke.
An alternate approach to accelerate BBB recovery after stroke may be to deliver, or enhance the expression of proteins that restore/stabilize BBB permeability, such as Ang-1. One potential problem with this approach is that Ang-1 may act via abluminal Tie-2 receptors requiring delivery of Ang-1 across the BBB.
One potential way of obviating the need to deliver agents, such as Ang-1, to the endothelial abluminal membrane is to target downstream signaling within the endothelium. For example, phosphorylation of TJ proteins plays an important role in increased BBB permeability after stroke and neuroinflammation (see Section 3.2.1). Current studies have generally focused on acute treatment after stroke (e.g. (Takenaga et al., 2009; Willis et al., 2010)) and whether inhibiting such TJ modification can accelerate BBB recovery after stroke merits more investigation. One mediator of TJ phosphorylation are the Rho kinases (Stamatovic et al., 2006) and a Rho kinase inhibitor, fasudil, inhibits BBB disruption after MCAO in mice (Gibson et al., 2014). Interestingly, fasudil has been used to improve long-term barrier characteristics in the setting of cerebral cavernous malformations (McDonald et al., 2012; Stockton et al., 2010).
Another approach would be to try and modify the TJ protein expression directly. Tian et al. have overexpressed claudin-5 in retinal endothelial cells in culture using lentivirus and enhanced barrier properties (Tian et al., 2014). Whether such an approach could be used in vivo to accelerate BBB repair after stroke is, as yet, uncertain.
As noted above, there has also been interest in using progenitor cells to promote both angiogenesis and barrier repair after stroke (Pena and Borlongan, 2015; Tenreiro et al., 2016; Yan et al., 2014). The pluripotent effects of such cells and the fact that they may be replicating/enhancing an endogenous response are advantages with such an approach. If these cells do integrate into the endothelium long-term, the extent to which they fully replicate the endothelium of the mature BBB should be investigated.
In summary, studies have begun to delineate important pathways regulating BBB restoration following stroke. However, several factors complicate manipulating those pathways to improve outcome. There is a need to balance barrier restoration and angiogenesis. In addition, many of the pathways have beneficial as well as detrimental aspects after stroke. Therapeutic approaches will likely have to vary with time and stroke severity.
7. Novel tools for blood-brain barrier research
The pathogenesis of BBB dysfunction and therapeutic strategies targeting the BBB have received increasing attention in stroke research, and emerging novel research tools may greatly facilitate such investigations. Current advanced imaging techniques enable non-invasive, real-time quantitative and functional assessments of the BBB with improved spatial resolution. More accurate spatial and temporal gene manipulation can be achieved in vivo using various genetic mouse models, while in vitro model systems are being refined to better reflect the in vivo BBB interface. Certain tools have already been employed in stroke BBB research, whereas others may launch a new era of BBB research with their application.
7.1. Advanced imaging techniques
Non-invasive molecular imaging modalities, such as MRI and positron emission tomography (PET), are widely used in animal-based studies as well as clinical applications to assess BBB structure and function. Changes in BBB integrity can be identified at an early stage after ischemic stroke using these techniques, which serve as not only a diagnostic indicator but also a predictor of later outcome (e.g. hemorrhagic transformation), thereby instructing the thrombolytic therapies (Kassner et al., 2009; Latour et al., 2004). The basis of imaging BBB integrity is to measure the brain uptake of a contrast agent or tracer that does not pass the BBB under physiological conditions. A commonly used MRI approach is T1-weighted imaging to follow the extravasation of paramagnetic gadolinium-based contrast agents across the impaired BBB (Grillon et al., 2008; Janowski et al., 2016; Knight et al., 2009; Song et al., 2016). High-resolution dynamic contrast-enhanced MRI further allows more sensitive and quantitative detection of BBB opening (Huang et al., 2016b). Researchers are now able to identify BBB hyperpermeability in different hippocampal subregions associated with normal human aging (Montagne et al., 2015). PET determines BBB permeability by the tissue uptake of tracers radiolabeled with short-lived positron emitting isotopes (Okada et al., 2015a), featuring higher sensitivity but lower spatial resolution compared to MRI (Venneti et al., 2013). Using appropriate substrates, PET can also non-invasively measure the function of BBB P-gp in patients (Bauer et al., 2015; Syvanen and Eriksson, 2013; Toyohara, 2016).
In animals, real-time in vivo imaging can be acquired by two-photon microscopy, which captures time-dependent vascular activities and dynamic cell-cell interactions that occur at the neurovascular network. Application of two-photon microscopy at the BBB interface is usually based on fluorescent labeling of brain microvessels by intravascular fluorescent tracers injected into the vascular lumen. Depending on their molecular weight, fluorescent tracers can either be confined within the vascular lumen and outline the vessels, or leak into the brain parenchyma with BBB disruption (Verant et al., 2008). Alternatively, brain microvessels can be imaged in transgenic mice expressing EC-specific reporters. Two commonly used strains being the Tie2-GFP mice (Harb et al., 2013) and eGFP-Claudin5 mice (Knowland et al., 2014).
Other recent methodologies for imaging the brain neurovascular network remain to be fully applied to BBB research. Clear Lipid-exchanged Acrylamide-hybridized Rigid Imaging compatible Tissue hYdrogel (CLARITY) is a newly developed method which converts large volumes of intact biological tissues into a hybrid form in which tissue components are replaced with exogenous elements for improved chemical and optical accessibility (Chung et al., 2013). The cleared brain facilitates fluorescent visualization, which, combined with endogenous fluorescent protein or immunostaining, extracts high-resolution three-dimensional architecture of the vascular network from millimeter-thick tissue blocks (Phillips et al., 2016). Future application of this method could shed light on the abnormalities of brain microvasculature associated with stroke or its comorbid conditions.
Another recently introduced approach, optogenetics, allows temporally precise manipulation of defined cell populations in the neurovascular network in vivo. Recent studies have successfully manipulated EC function in mice expressing channelrhodopsin in ECs, where optical stimulation of heart and kidney ECs produced fast, reproducible and long-lasting vasoconstriction (Zhang et al., 2015b). Photoacoustic imaging (PAI) is another novel non-invasive imaging technique which features supreme optical absorption and ultrasound spatial resolution and holds the potential for real-time biomedical imaging (Zhang et al., 2006). PAI can achieve several micrometers spatial resolution, and the maximum imaging depth is 500–600 μm from the surface of an intact skull, slightly less than the typical imaging depth of two-photon microscopy (600–800 μm) through a skull window (Hu et al., 2009). In vivo volumetric imaging of brain microvascular morphology down to single capillaries can be done using PAI (Hu et al., 2009). With different kinds of gold nanoparticles as contrast agents, PAI also provides a unique opportunity to detect the contrast enhancement caused by BBB hyperpermeability (Wang et al., 2012).
7.2. Genetically engineered mouse models
The availability of various genetic mouse models has greatly facilitated understanding of developmental, physiological and pathophysiological BBB regulation. In earlier studies, conventional genetic mouse models were widely used for gene silencing and revealed the participation of different mechanisms in ischemia-induced BBB dysfunction. Examples include the role of TJ proteins (Nitta et al., 2003; Saitou et al., 2000), MMPs (Asahi et al., 2000), transcytosis (Drab et al., 2001; Knowland et al., 2014), EC-leukocyte interactions (Jin et al., 2010) and inflammation (Strecker et al., 2013). However, conventional gene mutation can lead to embryonic lethality or infertility. It lacks temporal and cell type-specific control, which is improved in models based on Cre-Lox recombination (Nagy, 2000). Often used Cre mouse strains with EC-specific promoters include Tie2-Cre, Tie1-Cre, VE-cadherin-Cre (Alva et al., 2006; Gustafsson et al., 2001; Kisanuki et al., 2001). The main differences among these strains are in the expression time of Cre during embryonic development, as well as the targeted cell populations besides ECs. A commonly encountered problem with these strains is the “leaky” expression of Cre in cells of hematopoietic lineage (Gustafsson et al., 2001; Kisanuki et al., 2001); caution should be taken when drawing conclusions about EC-specific mechanisms, where an observed phenomenon could result from the irreversible deletion of floxed alleles in hematopoietic precursors at embryonic stages. This problem is partially resolved with the development of CreERT2 strains, e.g. Tie2-CreERT2 (Forde et al., 2002) and VE-cadherin-CreERT2 (Monvoisin et al., 2006), in which Cre is fused to a modified estrogen receptor and only targeted to the nucleus upon injection of tamoxifen (Feil et al., 1997). Another Slco1c1-CreERT2 strain, using the locus of solute carrier organic anion transporter 1c1 (Slco1c1, SLC21A14) as the CreERT2 insertion site, was reported to selectively target brain ECs and choroid plexus epithelial cells but not ECs in peripheral organs (Ridder et al., 2011). One caveat remains with the CreERT2 system, however, that the efficiency of Cre recombination may be compromised when tamoxifen is given during adulthood compared to embryonic or neonatal mice (Monvoisin et al., 2006). There are also various reporter lines engineered to express endogenous fluorescent proteins under the control of EC-specific promoters, which allows imaging in live animals and fixed tissues (see Section 7.1). For a detailed inventory of Cre and reporter strains targeting ECs or other cell types of the NVU, see recent reviews (Hartmann et al., 2015; Sohet and Daneman, 2013).
7.3. In vitro model systems
In vitro BBB model systems are being improved to better reflect the BBB interface, taking into consideration the crosstalk among various types of cells in the NVU and the impact of shear stress on the BBB. Early in vitro BBB studies largely relied on simple models, such as isolated brain microvessels (Joo, 1985) or culture of brain-derived ECs (Deli et al., 2005). Transwell systems allowed coculture of ECs and other types of cells in the NVU (e.g. astrocytes and pericytes), as well as transmigration of peripheral cells (Hayashi et al., 2004; Hurwitz et al., 1993). However, these two-dimensional BBB models could not supply the three-dimensional cell-cell interactions needed for appropriate EC differentiation, including polarization and appropriate transporters expression (Hopkins et al., 2015; Lyck et al., 2009; Worzfeld and Schwaninger, 2016). This drawback is partially improved by the more recently devised three-dimensional models based on matrigel (Davis et al., 2007) or spheroids (Urich et al., 2013), with or without the support of ECM scaffolding, respectively. Other microfluidic systems allow cells to be perfusion-cultured, with the support of three-dimensional cell-cell and cell-matrix interactions and influence of shear stress (e.g. (Toh et al., 2007)).
Failures to translate promising animal therapeutic studies to successful clinical trials have raised questions over potential species differences. It is, therefore, important that human in vitro BBB models be developed. Recently, Shusta and colleagues have used human induced pluripotent stem cells (iPSCs) to generate brain endothelial cells and other NVU cells (Li et al., 2015b; Lippmann et al., 2014; Lippmann et al., 2012). They have been used to create humanized in vitro BBB models with TEERs close to those in vivo (Lippmann et al., 2014). The use of human iPSCs may also allow generation of patient-specific BBB models (e.g. to examine how genetic mutations alter the response to ischemic conditions).
Advances in in vitro modeling are expected to reproduce many of the key BBB properties, such as polarized ECs with luminal and abluminal transport systems, which exert functional efflux, metabolic and catalytic mechanisms as well as enhanced barrier tightness (Helms et al., 2016; Naik and Cucullo, 2012; Ruck et al., 2015). Such models will facilitate mechanistic understanding of how ischemia-like conditions impact cerebral ECs, interactions within the NVU and EC-leukocyte interactions.
8. Future perspectives and translation
Over the past decade, there has been marked increase in our understanding of normal BBB and NVU functions and how those are impacted by stroke. There remain, however, substantial facets of BBB endothelial biology that are poorly understood. For example, the dynamic behavior of TJ proteins has been intensively investigated in epithelial and endothelial cells of peripheral organs (Stamatovic et al., 2017). The internalization of TJ proteins from the cell membrane, and subsequent trafficking, recycling and degradation of these proteins, represent important regulation of TJ plasticity and barrier properties (Stamatovic et al., 2017). Cell-specific proteins and signaling pathways involved in such processes, and how these influence EC and barrier responses should be considered in future BBB studies, which may provide insights allowing modulation of BBB permeability and facilitation of drug delivery to the CNS.
Multiple potential therapeutic targets have been identified from the present BBB research, the ultimate goal of which is translation to the clinic. Many agents have shown preclinical therapeutic efficacy in protecting against ischemic stroke, including BBB protection, yet none of them has been successfully translated to clinical use. Multiple factors may contribute to this failure, including limitations in preclinical stroke models. The most widely used animal model, MCAO, does not cover all types of clinical ischemic stroke. Furthermore, only a small portion of studies employ post-ischemic reperfusion and the use of tPA. Suitable stroke models that mimic the cellular and molecular mechanisms of thrombosis and thrombolysis are warranted in future research. The use of the embolic cerebral ischemia model, using a fibrin-rich allogeneic clot to occlude the MCA followed by tPA thrombolysis (Zhang et al., 2015a), may help fill this gap. However, there remain pertinent disadvantages with this model. Intravascular introduction of emboli can result in multifocal ischemia with significant variability in infarct size and location because dried blood clots do not adhere to the blood vessel. Moreover, the thrombosis and aging of the thrombus in vitro differ from in vivo, which may affect the time window for thrombolysis (Ma et al., 2016).
Another reason that may underlie the failure to translate preclinical results to the clinic relates to stroke risk factors and comorbid conditions. These may either lessen therapeutic effects or provide new therapeutic opportunities. For example, VEGF-R2 inhibition appears to provide stroke recovery in type I diabetic mice but fails to protect nondiabetic mice (Reeson et al., 2015). These data stress the importance of developing personalized therapies for patients with different co-morbidities.
Future preclinical research should also take into consideration the multiple cell types in the NVU and the influence of peripheral systems on the NVU. For example, the gut microbiota has a profound impact on many biological functions in the body, such as metabolism and immune responses (Hooper et al., 2012), but also brain development and function (Diaz Heijtz et al., 2011) and the outcome of ischemic stroke (Benakis et al., 2016; Winek et al., 2016). Furthermore, Braniste et al. recently found that germ-free mice free display increased BBB permeability compared to mice with normal gut flora, beginning with intrauterine life and maintained until adulthood with reduced expression of occludin and claudin-5 (Braniste et al., 2014). Although there is no direct evidence of gut microbiota regulating BBB after ischemic stroke, studies on gut-brain communication may provide strategies for developing new therapeutics.
Finally, it is reasonable to propose that strategies that can promote BBB restoration and repair after ischemic stroke will improve the functional recovery and patient outcome. Long-term endothelial and BBB repair can be achieved through angiogenesis and division of ECs. The potential of other cells to facilitate vessel repair (e.g. microglia/macrophages (Liu et al., 2016a)) warrants further study.
Highlights.
Mechanisms of blood-brain barrier dysfunction after ischemic stroke
Regulation of blood-brain barrier integrity by cells in the neurovascular unit
Stroke comorbidities and blood-brain barrier dysfunction
Blood-brain barrier recovery and repair after ischemic stroke
Advanced tools to study the blood-brain barrier
Acknowledgments
This work was supported by the U.S. National Institutes of Health grants NS089534, NS045048, NS056118 (to J.C.), NS036736, NS095029 (to M.V.L.B. and J.C.), NS098066 (to A.V.A.), NS093399 (to R.F.K.), the U.S. Department of Veterans Affairs (VA) Merit Review award BX002495 (to J.C.), and the Chinese Natural Science Foundation grants 81529002 (to J.C.). J.C. is a recipient of the VA Senior Research Career Scientist Award, the Richard King Mellon Endowed Professorship, and the University of Pittsburgh Medical Center (UPMC) Endowed Professorship. M.V.L.B is the Sylvia and Robert S. Olnick Professor of Neuroscience and a recipient of the High-end Distinguished Professorship GDW20133100069 from the State Administration of Foreign Experts Affairs, China. Y.S. is supported by the American Heart Association grant 15POST22260011. A.V.A. is supported by the American Diabetes Association grant 1-16-IBS-008.
Abbreviations
- ABC
ATP-binding cassette
- AJ
adherens junction
- Ang
angiopoietin
- ApoE
apolipoprotein E
- AQP4
aquaporin 4
- BBB
blood-brain barrier
- cAMP
cyclic AMP
- CSF
cerebrospinal fluid
- EAAT
excitatory amino acid transporter
- EC
endothelial cell
- ECF
extracellular fluid
- ECM
extracellular matrix
- eNOS
endothelial nitric oxide synthase
- ER
estrogen receptors
- GDNF
glial cell-derived neurotrophic factor
- GLUT1
glucose transporter isoform 1
- GPER-1
G protein- coupled estrogen receptor 1
- HFD
high fat diet
- HIF
hypoxia-inducible factor
- HMGB1
high-mobility group box1
- ICAM
intercellular adhesion molecule
- IL
interleukin
- iPSC
induced pluripotent stem cell
- JAM
junctional adhesion molecule
- LAT
L-type amino acid transporter
- LDL
low-density lipoprotein
- L-NAME
Nω-Nitro-L-arginine methyl ester
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- MCAO
middle cerebral artery occlusion MCAO
- MCP1
monocyte chemoattractant protein 1
- MIF
Macrophage migration inhibitory factor
- MMP
metalloproteinase
- MRI
magnetic resonance imaging
- NO
nitric oxide
- NOS
nitric oxide synthase
- NVU
neurovascular unit
- OGD
oxygen glucose deprivation
- PAI
photoacoustic imaging
- PDGFR
platelet-derived growth factor receptor
- PECAM-1
platelet endothelial cell adhesion molecule 1
- PET
positron emission tomography
- P-gp
P-glycoprotein
- PI3K
phosphatidylinositide 3-kinase
- PKC
protein kinase C
- ROCK
Rho-associated protein kinase
- ROS
reactive oxygen species
- shh
Sonic hedgehog
- SHR
spontaneously hypertensive rat
- SHRSP
stroke-prone spontaneously hypertensive rat
- SOD
superoxide dismutase
- TEER
transendothelial electrical resistance
- TGFβ
Transforming growth factor beta
- TJ
tight junction
- TNF
tumor necrosis factor
- tPA
tissue plasminogen activator
- Treg
regulatory T-cells
- VCAM
vascular cell adhesion protein
- VEGF
vascular endothelial growth factor
- WKY
Wistar Kyoto
- ZO
zonula occludens
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
None.
