

Abstract
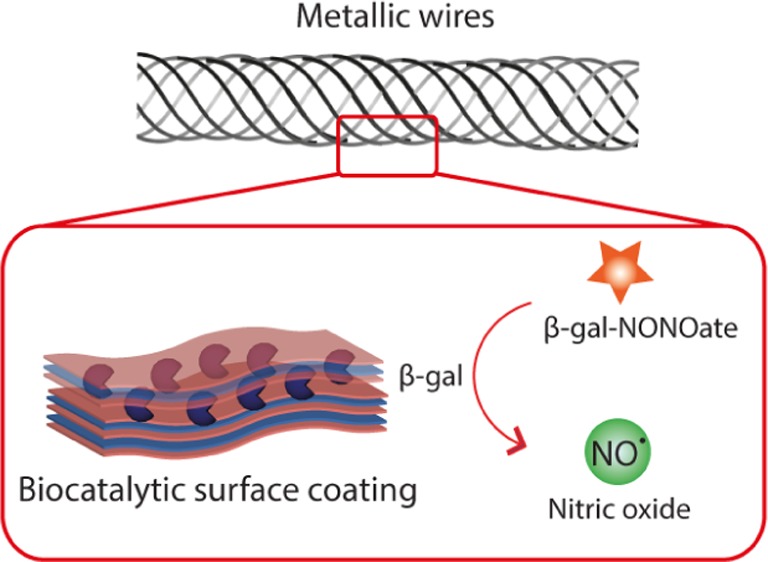
Nitric oxide (NO) is a highly potent but short-lived endogenous radical with a wide spectrum of physiological activities. In this work, we developed an enzymatic approach to the site-specific synthesis of NO mediated by biocatalytic surface coatings. Multilayered polyelectrolyte films were optimized as host compartments for the immobilized β-galactosidase (β-Gal) enzyme through a screen of eight polycations and eight polyanions. The lead composition was used to achieve localized production of NO through the addition of β-Gal–NONOate, a prodrug that releases NO following enzymatic bioconversion. The resulting coatings afforded physiologically relevant flux of NO matching that of the healthy human endothelium. The antiproliferative effect due to the synthesized NO in cell culture was site-specific: within a multiwell dish with freely shared media and nutrients, a 10-fold inhibition of cell growth was achieved on top of the biocatalytic coatings compared to the immediately adjacent enzyme-free microwells. The physiological effect of NO produced via the enzyme prodrug therapy was validated ex vivo in isolated arteries through the measurement of vasodilation. Biocatalytic coatings were deposited on wires produced using alloys used in clinical practice and successfully mediated a NONOate concentration-dependent vasodilation in the small arteries of rats. The results of this study present an exciting opportunity to manufacture implantable biomaterials with physiological responses controlled to the desired level for personalized treatment.
Keywords: enzyme-prodrug therapy, nitric oxide, biocatalytic coating, polyelectrolyte multilayers, galactosidase, stent, vasodilatation
Introduction
Nitric oxide (NO) is a molecule with an incredibly broad spectrum of physiological activity.1−4 This small but a highly potent molecule is implicated in the progression of and therapies for inflammation,5 cancer,6 and viral pathologies,7 among others. It was dubbed the “guardian of cardiovascular grafts”8 because of its proproliferative activity on the endothelium as well as antiadhesion and antiaggregation signaling to platelets in the circulating blood. Being a radical species, the lifetime of NO in human blood is very short—approximately 1 s, over which time it has the capacity to rapidly diffuse over a distance of approximately 100 μm, which is the length scale of the adjacent interacting cells. Because of the important biological functions of NO and its highly tissue-specific activity, strategies for site-specific delivery of this molecule for human therapy are highly desirable.4,9,10 Current methods for the site-specific delivery of NO rely on the release of the drug from its adduct depots.9,10 Although powerful in their own right, these methods are limited in their capacity to (a) engineer a constant, zero-order release of NO, (b) engineer NO depots into the existing biomaterials used for vascular tissue engineering, and (c) control the dosage of the drug within the biomaterial.9,10 The latter aspect is particularly important in that controlling the drug feed upon implantation of a therapeutic device is the necessary step toward personalized medical care such that the drug levels can be tuned to a desired level established individually for each patient.
