

Abstract
The substrates used to modify nucleic acids and chromatin are affected by nutrient availability and the activity of metabolic pathways. Thus, cellular metabolism constitutes a fundamental component of chromatin status and thereby of genome regulation. Here we describe the biochemical and genetic principles of how metabolism can influence chromatin biology and epigenetics, discuss the functional roles of this interplay in developmental and cancer biology, and present future directions in this rapidly emerging area.
Introduction
Organismal metabolism begins with the ingestion of nutrients from food sources. It continues with the processing of these nutrients in the gut, which then interacts with the microbiome, liver, pancreas, muscle, and many other organs to result in a set of compounds that circulate in the plasma1. Cells take up these plasma-supplied nutrients along with other components provided by their microenvironment2, 3, and use them to create cellular metabolic networks that are organized through interconnected chemical reactions with thousands of metabolites linked by commensurate numbers of reactions. Metabolic network activity is characterized by the concentrations of intermediate metabolites and the rates (i.e. fluxes) at which one metabolite is converted to another, and is mediated by genotype, epigenotype, and environmental inputs such as nutrient availability, and the engagement of signaling pathways3–6.
Chromatin is the intracellular structure that packages DNA in eukaryotic cells. The principal unit of chromatin organization is the nucleosome, which is formed by DNA wrapped around an octamer of histone proteins. Chromatin can exist in different stable states and is altered by covalent modifications on the histones along with the presence of many other factors such as long non-coding RNAs, protein chaperones, and chromatin remodeling enzymes7–10. These modifications influence chromatin structure and binding of chromatin remodeling enzymes and transcription factors in complex and often poorly understood ways7–11. They also can mark the existence of functional genomic elements (e.g. promoters, enhancers and exons)7–10, 12–14. Thus, there is tremendous potential for these posttranslational modifications to have profound effects on gene expression and substantial ongoing efforts aim to understand the structure and function of chromatin modifications10, 11, 15–19. Chromatin and nucleic acid modifications, when inherited after cell division, or in offspring after reproduction (e.g. genomic imprinting), are often referred to as epigenetics18, 20.
Because metabolites are the substrates used to generate chromatin modifications, there exists an intriguing but complex connection between metabolism and epigenetics. In this review, we first introduce biochemical principles that enable the epigenome to respond to metabolic variation and then discuss the genetic basis for how this interaction may generate stable phenotypes. We next discuss recent advances in our understanding of this connection with particular emphasis on stem cell biology and tumorigenesis. Our aim is to provide both a foundation of the principles that govern the interaction between metabolism and chromatin state and a discussion of ongoing developments that are shaping our understanding of its role in biology.
Biochemical principles of the link from metabolism to chromatin
More than 100 distinct covalent modifications have been identified on chromatin, DNA and RNA with many having substantially documented or emerging functional annotation21–23. Among them, methylation, phosphorylation, ubiquitination, and acetylation are the most well understood, but less studied modifications including glycosylation, crotonylation, succinylation, are also known to be functionally important24, 25. Addition and removal of these modifications are, with some exceptions26, catalyzed by enzymes of which the activities are mediated by the availability of substrates, cofactors and allosteric regulators that are derived from metabolic pathways. A key characteristic that defines the crosstalk between metabolism and chromatin is that the kinetic (e.g. Km values) and thermodynamic (e.g. Kd values) properties of these interactions are commensurate with the dynamic range of physiological concentrations of the corresponding intermediates in metabolism (Table 1). For example, methylation and acetylation reactions often have substrates that have typical cellular concentrations that are commensurate with enzyme Km values and thus are responsive to changes in metabolism (Table 1, Fig 1)19, 26–31. In contrast, modifications such as phosphorylation and ubiquitination do not respond to changes in metabolism because kinases and E3 ligases that carry out phosphorylation and ubiquitination reactions use ATP as a metabolic substrate27. ATP levels (~mM in cells) do not reach physiological levels that limit the activities of these enzymes (Km ~ uM) (Fig 1a).
Table 1.
Ranges of kinetic parameters and concentrations of substrates and cofactors of chromatin-modifying enzymes
| Enzyme | Substrates and cofactors | Kinetic parameter range [mM] | Substrate concentration range [mM] | [S]/Km range | Refs |
|---|---|---|---|---|---|
| Histone Acetyltransferase s (e.g. HATs, KATs, GCN5, CBP, p300) |
|
|
|
|
82, 134–137 |
| Histone Methyltransferas es (e.g. COMPASS, MLLs, EZH2/PRC2, SETs, DOT1L) |
|
|
|
|
138–142 |
| DNA Methyltransferas es (e.g. DNMTs) |
|
|
|
|
141–146 |
| Histone Deacetylases (e.g. SIRTs, HDACs) |
|
|
|
|
147–152 |
| Histone |
|
|
|
|
107, 153–158 |
| Demethylases (e.g. LSD1, JHDMs, JMJDs, JARIDs, UTX) |
|
|
|
(Oxygen) | |
| DNA Demethylases (e.g. TETs) |
|
|
|
|
156–160 |
HATs, histone acetyltransferases; KATs, lysine acetyltransferases; GCN5, GCN5-related N-acetyltransferase; CBP, CREB binding protein; p300; E1A binding protein p300; COMPASS, complex proteins associated with Set1; MLL, mixed-lineage leukemia histone methyltransferases; EZH2/PRC2, enhancer of zeste 2 polycomb repressive complex 2; SETs, SET domain-containing methyltransferases; DOT1L, DOT1 like histone lysine methyltransferase; DNMTs, DNA methyltransferases; SIRTs, Sirtuins; HDACs, histone deacetylases; LSD1, lysine demethylase 1; JHDMs; jumonji domain-containing histone demethylases; JMJDs, jumonji C domain-containing histone demethylases; JARIDs, jumonji and AT-rich interaction domain-containing histone demethylases; UTX, ubiquitously-transcribed X chromosome histone demethylase; TETs, ten-eleven translocation DNA demethylases; SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; MTA, methylthioadenosine; NAD+, nicotinamide adenine dinucleotide (oxidized); αKG, alpha-ketoglutarate; FAD, flavin adenine dinucleotide (oxidized).
Figure 1. Biochemical basis of metabolite interaction with chromatin and metabolic pathways that contribute.