References
- Abbott NJ. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell Mol Neurobiol. 2000;20:131–147. doi: 10.1023/A:1007074420772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Abumiya T, Lucero J, Heo JH, Tagaya M, Koziol JA, Copeland BR, del Zoppo GJ. Activated microvessels express vascular endothelial growth factor and integrin alpha(v)beta3 during focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:1038–1050. doi: 10.1097/00004647-199909000-00012. [DOI] [PubMed] [Google Scholar]
- Aggarwal A, Khera A, Singh I, Sandhir R. S-nitrosoglutathione prevents blood-brain barrier disruption associated with increased matrix metalloproteinase-9 activity in experimental diabetes. J Neurochem. 2015;132:595–608. doi: 10.1111/jnc.12939. [DOI] [PubMed] [Google Scholar]
- Agundez JA, Jimenez-Jimenez FJ, Alonso-Navarro H, Garcia-Martin E. Drug and xenobiotic biotransformation in the blood-brain barrier: a neglected issue. Front Cell Neurosci. 2014;8:335. doi: 10.3389/fncel.2014.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahnstedt H, McCullough LD, Cipolla MJ. The Importance of Considering Sex Differences in Translational Stroke Research. Transl Stroke Res. 2016;7:261–273. doi: 10.1007/s12975-016-0450-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkayed NJ, Goto S, Sugo N, Joh HD, Klaus J, Crain BJ, Bernard O, Traystman RJ, Hurn PD. Estrogen and Bcl-2: gene induction and effect of transgene in experimental stroke. J Neurosci. 2001;21:7543–7550. doi: 10.1523/JNEUROSCI.21-19-07543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen C, Srivastava K, Bayraktutan U. Small GTPase RhoA and its effector rho kinase mediate oxygen glucose deprivation-evoked in vitro cerebral barrier dysfunction. Stroke. 2010;41:2056–2063. doi: 10.1161/STROKEAHA.109.574939. [DOI] [PubMed] [Google Scholar]
- Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, Carmeliet P, Iruela-Arispe ML. VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn. 2006;235:759–767. doi: 10.1002/dvdy.20643. [DOI] [PubMed] [Google Scholar]
- Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, Sabbagh M, Wosik K, Bourbonniere L, Bernard M, van Horssen J, de Vries HE, Charron F, Prat A. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011;334:1727–1731. doi: 10.1126/science.1206936. [DOI] [PubMed] [Google Scholar]
- Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia. 2013;61:1939–1958. doi: 10.1002/glia.22575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano S, Yamagishi S, Inagaki Y, Nakamura K, Takeuchi M, Inoue H, Imaizumi T. Pigment epithelium-derived factor inhibits oxidative stress-induced apoptosis and dysfunction of cultured retinal pericytes. Microvasc Res. 2005;69:45–55. doi: 10.1016/j.mvr.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Andjelkovic AV, Stamatovic SM, Keep RF. The protective effects of preconditioning on cerebral endothelial cells in vitro. J Cereb Blood Flow Metab. 2003;23:1348–1355. doi: 10.1097/01.WCB.0000091762.61714.FE. [DOI] [PubMed] [Google Scholar]
- Andras IE, Deli MA, Veszelka S, Hayashi K, Hennig B, Toborek M. The NMDA and AMPA/KA receptors are involved in glutamate-induced alterations of occludin expression and phosphorylation in brain endothelial cells. J Cereb Blood Flow Metab. 2007;27:1431–1443. doi: 10.1038/sj.jcbfm.9600445. [DOI] [PubMed] [Google Scholar]
- Andriopoulou P, Navarro P, Zanetti A, Lampugnani MG, Dejana E. Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler Thromb Vasc Biol. 1999;19:2286–2297. doi: 10.1161/01.atv.19.10.2286. [DOI] [PubMed] [Google Scholar]
- Antonetti DA, Barber AJ, Hollinger LA, Wolpert EB, Gardner TW. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J Biol Chem. 1999;274:23463–23467. doi: 10.1074/jbc.274.33.23463. [DOI] [PubMed] [Google Scholar]
- Arbo BD, Bennetti F, Ribeiro MF. Astrocytes as a target for neuroprotection: Modulation by progesterone and dehydroepiandrosterone. Prog Neurobiol. 2016;144:27–47. doi: 10.1016/j.pneurobio.2016.03.010. [DOI] [PubMed] [Google Scholar]
- Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Arnold T, Betsholtz C. The importance of microglia in the development of the vasculature in the central nervous system. Vasc Cell. 2013;5:4. doi: 10.1186/2045-824X-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20:1681–1689. doi: 10.1097/00004647-200012000-00007. [DOI] [PubMed] [Google Scholar]
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslam M, Ahmad N, Srivastava R, Hemmer B. TNF-alpha induced NFkappaB signaling and p65 (RelA) overexpression repress Cldn5 promoter in mouse brain endothelial cells. Cytokine. 2012;57:269–275. doi: 10.1016/j.cyto.2011.10.016. [DOI] [PubMed] [Google Scholar]
- Attwell D, Mishra A, Hall CN, O’Farrell FM, Dalkara T. What is a pericyte? J Cereb Blood Flow Metab. 2016;36:451–455. doi: 10.1177/0271678X15610340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurrand-Lions M, Johnson-Leger C, Wong C, Du Pasquier L, Imhof BA. Heterogeneity of endothelial junctions is reflected by differential expression and specific subcellular localization of the three JAM family members. Blood. 2001;98:3699–3707. doi: 10.1182/blood.v98.13.3699. [DOI] [PubMed] [Google Scholar]
- Ayata C, Shin HK, Dilekoz E, Atochin DN, Kashiwagi S, Eikermann-Haerter K, Huang PL. Hyperlipidemia disrupts cerebrovascular reflexes and worsens ischemic perfusion defect. J Cereb Blood Flow Metab. 2013;33:954–962. doi: 10.1038/jcbfm.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird TA, Parsons MW, Phan T, Butcher KS, Desmond PM, Tress BM, Colman PG, Chambers BR, Davis SM. Persistent poststroke hyperglycemia is independently associated with infarct expansion and worse clinical outcome. Stroke. 2003;34:2208–2214. doi: 10.1161/01.STR.0000085087.41330.FF. [DOI] [PubMed] [Google Scholar]
- Bake S, Sohrabji F. 17beta-estradiol differentially regulates blood-brain barrier permeability in young and aging female rats. Endocrinology. 2004;145:5471–5475. doi: 10.1210/en.2004-0984. [DOI] [PubMed] [Google Scholar]
- Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Barreto GE. Targeting astrocytes in brain injuries: A translational research approach. Prog Neurobiol. 2016;144:1–4. doi: 10.1016/j.pneurobio.2016.09.001. [DOI] [PubMed] [Google Scholar]
- Basuroy S, Bhattacharya S, Tcheranova D, Qu Y, Regan RF, Leffler CW, Parfenova H. HO-2 provides endogenous protection against oxidative stress and apoptosis caused by TNF-alpha in cerebral vascular endothelial cells. Am J Physiol Cell Physiol. 2006;291:C897–908. doi: 10.1152/ajpcell.00032.2006. [DOI] [PubMed] [Google Scholar]
- Bauer M, Karch R, Zeitlinger M, Philippe C, Romermann K, Stanek J, Maier-Salamon A, Wadsak W, Jager W, Hacker M, Muller M, Langer O. Approaching complete inhibition of P-glycoprotein at the human blood-brain barrier: an (R)-[11C]verapamil PET study. J Cereb Blood Flow Metab. 2015;35:743–746. doi: 10.1038/jcbfm.2015.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H, Plate KH. Angiogenesis after cerebral ischemia. Acta Neuropathol. 2009;117:481–496. doi: 10.1007/s00401-009-0483-6. [DOI] [PubMed] [Google Scholar]
- Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb Haemost. 2010;103:40–55. doi: 10.1160/TH09-06-0403. [DOI] [PubMed] [Google Scholar]
- Bell GI, Burant CF, Takeda J, Gould GW. Structure and function of mammalian facilitative sugar transporters. J Biol Chem. 1993;268:19161–19164. [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, Sita G, Racchumi G, Ling L, Pamer EG, Iadecola C, Anrather J. Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat Med. 2016;22:516–523. doi: 10.1038/nm.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz AL, Coester HC. Effect of steroids on edema and sodium uptake of the brain during focal ischemia in rats. Stroke. 1990;21:1199–1204. doi: 10.1161/01.str.21.8.1199. [DOI] [PubMed] [Google Scholar]
- Betz AL, Firth JA, Goldstein GW. Polarity of the blood-brain barrier: distribution of enzymes between the luminal and antiluminal membranes of brain capillary endothelial cells. Brain Res. 1980;192:17–28. doi: 10.1016/0006-8993(80)91004-5. [DOI] [PubMed] [Google Scholar]
- Borlongan CV, Emerich DF. Facilitation of drug entry into the CNS via transient permeation of blood brain barrier: laboratory and preliminary clinical evidence from bradykinin receptor agonist, Cereport. Brain Res Bull. 2003;60:297–306. doi: 10.1016/s0361-9230(03)00043-1. [DOI] [PubMed] [Google Scholar]
- Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Toth M, Korecka A, Bakocevic N, Ng LG, Kundu P, Gulyas B, Halldin C, Hultenby K, Nilsson H, Hebert H, Volpe BT, Diamond B, Pettersson S. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med. 2014;6:263ra158. doi: 10.1126/scitranslmed.3009759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RC, Davis TP. Calcium modulation of adherens and tight junction function: a potential mechanism for blood-brain barrier disruption after stroke. Stroke. 2002;33:1706–1711. doi: 10.1161/01.str.0000016405.06729.83. [DOI] [PubMed] [Google Scholar]
- Bruno A, Biller J, Adams HP, Jr, Clarke WR, Woolson RF, Williams LS, Hansen MD. Acute blood glucose level and outcome from ischemic stroke. Trial of ORG 10172 in Acute Stroke Treatment (TOAST) Investigators. Neurology. 1999;52:280–284. doi: 10.1212/wnl.52.2.280. [DOI] [PubMed] [Google Scholar]
- Burek M, Arias-Loza PA, Roewer N, Forster CY. Claudin-5 as a novel estrogen target in vascular endothelium. Arterioscler Thromb Vasc Biol. 2010;30:298–304. doi: 10.1161/ATVBAHA.109.197582. [DOI] [PubMed] [Google Scholar]
- Burridge K, Wittchen ES. The tension mounts: stress fibers as force-generating mechanotransducers. J Cell Biol. 2013;200:9–19. doi: 10.1083/jcb.201210090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos-Bedolla P, Walter FR, Veszelka S, Deli MA. Role of the blood-brain barrier in the nutrition of the central nervous system. Arch Med Res. 2014;45:610–638. doi: 10.1016/j.arcmed.2014.11.018. [DOI] [PubMed] [Google Scholar]
- Cao XL, Du J, Zhang Y, Yan JT, Hu XM. Hyperlipidemia exacerbates cerebral injury through oxidative stress, inflammation and neuronal apoptosis in MCAO/reperfusion rats. Exp Brain Res. 2015;233:2753–2765. doi: 10.1007/s00221-015-4269-x. [DOI] [PubMed] [Google Scholar]
- Cederberg HH, Uhd NC, Brodin B. Glutamate efflux at the blood-brain barrier: cellular mechanisms and potential clinical relevance. Arch Med Res. 2014;45:639–645. doi: 10.1016/j.arcmed.2014.11.004. [DOI] [PubMed] [Google Scholar]
- Chao AC, Lee TC, Juo SH, Yang DI. Hyperglycemia Increases the Production of Amyloid Beta-Peptide Leading to Decreased Endothelial Tight Junction. CNS Neurosci Ther. 2016;22:291–297. doi: 10.1111/cns.12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zacharek A, Zhang C, Jiang H, Li Y, Roberts C, Lu M, Kapke A, Chopp M. Endothelial nitric oxide synthase regulates brain-derived neurotrophic factor expression and neurogenesis after stroke in mice. J Neurosci. 2005;25:2366–2375. doi: 10.1523/JNEUROSCI.5071-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JT, Lin YL, Chen TL, Tai YT, Chen CY, Chen RM. Ketamine alleviates bradykinin-induced disruption of the mouse cerebrovascular endothelial cell-constructed tight junction barrier via a calcium-mediated redistribution of occludin polymerization. Toxicology. 2016:368–369. 142–151. doi: 10.1016/j.tox.2016.09.004. [DOI] [PubMed] [Google Scholar]
- Chen W, Jadhav V, Tang J, Zhang JH. HIF-1alpha inhibition ameliorates neonatal brain injury in a rat pup hypoxic-ischemic model. Neurobiol Dis. 2008;31:433–441. doi: 10.1016/j.nbd.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Wallace BK, Yuen N, Jenkins DP, Wulff H, O’Donnell ME. Blood-brain barrier KCa3.1 channels: evidence for a role in brain Na uptake and edema in ischemic stroke. Stroke. 2015;46:237–244. doi: 10.1161/STROKEAHA.114.007445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi EY, Santoso S, Chavakis T. Mechanisms of neutrophil transendothelial migration. Front Biosci (Landmark Ed) 2009;14:1596–1605. doi: 10.2741/3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JY, Cui Y, Kim BG. Interaction between hypertension and cerebral hypoperfusion in the development of cognitive dysfunction and white matter pathology in rats. Neuroscience. 2015;303:115–125. doi: 10.1016/j.neuroscience.2015.06.056. [DOI] [PubMed] [Google Scholar]
- Choi KH, Kim HS, Park MS, Kim JT, Kim JH, Cho KA, Lee MC, Lee HJ, Cho KH. Regulation of Caveolin-1 Expression Determines Early Brain Edema After Experimental Focal Cerebral Ischemia. Stroke. 2016;47:1336–1343. doi: 10.1161/STROKEAHA.116.013205. [DOI] [PubMed] [Google Scholar]
- Chung K, Wallace J, Kim SY, Kalyanasundaram S, Andalman AS, Davidson TJ, Mirzabekov JJ, Zalocusky KA, Mattis J, Denisin AK, Pak S, Bernstein H, Ramakrishnan C, Grosenick L, Gradinaru V, Deisseroth K. Structural and molecular interrogation of intact biological systems. Nature. 2013;497:332–337. doi: 10.1038/nature12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolla MJ, Huang Q, Sweet JG. Inhibition of protein kinase Cbeta reverses increased blood-brain barrier permeability during hyperglycemic stroke and prevents edema formation in vivo. Stroke. 2011;42:3252–3257. doi: 10.1161/STROKEAHA.111.623991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio L, Kress Y, Norton WT, Brosnan CF. Increased vesicular transport and decreased mitochondrial content in blood-brain barrier endothelial cells during experimental autoimmune encephalomyelitis. Am J Pathol. 1989;135:1157–1168. [PMC free article] [PubMed] [Google Scholar]
- Cui J, Chen S, Zhang C, Meng F, Wu W, Hu R, Hadass O, Lehmidi T, Blair GJ, Lee M, Chang M, Mobashery S, Sun GY, Gu Z. Inhibition of MMP-9 by a selective gelatinase inhibitor protects neurovasculature from embolic focal cerebral ischemia. Mol Neurodegener. 2012;7:21. doi: 10.1186/1750-1326-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins PM. Occludin: one protein, many forms. Mol Cell Biol. 2012;32:242–250. doi: 10.1128/MCB.06029-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio M, Salazar IL, Mele M, Canzoniero LM, Duarte CB. Calpains and neuronal damage in the ischemic brain: The swiss knife in synaptic injury. Prog Neurobiol. 2016;143:1–35. doi: 10.1016/j.pneurobio.2016.06.001. [DOI] [PubMed] [Google Scholar]
- da Fonseca AC, Matias D, Garcia C, Amaral R, Geraldo LH, Freitas C, Lima FR. The impact of microglial activation on blood-brain barrier in brain diseases. Front Cell Neurosci. 2014;8:362. doi: 10.3389/fncel.2014.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalkara T, Gursoy-Ozdemir Y, Yemisci M. Brain microvascular pericytes in health and disease. Acta Neuropathol. 2011;122:1–9. doi: 10.1007/s00401-011-0847-6. [DOI] [PubMed] [Google Scholar]
- Dallas S, Miller DS, Bendayan R. Multidrug resistance-associated proteins: expression and function in the central nervous system. Pharmacol Rev. 2006;58:140–161. doi: 10.1124/pr.58.2.3. [DOI] [PubMed] [Google Scholar]
- Dallerac G, Rouach N. Astrocytes as new targets to improve cognitive functions. Prog Neurobiol. 2016;144:48–67. doi: 10.1016/j.pneurobio.2016.01.003. [DOI] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GE, Koh W, Stratman AN. Mechanisms controlling human endothelial lumen formation and tube assembly in three-dimensional extracellular matrices. Birth Defects Res C Embryo Today. 2007;81:270–285. doi: 10.1002/bdrc.20107. [DOI] [PubMed] [Google Scholar]
- De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med. 1991;325:703–709. doi: 10.1056/NEJM199109053251006. [DOI] [PubMed] [Google Scholar]
- Deli MA, Abraham CS, Kataoka Y, Niwa M. Permeability studies on in vitro blood-brain barrier models: physiology, pathology, and pharmacology. Cell Mol Neurobiol. 2005;25:59–127. doi: 10.1007/s10571-004-1377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demchuk AM, Morgenstern LB, Krieger DW, Linda Chi T, Hu W, Wein TH, Hardy RJ, Grotta JC, Buchan AM. Serum glucose level and diabetes predict tissue plasminogen activator-related intracerebral hemorrhage in acute ischemic stroke. Stroke. 1999;30:34–39. doi: 10.1161/01.str.30.1.34. [DOI] [PubMed] [Google Scholar]
- Deng J, Zhang J, Feng C, Xiong L, Zuo Z. Critical role of matrix metalloprotease-9 in chronic high fat diet-induced cerebral vascular remodelling and increase of ischaemic brain injury in micedagger. Cardiovasc Res. 2014;103:473–484. doi: 10.1093/cvr/cvu154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai TR, Leeper NJ, Hynes KL, Gewertz BL. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J Surg Res. 2002;104:118–123. doi: 10.1006/jsre.2002.6415. [DOI] [PubMed] [Google Scholar]
- Deutsch C, Portik-Dobos V, Smith AD, Ergul A, Dorrance AM. Diet-induced obesity causes cerebral vessel remodeling and increases the damage caused by ischemic stroke. Microvasc Res. 2009;78:100–106. doi: 10.1016/j.mvr.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmasaroja PA. Fluid Intake Related to Brain Edema in Acute Middle Cerebral Artery Infarction. Transl Stroke Res. 2016;7:49–53. doi: 10.1007/s12975-015-0439-1. [DOI] [PubMed] [Google Scholar]