Inspired by nature, we hypothesized that localized synthesis is the most appropriate approach toward the generation and site-specific delivery of controlled amounts of NO. We envision that this can be accomplished with the use of substrate-mediated enzyme prodrug therapy (SMEPT).11 In our past studies, we engineered biocatalytic (enzyme-containing) hydrogel matrices12,13 and multilayered polymer coatings14,15 to illustrate the fundamental advantages of SMEPT over the conventional implant-mediated drug delivery. Specifically, SMEPT affords an on-demand drug delivery15 because localized drug synthesis relies on externally administered benign prodrugs. The same biomaterial can be tuned to release the desired amount of the drug in unit time simply through the choice of concentration of the administered prodrug.12 Using SMEPT, the same biomaterial is capable of synthesizing a range of therapeutic molecules at their nominated concentration and time of administration—taken individually, in sequence, or in combination.13 Within the lifetime of the enzyme, drug release is sustained and follows a highly beneficial zero-order, linear release pattern.14 These features make SMEPT well-suited to accomplish the localized synthesis needed to deliver NO.
Several research groups have recently and independently developed SMEPT-like systems for localized synthesis of NO. The original report from Cha and Meyerhoff revealed that selenium-containing organic compounds mimic selenium-containing enzyme, glutathione peroxidase, and efficiently mediate the release of NO from S-nitrosothiols (RSNO).16 On the basis of this finding, several groups have performed substrate-mediated synthesis of NO achieved by the biomaterials that are surface-modified with selenocystamine and/or diselenodipropionic acid17,18 and organotellurium compounds.19 These systems produced highly favorable results upon in vivo validation and provided inspiration for a broader development of this approach. An impressive achievement was reported by Yang et al.,20 whereby NO was generated using endogenous donors of NO (natural RSNO), thus avoiding reliance on the external administration of prodrugs.
Being highly important in their own right, enzyme mimics are limited in that, as described, these catalysts can only mediate the synthesis of NO, that is, they are only suited to mediate a monotherapy. In contrast, natural enzymes are capable of converting a wide range of substrates, and within the same family of prodrugs (e.g., glucuronides, phosphates), one enzyme performs bioconversion to synthesize multiple drugs—providing for the flexibility of drug choice and to mediate combination therapy, as is highly desired for a range of drug delivery applications.13 Of the enzymes that are typically used for enzyme–prodrug therapies,21 only β-galactosidase (β-Gal) has a readily available corresponding prodrug for the synthesis of NO, namely, β-Gal–NONOate.22,23 On the basis of the above discussion, we chose β-Gal as the enzyme and β-Gal–NONOate as the prodrug.
The overall goal of the work presented herein is to engineer SMEPT onto the surface of the metallic substrates used for the production of diverse medicinal implants such as to achieve an enzyme-mediated localized synthesis of NO. To engineer SMEPT onto the surface of the metallic wires, we used the sequential polymer deposition technique (also known as “layer-by-layer” deposition, LbL) to form biocatalytic β-Gal-containing multilayered films (Figure 1). The prime advantage of this surface modification method is that it is an all-aqueous, solution-based approach. This technique accommodates modification of any substrate with no restriction on surface geometry and topography. Multilayered thin films have previously been deposited onto the surface of cardiovascular stents for surface-mediated gene delivery.24 For the production of NO, multilayered films have previously been assembled to contain arginine, a natural precursor for the synthesis of NO by NO synthase,25 and to contain selenium-based enzyme mimics.26 However, to our knowledge, there are no prior reports of multilayered thin films for the enzyme-mediated synthesis of NO. A prime consideration for the biocatalytic performance of such coatings is the choice of the polyelectrolyte pair used to assemble the multilayered thin film. The existing examples of enzyme-containing LbL films27 do not provide predictive power to nominate an optimal coating for the catalytic output of the immobilized enzyme. Therefore, the first objective of this work was to conduct a broad screen of polyelectrolyte multilayered coatings, focusing on the catalytic output of the film as a criterion of selection. The second objective of this work was to establish control over the synthesis of NO by the biocatalytic coatings and to validate if a surface-mediated approach to the delivery of NO is site-specific. Finally, the ultimate goal of this study was to provide biomedical characterization of the physiological activity of NO as produced via localized enzymatic biocatalytic conversion. We envisioned that an ex vivo wire myograph model presents a favorable setting for this test, in that it uses mammalian tissue and records native physiological responses and it is readily suitable for the systematic variation of experimental conditions (such as recording dose–response curves). In doing so, ex vivo tissue-based studies minimize the use of laboratory animals, yet provide the sought-after validation of physiological effects mediated by implantable biomaterials.