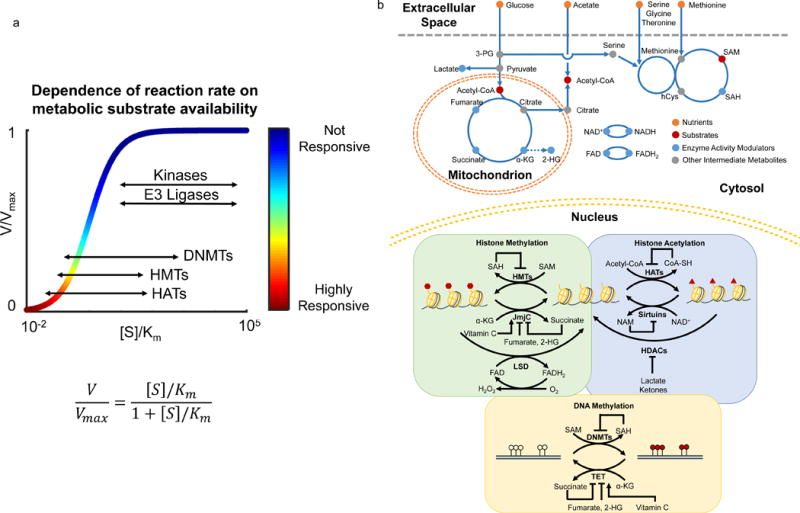
a) In contrast to kinases and E3 ligases, the physiological concentrations of substrates of chromatin modifying enzymes such as DNA methyltransferase (DNMTs), histone methyltransferase (HMTs), and histone acetyltransferases (HATs) are much lower thus limiting enzymatic activities. Thus, the reaction rates of these enzymes are highly responsive to local changes in substrate availability. x axis: ratio of substrate concentration to Km value; y axis: relative reaction rate. Ranges of [S]/Km for all five types of enzymes were estimated from Km values in the BRENDA database (www.brenda-enzymes.org). b) Uptake and catabolism of macronutrients such as glucose and amino acids generate substrates such as acetyl-CoA and S-adenosylmethionine (SAM), and activity modulators such as alpha-ketoglutarate (αKG), (R)-2-hydroxyglutarate (2-HG), succinate, fumarate, lactate, S-adenosylhomocysteine (SAH), oxidized and reduced nicotinamide adenine dinucleotide (NAD+, NADH), and oxidized and reduced flavin adenine dinucleotide (FAD, FADH2) used by enzymes that modify chromatin. SAM is the major methyl donor for methylation of cytosine bases in DNA and histone residues by DNA methyltransferase (DNMTs) and histone methyltransferases (HMTs), respectively. Acetyl-CoA is an essential substrate for acetylation of histone residues carried out by histone acetyltransferases (HATs). Other metabolites such as αKG, NAD+, and FAD are critical co-factors for the activity of chromatin modifying enzymes. αKG is used by TET-family DNA demethylases (TET) and JmjC-family histone demethylases (JmjC) to facilitate removal of methyl groups from cytosine bases and histone residues, respectively. LSD-family histone demethylases (LSD) require FAD to demethylate histone residues. Sirtuins and other histone deacetylaces (HDACs) require NAD+ to deacetylate histone residues. Additionally, metabolites such as 2-HG, succinate, fumarate, lactate and SAH can inhibit the activity of chromatin modifying enzymes.
There are numerous chromatin and nucleic acid-modifying enzymes (Fig 1b). Some examples which are non-exhaustive and have been reviewed extensively elsewhere7–9, 17, 25, 32–38 include histone methyltransferases, glycosyltransferases, demethylases, acetyltransferases, deacetylases, deacylases, DNA and RNA methyltransferases and demethylases. As has been reviewed extensively3, 9, 19, 28, 30, 31, 39–44, these enzymes utilize, as substrates and cofactors, metabolites derived from diverse metabolic pathways including serine-glycine one carbon (SGOC) metabolism and particularly the methionine cycle, the tricarboxylic acid cycle, beta oxidation, glycolysis, and hexosamine biosynthesis. In these metabolic networks, intermediate metabolites including S-adenosylmethionine (SAM), Acetyl-coenzyme A (Acetyl-CoA), NAD+, alpha-ketoglutarate (αKG), and Uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), and others, serve as substrates for enzymes that modify chromatin and nucleic acids. Additionally, metabolites such as S-adenosylhomocysteine (SAH), Coenzyme A (coA), beta-hydroxybutyrate, fumarate, succinate, lactate, and S and R enantiomeric forms of 2-hydroxyglutarate modify enzyme activity often by competitively inhibiting substrate utilization. There is also emerging evidence that vitamin C may act as a cofactor for dioxygenases that modify chromatin and DNA45–47. Thus, each modification can be affected by metabolites from multiple metabolic pathways – for instance, enzymes involved in histone and DNA methylation and demethylation can be regulated by both methionine metabolism and the TCA cycle – thus enabling the epigenome to respond to the status of the whole metabolic network.
Principles for the influence of metabolism on epigenomics and phenotypic outcomes
Epigenetic modifications are maintained over cellular and organismal generations even when the environmental stimuli triggering a particular epigenetic reprogramming mechanism are removed. For example, individuals exposed to famine during the Dutch Hunger Winter displayed altered DNA methylation patterns for over six decades48. Moreover, factors such as diet49–52, microbiome53, temperature54, malnutrition55, chemical exposure56 and many others are able to induce heritable alterations in nucleic acid or histone methylation profiles that can be stably transmitted through more than 10 generations54. How epigenomic states are stably inherited is unknown, but perturbations to chromatin-modifying enzymes in the germline in controlled laboratory settings have yielded comparable heritable effects to what is observed in settings of human exposure57. This suggests that changes to the activity of chromatin-modifying enzymes that are known to be affected by metabolism, may be possible transient events that contribute to these phenotypic changes.
Furthermore, the epigenome can be used to characterize cellular state or type. Comparative analysis of epigenomic profiles have identified cell- and tissue-type-specific chromatin and DNA methylation features58. Alterations to chromatin marks have been shown to serve as limiting steps to cell fate transitions such as those occurring during nuclear transfer59, 60, indicating that the epigenome is causally implicated in the establishment of cellular states. A conceptual framework known as Waddington’s Landscape is often used to illustrate the relationship between the epigenome and cell states61–66. In the 1940s, Conrad Waddington developed the concept of an epigenomic landscape as a blueprint for the differentiation program during development65–68. Waddington’s landscape is composed of valleys and summits, with valleys representing epigenotypes (in modern terms, stable chromatin modification profiles that define a phenotype) and summits corresponding to the barriers required for the maintenance of stable, heritable epigenomic states that prevent transitions between epigenotypes. Thus valleys can represent different cell states (e.g. pluripotent versus differentiated, normal versus cancerous), the transitions between which are limited by changes in chromatin status (Figs 2a,b).
Figure 2. Metabolic reprograming and Waddington’s epigenomic landscape.