- Dias IH, Polidori MC, Griffiths HR. Hypercholesterolaemia-induced oxidative stress at the blood-brain barrier. Biochem Soc Trans. 2014;42:1001–1005. doi: 10.1042/BST20140164. [DOI] [PubMed] [Google Scholar]
- Diaz-Otero JM, Garver H, Fink GD, Jackson WF, Dorrance AM. Aging is associated with changes to the biomechanical properties of the posterior cerebral artery and parenchymal arterioles. Am J Physiol Heart Circ Physiol. 2016;310:H365–375. doi: 10.1152/ajpheart.00562.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz Heijtz R, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A, Hibberd ML, Forssberg H, Pettersson S. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci U S A. 2011;108:3047–3052. doi: 10.1073/pnas.1010529108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick AP, Harik SI, Klip A, Walker DM. Identification and characterization of the glucose transporter of the blood-brain barrier by cytochalasin B binding and immunological reactivity. Proc Natl Acad Sci U S A. 1984;81:7233–7237. doi: 10.1073/pnas.81.22.7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich WD, Alonso O, Busto R. Moderate hyperglycemia worsens acute blood-brain barrier injury after forebrain ischemia in rats. Stroke. 1993;24:111–116. doi: 10.1161/01.str.24.1.111. [DOI] [PubMed] [Google Scholar]
- Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Effects of the chemokine CCL2 on blood-brain barrier permeability during ischemia-reperfusion injury. J Cereb Blood Flow Metab. 2006;26:797–810. doi: 10.1038/sj.jcbfm.9600229. [DOI] [PubMed] [Google Scholar]
- DiNapoli VA, Huber JD, Houser K, Li X, Rosen CL. Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol Aging. 2008;29:753–764. doi: 10.1016/j.neurobiolaging.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgodilina E, Imobersteg S, Laczko E, Welt T, Verrey F, Makrides V. Brain interstitial fluid glutamine homeostasis is controlled by blood-brain barrier SLC7A5/LAT1 amino acid transporter. J Cereb Blood Flow Metab. 2016;36:1929–1941. doi: 10.1177/0271678X15609331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfel MJ, Huber O. A phosphorylation hotspot within the occludin C-terminal domain. Ann N Y Acad Sci. 2012;1257:38–44. doi: 10.1111/j.1749-6632.2012.06536.x. [DOI] [PubMed] [Google Scholar]
- Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- Duz B, Oztas E, Erginay T, Erdogan E, Gonul E. The effect of moderate hypothermia in acute ischemic stroke on pericyte migration: an ultrastructural study. Cryobiology. 2007;55:279–284. doi: 10.1016/j.cryobiol.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Eira J, Silva CS, Sousa MM, Liz MA. The cytoskeleton as a novel therapeutic target for old neurodegenerative disorders. Prog Neurobiol. 2016;141:61–82. doi: 10.1016/j.pneurobio.2016.04.007. [DOI] [PubMed] [Google Scholar]
- Elahy M, Jackaman C, Mamo JC, Lam V, Dhaliwal SS, Giles C, Nelson D, Takechi R. Blood-brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immun Ageing. 2015;12:2. doi: 10.1186/s12979-015-0029-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElAli A, Doeppner TR, Zechariah A, Hermann DM. Increased blood-brain barrier permeability and brain edema after focal cerebral ischemia induced by hyperlipidemia: role of lipid peroxidation and calpain-1/2, matrix metalloproteinase-2/9, and RhoA overactivation. Stroke. 2011;42:3238–3244. doi: 10.1161/STROKEAHA.111.615559. [DOI] [PubMed] [Google Scholar]
- Ennis SR, Keep RF. Effect of sustained-mild and transient-severe hyperglycemia on ischemia-induced blood-brain barrier opening. J Cereb Blood Flow Metab. 2007;27:1573–1582. doi: 10.1038/sj.jcbfm.9600454. [DOI] [PubMed] [Google Scholar]
- Ezan P, Andre P, Cisternino S, Saubamea B, Boulay AC, Doutremer S, Thomas MA, Quenech’du N, Giaume C, Cohen-Salmon M. Deletion of astroglial connexins weakens the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32:1457–1467. doi: 10.1038/jcbfm.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan SC, Hess DC, Hohnadel EJ, Pollock DM, Ergul A. Targets for vascular protection after acute ischemic stroke. Stroke. 2004;35:2220–2225. doi: 10.1161/01.STR.0000138023.60272.9e. [DOI] [PubMed] [Google Scholar]
- Fan X, Jiang Y, Yu Z, Yuan J, Sun X, Xiang S, Lo EH, Wang X. Combination approaches to attenuate hemorrhagic transformation after tPA thrombolytic therapy in patients with poststroke hyperglycemia/diabetes. Adv Pharmacol. 2014;71:391–410. doi: 10.1016/bs.apha.2014.06.007. [DOI] [PubMed] [Google Scholar]
- Fan Y, Lan L, Zheng L, Ji X, Lin J, Zeng J, Huang R, Sun J. Spontaneous white matter lesion in brain of stroke-prone renovascular hypertensive rats: a study from MRI, pathology and behavior. Metab Brain Dis. 2015a;30:1479–1486. doi: 10.1007/s11011-015-9722-9. [DOI] [PubMed] [Google Scholar]
- Fan Y, Yang X, Tao Y, Lan L, Zheng L, Sun J. Tight junction disruption of blood-brain barrier in white matter lesions in chronic hypertensive rats. Neuroreport. 2015b;26:1039–1043. doi: 10.1097/WNR.0000000000000464. [DOI] [PubMed] [Google Scholar]
- Fang W, Sha L, Kodithuwakku ND, Wei J, Zhang R, Han D, Mao L, Li Y. Attenuated Blood-Brain Barrier Dysfunction by XQ-1H Following Ischemic Stroke in Hyperlipidemic Rats. Mol Neurobiol. 2015;52:162–175. doi: 10.1007/s12035-014-8851-1. [DOI] [PubMed] [Google Scholar]
- Fantin A, Vieira JM, Gestri G, Denti L, Schwarz Q, Prykhozhij S, Peri F, Wilson SW, Ruhrberg C. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood. 2010;116:829–840. doi: 10.1182/blood-2009-12-257832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraco G, Iadecola C. Hypertension: a harbinger of stroke and dementia. Hypertension. 2013;62:810–817. doi: 10.1161/HYPERTENSIONAHA.113.01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas E, De Jong GI, Apro E, Keuker JI, Luiten PG. Calcium antagonists decrease capillary wall damage in aging hypertensive rat brain. Neurobiol Aging. 2001;22:299–309. doi: 10.1016/s0197-4580(00)00225-6. [DOI] [PubMed] [Google Scholar]
- Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol. 2001;64:575–611. doi: 10.1016/s0301-0082(00)00068-x. [DOI] [PubMed] [Google Scholar]
- Farrall AJ, Wardlaw JM. Blood-brain barrier: ageing and microvascular disease–systematic review and meta-analysis. Neurobiol Aging. 2009;30:337–352. doi: 10.1016/j.neurobiolaging.2007.07.015. [DOI] [PubMed] [Google Scholar]
- Farrell CL, Yang J, Pardridge WM. GLUT-1 glucose transporter is present within apical and basolateral membranes of brain epithelial interfaces and in microvascular endothelia with and without tight junctions. J Histochem Cytochem. 1992;40:193–199. doi: 10.1177/40.2.1552163. [DOI] [PubMed] [Google Scholar]
- Feil R, Wagner J, Metzger D, Chambon P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun. 1997;237:752–757. doi: 10.1006/bbrc.1997.7124. [DOI] [PubMed] [Google Scholar]
- Fernandez-Klett F, Potas JR, Hilpert D, Blazej K, Radke J, Huck J, Engel O, Stenzel W, Genove G, Priller J. Early loss of pericytes and perivascular stromal cell-induced scar formation after stroke. J Cereb Blood Flow Metab. 2013;33:428–439. doi: 10.1038/jcbfm.2012.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Klett F, Priller J. Diverse functions of pericytes in cerebral blood flow regulation and ischemia. J Cereb Blood Flow Metab. 2015;35:883–887. doi: 10.1038/jcbfm.2015.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Lopez D, Faustino J, Klibanov AL, Derugin N, Blanchard E, Simon F, Leib SL, Vexler ZS. Microglial Cells Prevent Hemorrhage in Neonatal Focal Arterial Stroke. J Neurosci. 2016;36:2881–2893. doi: 10.1523/JNEUROSCI.0140-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filous AR, Silver J. Targeting astrocytes in CNS injury and disease: A translational research approach. Prog Neurobiol. 2016;144:173–187. doi: 10.1016/j.pneurobio.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer S, Wobben M, Marti HH, Renz D, Schaper W. Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc Res. 2002;63:70–80. doi: 10.1006/mvre.2001.2367. [DOI] [PubMed] [Google Scholar]
- Fisher MJ. Brain regulation of thrombosis and hemostasis: from theory to practice. Stroke. 2013;44:3275–3285. doi: 10.1161/STROKEAHA.113.000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkow B. Physiological aspects of primary hypertension. Physiol Rev. 1982;62:347–504. doi: 10.1152/physrev.1982.62.2.347. [DOI] [PubMed] [Google Scholar]
- Forde A, Constien R, Grone HJ, Hammerling G, Arnold B. Temporal Cre-mediated recombination exclusively in endothelial cells using Tie2 regulatory elements. Genesis. 2002;33:191–197. doi: 10.1002/gene.10117. [DOI] [PubMed] [Google Scholar]
- Forster C, Burek M, Romero IA, Weksler B, Couraud PO, Drenckhahn D. Differential effects of hydrocortisone and TNFalpha on tight junction proteins in an in vitro model of the human blood-brain barrier. J Physiol. 2008;586:1937–1949. doi: 10.1113/jphysiol.2007.146852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4–9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- Franco R, Fernandez-Suarez D. Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol. 2015;131:65–86. doi: 10.1016/j.pneurobio.2015.05.003. [DOI] [PubMed] [Google Scholar]
- Frechou M, Zhang S, Liere P, Delespierre B, Soyed N, Pianos A, Schumacher M, Mattern C, Guennoun R. Intranasal delivery of progesterone after transient ischemic stroke decreases mortality and provides neuroprotection. Neuropharmacology. 2015;97:394–403. doi: 10.1016/j.neuropharm.2015.06.002. [DOI] [PubMed] [Google Scholar]
- Fredriksson K, Kalimo H, Westergren I, Kahrstrom J, Johansson BB. Blood-brain barrier leakage and brain edema in stroke-prone spontaneously hypertensive rats. Effect of chronic sympathectomy and low protein/high salt diet. Acta Neuropathol. 1987;74:259–268. doi: 10.1007/BF00688190. [DOI] [PubMed] [Google Scholar]
- Fujibe M, Chiba H, Kojima T, Soma T, Wada T, Yamashita T, Sawada N. Thr203 of claudin-1, a putative phosphorylation site for MAP kinase, is required to promote the barrier function of tight junctions. Exp Cell Res. 2004;295:36–47. doi: 10.1016/j.yexcr.2003.12.014. [DOI] [PubMed] [Google Scholar]
- Fujihara R, Chiba Y, Nakagawa T, Nishi N, Murakami R, Matsumoto K, Kawauchi M, Yamamoto T, Ueno M. Albumin microvascular leakage in brains with diabetes mellitus. Microsc Res Tech. 2016;79:833–837. doi: 10.1002/jemt.22708. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Nakagawa S, Tatsumi R, Morofuji Y, Takeshita T, Hayashi K, Tanaka K, Matsuo T, Niwa M. Glucagon-Like Peptide-1 Strengthens the Barrier Integrity in Primary Cultures of Rat Brain Endothelial Cells Under Basal and Hyperglycemia Conditions. J Mol Neurosci. 2016;59:211–219. doi: 10.1007/s12031-015-0696-1. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Yasu T, Kobayashi N, Ikeda N, Schmid-Schonbein GW. Contribution of fluid shear response in leukocytes to hemodynamic resistance in the spontaneously hypertensive rat. Circ Res. 2004;95:100–108. doi: 10.1161/01.RES.0000133677.77465.38. [DOI] [PubMed] [Google Scholar]
- Gandhi GK, Ball KK, Cruz NF, Dienel GA. Hyperglycaemia and diabetes impair gap junctional communication among astrocytes. ASN Neuro. 2010;2:e00030. doi: 10.1042/AN20090048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbuzova-Davis S, Rodrigues MC, Hernandez-Ontiveros DG, Tajiri N, Frisina-Deyo A, Boffeli SM, Abraham JV, Pabon M, Wagner A, Ishikawa H, Shinozuka K, Haller E, Sanberg PR, Kaneko Y, Borlongan CV. Blood-brain barrier alterations provide evidence of subacute diaschisis in an ischemic stroke rat model. PLoS One. 2013;8:e63553. doi: 10.1371/journal.pone.0063553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, Arumugam TV, Orthey E, Gerloff C, Tolosa E, Magnus T. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–1857. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- Gibson CL, Srivastava K, Sprigg N, Bath PM, Bayraktutan U. Inhibition of Rho-kinase protects cerebral barrier from ischaemia-evoked injury through modulations of endothelial cell oxidative stress and tight junctions. J Neurochem. 2014;129:816–826. doi: 10.1111/jnc.12681. [DOI] [PubMed] [Google Scholar]
- Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- Giraud M, Cho TH, Nighoghossian N, Maucort-Boulch D, Deiana G, Ostergaard L, Baron JC, Fiehler J, Pedraza S, Derex L, Berthezene Y. Early Blood Brain Barrier Changes in Acute Ischemic Stroke: A Sequential MRI Study. J Neuroimaging. 2015;25:959–963. doi: 10.1111/jon.12225. [DOI] [PubMed] [Google Scholar]
- Goldmann EE. Die äussere und innere Sekretion des gesunden und kranken Organismus im Lichte der ‘vitalen Farbung’. Beitr klin Chir. 1909;64:192–265. [Google Scholar]
- Goldmann EE. Vitalfarbung am Zentralnervensystem. Abh preuss Akad Wiss Phys-Math. 1913;1:1–60. [Google Scholar]
- Gonzalez-Mariscal L, Tapia R, Chamorro D. Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta. 2008;1778:729–756. doi: 10.1016/j.bbamem.2007.08.018. [DOI] [PubMed] [Google Scholar]
- Goritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisen J. A pericyte origin of spinal cord scar tissue. Science. 2011;333:238–242. doi: 10.1126/science.1203165. [DOI] [PubMed] [Google Scholar]
- Gorle N, Van Cauwenberghe C, Libert C, Vandenbroucke RE. The effect of aging on brain barriers and the consequences for Alzheimer’s disease development. Mamm Genome. 2016;27:407–420. doi: 10.1007/s00335-016-9637-8. [DOI] [PubMed] [Google Scholar]
- Grieb P, Forster RE, Strome D, Goodwin CW, Pape PC. O2 exchange between blood and brain tissues studied with 18O2 indicator-dilution technique. J Appl Physiol (1985) 1985;58:1929–1941. doi: 10.1152/jappl.1985.58.6.1929. [DOI] [PubMed] [Google Scholar]
- Grillon E, Provent P, Montigon O, Segebarth C, Remy C, Barbier EL. Blood-brain barrier permeability to manganese and to Gd-DOTA in a rat model of transient cerebral ischaemia. NMR Biomed. 2008;21:427–436. doi: 10.1002/nbm.1206. [DOI] [PubMed] [Google Scholar]
- Gu Y, Zheng G, Xu M, Li Y, Chen X, Zhu W, Tong Y, Chung SK, Liu KJ, Shen J. Caveolin-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood-brain barrier permeability in focal cerebral ischemia and reperfusion injury. J Neurochem. 2012;120:147–156. doi: 10.1111/j.1471-4159.2011.07542.x. [DOI] [PubMed] [Google Scholar]
- Guell K, Bix GJ. Brain endothelial cell specific integrins and ischemic stroke. Expert Rev Neurother. 2014;14:1287–1292. doi: 10.1586/14737175.2014.964210. [DOI] [PubMed] [Google Scholar]
- Guo J, Krause DN, Horne J, Weiss JH, Li X, Duckles SP. Estrogen-receptor-mediated protection of cerebral endothelial cell viability and mitochondrial function after ischemic insult in vitro. J Cereb Blood Flow Metab. 2010;30:545–554. doi: 10.1038/jcbfm.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gursoy-Ozdemir Y, Can A, Dalkara T. Reperfusion-induced oxidative/nitrative injury to neurovascular unit after focal cerebral ischemia. Stroke. 2004;35:1449–1453. doi: 10.1161/01.STR.0000126044.83777.f4. [DOI] [PubMed] [Google Scholar]
- Gustafsson E, Brakebusch C, Hietanen K, Fassler R. Tie-1-directed expression of Cre recombinase in endothelial cells of embryoid bodies and transgenic mice. J Cell Sci. 2001;114:671–676. doi: 10.1242/jcs.114.4.671. [DOI] [PubMed] [Google Scholar]
- Hafezi-Moghadam A, Thomas KL, Wagner DD. ApoE deficiency leads to a progressive age-dependent blood-brain barrier leakage. Am J Physiol Cell Physiol. 2007;292:C1256–1262. doi: 10.1152/ajpcell.00563.2005. [DOI] [PubMed] [Google Scholar]
- Haley MJ, Lawrence CB. The blood-brain barrier after stroke: Structural studies and the role of transcytotic vesicles. J Cereb Blood Flow Metab. 2017;37:456–470. doi: 10.1177/0271678X16629976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O’Farrell FM, Buchan AM, Lauritzen M, Attwell D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508:55–60. doi: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harb R, Whiteus C, Freitas C, Grutzendler J. In vivo imaging of cerebral microvascular plasticity from birth to death. J Cereb Blood Flow Metab. 2013;33:146–156. doi: 10.1038/jcbfm.2012.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann DA, Underly RG, Watson AN, Shih AY. A murine toolbox for imaging the neurovascular unit. Microcirculation. 2015;22:168–182. doi: 10.1111/micc.12176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BT, Gu YH, Izawa Y, del Zoppo GJ. Dabigatran abrogates brain endothelial cell permeability in response to thrombin. J Cereb Blood Flow Metab. 2015;35:985–992. doi: 10.1038/jcbfm.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BT, Lundeen TF, Norwood KM, Brooks HL, Egleton RD. Increased blood-brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia. 2007;50:202–211. doi: 10.1007/s00125-006-0485-z. [DOI] [PubMed] [Google Scholar]