Figure 1.
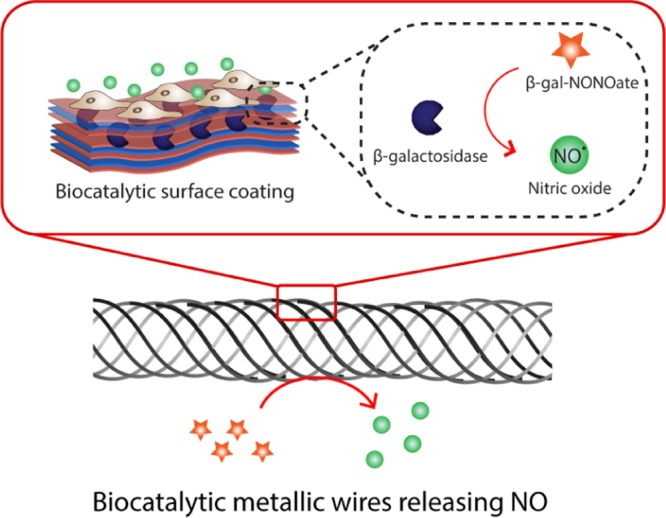
Enzymatic synthesis of NO is engineered in this work into multilayered polyelectrolyte coatings. When used as substrates for cell culture, these biocatalytic coatings provide localized synthesis of NO for localized delivery to the adhering cells.
Materials and Methods
Materials and Instruments
Unless stated otherwise, all materials were purchased from Sigma-Aldrich. Pyrogallol and the enzyme β-Gal (derived from Escherichia coli, 465 kDa) were purchased from Merck, NO gas was obtained from Air Liquide Danmark AS, and β-Gal–NONOate was obtained from Cayman Chemical. Fluorescein diacetate (FDA) and propidium iodide (PI) were used as live/dead stains. PrestoBlue cell viability reagent and Quant-iT PicoGreen dsDNA assay kit were both obtained from Life Technologies. Round metal wires with a 200 μm diameter (alloys 35N LT, 316L, and L605) were manufactured by Fort Wayne Metals and processed as reported elsewhere.28
Quartz crystal microbalance (QCM) experiments were conducted on QSense E4 (Biolin Scientific). Quantitative absorbance and fluorescence measurements were conducted using an EnSpire PerkinElmer multilabel plate reader and a Tecan infinite M200 PRO multimode reader. Imaging was performed on a Zeiss Axio Observer Z1 microscope. Ultrapure water (MQ) with a resistivity of 18.2 MΩ cm–1 obtained from a Milli Q direct 8 system (Millipore) was used for all solutions.
Atomic force microscopy (AFM) characterization of the samples was carried out in a tapping mode (MultiMode VIII, Bruker, USA). A cantilever (ScanAsyst-Air, Bruker, USA) with a sharp tip (nominal tip height 2.5–8 μm and nominal tip radius 2 nm) and triangular geometry (nominal resonant frequency 70 kHz, spring constant 0.4 N/m, length 115 μm, and width 25 μm) was used to conduct the experiment. Operational parameters such as tapping amplitude and gains were adjusted to optimize the resolution and prevent the tip–sample damage. The scan rate was set to 1 Hz. The AFM experiment was conducted in air at 21 °C and 54% humidity. Raw data were processed using open-source software Gwyddion for tilt correction and adequate polynomial leveling. To calculate the root-mean-square (rms) roughness, images were acquired from five different locations at various scan sizes.
Polymers used in this work were poly(ethyleneimine) (PEI, ∼25 000 Da, branched), poly(sodium-4-styrene sulfonate) (PSS, ∼70 000 Da), poly(methacrylic acid) (PMA, ∼18 500 Da), poly(acrylic acid) (PAA, ∼28 300 Da), dextran sulfate (200 000 Da), chrondoitin sulfate (5–100 kDa), hyaluronic acid (HA, low: 15 000–30 000 Da, high: 1.5–1.8 × 106 Da), alginate (ALG, 12 000–40 000 Da), DNA (from herring testes), poly(diallyldimethylammonium chloride) (100 000–200 000 Da), poly-l-lysine (30 000–70 000 Da), poly-l-arginine hydrochloride (PLA, 15 000–70 000 Da), poly-l-histidine (PLH, 5000–25 000 Da), poly(allylamine hydrochloride) (PAH, ∼15 000 and 17 500 Da), chitosan (Chi, 190 000–310 000 Da), biodegradable polyamidoester29 (∼8300 Da), and protamine sulfate (PRT, ∼5100 Da). Phosphate-buffered saline (PBS) and 4-(2-hydroxyethyl)piperazine-1-ethane-sulfonic acid (HEPES) of 10 mM containing 150 mM NaCl in MQ with pH 7.4 were used as buffers.