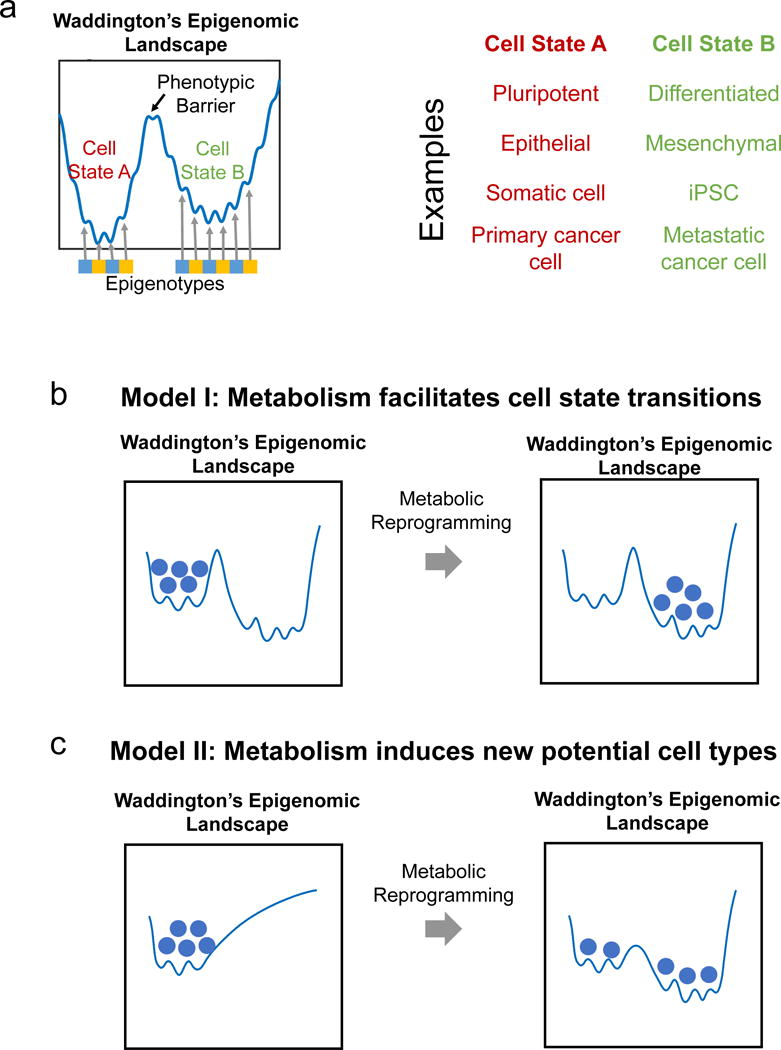
a) Schematic representation of Waddington’s Landscape depicting cell states existing in valleys maintained by epigenotypes and the phenotypic barrier between two cell states such as pluripotent and differentiated, epithelial and mesenchymal, somatic and induced pluripotent (iPSC), and primary and metastatic cancer cells. b) Model of how metabolism could facilitate cell state transitions without affecting the shape of the epigenomic landscape such as a change in metabolite level allowing for reorganization of specific chromatin marks. c) Model of how metabolic reprograming could reshape the entire epigenomic landscape leading to new cell states in a case where a cell type has different metabolic requirements. Balls represent cells transitioning from one state to another after changes in metabolism-dependent chromatin remodeling alters the phenotypic barrier.
Two models are proposed for how epigenetics could be affected by metabolic alterations in the context of Waddington’s Landscape (Figs 2c,d). One possibility (Model I) is that metabolic reprogramming facilitates the transition from one to another cell type (e.g. differentiation) by changing specific chromatin modifications. The stability of these cell states and thus how likely it is for a cell that has transitioned to a new cell state, to return to its previous cell type, depends on the height of the barrier. For example a change in the levels of metabolites such as methionine or αKG would modulate the activity of methyltransferase or demethylase enzymes, thereby promoting the reorganization of specific epigenetic marks and facilitating cell differentiation across a barrier (Fig 2b). Another possibility (Model II) is that metabolic reprogramming reshapes Waddington’s Landscape and induces the formation of new stable epigenetic states. In this model, a change in cellular metabolism could either induce gene expression programs related to chromatin remodeling through any number of mechanisms known to allow for such effects, or could directly affect the availability of substrates and cofactors for chromatin-modifying enzymes. In both instances, the cell state transition would be irreversible since Waddington’s Landscape has changed. This may occur during germline transmission of an epigenomic phenotype due to a parental diet or a germline mutation in a chromatin-modifying enzyme49, 57, 69 (Fig 2c). Although these proposed models are intriguing, more research is needed to reconstruct the structure of the epigenetic landscape under different metabolic conditions to investigate and distinguish between different possibilities.
The nexus of metabolism and epigenetics in cell fate and development
In recent years the link between cellular metabolism, cell fate and early organismal development has been an area of intense study. Early events in stem cell differentiation occur rapidly, and thus changes in metabolism as a driver of such events is an attractive hypothesis40. Indeed the influence of metabolism in the maintenance of stem cell pluripotency has been extensively explored5, 40, 70–74. The effects of metabolism on stem cell fate have been recently reviewed in detail elsewhere3, 5, 75 and thus here we will touch on specific aspects that highlight their interplay.
Two subsets of embryonic stem cells (ESCs), termed naïve and primed, have been defined based on their distinct pluripotency versus differentiation properties, and are also characterized by epigenetically distinct states5, 40. Naïve pluripotent stem cells are characterized by their ability to form all cell types without bias, whereas primed pluripotent stem cells are considered to be poised for lineage differentiation5, 40. In general, high rates of glycolysis even in the presence of oxygen, a phenomenon known as the Warburg Effect28, is prevalent in pluripotency and appears to be dynamically regulated in order to facilitate the differentiation process76. For example, human naïve pluripotent stem cells (PSCs) were shown to have higher glycolytic metabolism than human primed PSCs or differentiated cells71, and loss of mitochondrial oxidative metabolism was shown to cause defects in mouse hematopoietic stem cell differentiation77. Of note, other reports have shown increased oxidative phosphorylation in human and mouse naïve PSCs compared to their primed counterparts78. Together, these studies emphasize the temporal complexities of cellular metabolism in driving cell fate, and that glycolysis and oxidative metabolism may not be mutually exclusive when it comes to regulating pluripotency. Although metabolism unquestionably influences the pluripotent state, additional studies are needed to elucidate the exact mechanisms for how metabolic features contribute to pluripotency or differentiation.
In addition to the metabolic changes that occur during these cell fate transitions, it is also now widely appreciated that changes in metabolism are directly linked to changes in chromatin and DNA state. The levels of acetyl-CoA, the substrate for histone acetylation79–83, have been shown to be critical for the maintenance of human and mouse stem cell pluripotency70. Reduced NAD+ levels due to increased glycolytic metabolism have been shown to decrease NAD+-dependent histone deacetylase activity and to promote mouse muscle stem cell differentiation84. αKG was shown to maintain mouse naïve stem cell self-renewal by promoting histone and DNA demethylation through the activity of JmjC-family histone demethylases and TET-family DNA demethylases85. Increased αKG levels were also found to promote early differentiation of human primed PSCs and mouse epiblast stem cells86. Of significance, reduction of the αKG/succinate ratio was able to reverse the observed effects85, 86, indicating that the alterations in metabolic pathways drive chromatin dynamics.
The methionine cycle has also been shown to regulate histone methylation in mouse ESCs74. As with αKG, methionine-derived SAM appears to play multiple roles in mediating cell fate depending on context. Depletion of SAM through short-term methionine deprivation triggered differentiation of human primed ESCs73. Additionally, Nicotinamide N-methyltransferase upregulation in human naïve ESCs depleted SAM pools and maintained self-renewal, thus preventing the differentiation process76. Beyond the literature on pluripotency and metabolism, metabolism has been recently shown to maintain or induce specific adult stem cell lineages87, 88, however whether epigenetics may play a role in these settings remains to be determined. Together, these reports highlight that differing metabolomes are found in distinct cell states, and demonstrate the functional consequence of how changes in metabolism can affect and possibly specify cell fate.
The hematopoietic lineage is another well-studied system that exhibits cell state transitions. Recent reports highlight a critical role for metabolism in driving immune cell activation and differentiation89, 90. For example, T-cells undergo rapid changes in glycolysis upon activation91. There are additionally well established roles for epigenetics in immune cell fate92: enhanced glycolysis-dependent acetyl-CoA production in regulatory T-cells has been shown to promote differentiation through increased histone acetylation93, and αKG has been reported to regulate context-specific gene reprograming for helper T-cell differentiation94. Another example was observed in a C. elegans pathogen feeding model, in which deletion of methionine synthase reduced the immune response to pathogens by preventing expression of protective genes dependent on histone methylation95. Future studies will undoubtedly uncover more links, and it will be interesting to examine how changes in nutrient availability as a result of metabolic competition with other cells96, 97 or metabolite exchange affects immune cell metabolism, chromatin biology, and function.