- Hawkins RA, O’Kane RL, Simpson IA, Vina JR. Structure of the blood-brain barrier and its role in the transport of amino acids. J Nutr. 2006;136:218S–226S. doi: 10.1093/jn/136.1.218S. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Nakao S, Nakaoke R, Nakagawa S, Kitagawa N, Niwa M. Effects of hypoxia on endothelial/pericytic co-culture model of the blood-brain barrier. Regul Pept. 2004;123:77–83. doi: 10.1016/j.regpep.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- Helms HC, Abbott NJ, Burek M, Cecchelli R, Couraud PO, Deli MA, Forster C, Galla HJ, Romero IA, Shusta EV, Stebbins MJ, Vandenhaute E, Weksler B, Brodin B. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J Cereb Blood Flow Metab. 2016;36:862–890. doi: 10.1177/0271678X16630991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, del Zoppo GJ. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:624–633. doi: 10.1097/00004647-199906000-00005. [DOI] [PubMed] [Google Scholar]
- Hermida RC, Ayala DE, Smolensky MH, Fernandez JR, Mojon A, Portaluppi F. Chronotherapy with conventional blood pressure medications improves management of hypertension and reduces cardiovascular and stroke risks. Hypertens Res. 2016;39:277–292. doi: 10.1038/hr.2015.142. [DOI] [PubMed] [Google Scholar]
- Heyes S, Pratt WS, Rees E, Dahimene S, Ferron L, Owen MJ, Dolphin AC. Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog Neurobiol. 2015;134:36–54. doi: 10.1016/j.pneurobio.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hladky SB, Barrand MA. Fluid and ion transfer across the blood-brain and blood-cerebrospinal fluid barriers; a comparative account of mechanisms and roles. Fluids Barriers CNS. 2016;13:19. doi: 10.1186/s12987-016-0040-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hom S, Fleegal MA, Egleton RD, Campos CR, Hawkins BT, Davis TP. Comparative changes in the blood-brain barrier and cerebral infarction of SHR and WKY rats. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1881–1892. doi: 10.1152/ajpregu.00761.2005. [DOI] [PubMed] [Google Scholar]
- Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins AM, DeSimone E, Chwalek K, Kaplan DL. 3D in vitro modeling of the central nervous system. Prog Neurobiol. 2015;125:1–25. doi: 10.1016/j.pneurobio.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz A, Seerapu HR. Regulation of VEGF signaling by membrane traffic. Cell Signal. 2012;24:1810–1820. doi: 10.1016/j.cellsig.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostenbach S, D’Haeseleer M, Kooijman R, De Keyser J. The pathophysiological role of astrocytic endothelin-1. Prog Neurobiol. 2016;144:88–102. doi: 10.1016/j.pneurobio.2016.04.009. [DOI] [PubMed] [Google Scholar]
- Hu S, Maslov K, Tsytsarev V, Wang LV. Functional transcranial brain imaging by optical-resolution photoacoustic microscopy. J Biomed Opt. 2009;14:040503. doi: 10.1117/1.3194136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, Chen J. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol. 2015;11:56–64. doi: 10.1038/nrneurol.2014.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liu B, Yang C, Chen H, Eunice D, Yuan Z. Acute hyperglycemia worsens ischemic stroke-induced brain damage via high mobility group box-1 in rats. Brain Res. 2013;1535:148–155. doi: 10.1016/j.brainres.2013.08.057. [DOI] [PubMed] [Google Scholar]
- Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol. 2006;66:232–245. doi: 10.1016/j.surneu.2005.12.028. [DOI] [PubMed] [Google Scholar]
- Huang T, Gao D, Hei Y, Zhang X, Chen X, Fei Z. D-allose protects the blood brain barrier through PPARgamma-mediated anti-inflammatory pathway in the mice model of ischemia reperfusion injury. Brain Res. 2016a;1642:478–486. doi: 10.1016/j.brainres.2016.04.038. [DOI] [PubMed] [Google Scholar]
- Huang WY, Wu G, Li JJ, Geng DY, Tan WL, Yu XR. Early prediction of functional outcome using dynamic contrast enhanced magnetic resonance imaging in experimental stroke. Magn Reson Imaging. 2016b;34:1000–1007. doi: 10.1016/j.mri.2016.04.015. [DOI] [PubMed] [Google Scholar]
- Huber JD, VanGilder RL, Houser KA. Streptozotocin-induced diabetes progressively increases blood-brain barrier permeability in specific brain regions in rats. Am J Physiol Heart Circ Physiol. 2006;291:H2660–2668. doi: 10.1152/ajpheart.00489.2006. [DOI] [PubMed] [Google Scholar]
- Hun Lee J, Won S, Stein DG. Progesterone attenuates thrombin-induced endothelial barrier disruption in the brain endothelial cell line bEnd.3: The role of tight junction proteins and the endothelial protein C receptor. Brain Res. 2015;1613:73–80. doi: 10.1016/j.brainres.2015.04.002. [DOI] [PubMed] [Google Scholar]
- Hung VK, Yeung PK, Lai AK, Ho MC, Lo AC, Chan KC, Wu EX, Chung SS, Cheung CW, Chung SK. Selective astrocytic endothelin-1 overexpression contributes to dementia associated with ischemic stroke by exaggerating astrocyte-derived amyloid secretion. J Cereb Blood Flow Metab. 2015;35:1687–1696. doi: 10.1038/jcbfm.2015.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurn PD, Littleton-Kearney MT, Kirsch JR, Dharmarajan AM, Traystman RJ. Postischemic cerebral blood flow recovery in the female: effect of 17 beta-estradiol. J Cereb Blood Flow Metab. 1995;15:666–672. doi: 10.1038/jcbfm.1995.82. [DOI] [PubMed] [Google Scholar]
- Hurwitz AA, Berman JW, Rashbaum WK, Lyman WD. Human fetal astrocytes induce the expression of blood-brain barrier specific proteins by autologous endothelial cells. Brain Res. 1993;625:238–243. doi: 10.1016/0006-8993(93)91064-y. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- Igarashi Y, Utsumi H, Chiba H, Yamada-Sasamori Y, Tobioka H, Kamimura Y, Furuuchi K, Kokai Y, Nakagawa T, Mori M, Sawada N. Glial cell line-derived neurotrophic factor induces barrier function of endothelial cells forming the blood-brain barrier. Biochem Biophys Res Commun. 1999;261:108–112. doi: 10.1006/bbrc.1999.0992. [DOI] [PubMed] [Google Scholar]
- Ishida H, Takemori K, Dote K, Ito H. Expression of glucose transporter-1 and aquaporin-4 in the cerebral cortex of stroke-prone spontaneously hypertensive rats in relation to the blood-brain barrier function. Am J Hypertens. 2006;19:33–39. doi: 10.1016/j.amjhyper.2005.06.023. [DOI] [PubMed] [Google Scholar]
- Ishrat T, Sayeed I, Atif F, Hua F, Stein DG. Progesterone and allopregnanolone attenuate blood-brain barrier dysfunction following permanent focal ischemia by regulating the expression of matrix metalloproteinases. Exp Neurol. 2010;226:183–190. doi: 10.1016/j.expneurol.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata A, Koike F, Arasaki K, Tamaki M. Blood brain barrier destruction in hyperglycemic chorea in a patient with poorly controlled diabetes. J Neurol Sci. 1999;163:90–93. doi: 10.1016/s0022-510x(98)00325-6. [DOI] [PubMed] [Google Scholar]
- Jaerve A, Muller HW. Chemokines in CNS injury and repair. Cell Tissue Res. 2012;349:229–248. doi: 10.1007/s00441-012-1427-3. [DOI] [PubMed] [Google Scholar]
- Janowski M, Walczak P, Pearl MS. Predicting and optimizing the territory of blood-brain barrier opening by superselective intra-arterial cerebral infusion under dynamic susceptibility contrast MRI guidance. J Cereb Blood Flow Metab. 2016;36:569–575. doi: 10.1177/0271678X15615875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson D, Rustenhoven J, Feng S, Hurley D, Oldfield RL, Bergin PS, Mee EW, Faull RL, Dragunow M. A role for human brain pericytes in neuroinflammation. J Neuroinflammation. 2014;11:104. doi: 10.1186/1742-2094-11-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325:253–257. doi: 10.1038/325253a0. [DOI] [PubMed] [Google Scholar]
- Jasmin JF, Malhotra S, Singh Dhallu M, Mercier I, Rosenbaum DM, Lisanti MP. Caveolin-1 deficiency increases cerebral ischemic injury. Circ Res. 2007;100:721–729. doi: 10.1161/01.RES.0000260180.42709.29. [DOI] [PubMed] [Google Scholar]
- Jeynes B. Reactions of granular pericytes in a rabbit cerebrovascular ischemia model. Stroke. 1985;16:121–125. doi: 10.1161/01.str.16.1.121. [DOI] [PubMed] [Google Scholar]
- Jiang X, Pu H, Hu X, Wei Z, Hong D, Zhang W, Gao Y, Chen J, Shi Y. A Post-stroke Therapeutic Regimen with Omega-3 Polyunsaturated Fatty Acids that Promotes White Matter Integrity and Beneficial Microglial Responses after Cerebral Ischemia. Transl Stroke Res. 2016;7:548–561. doi: 10.1007/s12975-016-0502-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, Sharp FR. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab. 2015;35:888–901. doi: 10.1038/jcbfm.2015.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jickling GC, Liu D, Stamova B, Ander BP, Zhan X, Lu A, Sharp FR. Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab. 2014;34:185–199. doi: 10.1038/jcbfm.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin AY, Tuor UI, Rushforth D, Kaur J, Muller RN, Petterson JL, Boutry S, Barber PA. Reduced blood brain barrier breakdown in P-selectin deficient mice following transient ischemic stroke: a future therapeutic target for treatment of stroke. BMC Neurosci. 2010;11:12. doi: 10.1186/1471-2202-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Liu J, Liu W. Early ischemic blood brain barrier damage: a potential indicator for hemorrhagic transformation following tissue plasminogen activator (tPA) thrombolysis? Curr Neurovasc Res. 2014;11:254–262. doi: 10.2174/1567202611666140530145643. [DOI] [PubMed] [Google Scholar]
- Jin X, Sun Y, Xu J, Liu W. Caveolin-1 mediates tissue plasminogen activator-induced MMP-9 up-regulation in cultured brain microvascular endothelial cells. J Neurochem. 2015;132:724–730. doi: 10.1111/jnc.13065. [DOI] [PubMed] [Google Scholar]
- Johann S, Beyer C. Neuroprotection by gonadal steroid hormones in acute brain damage requires cooperation with astroglia and microglia. J Steroid Biochem Mol Biol. 2013;137:71–81. doi: 10.1016/j.jsbmb.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Joo F. The blood-brain barrier in vitro: Ten years of research on microvessels isolated from the brain. Neurochem Int. 1985;7:1–25. doi: 10.1016/0197-0186(85)90002-6. [DOI] [PubMed] [Google Scholar]
- Justicia C, Panes J, Sole S, Cervera A, Deulofeu R, Chamorro A, Planas AM. Neutrophil infiltration increases matrix metalloproteinase-9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J Cereb Blood Flow Metab. 2003;23:1430–1440. doi: 10.1097/01.WCB.0000090680.07515.C8. [DOI] [PubMed] [Google Scholar]
- Kago T, Takagi N, Date I, Takenaga Y, Takagi K, Takeo S. Cerebral ischemia enhances tyrosine phosphorylation of occludin in brain capillaries. Biochem Biophys Res Commun. 2006;339:1197–1203. doi: 10.1016/j.bbrc.2005.11.133. [DOI] [PubMed] [Google Scholar]
- Kalayci R, Kaya M, Uzun H, Bilgic B, Ahishali B, Arican N, Elmas I, Kucuk M. Influence of hypercholesterolemia and hypertension on the integrity of the blood-brain barrier in rats. Int J Neurosci. 2009;119:1881–1904. doi: 10.1080/14647270802336650. [DOI] [PubMed] [Google Scholar]
- Kale G, Naren AP, Sheth P, Rao RK. Tyrosine phosphorylation of occludin attenuates its interactions with ZO-1, ZO-2, and ZO-3. Biochem Biophys Res Commun. 2003;302:324–329. doi: 10.1016/s0006-291x(03)00167-0. [DOI] [PubMed] [Google Scholar]
- Kamada H, Yu F, Nito C, Chan PH. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats: relation to blood-brain barrier dysfunction. Stroke. 2007;38:1044–1049. doi: 10.1161/01.STR.0000258041.75739.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassner A, Roberts TP, Moran B, Silver FL, Mikulis DJ. Recombinant tissue plasminogen activator increases blood-brain barrier disruption in acute ischemic stroke: an MR imaging permeability study. AJNR Am J Neuroradiol. 2009;30:1864–1869. doi: 10.3174/ajnr.A1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur J, Tuor UI, Zhao Z, Barber PA. Quantitative MRI reveals the elderly ischemic brain is susceptible to increased early blood-brain barrier permeability following tissue plasminogen activator related to claudin 5 and occludin disassembly. J Cereb Blood Flow Metab. 2011;31:1874–1885. doi: 10.1038/jcbfm.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai N, Keep RF, Betz AL. Hyperglycemia and the vascular effects of cerebral ischemia. Stroke. 1997;28:149–154. doi: 10.1161/01.str.28.1.149. [DOI] [PubMed] [Google Scholar]
- Kawai N, Keep RF, Betz AL, Nagao S. Hyperglycemia induces progressive changes in the cerebral microvasculature and blood-brain barrier transport during focal cerebral ischemia. Acta Neurochir Suppl. 1998;71:219–221. doi: 10.1007/978-3-7091-6475-4_63. [DOI] [PubMed] [Google Scholar]
- Kawai N, Stummer W, Ennis SR, Betz AL, Keep RF. Blood-brain barrier glutamine transport during normoglycemic and hyperglycemic focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:79–86. doi: 10.1097/00004647-199901000-00009. [DOI] [PubMed] [Google Scholar]
- Kawamura K, Takahashi T, Kanazawa M, Igarashi H, Nakada T, Nishizawa M, Shimohata T. Effects of angiopoietin-1 on hemorrhagic transformation and cerebral edema after tissue plasminogen activator treatment for ischemic stroke in rats. PLoS One. 2014;9:e98639. doi: 10.1371/journal.pone.0098639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keaney J, Campbell M. The dynamic blood-brain barrier. FEBS J. 2015;282:4067–4079. doi: 10.1111/febs.13412. [DOI] [PubMed] [Google Scholar]
- Keep RF, Andjelkovic AV, Stamatovic SM, Shakui P, Ennis SR. Ischemia-induced endothelial cell dysfunction. Acta Neurochir Suppl. 2005;95:399–402. doi: 10.1007/3-211-32318-x_81. [DOI] [PubMed] [Google Scholar]
- Keep RF, Xiang J, Ennis SR, Andjelkovic A, Hua Y, Xi G, Hoff JT. Blood-brain barrier function in intracerebral hemorrhage. Acta Neurochir Suppl. 2008;105:73–77. doi: 10.1007/978-3-211-09469-3_15. [DOI] [PubMed] [Google Scholar]
- Keep RF, Zhou N, Xiang J, Andjelkovic AV, Hua Y, Xi G. Vascular disruption and blood-brain barrier dysfunction in intracerebral hemorrhage. Fluids Barriers CNS. 2014;11:18. doi: 10.1186/2045-8118-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MA, Shuaib A, Todd KG. Matrix metalloproteinase activation and blood-brain barrier breakdown following thrombolysis. Exp Neurol. 2006;200:38–49. doi: 10.1016/j.expneurol.2006.01.032. [DOI] [PubMed] [Google Scholar]
- Kent DM, Price LL, Ringleb P, Hill MD, Selker HP. Sex-based differences in response to recombinant tissue plasminogen activator in acute ischemic stroke: a pooled analysis of randomized clinical trials. Stroke. 2005;36:62–65. doi: 10.1161/01.STR.0000150515.15576.29. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Kim CH. Chemokine-chemokine receptor network in immune cell trafficking. Curr Drug Targets Immune Endocr Metabol Disord. 2004;4:343–361. doi: 10.2174/1568008043339712. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK. Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia. 2005;50:389–397. doi: 10.1002/glia.20174. [DOI] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- Kleinschnitz C, Blecharz K, Kahles T, Schwarz T, Kraft P, Gobel K, Meuth SG, Burek M, Thum T, Stoll G, Forster C. Glucocorticoid insensitivity at the hypoxic blood-brain barrier can be reversed by inhibition of the proteasome. Stroke. 2011;42:1081–1089. doi: 10.1161/STROKEAHA.110.592238. [DOI] [PubMed] [Google Scholar]
- Kleinschnitz C, Kraft P, Dreykluft A, Hagedorn I, Gobel K, Schuhmann MK, Langhauser F, Helluy X, Schwarz T, Bittner S, Mayer CT, Brede M, Varallyay C, Pham M, Bendszus M, Jakob P, Magnus T, Meuth SG, Iwakura Y, Zernecke A, Sparwasser T, Nieswandt B, Stoll G, Wiendl H. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood. 2013;121:679–691. doi: 10.1182/blood-2012-04-426734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight RA, Karki K, Ewing JR, Divine GW, Fenstermacher JD, Patlak CS, Nagaraja TN. Estimating blood and brain concentrations and blood-to-brain influx by magnetic resonance imaging with step-down infusion of Gd-DTPA in focal transient cerebral ischemia and confirmation by quantitative autoradiography with Gd-[(14)C]DTPA. J Cereb Blood Flow Metab. 2009;29:1048–1058. doi: 10.1038/jcbfm.2009.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowland D, Arac A, Sekiguchi KJ, Hsu M, Lutz SE, Perrino J, Steinberg GK, Barres BA, Nimmerjahn A, Agalliu D. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron. 2014;82:603–617. doi: 10.1016/j.neuron.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox CA, Yates RD, Chen I, Klara PM. Effects of aging on the structural and permeability characteristics of cerebrovasculature in normotensive and hypertensive strains of rats. Acta Neuropathol. 1980;51:1–13. doi: 10.1007/BF00688844. [DOI] [PubMed] [Google Scholar]
- Kokovay E, Li L, Cunningham LA. Angiogenic recruitment of pericytes from bone marrow after stroke. J Cereb Blood Flow Metab. 2006;26:545–555. doi: 10.1038/sj.jcbfm.9600214. [DOI] [PubMed] [Google Scholar]
- Kotsarini C, Griffiths PD, Wilkinson ID, Hoggard N. A systematic review of the literature on the effects of dexamethasone on the brain from in vivo human-based studies: implications for physiological brain imaging of patients with intracranial tumors. Neurosurgery. 2010;67:1799–1815. doi: 10.1227/NEU.0b013e3181fa775b. discussion 1815. [DOI] [PubMed] [Google Scholar]