Polymer Screen
For the assembly of polyelectrolyte multilayered coatings, the polymers were dissolved in PBS to 0.1 g/L. Chi was dissolved in acetic acid to 10 g/L and diluted in PBS to 0.1 g/L, whereas PLA and PLH were dissolved in 0.1 M HCl to 10 g/L and diluted to 0.1 g/L with PBS. All multilayered films were fabricated in standard tissue culture polystyrene (TCPS) 96-well plates, unless stated otherwise. To ensure film buildup, a primary layer of PEI was applied, followed by the alternating layers of polyanions and polycations (100 μL per well). Each layer was left to adsorb for 5 min, followed by a washing step two times with MQ. The enzyme β-Gal was added at a concentration of 20 mg/L (100 μL), unless stated otherwise, and allowed to incubate for 1 h prior to a single washing step. Additional film buildup was performed as described above, resulting in a final architecture of PEI–(polyanion/polycation)3–β-Gal–(polycation/polyanion)2.5. For quantitative evaluations, fluorogenic prodrug resorufin galactopyranoside was added together with fresh media yielding 5 mg/L and allowed to incubate for 30 min prior to readout.
Film Assembly Using PSS/PAH
The multilayered films of PSS and PAH were prepared with the polymers dissolved in HEPES buffer to a concentration of 0.1 g/L. To ensure film buildup, a primary layer of PEI was applied, followed by the alternating layers of PSS and PAH, as described previously. The resulting multilayered films had the final architecture of PEI–(PSS/PAH)3–β-Gal–(PAH/PSS)2.5. The multilayered films were then incubated in a 100 μL HEPES buffer at 4 °C until usage.
Multilayer Film Assembly on Metal Wires
The films deposited on metal wires consisted of the polymers PEI/PSS/PAH dissolved in PBS to a concentration of 1 g/L. PEI was dissolved in MQ water and left to adsorb for 30 min, all PSS/PAH layers were left to adsorb for 10 min, and the enzyme β-Gal was allowed to incubate for 2 h. Prior to film assembly, the wires were cleaned by consecutive immersions in 20 w/v % citric acid, demineralized water, and 70% ethanol using sonication. The resulting multilayered films had the final architecture of PEI–(PSS/PAH)3–β-Gal–(PAH/PSS)1.5. The durability of the enzyme activity deposited onto the wires was evaluated after 7, 14, 21, and 28 days of incubation in PBS at 37 °C using resorufin-β-d-galactopyranoside at a concentration of 5 mg/L, which was allowed to incubate for 30 min prior to readout.
Quantification of NO
Deoxygenated water was prepared using argon gas. The latter was first led through a vial containing 10 mM pyrogallol (to remove traces of oxygen) and then bubbled through water for 1 h to undergo deoxygenation. NO gas was led through 10 mM NaOH in water to remove traces of higher nitrogen oxides and then through deoxygenated water to prepare a saturated solution of NO (with a concentration of 2 mM, ref (30)). Serial dilutions of NO were then made using deoxygenated water using a gastight Hamilton syringe. The solutions of NO were mixed with DAF-FM in a black 96-well plate to the final concentration of the dye of 8 μM. The fluorescence of the solutions was recorded for 40 min (at λex/λem 495/515 nm) during which time the fluorescence readings reached a constant level. The final attained values of fluorescence were used to plot the fluorescence intensity versus NO concentration calibration curve, which resulted in a linear fit with the rms correlation coefficient of 0.92. The multilayered PSS/PAH coatings containing β-Gal were assembled in the wells of the black 96-well plates as described above. The wells were filled with fresh physiological saline solution containing 8 μM DAF-FM and varied concentrations of β-Gal–NONOate (5, 10, 15, and 20 μM). The fluorescence of the solutions was recorded over 30 min on a plate reader (λex/λem 495/515 nm). All experiments were carried out thrice in triplicates.