A remaining question concerns how metabolism-dependent cell state changes affect the overall development of the organism. Tissue-type specific mechanisms for how metabolic changes affect development have started emerging98, 99, and future studies will uncover how these may control cell fate, tissue morphogenesis and development through epigenetic mechanisms. Given that recent reports indicate systemic and distinct changes in histone methylation in early mammalian development100–102, it will be exciting to explore the role metabolism and perhaps diet has in these contexts.
The influence of metabolism and epigenetics in cancer biology and therapeutic potential
Altered metabolism is a hallmark of cancer2, 4. For almost a century malignant cells have been known to exhibit nutritional differences compared to normal cells28, and recent evidence supports that they also harbor epigenetic changes driven by their rewired cellular metabolism39, 41, 103. A major breakthrough in our understanding of the connection between metabolism and epigenetics in cancer was the discovery of gain-of-function mutations to the genes encoding isocitrate dehydrogenase (IDH) 1 and 2 that cause an altered enzymatic activity resulting in the production of the (R)-2-hydroxyglutarate (2-HG) metabolite104, 105. These mutations are recurrent and their consequences in tumorigenesis have been reviewed extensively elsewhere44. In brief, cells harboring IDH1/2 mutations display DNA and histone hypermethylation as accumulation of 2-HG inhibits the activity of TET-family DNA and JmjC-family histone demethylase enzymes106. These mutations have been linked to the pathogenesis of glioblastoma multiforme, acute myeloid leukemia, chondrosarcoma, cholangiocarcinoma, and other human malignancies44.
Furthermore, mutations in the genes encoding fumarate hydratase (FH) and succinate dehydrogenase (SDH), the enzymes that catabolize fumarate and succinate, have been identified in several sporadic and hereditary cancers and cause accumulation of their substrates107. High levels of fumarate and succinate can also inhibit αKG-dependent DNA and histone demethylases and loss of FH and SDH activity was shown to lead to hypermethylation of DNA and histone residues107, 108. A recent report demonstrated that loss-of-function mutations in FH and the subsequent accumulation of fumarate promotes epithelial-to-mesenchymal-transition (EMT) through fumarate-dependent inhibition of TET demethylases and subsequent induction of genes necessary for EMT109. FH was also found to be O-GlcNAcylated, which caused changes in histone methylation110, and provides evidence for additional layers of metabolic regulation of chromatin. Indeed a recent study proposed that a substantial portion of variation in DNA methylation profiles across all human cancers could be explained by differences in the expression of enzymes related to methionine and the one carbon network111. Taken together these studies define clear and sometimes quantitative roles for metabolism in specifying aspects of the epigenome.
Cancer-specific deletions of other metabolic enzymes with implications in epigenetic regulation have also been reported. The gene encoding 5-methylthioadenosine phosphorylase (MTAP), a key enzyme in the methionine salvage pathway, is located near the ubiquitous tumor suppressor gene CDKN2A and the two are commonly co-deleted112, 113, with the loss of MTAP thought to be a passenger event in cancer progression. However, recent reports have established a collateral dependency in CDKN2A-deleted tumors in which loss of MTAP causes accumulation of methylthioadenosine (MTA), the metabolite cleaved by MTAP, leading to inhibition of the PRMT5 protein arginine methyltransferase which was required for tumor growth112, 113. The importance for methionine-derived SAM in regulating cell state and epigenetics73, 74 suggests that there could be instances where changes in SAM levels due to loss of the methionine salvage pathway could have dramatic effects on chromatin state, which would support a more active role for MTAP in cancer development. However, whether MTAP/CDKN2A deleted cancers display an altered chromatin state remains to be determined.
Although as discussed above alterations in genes encoding metabolic enzymes have been identified in cancer, they are overall rare. In contrast, lesions in genes related to cancer-associated growth signaling pathways and downstream transcription factors are common114. Indeed, the ability of cancer cells to obtain growth factor independence by acquiring mutations that allow them to constitutively engage signaling pathways that control cell growth, survival, and proliferation is a recognized hallmark of oncogenesis. Mutations in enzymes that modify chromatin and DNA are recurrent and constitute a more recently defined class of cancer-associated mutations66 that often result in aberrant chromatin and DNA epigenomic profiles. It is tempting to speculate that the function of these mutations is to uncouple their status from the interaction with metabolism and thus subvert this normal epigenetic regulation by nutrition and metabolism (Fig 3), however further studies are needed to better define the relationship between the normal metabolic regulation of chromatin and the cancer-associated chromatin mutations.
Figure 3. Analogy of cancer-associated mutations found in growth signaling with those in metabolism-dependent chromatin modifying processes.
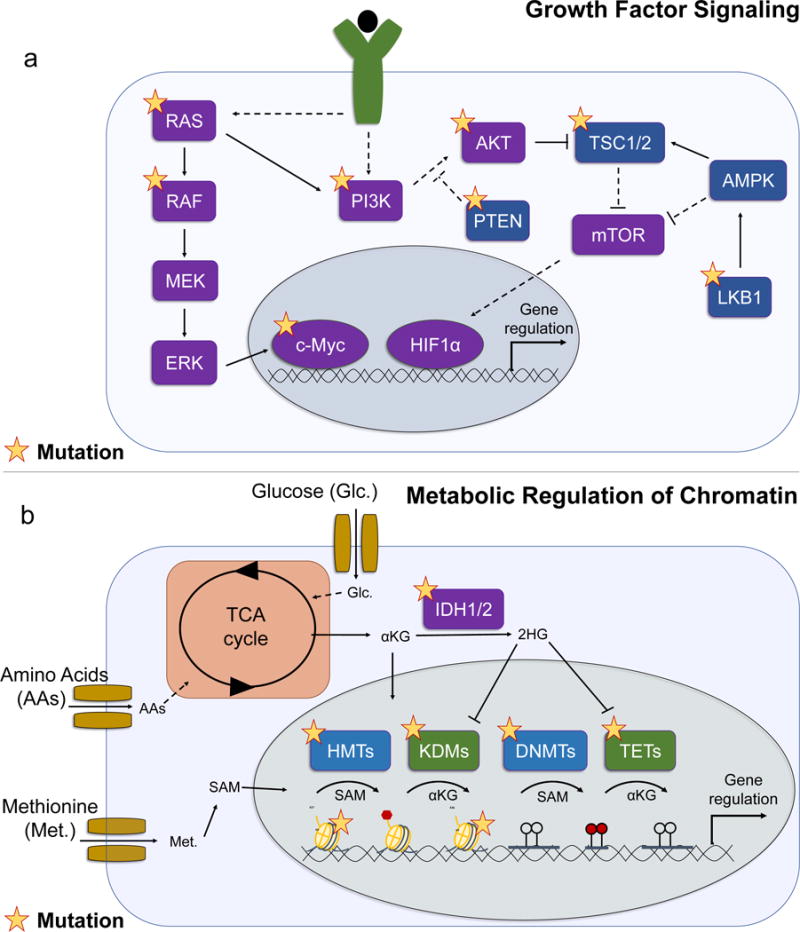
a) During oncogenesis, cells gain growth factor independence by frequently acquiring mutations that co-opt normal growth signaling. RAS and RAF are commonly mutated in cancer and drive downstream signaling through MEK and ERK, which can lead to gene regulation by c-Myc. RAS and growth factor signaling can activate the PI3K/AKT/mTOR signaling axis to promote cell growth and survival through downstream transcription factors such as HIF1α. Mutations to PI3K, AKT, PTEN, TSC, and LKB1 are also common in cancer. Purple indicates oncogenes; blue indicates tumor suppressors; yellow star indicates common lesions in cancer; solid lines represent direct biochemical interactions; dotted lines represent indirect regulation. b) Metabolism regulates normal physiological activity of chromatin modifying enzymes, which are commonly mutated in cancer. Glucose (Glc.) and amino acids (AAs) feed into the TCA cycle, which generates regulators of chromatin modifying enzymes such as αKG. Methionine (Met.) produces the methyl donor SAM in the methionine cycle. With exception of Isocitrate dehydrogenase (IDH1/2), mutations in metabolic enzymes are uncommon in cancer, yet cancer-associated mutations in chromatin modifiers such as DNA methyltransferases (DNMTs), TET-family DNA demethylases (TET), histone methyltransferases (HMTs), histone lysine demethylases (KDMs), and histones (H3K27 and H3K36) are prevalent suggesting cells may subvert the normal regulation of these enzymes by metabolism during transformation. Blue indicates enzymes that perform methylation reactions; green indicates enzymes that perform demethylation reactions; yellow star indicates common lesions in cancer.