- Krizbai IA, Deli MA. Signalling pathways regulating the tight junction permeability in the blood-brain barrier. Cell Mol Biol (Noisy-le-grand) 2003;49:23–31. [PubMed] [Google Scholar]
- Kruyt ND, Biessels GJ, Devries JH, Roos YB. Hyperglycemia in acute ischemic stroke: pathophysiology and clinical management. Nat Rev Neurol. 2010;6:145–155. doi: 10.1038/nrneurol.2009.231. [DOI] [PubMed] [Google Scholar]
- Kumagai N, Chiba Y, Hosono M, Fujii M, Kawamura N, Keino H, Yoshikawa K, Ishii S, Saitoh Y, Satoh M, Shimada A, Hosokawa M. Involvement of pro-inflammatory cytokines and microglia in an age-associated neurodegeneration model, the SAMP10 mouse. Brain Res. 2007;1185:75–85. doi: 10.1016/j.brainres.2007.09.021. [DOI] [PubMed] [Google Scholar]
- Kumari R, Willing LB, Patel SD, Baskerville KA, Simpson IA. Increased cerebral matrix metalloprotease-9 activity is associated with compromised recovery in the diabetic db/db mouse following a stroke. J Neurochem. 2011;119:1029–1040. doi: 10.1111/j.1471-4159.2011.07487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz M, Mysiorek C, Petrault O, Boucau MC, Aijjou R, Uzbekov R, Berezowski V. Transient oxygen-glucose deprivation sensitizes brain capillary endothelial cells to rtPA at 4h of reoxygenation. Microvasc Res. 2014a;91:44–57. doi: 10.1016/j.mvr.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Kuntz M, Mysiorek C, Petrault O, Petrault M, Uzbekov R, Bordet R, Fenart L, Cecchelli R, Berezowski V. Stroke-induced brain parenchymal injury drives blood-brain barrier early leakage kinetics: a combined in vivo/in vitro study. J Cereb Blood Flow Metab. 2014b;34:95–107. doi: 10.1038/jcbfm.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam TI, Wise PM, O’Donnell ME. Cerebral microvascular endothelial cell Na/H exchange: evidence for the presence of NHE1 and NHE2 isoforms and regulation by arginine vasopressin. Am J Physiol Cell Physiol. 2009;297:C278–289. doi: 10.1152/ajpcell.00093.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamagna C, Bergers G. The bone marrow constitutes a reservoir of pericyte progenitors. J Leukoc Biol. 2006;80:677–681. doi: 10.1189/jlb.0506309. [DOI] [PubMed] [Google Scholar]
- Landis DM. The early reactions of non-neuronal cells to brain injury. Annu Rev Neurosci. 1994;17:133–151. doi: 10.1146/annurev.ne.17.030194.001025. [DOI] [PubMed] [Google Scholar]
- Langdon KD, Clarke J, Corbett D. Long-term exposure to high fat diet is bad for your brain: exacerbation of focal ischemic brain injury. Neuroscience. 2011;182:82–87. doi: 10.1016/j.neuroscience.2011.03.028. [DOI] [PubMed] [Google Scholar]
- Lapi D, Colantuoni A. Remodeling of Cerebral Microcirculation after Ischemia-Reperfusion. J Vasc Res. 2015;52:22–31. doi: 10.1159/000381096. [DOI] [PubMed] [Google Scholar]
- Lapi D, Vagnani S, Sapio D, Mastantuono T, Sabatino L, Paterni M, Colantuoni A. Long-term remodeling of rat pial microcirculation after transient middle cerebral artery occlusion and reperfusion. J Vasc Res. 2013;50:332–345. doi: 10.1159/000353295. [DOI] [PubMed] [Google Scholar]
- Lassen NA. Autoregulation of Cerebral Blood Flow. Circ Res. 1964;15(SUPPL):201–204. [PubMed] [Google Scholar]
- Latour LL, Kang DW, Ezzeddine MA, Chalela JA, Warach S. Early blood-brain barrier disruption in human focal brain ischemia. Ann Neurol. 2004;56:468–477. doi: 10.1002/ana.20199. [DOI] [PubMed] [Google Scholar]
- Leung JW, Chung SS, Chung SK. Endothelial endothelin-1 over-expression using receptor tyrosine kinase tie-1 promoter leads to more severe vascular permeability and blood brain barrier breakdown after transient middle cerebral artery occlusion. Brain Res. 2009;1266:121–129. doi: 10.1016/j.brainres.2009.01.070. [DOI] [PubMed] [Google Scholar]
- Li H, Gao A, Feng D, Wang Y, Zhang L, Cui Y, Li B, Wang Z, Chen G. Evaluation of the protective potential of brain microvascular endothelial cell autophagy on blood-brain barrier integrity during experimental cerebral ischemia-reperfusion injury. Transl Stroke Res. 2014a;5:618–626. doi: 10.1007/s12975-014-0354-x. [DOI] [PubMed] [Google Scholar]
- Li L, Tao Y, Tang J, Chen Q, Yang Y, Feng Z, Chen Y, Yang L, Yang Y, Zhu G, Feng H, Chen Z. A Cannabinoid Receptor 2 Agonist Prevents Thrombin-Induced Blood-Brain Barrier Damage via the Inhibition of Microglial Activation and Matrix Metalloproteinase Expression in Rats. Transl Stroke Res. 2015a;6:467–477. doi: 10.1007/s12975-015-0425-7. [DOI] [PubMed] [Google Scholar]
- Li P, Gan Y, Sun BL, Zhang F, Lu B, Gao Y, Liang W, Thomson AW, Chen J, Hu X. Adoptive regulatory T-cell therapy protects against cerebral ischemia. Ann Neurol. 2013;74:458–471. doi: 10.1002/ana.23815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Mao L, Liu X, Gan Y, Zheng J, Thomson AW, Gao Y, Chen J, Hu X. Essential role of program death 1-ligand 1 in regulatory T-cell-afforded protection against blood-brain barrier damage after stroke. Stroke. 2014b;45:857–864. doi: 10.1161/STROKEAHA.113.004100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Chen S, Li JY. Human induced pluripotent stem cells in Parkinson’s disease: A novel cell source of cell therapy and disease modeling. Prog Neurobiol. 2015b;134:161–177. doi: 10.1016/j.pneurobio.2015.09.009. [DOI] [PubMed] [Google Scholar]
- Li X, Bai R, Zhang J, Wang X. Effect of progesterone intervention on the dynamic changes of AQP-4 in hypoxic-ischaemic brain damage. Int J Clin Exp Med. 2015c;8:18831–18836. [PMC free article] [PubMed] [Google Scholar]
- Li YN, Pan R, Qin XJ, Yang WL, Qi Z, Liu W, Liu KJ. Ischemic neurons activate astrocytes to disrupt endothelial barrier via increasing VEGF expression. J Neurochem. 2014c;129:120–129. doi: 10.1111/jnc.12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Wu G, Fan C, Xu J, Jiang S, Yan X, Di S, Ma Z, Hu W, Yang Y. The emerging role of signal transducer and activator of transcription 3 in cerebral ischemic and hemorrhagic stroke. Prog Neurobiol. 2016;137:1–16. doi: 10.1016/j.pneurobio.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Liao YJ, Ueno M, Nakagawa T, Huang C, Kanenishi K, Onodera M, Sakamoto H. Oxidative damage in cerebral vessels of diabetic db/db mice. Diabetes Metab Res Rev. 2005;21:554–559. doi: 10.1002/dmrr.579. [DOI] [PubMed] [Google Scholar]
- Liesz A, Hu X, Kleinschnitz C, Offner H. Functional role of regulatory lymphocytes in stroke: facts and controversies. Stroke. 2015;46:1422–1430. doi: 10.1161/STROKEAHA.114.008608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A, Kleinschnitz C. Regulatory T Cells in Post-stroke Immune Homeostasis. Transl Stroke Res. 2016;7:313–321. doi: 10.1007/s12975-016-0465-7. [DOI] [PubMed] [Google Scholar]
- Lin CY, Chang C, Cheung WM, Lin MH, Chen JJ, Hsu CY, Chen JH, Lin TN. Dynamic changes in vascular permeability, cerebral blood volume, vascular density, and size after transient focal cerebral ischemia in rats: evaluation with contrast-enhanced magnetic resonance imaging. J Cereb Blood Flow Metab. 2008;28:1491–1501. doi: 10.1038/jcbfm.2008.42. [DOI] [PubMed] [Google Scholar]
- Lindsberg PJ, Strbian D, Karjalainen-Lindsberg ML. Mast cells as early responders in the regulation of acute blood-brain barrier changes after cerebral ischemia and hemorrhage. J Cereb Blood Flow Metab. 2010;30:689–702. doi: 10.1038/jcbfm.2009.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, Shusta EV. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci Rep. 2014;4:4160. doi: 10.1038/srep04160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, Al-Ahmad A, Palecek SP, Shusta EV. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol. 2012;30:783–791. doi: 10.1038/nbt.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisabeth L, Bushnell C. Stroke risk in women: the role of menopause and hormone therapy. Lancet Neurol. 2012;11:82–91. doi: 10.1016/S1474-4422(11)70269-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Wu C, Yang Q, Gao J, Li L, Yang D, Luo L. Macrophages Mediate the Repair of Brain Vascular Rupture through Direct Physical Adhesion and Mechanical Traction. Immunity. 2016a;44:1162–1176. doi: 10.1016/j.immuni.2016.03.008. [DOI] [PubMed] [Google Scholar]
- Liu DZ, Ander BP, Xu H, Shen Y, Kaur P, Deng W, Sharp FR. Blood-brain barrier breakdown and repair by Src after thrombin-induced injury. Ann Neurol. 2010;67:526–533. doi: 10.1002/ana.21924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Wang Y, Xiao Y, Hua Z, Cheng J, Jia J. Hydrogen Sulfide Attenuates Tissue Plasminogen Activator-Induced Cerebral Hemorrhage Following Experimental Stroke. Transl Stroke Res. 2016b;7:209–219. doi: 10.1007/s12975-016-0459-5. [DOI] [PubMed] [Google Scholar]
- Liu HS, Chung HW, Chou MC, Liou M, Wang CY, Kao HW, Chiang SW, Juan CJ, Huang GS, Chen CY. Effects of microvascular permeability changes on contrast-enhanced T1 and pharmacokinetic MR imagings after ischemia. Stroke. 2013;44:1872–1877. doi: 10.1161/STROKEAHA.113.001558. [DOI] [PubMed] [Google Scholar]
- Liu J, Jin X, Liu KJ, Liu W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci. 2012;32:3044–3057. doi: 10.1523/JNEUROSCI.6409-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Wen Y, Perez E, Wang X, Day AL, Simpkins JW, Yang SH. 17beta-Estradiol attenuates blood-brain barrier disruption induced by cerebral ischemia-reperfusion injury in female rats. Brain Res. 2005;1060:55–61. doi: 10.1016/j.brainres.2005.08.048. [DOI] [PubMed] [Google Scholar]
- Liu Z, Chopp M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog Neurobiol. 2016;144:103–120. doi: 10.1016/j.pneurobio.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou N, Takano T, Pei Y, Xavier AL, Goldman SA, Nedergaard M. Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proc Natl Acad Sci U S A. 2016;113:1074–1079. doi: 10.1073/pnas.1520398113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan H, Kan Z, Xu Y, Lv C, Jiang W. Rosmarinic acid protects against experimental diabetes with cerebral ischemia: relation to inflammation response. J Neuroinflammation. 2013;10:28. doi: 10.1186/1742-2094-10-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucke-Wold BP, Logsdon AF, Turner RC, Rosen CL, Huber JD. Aging, the metabolic syndrome, and ischemic stroke: redefining the approach for studying the blood-brain barrier in a complex neurological disease. Adv Pharmacol. 2014;71:411–449. doi: 10.1016/bs.apha.2014.07.001. [DOI] [PubMed] [Google Scholar]
- Lyck R, Ruderisch N, Moll AG, Steiner O, Cohen CD, Engelhardt B, Makrides V, Verrey F. Culture-induced changes in blood-brain barrier transcriptome: implications for amino-acid transporters in vivo. J Cereb Blood Flow Metab. 2009;29:1491–1502. doi: 10.1038/jcbfm.2009.72. [DOI] [PubMed] [Google Scholar]
- Lynch JR, Pineda JA, Morgan D, Zhang L, Warner DS, Benveniste H, Laskowitz DT. Apolipoprotein E affects the central nervous system response to injury and the development of cerebral edema. Ann Neurol. 2002;51:113–117. doi: 10.1002/ana.10098. [DOI] [PubMed] [Google Scholar]
- Ma Y, Li L, Niu Z, Song J, Lin Y, Zhang H, Du G. Effect of recombinant plasminogen activator timing on thrombolysis in a novel rat embolic stroke model. Pharmacol Res. 2016;107:291–299. doi: 10.1016/j.phrs.2016.03.030. [DOI] [PubMed] [Google Scholar]
- Maggioli E, McArthur S, Mauro C, Kieswich J, Kusters DH, Reutelingsperger CP, Yaqoob M, Solito E. Estrogen protects the blood-brain barrier from inflammation-induced disruption and increased lymphocyte trafficking. Brain Behav Immun. 2016;51:212–222. doi: 10.1016/j.bbi.2015.08.020. [DOI] [PubMed] [Google Scholar]
- Mahringer A, Fricker G. ABC transporters at the blood-brain barrier. Expert Opin Drug Metab Toxicol. 2016;12:499–508. doi: 10.1517/17425255.2016.1168804. [DOI] [PubMed] [Google Scholar]
- Mallucci G, Peruzzotti-Jametti L, Bernstock JD, Pluchino S. The role of immune cells, glia and neurons in white and gray matter pathology in multiple sclerosis. Prog Neurobiol. 2015;127–128:1–22. doi: 10.1016/j.pneurobio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamo YA, Angus JA, Ziogas J, Soeding PF, Wright CE. The role of voltage-operated and non-voltage-operated calcium channels in endothelin-induced vasoconstriction of rat cerebral arteries. Eur J Pharmacol. 2014;742:65–73. doi: 10.1016/j.ejphar.2014.09.002. [DOI] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Marcos-Contreras OA, Martinez de Lizarrondo S, Bardou I, Orset C, Pruvost M, Anfray A, Frigout Y, Hommet Y, Lebouvier L, Montaner J, Vivien D, Gauberti M. Hyperfibrinolysis increases blood-brain barrier permeability by a plasmin- and bradykinin-dependent mechanism. Blood. 2016;128:2423–2434. doi: 10.1182/blood-2016-03-705384. [DOI] [PubMed] [Google Scholar]
- Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, Simmons D, Dejana E. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruo N, Morita I, Shirao M, Murota S. IL-6 increases endothelial permeability in vitro. Endocrinology. 1992;131:710–714. doi: 10.1210/endo.131.2.1639018. [DOI] [PubMed] [Google Scholar]
- Masrur S, Cox M, Bhatt DL, Smith EE, Ellrodt G, Fonarow GC, Schwamm L. Association of Acute and Chronic Hyperglycemia With Acute Ischemic Stroke Outcomes Post-Thrombolysis: Findings From Get With The Guidelines-Stroke. J Am Heart Assoc. 2015;4:e002193. doi: 10.1161/JAHA.115.002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP. The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia. 2010;58:1094–1103. doi: 10.1002/glia.20990. [DOI] [PubMed] [Google Scholar]
- Matsumoto J, Dohgu S, Takata F, Nishioku T, Sumi N, Machida T, Takahashi H, Yamauchi A, Kataoka Y. Lipopolysaccharide-activated microglia lower P-glycoprotein function in brain microvascular endothelial cells. Neurosci Lett. 2012;524:45–48. doi: 10.1016/j.neulet.2012.07.004. [DOI] [PubMed] [Google Scholar]
- McBride DW, Legrand J, Krafft PR, Flores J, Klebe D, Tang J, Zhang JH. Acute Hyperglycemia Is Associated with Immediate Brain Swelling and Hemorrhagic Transformation After Middle Cerebral Artery Occlusion in Rats. Acta Neurochir Suppl. 2016;121:237–241. doi: 10.1007/978-3-319-18497-5_42. [DOI] [PubMed] [Google Scholar]
- McCarron RM, Wang L, Racke MK, McFarlin DE, Spatz M. Cytokine-regulated adhesion between encephalitogenic T lymphocytes and cerebrovascular endothelial cells. J Neuroimmunol. 1993;43:23–30. doi: 10.1016/0165-5728(93)90071-6. [DOI] [PubMed] [Google Scholar]
- McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci. 2008;28:9451–9462. doi: 10.1523/JNEUROSCI.2674-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald DA, Shi C, Shenkar R, Stockton RA, Liu F, Ginsberg MH, Marchuk DA, Awad IA. Fasudil decreases lesion burden in a murine model of cerebral cavernous malformation disease. Stroke. 2012;43:571–574. doi: 10.1161/STROKEAHA.111.625467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina RJ, O’Neill CL, O’Doherty TM, Knott H, Guduric-Fuchs J, Gardiner TA, Stitt AW. Myeloid angiogenic cells act as alternative M2 macrophages and modulate angiogenesis through interleukin-8. Mol Med. 2011;17:1045–1055. doi: 10.2119/molmed.2011.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn ME, Karas RH. Estrogen and the blood vessel wall. Curr Opin Cardiol. 1994;9:619–626. doi: 10.1097/00001573-199409000-00018. [DOI] [PubMed] [Google Scholar]
- Methia N, Andre P, Hafezi-Moghadam A, Economopoulos M, Thomas KL, Wagner DD. ApoE deficiency compromises the blood brain barrier especially after injury. Mol Med. 2001;7:810–815. [PMC free article] [PubMed] [Google Scholar]
- Miguel-Hidalgo JJ, Nithuairisg S, Stockmeier C, Rajkowska G. Distribution of ICAM-1 immunoreactivity during aging in the human orbitofrontal cortex. Brain Behav Immun. 2007;21:100–111. doi: 10.1016/j.bbi.2006.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milatz S, Krug SM, Rosenthal R, Gunzel D, Muller D, Schulzke JD, Amasheh S, Fromm M. Claudin-3 acts as a sealing component of the tight junction for ions of either charge and uncharged solutes. Biochim Biophys Acta. 2010;1798:2048–2057. doi: 10.1016/j.bbamem.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Min LJ, Mogi M, Shudou M, Jing F, Tsukuda K, Ohshima K, Iwanami J, Horiuchi M. Peroxisome proliferator-activated receptor-gamma activation with angiotensin II type 1 receptor blockade is pivotal for the prevention of blood-brain barrier impairment and cognitive decline in type 2 diabetic mice. Hypertension. 2012;59:1079–1088. doi: 10.1161/HYPERTENSIONAHA.112.192401. [DOI] [PubMed] [Google Scholar]
- Mohammadi MT, Dehghani GA. Acute hypertension induces brain injury and blood-brain barrier disruption through reduction of claudins mRNA expression in rat. Pathol Res Pract. 2014;210:985–990. doi: 10.1016/j.prp.2014.05.007. [DOI] [PubMed] [Google Scholar]
- Moisan A, Favre IM, Rome C, Grillon E, Naegele B, Barbieux M, De Fraipont F, Richard MJ, Barbier EL, Remy C, Detante O. Microvascular plasticity after experimental stroke: a molecular and MRI study. Cerebrovasc Dis. 2014;38:344–353. doi: 10.1159/000368597. [DOI] [PubMed] [Google Scholar]