Cell Culture
The mouse myoblast cell line C2C12 was cultured in Dulbecco’s modified Eagle medium supplemented with 10 v/v % fetal bovine serum, 1 v/v % penicillin–streptomycin, and 1 mM sodium pyruvate. A 1/10 cell splitting was performed before reaching 70–80% confluence.
Myoblasts on Multilayered Coatings
The multilayered films produced as described above with an architecture of PEI–(PSS/PAH)3–β-Gal–(PAH/PSS)2.5 were UV-sterilized for 10 min prior to cell seeding. C2C12 myoblasts were seeded out at a starting density of 500 cells per well in 100 μL media and allowed to adhere overnight. NONOate (100–0 μM) was added together with fresh media, and the cells were left to incubate for 24 h at 37 °C and 5% CO2. For incubation of 48 and 72 h, the cells were administered fresh media with the respective (pro)drug every 24 h. The viability of the C2C12 myoblasts was evaluated using the PrestoBlue viability reagent, whereas quantitative DNA measurements were performed with Quant-iT PicoGreen.
Cell Imaging
C2C12 myoblasts were seeded out in 12-well tissue culture plates on 16 mm glass slides coated with multilayered films with or without the enzyme. The initial cell seeding density was 5000 cells per well in 1 mL media. Cells were allowed to adhere overnight, followed by the addition of 100 μM NONOate in fresh media. The samples were incubated for 24, 48, and 72 h, with fresh media and NONOate added every 24 h. Fresh media containing the LIVE/DEAD stains of FDA (5 mg/L) and PI (4 mg/L) were added to the samples and incubated for 5 min in the dark. After 2× washing with PBS, the cells were visualized.
Local Delivery Using Coculture μ-Slides
For the demonstration of local delivery, myoblast cells were seeded out into coculture μ-slides allowing nine individual subcultures in one major well (Ibidi GmbH). The designated wells were precoated with biocatalytic coatings as described above. The starting density of the cell was 700 cells in 50 μL media per minor well. The cells were allowed to adhere for 3–4 h before replenishing with 1 mL fresh media and incubated overnight. 100 μM solution of NONOate was subsequently administered in fresh media and replenished after 24 h. After a total of 48 h of incubation with NONOate, the samples were evaluated using LIVE/DEAD stain as described.
Ex Vivo Wire Myograph Study
Ethics Statement
All animal experiments in this study were approved by the Danish Animal Experiments Inspectorate (permission 2011/561-2011), and recommendations described in the Guide for the Care and Use of Laboratory Animals of the U.S. National Institutes of Health and the ARRIVE Guidelines were followed. Animals were housed in the animal facility in Universal Euro III type long with cages with standard wood bedding and space for two rats. There was a 12 h shift between light and darkness, and the animals had free access to food and drinking water.
Tissue
Male Wistar rats (9–11 weeks) with a weight of approximately 450–550 g were euthanized by cervical dislocation followed by exsanguination. The mesenteric bed was removed and placed in cold physiological saline solution (4.7 mM KCl, 1.17 mM MgSO4·7H2O, 119 mM NaCl, 25 mM NaHCO3, 1.18 mM KH2PO4, 0.026 mM ethylenediaminetetraacetic acid, 5.5 mM glucose, and 1.6 mM CaCl2). The first or second branch arteries with a diameter of around 300–450 μm were dissected using microforceps (Dumont no. 5) and a microsurgery scissor.
Mounting and Normalization
Arteries with a length of 1.5–2 mm were mounted on a dual wire myograph (model 410 A, Danish Myo Technology A/S, Denmark) using a 40 μm steel wire and cold physiological saline solution. The arteries were left to equilibrate at 37 °C while bubbled with a bioair (21% O2, 5% CO2, and 74% N2). To ensure comparable results, the arteries were normalized to an internal circumference corresponding to 90% of the internal circumference of a fully relaxed artery at a transmural pressure of 100 mmHg. Norepinephrine (NE) was used for contraction of the small mesenteric arteries. Iberiotoxin (IbTX) and 1H-[1,2,4]oxa-diazolo[4,3-a]quinoxalin-1-one (ODQ), both from Tocris Bioscience, were used as inhibitors of the NO-mediated vasodilation.