A major goal in studying metabolism-dependent epigenetic mechanisms in cancer is the hope of identifying targetable liabilities. Encouragingly, small molecules targeting mutant IDH1/2 are now very advanced clinically115. At the preclinical level a study on nutrient heterogeneity of the tumor microenvironment reported that the core regions of melanoma tumors had enhanced histone methylation as a result of reduced αKG levels, which led to resistance to BRAF inhibitor treatment, and the combination of histone methyltransferase and BRAF inhibitors was sufficient to overcome resistance116. Separately the SGOC network was shown to be upregulated in LKB1-deficient tumors with KRAS activation and could be coupled to SAM generation, causing enhanced DNA methyltransferase activity and elevated DNA methylation117. This study indicated that LKB1-deficiency could be a key vulnerability as DNA methyltransferase and serine metabolism inhibition reduced tumor growth117. A distinct line of work on the evolution of distant metastases of pancreatic ductal adenocarcinoma (PDAC) demonstrated that the oxidative branch of the pentose phosphate pathway (oxPPP) was a driving force for epigenome landscape reprogramming and the fitness of metastatic cells118, suggesting that targeting the oxPPP could be effective in metastatic PDAC. Together, these studies represent a few examples on how advances in our understanding of metabolic effects on epigenetics can be translated into potential therapies.
Future directions
Much remains unanswered in each of the areas discussed in this Review. The key element of the biochemistry is that enzymatic parameters such as Kms, Vmaxs, and allosteric and inhibitory binding constants must be tuned to values that can limit enzyme activity. Although there is ample evidence that this can be achieved in pathophysiological conditions such as the presence of IDH1/2 or FH mutations, resulting in millimolar concentrations of 2HG in the case of IDH1/2105, which is well above the inhibitory constant of dioxygenase enzymes, there is accumulating albeit far less evidence that such regulation occurs in physiological conditions (Table 1)119.
Additional complications that limit our understanding are potential differences in enzymatic parameters measured under conditions in vitro versus in vivo, and the difficulty in obtaining accurate measurements of exact concentrations in vivo, especially when the relevant concentration is compartmentalized in cells. Thus, more studies are needed to define physiological conditions in which the concentration dynamics of relevant substrates and cofactors causally underlie a change in chromatin state. It will also be necessary to clarify the extent to which environmental variables such as diet, which have profound effects on cancer outcome120 and cell fate121, can modulate epigenetics by altering levels of the relevant metabolites to the needed concentrations.
A further complexity is that enzymes for both activating and repressive histone marks require metabolites. Thus, the precise input of cellular metabolism into the complex arrangement of multiple modifications on histones and DNA that have distinct functions remains an open question. For example, how do changes in the levels of metabolites such as SAM lead to predictable changes in gene expression? Additional, poorly understood layers of regulation likely exist that define the specificity of the chromatin-metabolite link. For instance, the formation of multiprotein complexes in which enzyme activities are affected by dynamic protein interactions and their localization to specific sites of the genome would occur in parallel with metabolite changes that also affect enzyme activity increasing the level of regulatory complexity that further work is expected to elucidate in the coming years. Finally, although the expression levels of metabolic network components appear to be to some extent predictive of DNA methylation levels111, how predictive metabolite levels are of the overall chromatin state and epigenetic phenotype remains largely unknown. As we know, many other factors influence chromatin state such as gene expression and much of the study of epigenetics and chromatin biology was historically conducted without consideration of metabolic influences. How the magnitude of the metabolic effects on the activity of chromatin-modifying enzymes compares relative to transcriptional programs that control the expression of these enzymes remains unknown.
Moreover, our understanding of the genetic basis for how stable chromatin states or traits can be established through metabolic changes is very limited. In addition, although certain architectural aspects of chromatin modifications, such as peak shape, are known to encode information about phenotype12, 13, the specific aspects of genomic architecture that may be affected by metabolites remain unknown. Our current knowledge of metabolic regulation of chromatin structure focuses on individual covalent chromatin marks, but the effect on higher-level chromatin structure such as genome folding and chromatin accessibility remains to be elucidated.
In stem and developmental biology, there are numerous examples of cell type transitions that show concomitant changes in metabolism and the chromatin landscape. Nevertheless, there are few examples that show that a metabolic change leads to a biological outcome due to a specific effect on chromatin or DNA modifications and independently of all other effects that may occur alongside this change in metabolite levels. This complexity results from the fact that metabolites involved in epigenetics are also connected to larger metabolic networks that affect nearly all aspects of cellular physiology. New CRISPR-CAS9-based technologies that can engineer posttranslational modifications at specific genomic loci, when combined with defined metabolic perturbations, may address some of these challenges122–124.
In cancer, although there is much interest in targeting both altered metabolism and altered epigenetics, whether these two hallmarks confer dependencies in tumors synergistically is unknown, with the exception of a few examples115, 116, 117, 118. The same difficulties in establishing causal links apply also in this setting. In that respect exploring metabolic dependencies in settings where a genetic lesion modifies chromatin as in MLL-rearranged leukemias125, 126, or pediatric brain tumors and sarcomas with histone mutations127, 128, might prove fruitful as these cases could be particularly susceptible to a disruption in metabolism.
Although our understanding remains at a very early stage, rapid progress in our understanding is expected, especially considering the techniques that are available for chromatin and metabolic state characterization, and cell culture methods, including organoid systems, that can model and manipulate physiological metabolism more effectively119, 129–133. This wealth of technology available to probe and interpret both chromatin status and metabolism and the collective interest in both subjects, raise optimism that rapid progress will continue to be made.
Acknowledgments
We apologize to those whose work could not be cited due to space constraints. We thank members of the Locasale lab and Purushothama Rao Tata for constructive comments on the manuscript. J.W.L. acknowledges support from R01CA193256 and P30CA014236.