- Moller K, Posel C, Kranz A, Schulz I, Scheibe J, Didwischus N, Boltze J, Weise G, Wagner DC. Arterial Hypertension Aggravates Innate Immune Responses after Experimental Stroke. Front Cell Neurosci. 2015;9:461. doi: 10.3389/fncel.2015.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic BV. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296–302. doi: 10.1016/j.neuron.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monvoisin A, Alva JA, Hofmann JJ, Zovein AC, Lane TF, Iruela-Arispe ML. VE-cadherin-CreERT2 transgenic mouse: a model for inducible recombination in the endothelium. Dev Dyn. 2006;235:3413–3422. doi: 10.1002/dvdy.20982. [DOI] [PubMed] [Google Scholar]
- Mooradian AD. Blood-brain barrier transport of choline is reduced in the aged rat. Brain Res. 1988a;440:328–332. doi: 10.1016/0006-8993(88)91002-5. [DOI] [PubMed] [Google Scholar]
- Mooradian AD. Effect of aging on the blood-brain barrier. Neurobiol Aging. 1988b;9:31–39. doi: 10.1016/s0197-4580(88)80013-7. [DOI] [PubMed] [Google Scholar]
- Mooradian AD, Haas MJ, Batejko O, Hovsepyan M, Feman SS. Statins ameliorate endothelial barrier permeability changes in the cerebral tissue of streptozotocin-induced diabetic rats. Diabetes. 2005;54:2977–2982. doi: 10.2337/diabetes.54.10.2977. [DOI] [PubMed] [Google Scholar]
- Mooradian AD, Meredith KE. The effect of age on protein composition of rat cerebral microvessels. Neurochem Res. 1992;17:665–670. doi: 10.1007/BF00968002. [DOI] [PubMed] [Google Scholar]
- Mooradian AD, Smith TL. The effect of experimentally induced diabetes mellitus on the lipid order and composition of rat cerebral microvessels. Neurosci Lett. 1992;145:145–148. doi: 10.1016/0304-3940(92)90007-t. [DOI] [PubMed] [Google Scholar]
- Mori M, Tsukahara F, Yoshioka T, Irie K, Ohta H. Suppression by 17beta-estradiol of monocyte adhesion to vascular endothelial cells is mediated by estrogen receptors. Life Sci. 2004;75:599–609. doi: 10.1016/j.lfs.2003.12.023. [DOI] [PubMed] [Google Scholar]
- Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, American Heart Association Statistics, C., Stroke Statistics, S Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38–360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- Mun-Bryce S, Rosenberg GA. Matrix metalloproteinases in cerebrovascular disease. J Cereb Blood Flow Metab. 1998;18:1163–1172. doi: 10.1097/00004647-199811000-00001. [DOI] [PubMed] [Google Scholar]
- Murakami T, Felinski EA, Antonetti DA. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J Biol Chem. 2009;284:21036–21046. doi: 10.1074/jbc.M109.016766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Frey T, Lin C, Antonetti DA. Protein kinase cbeta phosphorylates occludin regulating tight junction trafficking in vascular endothelial growth factor-induced permeability in vivo. Diabetes. 2012;61:1573–1583. doi: 10.2337/db11-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na W, Lee JY, Kim WS, Yune TY, Ju BG. 17beta-Estradiol Ameliorates Tight Junction Disruption via Repression of MMP Transcription. Mol Endocrinol. 2015;29:1347–1361. doi: 10.1210/ME.2015-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag S, Kapadia A, Stewart DJ. Review: molecular pathogenesis of blood-brain barrier breakdown in acute brain injury. Neuropathol Appl Neurobiol. 2011;37:3–23. doi: 10.1111/j.1365-2990.2010.01138.x. [DOI] [PubMed] [Google Scholar]
- Nag S, Venugopalan R, Stewart DJ. Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta Neuropathol. 2007;114:459–469. doi: 10.1007/s00401-007-0274-x. [DOI] [PubMed] [Google Scholar]
- Nagai M, Terao S, Vital SA, Rodrigues SF, Yilmaz G, Granger DN. Role of blood cell-associated angiotensin II type 1 receptors in the cerebral microvascular response to ischemic stroke during angiotensin-induced hypertension. Exp Transl Stroke Med. 2011;3:15. doi: 10.1186/2040-7378-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraja TN, Karki K, Ewing JR, Croxen RL, Knight RA. Identification of variations in blood-brain barrier opening after cerebral ischemia by dual contrast-enhanced magnetic resonance imaging and T 1sat measurements. Stroke. 2008;39:427–432. doi: 10.1161/STROKEAHA.107.496059. [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Ottersen OP. Physiological roles of aquaporin-4 in brain. Physiol Rev. 2013;93:1543–1562. doi: 10.1152/physrev.00011.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000;26:99–109. [PubMed] [Google Scholar]
- Nahirney PC, Reeson P, Brown CE. Ultrastructural analysis of blood-brain barrier breakdown in the peri-infarct zone in young adult and aged mice. J Cereb Blood Flow Metab. 2016;36:413–425. doi: 10.1177/0271678X15608396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik P, Cucullo L. In vitro blood-brain barrier models: current and perspective technologies. J Pharm Sci. 2012;101:1337–1354. doi: 10.1002/jps.23022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagomi T, Kubo S, Nakano-Doi A, Sakuma R, Lu S, Narita A, Kawahara M, Taguchi A, Matsuyama T. Brain vascular pericytes following ischemia have multipotential stem cell activity to differentiate into neural and vascular lineage cells. Stem Cells. 2015;33:1962–1974. doi: 10.1002/stem.1977. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Arimura K, Nishimura A, Tachibana M, Yoshikawa Y, Makihara N, Wakisaka Y, Kuroda J, Kamouchi M, Ooboshi H, Kitazono T, Ago T. Possible involvement of basic FGF in the upregulation of PDGFRbeta in pericytes after ischemic stroke. Brain Res. 2016;1630:98–108. doi: 10.1016/j.brainres.2015.11.003. [DOI] [PubMed] [Google Scholar]
- Nelson RH. Hyperlipidemia as a risk factor for cardiovascular disease. Prim Care. 2013;40:195–211. doi: 10.1016/j.pop.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus J, Risau W, Wolburg H. Induction of blood-brain barrier characteristics in bovine brain endothelial cells by rat astroglial cells in transfilter coculture. Ann N Y Acad Sci. 1991;633:578–580. doi: 10.1111/j.1749-6632.1991.tb15667.x. [DOI] [PubMed] [Google Scholar]
- Ng KF, Anderson S, Mayo P, Aung HH, Walton JH, Rutledge JC. Characterizing blood-brain barrier perturbations after exposure to human triglyceride-rich lipoprotein lipolysis products using MRI in a rat model. Magn Reson Med. 2016;76:1246–1251. doi: 10.1002/mrm.25985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HX, O’Barr TJ, Anderson AJ. Polymorphonuclear leukocytes promote neurotoxicity through release of matrix metalloproteinases, reactive oxygen species, and TNF-alpha. J Neurochem. 2007;102:900–912. doi: 10.1111/j.1471-4159.2007.04643.x. [DOI] [PubMed] [Google Scholar]
- Nguyen M, Arkell J, Jackson CJ. Thrombin rapidly and efficiently activates gelatinase A in human microvascular endothelial cells via a mechanism independent of active MT1 matrix metalloproteinase. Lab Invest. 1999;79:467–475. [PubMed] [Google Scholar]
- Nishioku T, Matsumoto J, Dohgu S, Sumi N, Miyao K, Takata F, Shuto H, Yamauchi A, Kataoka Y. Tumor necrosis factor-alpha mediates the blood-brain barrier dysfunction induced by activated microglia in mouse brain microvascular endothelial cells. J Pharmacol Sci. 2010;112:251–254. doi: 10.1254/jphs.09292sc. [DOI] [PubMed] [Google Scholar]
- Nishitsuji K, Hosono T, Nakamura T, Bu G, Michikawa M. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J Biol Chem. 2011;286:17536–17542. doi: 10.1074/jbc.M111.225532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell ME. Blood-brain barrier Na transporters in ischemic stroke. Adv Pharmacol. 2014;71:113–146. doi: 10.1016/bs.apha.2014.06.011. [DOI] [PubMed] [Google Scholar]
- O’Donnell ME, Chen YJ, Lam TI, Taylor KC, Walton JH, Anderson SE. Intravenous HOE-642 reduces brain edema and Na uptake in the rat permanent middle cerebral artery occlusion model of stroke: evidence for participation of the blood-brain barrier Na/H exchanger. J Cereb Blood Flow Metab. 2013;33:225–234. doi: 10.1038/jcbfm.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell ME, Lam TI, Tran LQ, Foroutan S, Anderson SE. Estradiol reduces activity of the blood-brain barrier Na-K-Cl cotransporter and decreases edema formation in permanent middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2006;26:1234–1249. doi: 10.1038/sj.jcbfm.9600278. [DOI] [PubMed] [Google Scholar]
- O’Donnell ME, Tran L, Lam TI, Liu XB, Anderson SE. Bumetanide inhibition of the blood-brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J Cereb Blood Flow Metab. 2004;24:1046–1056. doi: 10.1097/01.WCB.0000130867.32663.90. [DOI] [PubMed] [Google Scholar]
- Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19:1584–1596. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offner H, Hurn PD. A novel hypothesis: regulatory B lymphocytes shape outcome from experimental stroke. Transl Stroke Res. 2012;3:324–330. doi: 10.1007/s12975-012-0187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Kikuchi T, Okamura T, Ikoma Y, Tsuji AB, Wakizaka H, Kamakura T, Aoki I, Zhang MR, Kato K. In-vivo imaging of blood-brain barrier permeability using positron emission tomography with 2-amino-[3–11C]isobutyric acid. Nucl Med Commun. 2015a;36:1239–1248. doi: 10.1097/MNM.0000000000000385. [DOI] [PubMed] [Google Scholar]
- Okada R, Wu Z, Zhu A, Ni J, Zhang J, Yoshimine Y, Peters C, Saftig P, Nakanishi H. Cathepsin D deficiency induces oxidative damage in brain pericytes and impairs the blood-brain barrier. Mol Cell Neurosci. 2015b;64:51–60. doi: 10.1016/j.mcn.2014.12.002. [DOI] [PubMed] [Google Scholar]
- Oldendorf WH, Cornford ME, Brown WJ. The large apparent work capability of the blood-brain barrier: a study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol. 1977;1:409–417. doi: 10.1002/ana.410010502. [DOI] [PubMed] [Google Scholar]
- Osborn LM, Kamphuis W, Wadman WJ, Hol EM. Astrogliosis: An integral player in the pathogenesis of Alzheimer’s disease. Prog Neurobiol. 2016;144:121–141. doi: 10.1016/j.pneurobio.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Ostermann G, Fraemohs L, Baltus T, Schober A, Lietz M, Zernecke A, Liehn EA, Weber C. Involvement of JAM-A in mononuclear cell recruitment on inflamed or atherosclerotic endothelium: inhibition by soluble JAM-A. Arterioscler Thromb Vasc Biol. 2005;25:729–735. doi: 10.1161/01.ATV.0000157154.14474.3b. [DOI] [PubMed] [Google Scholar]
- Ozen I, Deierborg T, Miharada K, Padel T, Englund E, Genove G, Paul G. Brain pericytes acquire a microglial phenotype after stroke. Acta Neuropathol. 2014;128:381–396. doi: 10.1007/s00401-014-1295-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, He C, Liu H, Liao X, Dai B, Chen Y, Yang Y, Zhao B, Bihl J, Ma X. Microvascular endothelial cells-derived microvesicles imply in ischemic stroke by modulating astrocyte and blood brain barrier function and cerebral blood flow. Mol Brain. 2016;9:63. doi: 10.1186/s13041-016-0243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelisch N, Hosomi N, Mori H, Masaki T, Nishiyama A. RAS inhibition attenuates cognitive impairment by reducing blood- brain barrier permeability in hypertensive subjects. Curr Hypertens Rev. 2013;9:93–98. doi: 10.2174/15734021113099990003. [DOI] [PubMed] [Google Scholar]
- Pena I, Borlongan CV. Translating G-CSF as an Adjunct Therapy to Stem Cell Transplantation for Stroke. Transl Stroke Res. 2015;6:421–429. doi: 10.1007/s12975-015-0430-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Heilman D, Haorah J, Zelivyanskaya M, Persidsky R, Weber GA, Shimokawa H, Kaibuchi K, Ikezu T. Rho-mediated regulation of tight junctions during monocyte migration across the blood-brain barrier in HIV-1 encephalitis (HIVE) Blood. 2006;107:4770–4780. doi: 10.1182/blood-2005-11-4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Hill J, Zhang M, Dykstra H, Winfield M, Reichenbach NL, Potula R, Mukherjee A, Ramirez SH, Rom S. Dysfunction of brain pericytes in chronic neuroinflammation. J Cereb Blood Flow Metab. 2016;36:794–807. doi: 10.1177/0271678X15606149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson EC, Wang Z, Britz G. Regulation of cerebral blood flow. Int J Vasc Med. 2011;2011:823525. doi: 10.1155/2011/823525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, Miranda C, Bedoya EA, Fischer KD, Armsen W, Wang J, Danbolt NC, Rotenberg A, Aoki CJ, Rosenberg PA. Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci. 2015;35:5187–5201. doi: 10.1523/JNEUROSCI.4255-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovic-Djergovic D, Goonewardena SN, Pinsky DJ. Inflammatory Disequilibrium in Stroke. Circ Res. 2016;119:142–158. doi: 10.1161/CIRCRESAHA.116.308022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J, Laude A, Lightowlers R, Morris CM, Turnbull DM, Lax NZ. Development of passive CLARITY and immunofluorescent labelling of multiple proteins in human cerebellum: understanding mechanisms of neurodegeneration in mitochondrial disease. Sci Rep. 2016;6:26013. doi: 10.1038/srep26013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- Poggesi A, Pasi M, Pescini F, Pantoni L, Inzitari D. Circulating biologic markers of endothelial dysfunction in cerebral small vessel disease: A review. J Cereb Blood Flow Metab. 2016;36:72–94. doi: 10.1038/jcbfm.2015.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulet R, Gentile MT, Vecchione C, Distaso M, Aretini A, Fratta L, Russo G, Echart C, Maffei A, De Simoni MG, Lembo G. Acute hypertension induces oxidative stress in brain tissues. J Cereb Blood Flow Metab. 2006;26:253–262. doi: 10.1038/sj.jcbfm.9600188. [DOI] [PubMed] [Google Scholar]
- Poungvarin N. Steroids have no role in stroke therapy. Stroke. 2004;35:229–230. doi: 10.1161/01.STR.0000105931.81723.26. [DOI] [PubMed] [Google Scholar]
- Prakash R, Carmichael ST. Blood-brain barrier breakdown and neovascularization processes after stroke and traumatic brain injury. Curr Opin Neurol. 2015;28:556–564. doi: 10.1097/WCO.0000000000000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad S, Sajja RK, Naik P, Cucullo L. Diabetes Mellitus and Blood-Brain Barrier Dysfunction: An Overview. J Pharmacovigil. 2014;2:125. doi: 10.4172/2329-6887.1000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu H, Jiang X, Hu X, Xia J, Hong D, Zhang W, Gao Y, Chen J, Shi Y. Delayed Docosahexaenoic Acid Treatment Combined with Dietary Supplementation of Omega-3 Fatty Acids Promotes Long-Term Neurovascular Restoration After Ischemic Stroke. Transl Stroke Res. 2016;7:521–534. doi: 10.1007/s12975-016-0498-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Z, Liang J, Pan R, Dong W, Shen J, Yang Y, Zhao Y, Shi W, Luo Y, Ji X, Liu KJ. Zinc contributes to acute cerebral ischemia-induced blood-brain barrier disruption. Neurobiol Dis. 2016;95:12–21. doi: 10.1016/j.nbd.2016.07.003. [DOI] [PubMed] [Google Scholar]
- Raiteri L, Raiteri M. Multiple functions of neuronal plasma membrane neurotransmitter transporters. Prog Neurobiol. 2015;134:1–16. doi: 10.1016/j.pneurobio.2015.08.002. [DOI] [PubMed] [Google Scholar]
- Rapoport SI. Osmotic opening of the blood-brain barrier: principles, mechanism, and therapeutic applications. Cell Mol Neurobiol. 2000;20:217–230. doi: 10.1023/A:1007049806660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeson P, Tennant KA, Gerrow K, Wang J, Weiser Novak S, Thompson K, Lockhart KL, Holmes A, Nahirney PC, Brown CE. Delayed inhibition of VEGF signaling after stroke attenuates blood-brain barrier breakdown and improves functional recovery in a comorbidity-dependent manner. J Neurosci. 2015;35:5128–5143. doi: 10.1523/JNEUROSCI.2810-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempe RG, Hartz AM, Bauer B. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J Cereb Blood Flow Metab. 2016;36:1481–1507. doi: 10.1177/0271678X16655551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner O, Tsimpas A, Kostin S, Valable S, Petit E, Schaper W, Marti HH. Time- and cell type-specific induction of platelet-derived growth factor receptor-beta during cerebral ischemia. Brain Res Mol Brain Res. 2003;113:44–51. doi: 10.1016/s0169-328x(03)00085-8. [DOI] [PubMed] [Google Scholar]
- Reuter B, Rodemer C, Grudzenski S, Couraud PO, Weksler B, Romero IA, Meairs S, Bugert P, Hennerici MG, Fatar M. Temporal profile of matrix metalloproteinases and their inhibitors in a human endothelial cell culture model of cerebral ischemia. Cerebrovasc Dis. 2013;35:514–520. doi: 10.1159/000350731. [DOI] [PubMed] [Google Scholar]
- Reuter B, Rodemer C, Grudzenski S, Meairs S, Bugert P, Hennerici MG, Fatar M. Effect of simvastatin on MMPs and TIMPs in human brain endothelial cells and experimental stroke. Transl Stroke Res. 2015;6:156–159. doi: 10.1007/s12975-014-0381-7. [DOI] [PubMed] [Google Scholar]
- Ridder DA, Lang MF, Salinin S, Roderer JP, Struss M, Maser-Gluth C, Schwaninger M. TAK1 in brain endothelial cells mediates fever and lethargy. J Exp Med. 2011;208:2615–2623. doi: 10.1084/jem.20110398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochfort KD, Cummins PM. The blood-brain barrier endothelium: a target for pro-inflammatory cytokines. Biochem Soc Trans. 2015;43:702–706. doi: 10.1042/BST20140319. [DOI] [PubMed] [Google Scholar]