Experimental Protocol
Before experimentation, the viability of the smooth muscle cells (SMCs) was tested by contracting with 10 μM NE. Only the arteries with a contraction corresponding to a transmural pressure above 75 mmHg were included in these studies. Stent wires with a diameter of 100 μm and a length of 9 mm were placed in the lumen of the arteries before testing the viability of the SMCs. To investigate if the prodrug NONOate could produce NO in the presence of the LbL-coated wire, the arteries were contracted with 3 μM NE, and when stabilized, NONOate was added in a cumulative manner to produce a concentration–response curve (CRC) ranging from 0.5 nM to 15 μM. The arteries were discarded if they developed a contraction of less than 60% of the maximum contraction. Furthermore, controls were made both with and without the presence of the uncoated wire. For inhibition studies, the arteries were incubated for 30 min with 3 μM ODQ and 0.1 μM IbTX before adding NONOate, as described above.
Data Analysis
Unless stated otherwise, the numerical data are presented as mean ± SD and calculated based on at least three independent experiments. All the data were analyzed using Microsoft Excel 2010 and plotted in OriginPro 8 or GraphPad Prism 7. Ex vivo wire myograph data were collected by the LabChart 5 software program (ADInstruments Ltd, Oxfordshire, UK), presented as mean ± SEM, and calculated based on at least five experiments. The statistics were conducted using Student’s t-test or one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test in Excel or GraphPad Prism 7. For the myograph experiments, two-way ANOVA was used. Statistical significance was defined as P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***).
Results and Discussion
The optimization of the architecture of the multilayered polyelectrolyte films was conducted toward selection of compositions that favored high catalytic output of the coating. A total of eight polyanions and eight polycations (Figure 2) were used to assemble the multilayered thin films in the wells of the standard 96-well plates. Polyelectrolyte assembly at the interface may proceed “linearly” and afford relatively dense coatings (e.g., PSS/PAH, refs (31) and (32)) or “exponentially” and afford hydrogel-like films (HA- and ALG-containing films and polypeptide-containing films, ref (33)). For polymers with pH-dependent ionization (e.g., PAA, PMA, and PAH), the thickness and density will also depend on the coating assembly conditions.34 Furthermore, the polymers differ in their capacity to support protein adsorption,35 and it is also important that the enzyme is not displaced upon the deposition of subsequent polymer layers. These combined effects define the catalytic performance of the assembled coating. Although in-depth analysis of these factors individually fell beyond the scope of this study, we aimed to identify the composition(s) that satisfy each of these conditions and in doing so afford the coatings suitable for subsequent applications in SMEPT.
Figure 2.

Chemical structure and abbreviations of polyanions and polycations used in this study (DNA and PRT not shown).
Multilayered polyelectrolyte coatings were assembled, starting with a priming layer of PEI followed by a total of five polyelectrolyte bilayers. Enzyme immobilization was conducted through the exposure of the three bilayer coatings (with a polycation surface layer) followed by the subsequent deposition of 2.5 more bilayers (polyanion top layer). The resulting coatings were evaluated in terms of their catalytic output using the fluorogenic galactosidase substrate, resorufin β-d-galactopyranoside. To probe the spontaneous release of the protein from the coatings, bioconversion was also tested in the supernatants aspirated from the coatings immediately prior to the evaluation of the coating.
The prime conclusion from this screen is that the overall majority of coatings supported well the activity of the immobilized β-Gal (Figure 3). Linearly growing multilayers based on synthetic polyelectrolytes (e.g., PSS/PAH)31,32 were active, rather similar to the exponentially growing coatings based on polysaccharides,33 revealing that the density of the coating may not be decisive for the activity of the immobilized enzyme. This includes coatings based on polypeptides with the potential to make gradually degradable coatings36 and coatings based on nondegradable polymers such as to assemble permanent material surface modification.37 With a view toward long-lasting coatings, subsequent work relied on the PSS/PAH multilayers.
Figure 3.

Results of quantification of the catalytic output for the multilayered coatings composed of different polyanion/polycation combinations and equipped with β-Gal: (A) multilayered coatings with PAH or Chi as polycations and a variation of polyanions; (B) multilayers with PSS or HA as polyanions and a variation of polycations. HA with high and low molar mass is denoted as HAH and HAL, respectively. Enzymatic catalysis was evaluated using a fluorogenic enzyme substrate, resorufin β-d-galactopyranoside.