Footnotes
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Chandel NS. Navigating metabolism. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2015. [Google Scholar]
- 2.Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell. 2017;168:657–669. doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chandel NS, Jasper H, Ho TT, Passegue E. Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat Cell Biol. 2016;18:823–832. doi: 10.1038/ncb3385. [DOI] [PubMed] [Google Scholar]
- 4.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu J, Ocampo A, Izpisua Belmonte JC. Cellular Metabolism and Induced Pluripotency. Cell. 2016;166:1371–1385. doi: 10.1016/j.cell.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 6.Dai Z, Locasale JW. Understanding metabolism with flux analysis: From theory to application. Metab Eng. 2016 doi: 10.1016/j.ymben.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- 8.Schubeler D. Function and information content of DNA methylation. Nature. 2015;517:321–326. doi: 10.1038/nature14192. [DOI] [PubMed] [Google Scholar]
- 9.Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. 2015;16:258–264. doi: 10.1038/nrm3931. [DOI] [PubMed] [Google Scholar]
- 10.Piunti A, Shilatifard A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science. 2016;352:aad9780. doi: 10.1126/science.aad9780. [DOI] [PubMed] [Google Scholar]
- 11.Yin Y, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356 doi: 10.1126/science.aaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benayoun BA, et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell. 2014;158:673–688. doi: 10.1016/j.cell.2014.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen K, et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet. 2015;47:1149–1157. doi: 10.1038/ng.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neri F, et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature. 2017;543:72–77. doi: 10.1038/nature21373. [DOI] [PubMed] [Google Scholar]
- 15.Coleman RT, Struhl G. Causal role for inheritance of H3K27me3 in maintaining the OFF state of a Drosophila HOX gene. Science. 2017;356 doi: 10.1126/science.aai8236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorighi KM, et al. Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Mol Cell. 2017;66:568–576 e564. doi: 10.1016/j.molcel.2017.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol. 2014;15:703–708. doi: 10.1038/nrm3890. [DOI] [PubMed] [Google Scholar]
- 18.Feinberg AP, Fallin MD. Epigenetics at the Crossroads of Genes and the Environment. JAMA. 2015;314:1129–1130. doi: 10.1001/jama.2015.10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaelin WG, Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153:56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan M, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pradeepa MM, et al. Histone H3 globular domain acetylation identifies a new class of enhancers. Nat Genet. 2016;48:681–686. doi: 10.1038/ng.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie Z, et al. Metabolic Regulation of Gene Expression by Histone Lysine beta-Hydroxybutyrylation. Mol Cell. 2016;62:194–206. doi: 10.1016/j.molcel.2016.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plongthongkum N, Diep DH, Zhang K. Advances in the profiling of DNA modifications: cytosine methylation and beyond. Nat Rev Genet. 2014;15:647–661. doi: 10.1038/nrg3772. [DOI] [PubMed] [Google Scholar]
- 25.Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta. 2014;1839:627–643. doi: 10.1016/j.bbagrm.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wagner GR, Hirschey MD. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol Cell. 2014;54:5–16. doi: 10.1016/j.molcel.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Locasale JW, Cantley LC. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011;14:443–451. doi: 10.1016/j.cmet.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci. 2016;41:211–218. doi: 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mentch SJ, Locasale JW. One-carbon metabolism and epigenetics: understanding the specificity. Ann N Y Acad Sci. 2016;1363:91–98. doi: 10.1111/nyas.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Etchegaray JP, Mostoslavsky R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol Cell. 2016;62:695–711. doi: 10.1016/j.molcel.2016.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Z, Cai L, Tu BP. Dietary control of chromatin. Curr Opin Cell Biol. 2015;34:69–74. doi: 10.1016/j.ceb.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sabari BR, Zhang D, Allis CD, Zhao Y. Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol. 2017;18:90–101. doi: 10.1038/nrm.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 34.Whetstine JR, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 35.Ito S, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 2017 doi: 10.1038/nrg.2017.33. [DOI] [PubMed] [Google Scholar]
- 38.Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017;169:1187–1200. doi: 10.1016/j.cell.2017.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol. 2012;13:270–276. doi: 10.1038/nrm3305. [DOI] [PubMed] [Google Scholar]
- 40.Ryall JG, Cliff T, Dalton S, Sartorelli V. Metabolic Reprogramming of Stem Cell Epigenetics. Cell Stem Cell. 2015;17:651–662. doi: 10.1016/j.stem.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kinnaird A, Zhao S, Wellen KE, Michelakis ED. Metabolic control of epigenetics in cancer. Nat Rev Cancer. 2016;16:694–707. doi: 10.1038/nrc.2016.82. [DOI] [PubMed] [Google Scholar]
- 42.Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Losman JA, Kaelin WG., Jr What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27:836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Minor EA, Court BL, Young JI, Wang G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J Biol Chem. 2013;288:13669–13674. doi: 10.1074/jbc.C113.464800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blaschke K, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500:222–226. doi: 10.1038/nature12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agathocleous M, et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature. 2017 doi: 10.1038/nature23876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heijmans BT, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–17049. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ost A, et al. Paternal diet defines offspring chromatin state and intergenerational obesity. Cell. 2014;159:1352–1364. doi: 10.1016/j.cell.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 50.Wei Y, et al. Paternally induced transgenerational inheritance of susceptibility to diabetes in mammals. Proc Natl Acad Sci U S A. 2014;111:1873–1878. doi: 10.1073/pnas.1321195111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Radford EJ, et al. In utero effects. In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science. 2014;345:1255903. doi: 10.1126/science.1255903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carone BR, et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell. 2010;143:1084–1096. doi: 10.1016/j.cell.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Romano KA, et al. Metabolic, Epigenetic, and Transgenerational Effects of Gut Bacterial Choline Consumption. Cell Host Microbe. 2017 doi: 10.1016/j.chom.2017.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klosin A, Casas E, Hidalgo-Carcedo C, Vavouri T, Lehner B. Transgenerational transmission of environmental information in C. elegans. Science. 2017;356:320–323. doi: 10.1126/science.aah6412. [DOI] [PubMed] [Google Scholar]
- 55.Hardikar AA, et al. Multigenerational Undernutrition Increases Susceptibility to Obesity and Diabetes that Is Not Reversed after Dietary Recuperation. Cell Metab. 2015;22:312–319. doi: 10.1016/j.cmet.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 56.Dias BG, Ressler KJ. Parental olfactory experience influences behavior and neural structure in subsequent generations. Nat Neurosci. 2014;17:89–96. doi: 10.1038/nn.3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han S, et al. Mono-unsaturated fatty acids link H3K4me3 modifiers to C. elegans lifespan. Nature. 2017;544:185–190. doi: 10.1038/nature21686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roadmap Epigenomics, C. et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matoba S, et al. Embryonic development following somatic cell nuclear transfer impeded by persisting histone methylation. Cell. 2014;159:884–895. doi: 10.1016/j.cell.2014.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hormanseder E, et al. H3K4 Methylation-Dependent Memory of Somatic Cell Identity Inhibits Reprogramming and Development of Nuclear Transfer Embryos. Cell Stem Cell. 2017 doi: 10.1016/j.stem.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Waddington CH. The strategy of the genes; a discussion of some aspects of theoretical biology. Allen & Unwin; London: 1957. [Google Scholar]