- Rodrigues SF, Granger DN. Blood cells and endothelial barrier function. Tissue Barriers. 2015;3:e978720. doi: 10.4161/21688370.2014.978720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez PL, Jiang S, Fu Y, Avraham S, Avraham HK. The proinflammatory peptide substance P promotes blood-brain barrier breaching by breast cancer cells through changes in microvascular endothelial cell tight junctions. Int J Cancer. 2014;134:1034–1044. doi: 10.1002/ijc.28433. [DOI] [PubMed] [Google Scholar]
- Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29:1020–1030. doi: 10.1161/01.str.29.5.1020. [DOI] [PubMed] [Google Scholar]
- Rosell A, Ortega-Aznar A, Alvarez-Sabin J, Fernandez-Cadenas I, Ribo M, Molina CA, Lo EH, Montaner J. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke. 2006;37:1399–1406. doi: 10.1161/01.STR.0000223001.06264.af. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA. Ischemic brain edema. Prog Cardiovasc Dis. 1999;42:209–216. doi: 10.1016/s0033-0620(99)70003-4. [DOI] [PubMed] [Google Scholar]
- Rosenthal R, Milatz S, Krug SM, Oelrich B, Schulzke JD, Amasheh S, Gunzel D, Fromm M. Claudin-2, a component of the tight junction, forms a paracellular water channel. J Cell Sci. 2010;123:1913–1921. doi: 10.1242/jcs.060665. [DOI] [PubMed] [Google Scholar]
- Rossi D. Astrocyte physiopathology: At the crossroads of intercellular networking, inflammation and cell death. Prog Neurobiol. 2015;130:86–120. doi: 10.1016/j.pneurobio.2015.04.003. [DOI] [PubMed] [Google Scholar]
- Roussel BD, Macrez R, Jullienne A, Agin V, Maubert E, Dauphinot L, Potier MC, Plawinski L, Castel H, Hommet Y, Munuera J, Montaner J, Yepes M, Ali C, Vivien D. Age and albumin D site-binding protein control tissue plasminogen activator levels: neurotoxic impact. Brain. 2009;132:2219–2230. doi: 10.1093/brain/awp162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio RG, Adamis AP. Ocular Angiogenesis: Vascular Endothelial Growth Factor and Other Factors. Dev Ophthalmol. 2016;55:28–37. doi: 10.1159/000431129. [DOI] [PubMed] [Google Scholar]
- Ruck T, Bittner S, Meuth SG. Blood-brain barrier modeling: challenges and perspectives. Neural Regen Res. 2015;10:889–891. doi: 10.4103/1673-5374.158342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowsky JM, Wallace BK, Wise PM, O’Donnell ME. Effects of estradiol on ischemic factor-induced astrocyte swelling and AQP4 protein abundance. Am J Physiol Cell Physiol. 2011;301:C204–212. doi: 10.1152/ajpcell.00399.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rymo SF, Gerhardt H, Wolfhagen Sand F, Lang R, Uv A, Betsholtz C. A two-way communication between microglial cells and angiogenic sprouts regulates angiogenesis in aortic ring cultures. PLoS One. 2011;6:e15846. doi: 10.1371/journal.pone.0015846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, Noda T, Tsukita S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell. 2000;11:4131–4142. doi: 10.1091/mbc.11.12.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma R, Kawahara M, Nakano-Doi A, Takahashi A, Tanaka Y, Narita A, Kuwahara-Otani S, Hayakawa T, Yagi H, Matsuyama T, Nakagomi T. Brain pericytes serve as microglia-generating multipotent vascular stem cells following ischemic stroke. J Neuroinflammation. 2016;13:57. doi: 10.1186/s12974-016-0523-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salameh TS, Banks WA. Delivery of therapeutic peptides and proteins to the CNS. Adv Pharmacol. 2014;71:277–299. doi: 10.1016/bs.apha.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayner SL, Alexeyev M, Dessauer CW, Stevens T. Soluble adenylyl cyclase reveals the significance of cAMP compartmentation on pulmonary microvascular endothelial cell barrier. Circ Res. 2006;98:675–681. doi: 10.1161/01.RES.0000209516.84815.3e. [DOI] [PubMed] [Google Scholar]
- Scherer PE, Lewis RY, Volonte D, Engelman JA, Galbiati F, Couet J, Kohtz DS, van Donselaar E, Peters P, Lisanti MP. Cell-type and tissue-specific expression of caveolin-2. Caveolins 1 and 2 co-localize and form a stable hetero-oligomeric complex in vivo. J Biol Chem. 1997;272:29337–29346. doi: 10.1074/jbc.272.46.29337. [DOI] [PubMed] [Google Scholar]
- Scherer PE, Okamoto T, Chun M, Nishimoto I, Lodish HF, Lisanti MP. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc Natl Acad Sci U S A. 1996;93:131–135. doi: 10.1073/pnas.93.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnoor M, Parkos CA. Disassembly of endothelial and epithelial junctions during leukocyte transmigration. Front Biosci. 2008;13:6638–6652. doi: 10.2741/3178. [DOI] [PubMed] [Google Scholar]
- Schreibelt G, Kooij G, Reijerkerk A, van Doorn R, Gringhuis SI, van der Pol S, Weksler BB, Romero IA, Couraud PO, Piontek J, Blasig IE, Dijkstra CD, Ronken E, de Vries HE. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007;21:3666–3676. doi: 10.1096/fj.07-8329com. [DOI] [PubMed] [Google Scholar]
- Schreihofer DA, Ma Y. Estrogen receptors and ischemic neuroprotection: who, what, where, and when? Brain Res. 2013;1514:107–122. doi: 10.1016/j.brainres.2013.02.051. [DOI] [PubMed] [Google Scholar]
- Schuhmann MK, Kraft P, Stoll G, Lorenz K, Meuth SG, Wiendl H, Nieswandt B, Sparwasser T, Beyersdorf N, Kerkau T, Kleinschnitz C. CD28 superagonist-mediated boost of regulatory T cells increases thrombo-inflammation and ischemic neurodegeneration during the acute phase of experimental stroke. J Cereb Blood Flow Metab. 2015;35:6–10. doi: 10.1038/jcbfm.2014.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scuteri A, Nilsson PM, Tzourio C, Redon J, Laurent S. Microvascular brain damage with aging and hypertension: pathophysiological consideration and clinical implications. J Hypertens. 2011;29:1469–1477. doi: 10.1097/HJH.0b013e328347cc17. [DOI] [PubMed] [Google Scholar]
- Shao B, Bayraktutan U. Hyperglycaemia promotes cerebral barrier dysfunction through activation of protein kinase C-beta. Diabetes Obes Metab. 2013;15:993–999. doi: 10.1111/dom.12120. [DOI] [PubMed] [Google Scholar]
- Shen S, Zhang W. ABC transporters and drug efflux at the blood-brain barrier. Rev Neurosci. 2010;21:29–53. doi: 10.1515/revneuro.2010.21.1.29. [DOI] [PubMed] [Google Scholar]
- Shi Y, Jiang X, Zhang L, Pu H, Hu X, Zhang W, Cai W, Gao Y, Leak RK, Keep RF, Bennett MV, Chen J. Endothelium-targeted overexpression of heat shock protein 27 ameliorates blood-brain barrier disruption after ischemic brain injury. Proc Natl Acad Sci U S A. 2017;114:E1243–E1252. doi: 10.1073/pnas.1621174114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Zhang L, Pu H, Mao L, Hu X, Jiang X, Xu N, Stetler RA, Zhang F, Liu X, Leak RK, Keep RF, Ji X, Chen J. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat Commun. 2016;7:10523. doi: 10.1038/ncomms10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HK, Huang PL, Ayata C. Rho-kinase inhibition improves ischemic perfusion deficit in hyperlipidemic mice. J Cereb Blood Flow Metab. 2014;34:284–287. doi: 10.1038/jcbfm.2013.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JA, Yoon JC, Kim M, Park EM. Activation of classical estrogen receptor subtypes reduces tight junction disruption of brain endothelial cells under ischemia/reperfusion injury. Free Radic Biol Med. 2016;92:78–89. doi: 10.1016/j.freeradbiomed.2016.01.010. [DOI] [PubMed] [Google Scholar]
- Siddiqui MR, Mayanil CS, Kim KS, Tomita T. Angiopoietin-1 Regulates Brain Endothelial Permeability through PTPN-2 Mediated Tyrosine Dephosphorylation of Occludin. PLoS One. 2015;10:e0130857. doi: 10.1371/journal.pone.0130857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascular interface. J Neurosci. 2003;23:9254–9262. doi: 10.1523/JNEUROSCI.23-27-09254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simionescu M, Popov D, Sima A. Endothelial transcytosis in health and disease. Cell Tissue Res. 2009;335:27–40. doi: 10.1007/s00441-008-0688-3. [DOI] [PubMed] [Google Scholar]
- Simpkins AN, Dias C, Leigh R, National Institutes of Health Natural History of Stroke, I Identification of Reversible Disruption of the Human Blood-Brain Barrier Following Acute Ischemia. Stroke. 2016;47:2405–2408. doi: 10.1161/STROKEAHA.116.013805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladojevic N, Stamatovic SM, Keep RF, Grailer JJ, Sarma JV, Ward PA, Andjelkovic AV. Inhibition of junctional adhesion molecule-A/LFA interaction attenuates leukocyte trafficking and inflammation in brain ischemia/reperfusion injury. Neurobiol Dis. 2014;67:57–70. doi: 10.1016/j.nbd.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soejima Y, Ostrowski RP, Manaenko A, Fujii M, Tang J, Zhang JH. Hyperbaric oxygen preconditioning attenuates hyperglycemia enhanced hemorrhagic transformation after transient MCAO in rats. Med Gas Res. 2012;2:9. doi: 10.1186/2045-9912-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohet F, Daneman R. Genetic mouse models to study blood-brain barrier development and function. Fluids Barriers CNS. 2013;10:3. doi: 10.1186/2045-8118-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soma T, Chiba H, Kato-Mori Y, Wada T, Yamashita T, Kojima T, Sawada N. Thr(207) of claudin-5 is involved in size-selective loosening of the endothelial barrier by cyclic AMP. Exp Cell Res. 2004;300:202–212. doi: 10.1016/j.yexcr.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Song KS, Scherer PE, Tang Z, Okamoto T, Li S, Chafel M, Chu C, Kohtz DS, Lisanti MP. Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J Biol Chem. 1996;271:15160–15165. doi: 10.1074/jbc.271.25.15160. [DOI] [PubMed] [Google Scholar]
- Song L, Ge S, Pachter JS. Caveolin-1 regulates expression of junction-associated proteins in brain microvascular endothelial cells. Blood. 2007;109:1515–1523. doi: 10.1182/blood-2006-07-034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Walczak P, He X, Yang X, Pearl M, Bulte JW, Pomper MG, McMahon MT, Janowski M. Salicylic acid analogues as chemical exchange saturation transfer MRI contrast agents for the assessment of brain perfusion territory and blood-brain barrier opening after intra-arterial infusion. J Cereb Blood Flow Metab. 2016;36:1186–1194. doi: 10.1177/0271678X16637882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatovic SM, Dimitrijevic OB, Keep RF, Andjelkovic AV. Protein kinase Calpha-RhoA cross-talk in CCL2-induced alterations in brain endothelial permeability. J Biol Chem. 2006;281:8379–8388. doi: 10.1074/jbc.M513122200. [DOI] [PubMed] [Google Scholar]
- Stamatovic SM, Johnson AM, Keep RF, Andjelkovic AV. Junctional proteins of the blood-brain barrier: New insights into function and dysfunction. Tissue Barriers. 2016;4:e1154641. doi: 10.1080/21688370.2016.1154641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatovic SM, Johnson AM, Sladojevic N, Keep RF, Andjelkovic AV. Endocytosis of tight junction proteins and the regulation of degradation and recycling. Ann N Y Acad Sci. 2017 doi: 10.1111/nyas.13346. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatovic SM, Keep RF, Wang MM, Jankovic I, Andjelkovic AV. Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. J Biol Chem. 2009;284:19053–19066. doi: 10.1074/jbc.M109.000521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatovic SM, Sladojevic N, Keep RF, Andjelkovic AV. Relocalization of junctional adhesion molecule A during inflammatory stimulation of brain endothelial cells. Mol Cell Biol. 2012;32:3414–3427. doi: 10.1128/MCB.06678-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, von Bruhl ML, Gartner F, Khandoga AG, Legate KR, Pless R, Hepper I, Lauber K, Walzog B, Massberg S. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and ‘instruct’ them with pattern-recognition and motility programs. Nat Immunol. 2013;14:41–51. doi: 10.1038/ni.2477. [DOI] [PubMed] [Google Scholar]
- Starr JM, Wardlaw J, Ferguson K, MacLullich A, Deary IJ, Marshall I. Increased blood-brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J Neurol Neurosurg Psychiatry. 2003;74:70–76. doi: 10.1136/jnnp.74.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockton RA, Shenkar R, Awad IA, Ginsberg MH. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J Exp Med. 2010;207:881–896. doi: 10.1084/jem.20091258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokum JA, Gerzanich V, Simard JM. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab. 2016;36:513–538. doi: 10.1177/0271678X15617172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Hao S, Dey A, Yu X, Baban B. Blood-brain barrier breakdown promotes macrophage infiltration and cognitive impairment in leptin receptor-deficient mice. J Cereb Blood Flow Metab. 2016;36:2108–2121. doi: 10.1177/0271678X16642233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strbian D, Durukan A, Pitkonen M, Marinkovic I, Tatlisumak E, Pedrono E, Abo-Ramadan U, Tatlisumak T. The blood-brain barrier is continuously open for several weeks following transient focal cerebral ischemia. Neuroscience. 2008;153:175–181. doi: 10.1016/j.neuroscience.2008.02.012. [DOI] [PubMed] [Google Scholar]
- Strecker JK, Minnerup J, Schutte-Nutgen K, Gess B, Schabitz WR, Schilling M. Monocyte chemoattractant protein-1-deficiency results in altered blood-brain barrier breakdown after experimental stroke. Stroke. 2013;44:2536–2544. doi: 10.1161/STROKEAHA.111.000528. [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Ishibashi T, Sawamura T, Inoue N, Kamioka M, Uekita H, Ohkawara H, Sakamoto T, Sakamoto N, Okamoto Y, Takuwa Y, Kakino A, Fujita Y, Tanaka T, Teramoto T, Maruyama Y, Takeishi Y. LOX-1-MT1-MMP axis is crucial for RhoA and Rac1 activation induced by oxidized low-density lipoprotein in endothelial cells. Cardiovasc Res. 2009;84:127–136. doi: 10.1093/cvr/cvp177. [DOI] [PubMed] [Google Scholar]
- Sumi N, Nishioku T, Takata F, Matsumoto J, Watanabe T, Shuto H, Yamauchi A, Dohgu S, Kataoka Y. Lipopolysaccharide-activated microglia induce dysfunction of the blood-brain barrier in rat microvascular endothelial cells co-cultured with microglia. Cell Mol Neurobiol. 2010;30:247–253. doi: 10.1007/s10571-009-9446-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumii T, Lo EH. Involvement of matrix metalloproteinase in thrombolysis-associated hemorrhagic transformation after embolic focal ischemia in rats. Stroke. 2002;33:831–836. doi: 10.1161/hs0302.104542. [DOI] [PubMed] [Google Scholar]
- Sun YN, Liu LB, Xue YX, Wang P. Effects of insulin combined with idebenone on blood-brain barrier permeability in diabetic rats. J Neurosci Res. 2015;93:666–677. doi: 10.1002/jnr.23511. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Nagai N, Umemura K. A Review of the Mechanisms of Blood-Brain Barrier Permeability by Tissue-Type Plasminogen Activator Treatment for Cerebral Ischemia. Front Cell Neurosci. 2016;10:2. doi: 10.3389/fncel.2016.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykova E. Glial diffusion barriers during aging and pathological states. Prog Brain Res. 2001;132:339–363. doi: 10.1016/S0079-6123(01)32087-3. [DOI] [PubMed] [Google Scholar]
- Syvanen S, Eriksson J. Advances in PET imaging of P-glycoprotein function at the blood-brain barrier. ACS Chem Neurosci. 2013;4:225–237. doi: 10.1021/cn3001729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaga Y, Takagi N, Murotomi K, Tanonaka K, Takeo S. Inhibition of Src activity decreases tyrosine phosphorylation of occludin in brain capillaries and attenuates increase in permeability of the blood-brain barrier after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2009;29:1099–1108. doi: 10.1038/jcbfm.2009.30. [DOI] [PubMed] [Google Scholar]
- Tayebati SK, Tomassoni D, Amenta F. Neuroinflammatory Markers in Spontaneously Hypertensive Rat Brain: An Immunohistochemical Study. CNS Neurol Disord Drug Targets. 2016;15:995–1000. doi: 10.2174/1871527315666160527155014. [DOI] [PubMed] [Google Scholar]
- Tenreiro MM, Ferreira R, Bernardino L, Brito MA. Cellular response of the blood-brain barrier to injury: Potential biomarkers and therapeutic targets for brain regeneration. Neurobiol Dis. 2016;91:262–273. doi: 10.1016/j.nbd.2016.03.014. [DOI] [PubMed] [Google Scholar]
- Theriault P, Le Behot A, ElAli A, Rivest S. Sub-acute systemic erythropoietin administration reduces ischemic brain injury in an age-dependent manner. Oncotarget. 2016;7:35552–35561. doi: 10.18632/oncotarget.9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R, Luo Y, Liu Q, Cai M, Li J, Sun W, Wang J, He C, Liu Y, Liu X. The effect of claudin-5 overexpression on the interactions of claudin-1 and -2 and barrier function in retinal cells. Curr Mol Med. 2014;14:1226–1237. doi: 10.2174/1566524014666141015160355. [DOI] [PubMed] [Google Scholar]
- Toh YC, Zhang C, Zhang J, Khong YM, Chang S, Samper VD, van Noort D, Hutmacher DW, Yu H. A novel 3D mammalian cell perfusion-culture system in microfluidic channels. Lab Chip. 2007;7:302–309. doi: 10.1039/b614872g. [DOI] [PubMed] [Google Scholar]
- Tomassoni D, Bramanti V, Amenta F. Expression of aquaporins 1 and 4 in the brain of spontaneously hypertensive rats. Brain Res. 2010;1325:155–163. doi: 10.1016/j.brainres.2010.02.023. [DOI] [PubMed] [Google Scholar]