We quantified the buildup of these multilayers and more importantly the amount of the enzyme immobilized within the coating. The analyses were performed using QCM (Figure 4A). This experiment revealed that under chosen conditions (20 mg/L enzyme concentration in HEPES buffer, pH 7.4), enzyme immobilization proceeds rather fast and is complete within minutes affording a protein coverage of 198 ± 20 ng/cm2. This enzyme coverage is within the same order of magnitude as what has previously been reported for PSS/PAH and immobilization of immunoglobulin38 or albumin39 and adsorption of other proteins onto the multilayered polyelectrolyte surface coatings.35 The resulting multilayered coatings were imaged using AFM (Figure 4B), illustrating a typical morphology of LbL coatings and an rms roughness of 5 ± 2 nm also typical for these coatings.40
Figure 4.

(A) QCM monitoring of the assembly of multilayered surface coatings based on PSS and PAH (PEI priming layer) and immobilization of β-Gal. Quantification of protein coverage is based on three independent experiments. For experimental details, see the Materials and Methods. (B) AFM image of the PSS/PAH coating with immobilized β-Gal. Scale bars: 300 nm (black, XY dimension) and 0–6 nm (Z-direction).
Next, we aimed to characterize the coatings in detail in terms of their biocatalytic output and specifically in the production of NO. To achieve this, we established a fluorescence-based readout to quantify the production of NO. DAF-FM is a dye that reacts with NO and upon doing so becomes fluorescent. Because of this property, it has been previously used to, for example, visualize the delivery of NO to cells.41 The calibration curve for fluorescence versus concentration of NO was obtained using NO gas and serial dilution of its saturated solution (2 mM, ref (30)) in deoxygenated water. This experiment afforded a linear calibration curve correlating fluorescence of DAF-FM and feed of NO in solution, thus providing a facile method to quantify the production of NO by the biocatalytic coatings (for details, see Materials and Methods).
To quantify the production of NO using biocatalytic coatings, the latter were prepared as discussed above in the wells of black 96-well plates and incubated with the β-Gal–NONOate prodrug in the presence of DAF-FM. The fluorescence of the solutions was continuously recorded on a plate reader (Figure 5A). These data demonstrate that in the absence of β-Gal in the surface coatings, no production of NO was observed. In contrast, enzyme-containing coatings afforded a steady evolution of fluorescence, that is, steady production of NO over at least 30 min. The values of fluorescence intensity were then converted into the concentrations of NO and NO flux, that is, the rate of production of NO by the biocatalytic surface coatings in unit time (Figure 5B). This analysis affords a highly important conclusion that under SMEPT conditions, the assembled coatings afford a flux of NO, which matches with that reported for healthy endothelium (0.05–0.4 nmol min–1 cm–2).42,43 Furthermore, prodrug concentration is a facile tool to fine-tune NO flux to the desired level, a unique opportunity for personalized medicine.
Figure 5.
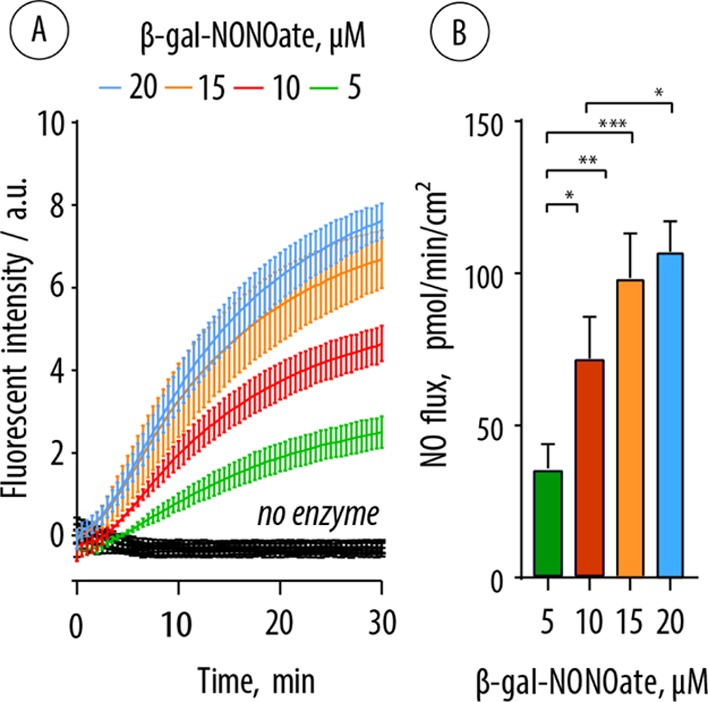
(A) Kinetic curves illustrating the evolution of fluorescence resulting from the biocatalytic production of NO by the multilayered surface coatings at varied concentrations of β-Gal–NONOate and subsequent conversion of DAF-FM into its fluorescent product. (B) Flux of NO afforded by the biocatalytic surface coatings at varied concentrations of β-Gal–NONOate (calculated from the linear part of the data curves in panel (A)).