- 62.Waddington CH. Canalization of Development and Genetic Assimilation of Acquired Characters. Nature. 1959;183:1654–1655. doi: 10.1038/1831654a0. [DOI] [PubMed] [Google Scholar]
- 63.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 64.Pujadas E, Feinberg AP. Regulated noise in the epigenetic landscape of development and disease. Cell. 2012;148:1123–1131. doi: 10.1016/j.cell.2012.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ladewig J, Koch P, Brustle O. Leveling Waddington: the emergence of direct programming and the loss of cell fate hierarchies. Nat Rev Mol Cell Biol. 2013;14:225–236. doi: 10.1038/nrm3543. [DOI] [PubMed] [Google Scholar]
- 66.Feinberg AP, Koldobskiy MA, Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17:284–299. doi: 10.1038/nrg.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moris N, Pina C, Arias AM. Transition states and cell fate decisions in epigenetic landscapes. Nat Rev Genet. 2016;17:693–703. doi: 10.1038/nrg.2016.98. [DOI] [PubMed] [Google Scholar]
- 68.Rajagopal J, Stanger BZ. Plasticity in the Adult: How Should the Waddington Diagram Be Applied to Regenerating Tissues? Dev Cell. 2016;36:133–137. doi: 10.1016/j.devcel.2015.12.021. [DOI] [PubMed] [Google Scholar]
- 69.Greer EL, et al. Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature. 2011;479:365–371. doi: 10.1038/nature10572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moussaieff A, et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015;21:392–402. doi: 10.1016/j.cmet.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 71.Gu W, et al. Glycolytic Metabolism Plays a Functional Role in Regulating Human Pluripotent Stem Cell State. Cell Stem Cell. 2016;19:476–490. doi: 10.1016/j.stem.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang J, et al. Dependence of mouse embryonic stem cells on threonine catabolism. Science. 2009;325:435–439. doi: 10.1126/science.1173288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shiraki N, et al. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 2014;19:780–794. doi: 10.1016/j.cmet.2014.03.017. [DOI] [PubMed] [Google Scholar]
- 74.Shyh-Chang N, et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science. 2013;339:222–226. doi: 10.1126/science.1226603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014;15:243–256. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sperber H, et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat Cell Biol. 2015;17:1523–1535. doi: 10.1038/ncb3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anso E, et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat Cell Biol. 2017;19:614–625. doi: 10.1038/ncb3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Takashima Y, et al. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell. 2014;158:1254–1269. doi: 10.1016/j.cell.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cluntun AA, et al. The rate of glycolysis quantitatively mediates specific histone acetylation sites. Cancer Metab. 2015;3:10. doi: 10.1186/s40170-015-0135-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wellen KE, et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell. 2011;42:426–437. doi: 10.1016/j.molcel.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee JV, et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014;20:306–319. doi: 10.1016/j.cmet.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mews P, et al. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature. 2017;546:381–386. doi: 10.1038/nature22405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ryall JG, et al. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell. 2015;16:171–183. doi: 10.1016/j.stem.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518:413–416. doi: 10.1038/nature13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.TeSlaa T, et al. Alpha-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metab. 2016;24:485–493. doi: 10.1016/j.cmet.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Flores A, et al. Lactate dehydrogenase activity drives hair follicle stem cell activation. Nat Cell Biol. 2017;19:1017–1026. doi: 10.1038/ncb3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schell JC, et al. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat Cell Biol. 2017;19:1027–1036. doi: 10.1038/ncb3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell. 2017;169:570–586. doi: 10.1016/j.cell.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gerriets VA, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. 2015;125:194–207. doi: 10.1172/JCI76012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Phan AT, Goldrath AW, Glass CK. Metabolic and Epigenetic Coordination of T Cell and Macrophage Immunity. Immunity. 2017;46:714–729. doi: 10.1016/j.immuni.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Peng M, et al. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science. 2016;354:481–484. doi: 10.1126/science.aaf6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chisolm DA, et al. CCCTC-Binding Factor Translates Interleukin 2- and alpha-Ketoglutarate-Sensitive Metabolic Changes in T Cells into Context-Dependent Gene Programs. Immunity. 2017;47:251–267 e257. doi: 10.1016/j.immuni.2017.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ding W, et al. s-Adenosylmethionine Levels Govern Innate Immunity through Distinct Methylation-Dependent Pathways. Cell Metab. 2015;22:633–645. doi: 10.1016/j.cmet.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chang CH, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162:1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ho PC, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell. 2015;162:1217–1228. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Oginuma M, et al. A Gradient of Glycolytic Activity Coordinates FGF and Wnt Signaling during Elongation of the Body Axis in Amniote Embryos. Dev Cell. 2017;40:342–353 e310. doi: 10.1016/j.devcel.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Agathocleous M, et al. Metabolic differentiation in the embryonic retina. Nat Cell Biol. 2012;14:859–864. doi: 10.1038/ncb2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang B, et al. Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature. 2016;537:553–557. doi: 10.1038/nature19361. [DOI] [PubMed] [Google Scholar]
- 101.Liu X, et al. Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature. 2016;537:558–562. doi: 10.1038/nature19362. [DOI] [PubMed] [Google Scholar]
- 102.Dahl JA, et al. Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature. 2016;537:548–552. doi: 10.1038/nature19360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gao X, Reid MA, Kong M, Locasale JW. Metabolic interactions with cancer epigenetics. Mol Aspects Med. 2017;54:50–57. doi: 10.1016/j.mam.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yan H, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dang L, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Figueroa ME, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xiao M, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jiang Y, et al. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nat Cell Biol. 2015;17:1158–1168. doi: 10.1038/ncb3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sciacovelli M, et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature. 2016;537:544–547. doi: 10.1038/nature19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang T, et al. O-GlcNAcylation of fumarase maintains tumour growth under glucose deficiency. Nat Cell Biol. 2017;19:833–843. doi: 10.1038/ncb3562. [DOI] [PubMed] [Google Scholar]
- 111.Mehrmohamadi M, Mentch LK, Clark AG, Locasale JW. Integrative modelling of tumour DNA methylation quantifies the contribution of metabolism. Nat Commun. 2016;7:13666. doi: 10.1038/ncomms13666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kryukov GV, et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science. 2016;351:1214–1218. doi: 10.1126/science.aad5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mavrakis KJ, et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science. 2016;351:1208–1213. doi: 10.1126/science.aad5944. [DOI] [PubMed] [Google Scholar]
- 114.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yen K, et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017;7:478–493. doi: 10.1158/2159-8290.CD-16-1034. [DOI] [PubMed] [Google Scholar]