- Tourdias T, Mori N, Dragonu I, Cassagno N, Boiziau C, Aussudre J, Brochet B, Moonen C, Petry KG, Dousset V. Differential aquaporin 4 expression during edema build-up and resolution phases of brain inflammation. J Neuroinflammation. 2011;8:143. doi: 10.1186/1742-2094-8-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyohara J. Importance of P-gp PET Imaging in Pharmacology. Curr Pharm Des. 2016;22:5830–5836. doi: 10.2174/1381612822666160804092258. [DOI] [PubMed] [Google Scholar]
- Turner RJ, Helps SC, Thornton E, Vink R. A substance P antagonist improves outcome when administered 4 h after onset of ischaemic stroke. Brain Res. 2011;1393:84–90. doi: 10.1016/j.brainres.2011.03.066. [DOI] [PubMed] [Google Scholar]
- Turner RJ, Sharp FR. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front Cell Neurosci. 2016;10:56. doi: 10.3389/fncel.2016.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Sakamoto H, Tomimoto H, Akiguchi I, Onodera M, Huang CL, Kanenishi K. Blood-brain barrier is impaired in the hippocampus of young adult spontaneously hypertensive rats. Acta Neuropathol. 2004;107:532–538. doi: 10.1007/s00401-004-0845-z. [DOI] [PubMed] [Google Scholar]
- Unterberg A, Wahl M, Baethmann A. Effects of bradykinin on permeability and diameter of pial vessels in vivo. J Cereb Blood Flow Metab. 1984;4:574–585. doi: 10.1038/jcbfm.1984.82. [DOI] [PubMed] [Google Scholar]
- Urich E, Patsch C, Aigner S, Graf M, Iacone R, Freskgard PO. Multicellular self-assembled spheroidal model of the blood brain barrier. Sci Rep. 2013;3:1500. doi: 10.1038/srep01500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnerova K, Koerner IP, Hurn PD. Gender and the injured brain. Anesth Analg. 2008;107:201–214. doi: 10.1213/ane.0b013e31817326a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bruggen N, Thibodeaux H, Palmer JT, Lee WP, Fu L, Cairns B, Tumas D, Gerlai R, Williams SP, van Lookeren Campagne M, Ferrara N. VEGF antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J Clin Invest. 1999;104:1613–1620. doi: 10.1172/JCI8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltkamp R, Rajapakse N, Robins G, Puskar M, Shimizu K, Busija D. Transient focal ischemia increases endothelial nitric oxide synthase in cerebral blood vessels. Stroke. 2002;33:2704–2710. doi: 10.1161/01.str.0000033132.85123.6a. [DOI] [PubMed] [Google Scholar]
- Venneti S, Lopresti BJ, Wiley CA. Molecular imaging of microglia/macrophages in the brain. Glia. 2013;61:10–23. doi: 10.1002/glia.22357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verant P, Serduc R, van der Sanden B, Chantal R, Ricard C, Coles JA, Vial JC. Subtraction method for intravital two-photon microscopy: intraparenchymal imaging and quantification of extravasation in mouse brain cortex. J Biomed Opt. 2008;13:011002. doi: 10.1117/1.2870083. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Steardo L, Parpura V, Montana V. Translational potential of astrocytes in brain disorders. Prog Neurobiol. 2016;144:188–205. doi: 10.1016/j.pneurobio.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waki H, Liu B, Miyake M, Katahira K, Murphy D, Kasparov S, Paton JF. Junctional adhesion molecule-1 is upregulated in spontaneously hypertensive rats: evidence for a prohypertensive role within the brain stem. Hypertension. 2007;49:1321–1327. doi: 10.1161/HYPERTENSIONAHA.106.085589. [DOI] [PubMed] [Google Scholar]
- Wang D, Pascual JM, Yang H, Engelstad K, Mao X, Cheng J, Yoo J, Noebels JL, De Vivo DC. A mouse model for Glut-1 haploinsufficiency. Hum Mol Genet. 2006;15:1169–1179. doi: 10.1093/hmg/ddl032. [DOI] [PubMed] [Google Scholar]
- Wang PH, Liu HL, Hsu PH, Lin CY, Wang CR, Chen PY, Wei KC, Yen TC, Li ML. Gold-nanorod contrast-enhanced photoacoustic micro-imaging of focused-ultrasound induced blood-brain-barrier opening in a rat model. J Biomed Opt. 2012;17:061222. doi: 10.1117/1.JBO.17.6.061222. [DOI] [PubMed] [Google Scholar]
- Wang Q, Doerschuk CM. The signaling pathways induced by neutrophil-endothelial cell adhesion. Antioxid Redox Signal. 2002;4:39–47. doi: 10.1089/152308602753625843. [DOI] [PubMed] [Google Scholar]
- Wang Y, Jin S, Sonobe Y, Cheng Y, Horiuchi H, Parajuli B, Kawanokuchi J, Mizuno T, Takeuchi H, Suzumura A. Interleukin-1beta induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS One. 2014;9:e110024. doi: 10.1371/journal.pone.0110024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Yang S, Liu R, Perez E, Yi KD, Koulen P, Simpkins JW. Estrogen attenuates nuclear factor-kappa B activation induced by transient cerebral ischemia. Brain Res. 2004;1008:147–154. doi: 10.1016/j.brainres.2004.02.019. [DOI] [PubMed] [Google Scholar]
- Wenger NK, Speroff L, Packard B. Cardiovascular health and disease in women. N Engl J Med. 1993;329:247–256. doi: 10.1056/NEJM199307223290406. [DOI] [PubMed] [Google Scholar]
- Wilkinson M, Hume R, Strange R, Bell JE. Glial and neuronal differentiation in the human fetal brain 9–23 weeks of gestation. Neuropathol Appl Neurobiol. 1990;16:193–204. doi: 10.1111/j.1365-2990.1990.tb01156.x. [DOI] [PubMed] [Google Scholar]
- Willenborg S, Lucas T, van Loo G, Knipper JA, Krieg T, Haase I, Brachvogel B, Hammerschmidt M, Nagy A, Ferrara N, Pasparakis M, Eming SA. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood. 2012;120:613–625. doi: 10.1182/blood-2012-01-403386. [DOI] [PubMed] [Google Scholar]
- Willis CL, Meske DS, Davis TP. Protein kinase C activation modulates reversible increase in cortical blood-brain barrier permeability and tight junction protein expression during hypoxia and posthypoxic reoxygenation. J Cereb Blood Flow Metab. 2010;30:1847–1859. doi: 10.1038/jcbfm.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AC, Clemente L, Liu T, Bowen RL, Meethal SV, Atwood CS. Reproductive hormones regulate the selective permeability of the blood-brain barrier. Biochim Biophys Acta. 2008;1782:401–407. doi: 10.1016/j.bbadis.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Winek K, Meisel A, Dirnagl U. Gut microbiota impact on stroke outcome: Fad or fact? J Cereb Blood Flow Metab. 2016;36:891–898. doi: 10.1177/0271678X16636890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter MC, Shasby SS, Ries DR, Shasby DM. Histamine selectively interrupts VE-cadherin adhesion independently of capacitive calcium entry. Am J Physiol Lung Cell Mol Physiol. 2004;287:L816–823. doi: 10.1152/ajplung.00056.2004. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Lippoldt A. Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul Pharmacol. 2002;38:323–337. doi: 10.1016/s1537-1891(02)00200-8. [DOI] [PubMed] [Google Scholar]
- Won S, Lee JH, Wali B, Stein DG, Sayeed I. Progesterone attenuates hemorrhagic transformation after delayed tPA treatment in an experimental model of stroke in rats: involvement of the VEGF-MMP pathway. J Cereb Blood Flow Metab. 2014;34:72–80. doi: 10.1038/jcbfm.2013.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won SJ, Tang XN, Suh SW, Yenari MA, Swanson RA. Hyperglycemia promotes tissue plasminogen activator-induced hemorrhage by Increasing superoxide production. Ann Neurol. 2011;70:583–590. doi: 10.1002/ana.22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HL, Wu XY, Bendayan R. Nanotechnological advances for the delivery of CNS therapeutics. Adv Drug Deliv Rev. 2012;64:686–700. doi: 10.1016/j.addr.2011.10.007. [DOI] [PubMed] [Google Scholar]
- Worzfeld T, Schwaninger M. Apicobasal polarity of brain endothelial cells. J Cereb Blood Flow Metab. 2016;36:340–362. doi: 10.1177/0271678X15608644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Walas S, Leung W, Sykes DB, Wu J, Lo EH, Lok J. Neuregulin1-beta decreases IL-1beta-induced neutrophil adhesion to human brain microvascular endothelial cells. Transl Stroke Res. 2015;6:116–124. doi: 10.1007/s12975-014-0347-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G, Gan LS. Receptor-mediated endocytosis and brain delivery of therapeutic biologics. Int J Cell Biol. 2013;2013:703545. doi: 10.1155/2013/703545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong XY, Liu L, Yang QW. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog Neurobiol. 2016;142:23–44. doi: 10.1016/j.pneurobio.2016.05.001. [DOI] [PubMed] [Google Scholar]
- Xu Y, Zhang W, Klaus J, Young J, Koerner I, Sheldahl LC, Hurn PD, Martinez-Murillo F, Alkayed NJ. Role of cocaine- and amphetamine-regulated transcript in estradiol-mediated neuroprotection. Proc Natl Acad Sci U S A. 2006;103:14489–14494. doi: 10.1073/pnas.0602932103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Zeng W, Sun J, Chen W, Zhang R, Yang Z, Yao Z, Wang L, Song L, Chen Y, Zhang Y, Wang C, Gong L, Wu B, Wang T, Zheng J, Gao F. The quantification of blood-brain barrier disruption using dynamic contrast-enhanced magnetic resonance imaging in aging rhesus monkeys with spontaneous type 2 diabetes mellitus. Neuroimage. 2016 doi: 10.1016/j.neuroimage.2016.07.017. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Ramirez SH, Sato S, Kiyota T, Cerny RL, Kaibuchi K, Persidsky Y, Ikezu T. Phosphorylation of claudin-5 and occludin by rho kinase in brain endothelial cells. Am J Pathol. 2008;172:521–533. doi: 10.2353/ajpath.2008.070076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Zhang Z, Shi H. HIF-1 is involved in high glucose-induced paracellular permeability of brain endothelial cells. Cell Mol Life Sci. 2012;69:115–128. doi: 10.1007/s00018-011-0731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan T, Venkat P, Ye X, Chopp M, Zacharek A, Ning R, Cui Y, Roberts C, Kuzmin-Nichols N, Sanberg CD, Chen J. HUCBCs increase angiopoietin 1 and induce neurorestorative effects after stroke in T1DM rats. CNS Neurosci Ther. 2014;20:935–944. doi: 10.1111/cns.12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Zhao K, Zhang X, Zhang J, Xu B. ATP Induces Disruption of Tight Junction Proteins via IL-1 Beta-Dependent MMP-9 Activation of Human Blood-Brain Barrier In Vitro. Neural Plast. 2016a;2016:8928530. doi: 10.1155/2016/8928530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Qian C, Wang N, Lin C, Wang Y, Wang G, Piao X. Tetramethylpyrazine Protects Against Oxygen-Glucose Deprivation-Induced Brain Microvascular Endothelial Cells Injury via Rho/Rho-kinase Signaling Pathway. Cell Mol Neurobiol. 2017;37:619–633. doi: 10.1007/s10571-016-0398-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Chu H, Tang Y, Dong Q. The role of connexin43 in hemorrhagic transformation after thrombolysis in vivo and in vitro. Neuroscience. 2016b;329:54–65. doi: 10.1016/j.neuroscience.2016.04.040. [DOI] [PubMed] [Google Scholar]
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- Yang Y, Salayandia VM, Thompson JF, Yang LY, Estrada EY, Yang Y. Attenuation of acute stroke injury in rat brain by minocycline promotes blood-brain barrier remodeling and alternative microglia/macrophage activation during recovery. J Neuroinflammation. 2015;12:26. doi: 10.1186/s12974-015-0245-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Thompson JF, Taheri S, Salayandia VM, McAvoy TA, Hill JW, Yang Y, Estrada EY, Rosenberg GA. Early inhibition of MMP activity in ischemic rat brain promotes expression of tight junction proteins and angiogenesis during recovery. J Cereb Blood Flow Metab. 2013;33:1104–1114. doi: 10.1038/jcbfm.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yemisci M, Caban S, Gursoy-Ozdemir Y, Lule S, Novoa-Carballal R, Riguera R, Fernandez-Megia E, Andrieux K, Couvreur P, Capan Y, Dalkara T. Systemically administered brain-targeted nanoparticles transport peptides across the blood-brain barrier and provide neuroprotection. J Cereb Blood Flow Metab. 2015;35:469–475. doi: 10.1038/jcbfm.2014.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yemisci M, Gursoy-Ozdemir Y, Vural A, Can A, Topalkara K, Dalkara T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–1037. doi: 10.1038/nm.2022. [DOI] [PubMed] [Google Scholar]
- Yoo DY, Yim HS, Jung HY, Nam SM, Kim JW, Choi JH, Seong JK, Yoon YS, Kim DW, Hwang IK. Chronic type 2 diabetes reduces the integrity of the blood-brain barrier by reducing tight junction proteins in the hippocampus. J Vet Med Sci. 2016;78:957–962. doi: 10.1292/jvms.15-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Yamada M, Wakabayashi K, Ikuta F, Kumanishi T. Endothelial basement membrane and seamless-type endothelium in the repair process of cerebral infarction in rats. Virchows Arch A Pathol Anat Histopathol. 1989;414:385–392. doi: 10.1007/BF00718621. [DOI] [PubMed] [Google Scholar]
- Yu AS, Cheng MH, Coalson RD. Calcium inhibits paracellular sodium conductance through claudin-2 by competitive binding. J Biol Chem. 2010;285:37060–37069. doi: 10.1074/jbc.M110.146621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Xu X, Jackson A, Sun J, Huang P, Mao Y, Chen Z, Lou M, Jiang Q, Zhang M. Blood Brain Barrier Disruption in Diabetic Stroke Related to Unfavorable Outcome. Cerebrovasc Dis. 2016;42:49–56. doi: 10.1159/000444809. [DOI] [PubMed] [Google Scholar]
- Zajac E, Schweighofer B, Kupriyanova TA, Juncker-Jensen A, Minder P, Quigley JP, Deryugina EI. Angiogenic capacity of M1- and M2-polarized macrophages is determined by the levels of TIMP-1 complexed with their secreted proMMP-9. Blood. 2013;122:4054–4067. doi: 10.1182/blood-2013-05-501494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotto C, Simao F, Gasparin MS, Biasibetti R, Tortorelli LS, Nardin P, Goncalves CA. Exendin-4 Reverses Biochemical and Functional Alterations in the Blood-Brain and Blood-CSF Barriers in Diabetic Rats. Mol Neurobiol. 2017;54:2154–2166. doi: 10.1007/s12035-016-9798-1. [DOI] [PubMed] [Google Scholar]
- Zechariah A, ElAli A, Hagemann N, Jin F, Doeppner TR, Helfrich I, Mies G, Hermann DM. Hyperlipidemia attenuates vascular endothelial growth factor-induced angiogenesis, impairs cerebral blood flow, and disturbs stroke recovery via decreased pericyte coverage of brain endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:1561–1567. doi: 10.1161/ATVBAHA.112.300749. [DOI] [PubMed] [Google Scholar]
- Zhang GS, Tian Y, Huang JY, Tao RR, Liao MH, Lu YM, Ye WF, Wang R, Fukunaga K, Lou YJ, Han F. The gamma-secretase blocker DAPT reduces the permeability of the blood-brain barrier by decreasing the ubiquitination and degradation of occludin during permanent brain ischemia. CNS Neurosci Ther. 2013a;19:53–60. doi: 10.1111/cns.12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HF, Maslov K, Stoica G, Wang LV. Functional photoacoustic microscopy for high-resolution and noninvasive in vivo imaging. Nat Biotechnol. 2006;24:848–851. doi: 10.1038/nbt1220. [DOI] [PubMed] [Google Scholar]
- Zhang L, Chopp M, Zhang Y, Xiong Y, Li C, Sadry N, Rhaleb I, Lu M, Zhang ZG. Diabetes Mellitus Impairs Cognitive Function in Middle-Aged Rats and Neurological Recovery in Middle-Aged Rats After Stroke. Stroke. 2016a;47:2112–2118. doi: 10.1161/STROKEAHA.115.012578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang RL, Jiang Q, Ding G, Chopp M, Zhang ZG. Focal embolic cerebral ischemia in the rat. Nat Protoc. 2015a;10:539–547. doi: 10.1038/nprot.2015.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Mao Y, Ramirez SH, Tuma RF, Chabrashvili T. Angiotensin II induced cerebral microvascular inflammation and increased blood-brain barrier permeability via oxidative stress. Neuroscience. 2010;171:852–858. doi: 10.1016/j.neuroscience.2010.09.029. [DOI] [PubMed] [Google Scholar]
- Zhang S, Cui N, Wu Y, Zhong W, Johnson CM, Jiang C. Optogenetic intervention to the vascular endothelium. Vascul Pharmacol. 2015b;74:122–129. doi: 10.1016/j.vph.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WC, Wang YG, Zhu ZF, Wu FQ, Peng YD, Chen ZY, Yang JH, Wu JJ, Lian YT, He MA, Wu TC, Cheng LX. Regulatory T cells protect fine particulate matter-induced inflammatory responses in human umbilical vein endothelial cells. Mediators Inflamm. 2014;2014:869148. doi: 10.1155/2014/869148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yeung PK, McAlonan GM, Chung SS, Chung SK. Transgenic mice over-expressing endothelial endothelin-1 show cognitive deficit with blood-brain barrier breakdown after transient ischemia with long-term reperfusion. Neurobiol Learn Mem. 2013b;101:46–54. doi: 10.1016/j.nlm.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang T, Yang K, Xu J, Ren L, Li W, Liu W. Cerebral Microvascular Endothelial Cell Apoptosis after Ischemia: Role of Enolase-Phosphatase 1 Activation and Aci-Reductone Dioxygenase 1 Translocation. Front Mol Neurosci. 2016b;9:79. doi: 10.3389/fnmol.2016.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yan J, Shi H. Hyperglycemia as a Risk Factor of Ischemic Stroke. J Drug Metab Toxicol. 2013c;4 doi: 10.4172/2157-7609.1000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yan J, Shi H. Role of Hypoxia Inducible Factor 1 in Hyperglycemia-Exacerbated Blood-Brain Barrier Disruption in Ischemic Stroke. Neurobiol Dis. 2016c;95:82–92. doi: 10.1016/j.nbd.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, Bruggen N, Chopp M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–838. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]