- 116.Pan M, et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol. 2016;18:1090–1101. doi: 10.1038/ncb3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kottakis F, et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature. 2016;539:390–395. doi: 10.1038/nature20132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McDonald OG, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet. 2017;49:367–376. doi: 10.1038/ng.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mentch SJ, et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015;22:861–873. doi: 10.1016/j.cmet.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Maddocks ODK, et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature. 2017;544:372–376. doi: 10.1038/nature22056. [DOI] [PubMed] [Google Scholar]
- 121.Taya Y, et al. Depleting dietary valine permits nonmyeloablative mouse hematopoietic stem cell transplantation. Science. 2016;354:1152–1155. doi: 10.1126/science.aag3145. [DOI] [PubMed] [Google Scholar]
- 122.Stricker SH, Koferle A, Beck S. From profiles to function in epigenomics. Nat Rev Genet. 2017;18:51–66. doi: 10.1038/nrg.2016.138. [DOI] [PubMed] [Google Scholar]
- 123.Liu XS, et al. Editing DNA Methylation in the Mammalian Genome. Cell. 2016;167:233–247 e217. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hilton IB, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–517. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Balbach ST, Orkin SH. An Achilles’ Heel for MLL-Rearranged Leukemias: Writers and Readers of H3 Lysine 36 Dimethylation. Cancer Discov. 2016;6:700–702. doi: 10.1158/2159-8290.CD-16-0564. [DOI] [PubMed] [Google Scholar]
- 126.Gilan O, et al. Functional interdependence of BRD4 and DOT1L in MLL leukemia. Nat Struct Mol Biol. 2016;23:673–681. doi: 10.1038/nsmb.3249. [DOI] [PubMed] [Google Scholar]
- 127.Lewis PW, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lu C, et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science. 2016;352:844–849. doi: 10.1126/science.aac7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu X, Romero IL, Litchfield LM, Lengyel E, Locasale JW. Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell Metab. 2016;24:728–739. doi: 10.1016/j.cmet.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu X, Locasale JW. Metabolomics: A Primer. Trends Biochem Sci. 2017;42:274–284. doi: 10.1016/j.tibs.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tardito S, et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol. 2015;17:1556–1568. doi: 10.1038/ncb3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cantor JR, et al. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell. 2017;169:258–272 e217. doi: 10.1016/j.cell.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dutta D, Heo I, Clevers H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol Med. 2017;23:393–410. doi: 10.1016/j.molmed.2017.02.007. [DOI] [PubMed] [Google Scholar]
- 134.Bohm J, Schlaeger EJ, Knippers R. Acetylation of nucleosomal histones in vitro. Eur J Biochem. 1980;112:353–362. doi: 10.1111/j.1432-1033.1980.tb07212.x. [DOI] [PubMed] [Google Scholar]
- 135.Lau OD, et al. p300/CBP-associated factor histone acetyltransferase processing of a peptide substrate. Kinetic analysis of the catalytic mechanism. J Biol Chem. 2000;275:21953–21959. doi: 10.1074/jbc.M003219200. [DOI] [PubMed] [Google Scholar]
- 136.Wiktorowicz JE, Campos KL, Bonner J. Substrate and product inhibition initial rate kinetics of histone acetyltransferase. Biochemistry. 1981;20:1464–1467. doi: 10.1021/bi00509a009. [DOI] [PubMed] [Google Scholar]
- 137.Tanner KG, Langer MR, Denu JM. Kinetic mechanism of human histone acetyltransferase P/CAF. Biochemistry. 2000;39:15652. doi: 10.1021/bi005121q. [DOI] [PubMed] [Google Scholar]
- 138.Tuck MT, Farooqui JZ, Paik WK. Two histone H1-specific protein-lysine N-methyltransferases from Euglena gracilis. Purification and characterization. J Biol Chem. 1985;260:7114–7121. [PubMed] [Google Scholar]
- 139.Rathert P, Zhang X, Freund C, Cheng X, Jeltsch A. Analysis of the substrate specificity of the Dim-5 histone lysine methyltransferase using peptide arrays. Chem Biol. 2008;15:5–11. doi: 10.1016/j.chembiol.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lobet Y, Lhoest J, Colson C. Partial purification and characterization of the specific protein-lysine N-methyltransferase of YL32, a yeast ribosomal protein. Biochim Biophys Acta. 1989;997:224–231. doi: 10.1016/0167-4838(89)90191-x. [DOI] [PubMed] [Google Scholar]
- 141.Oden KL, Clarke S. S-adenosyl-L-methionine synthetase from human erythrocytes: role in the regulation of cellular S-adenosylmethionine levels. Biochemistry. 1983;22:2978–2986. doi: 10.1021/bi00281a030. [DOI] [PubMed] [Google Scholar]
- 142.Melnyk S, Pogribna M, Pogribny IP, Yi P, James SJ. Measurement of plasma and intracellular S-adenosylmethionine and S-adenosylhomocysteine utilizing coulometric electrochemical detection: alterations with plasma homocysteine and pyridoxal 5'-phosphate concentrations. Clin Chem. 2000;46:265–272. [PubMed] [Google Scholar]
- 143.Kossykh VG, Schlagman SL, Hattman S. Phage T4 DNA [N6-adenine]methyltransferase. Overexpression, purification, and characterization. J Biol Chem. 1995;270:14389–14393. doi: 10.1074/jbc.270.24.14389. [DOI] [PubMed] [Google Scholar]
- 144.del Gaudio R, et al. Characterization of a new variant DNA (cytosine-5)-methyltransferase unable to methylate double stranded DNA isolated from the marine annelid worm Chaetopterus variopedatus. FEBS Lett. 1999;460:380–384. doi: 10.1016/s0014-5793(99)01379-4. [DOI] [PubMed] [Google Scholar]
- 145.Simon D, Grunert F, von Acken U, Doring HP, Kroger H. DNA-methylase from regenerating rat liver: purification and characterisation. Nucleic Acids Res. 1978;5:2153–2167. doi: 10.1093/nar/5.6.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cohen HM, Griffiths AD, Tawfik DS, Loakes D. Determinants of cofactor binding to DNA methyltransferases: insights from a systematic series of structural variants of S-adenosylhomocysteine. Org Biomol Chem. 2005;3:152–161. doi: 10.1039/b415446k. [DOI] [PubMed] [Google Scholar]
- 147.Guan X, Lin P, Knoll E, Chakrabarti R. Mechanism of inhibition of the human sirtuin enzyme SIRT3 by nicotinamide: computational and experimental studies. PLoS One. 2014;9:e107729. doi: 10.1371/journal.pone.0107729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Borra MT, Langer MR, Slama JT, Denu JM. Substrate specificity and kinetic mechanism of the Sir2 family of NAD+-dependent histone/protein deacetylases. Biochemistry. 2004;43:9877–9887. doi: 10.1021/bi049592e. [DOI] [PubMed] [Google Scholar]
- 149.Shimazu T, et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yamada K, Hara N, Shibata T, Osago H, Tsuchiya M. The simultaneous measurement of nicotinamide adenine dinucleotide and related compounds by liquid chromatography/electrospray ionization tandem mass spectrometry. Anal Biochem. 2006;352:282–285. doi: 10.1016/j.ab.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 151.Belenky P, et al. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+ Cell. 2007;129:473–484. doi: 10.1016/j.cell.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 152.Yang H, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cascella B, Mirica LM. Kinetic analysis of iron-dependent histone demethylases: alpha-ketoglutarate substrate inhibition and potential relevance to the regulation of histone demethylation in cancer cells. Biochemistry. 2012;51:8699–8701. doi: 10.1021/bi3012466. [DOI] [PubMed] [Google Scholar]
- 154.Chowdhury R, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rose NR, et al. Inhibitor scaffolds for 2-oxoglutarate-dependent histone lysine demethylases. J Med Chem. 2008;51:7053–7056. doi: 10.1021/jm800936s. [DOI] [PubMed] [Google Scholar]
- 156.Pritchard JB. Intracellular alpha-ketoglutarate controls the efficacy of renal organic anion transport. J Pharmacol Exp Ther. 1995;274:1278–1284. [PubMed] [Google Scholar]
- 157.Chin RM, et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature. 2014;510:397–401. doi: 10.1038/nature13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shen J, et al. Oxygen consumption rates and oxygen concentration in molt-4 cells and their mtDNA depleted (rho0) mutants. Biophys J. 2003;84:1291–1298. doi: 10.1016/S0006-3495(03)74944-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Laukka T, et al. Fumarate and Succinate Regulate Expression of Hypoxia-inducible Genes via TET Enzymes. J Biol Chem. 2016;291:4256–4265. doi: 10.1074/jbc.M115.688762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Koivunen P, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]