From a different perspective, we hypothesized that NO flux can also be optimized through the variation of the conditions of assembly of biocatalytic coatings, leading to a variation of enzyme content in the multilayered polyelectrolyte film. To test this, PSS/PAH coatings were assembled as discussed above, except that the enzyme immobilization step was performed using protein solutions with varied concentrations. QCM measurements revealed that with 20 mg/L feed, the enzyme surface immobilization was 2.0 ± 0.2 mg/m2 (Figure 4). With a decreased enzyme feed, the surface immobilization decreased accordingly, and at 2 mg/L, the feed was 0.38 ± 0.12 mg/m2, and at 0.2 mg/L, the feed was 0.10 ± 0.04 mg/m2. The resulting biocatalytic coatings were incubated with β-Gal–NONOate in the presence of DAF-FM with continuous recording of the fluorescence of the solution. With an excess prodrug, the biocatalytic production affords a highly desired linear profile of the evolution of fluorescence (Figure 6A). Furthermore, variation of the enzyme feed in the assembly solution affords a facile means of control over the biocatalytic output of the coating in a wide range of physiologically relevant flux of NO (Figure 6B). Over several days of analysis, assembled coatings revealed a minor decrease in the biocatalytic performance, indicating loss of enzyme activity; however, NO flux remained well within the physiologically relevant range42,43 (Figure 6C).
Figure 6.

(A) Kinetic curves illustrating the evolution of fluorescence resulting from the reaction of DAF-FM with NO produced by the biocatalytic surface coatings. Coatings were assembled using enzyme feed solutions, with the protein content from 0.2 to 20 mg/L; 100 μM β-Gal–NONOate. (B) NO flux sustained by the biocatalytic coatings (calculated from the linear part of the curves in panel (A)). (C) NO flux sustained by the biocatalytic surface coatings assembled using 20 mg/L enzyme feed solution in the presence of 100 μM β-Gal–NONOate as measured at the time points from 1 to 4 days.
Initial cell culture characterization of the biocatalytic coatings was carried out using myoblast cells. In the context of atherosclerosis and cardiovascular stenting, the proliferation of muscle cells is a highly undesirable event that can lead to the restenosis cascade.8 Current stents on the market are designed to gradually release cytotoxins such as paclitaxel specifically to prevent proliferation of muscle cells.44 NO in high concentrations is also known to elicit an antiproliferative activity on muscle cells,45 providing a convenient reporter system for the initial evaluation of surface coatings, releasing controlled quantities of NO. Multilayered surface coatings were assembled within the wells of the standard 96-well cell culture plate. Myoblasts were seeded and cultured directly on top of the coatings over 72 h. To quantify cell growth, two assays were performed, namely, viability screen using the standard commercially available viability kit (PrestoBlue) as well as direct quantification of DNA, the latter being proportional to the number of cells in the well. The effects of NO were quantified at 24, 48, and 72 h time points (Figure 7). Thin films prepared using 0.2 mg/L enzyme feed solution revealed no change in cell proliferation—readily explained by the low levels of generated NO. In contrast, higher enzyme content (2 and 20 mg/L) in the thin films endowed the coatings with a high level of biocatalytic activity, and at each time point, the surface coatings provided effective antiproliferative activity. Evaluation of cell growth through quantification of DNA proved to be a more sensitive readout, and even at 24 h of incubation, the decrease in cell numbers was statistically significant. Incubation of cells on biocatalytic films in the presence of the NONOate for 48 h suppressed the proliferation of myoblasts effectively to zero. Quantitative data were well-supported by the microscopic visualization of adhered cells (Figure 8). The images illustrate that in the absence of the enzyme, cells proliferate and increase in number over time. The cell stain used herein, nonfluorescent FDA, is converted by the intracellular esterases into its highly fluorescent product, fluorescein, which highlights that the cells are metabolically active and proliferating. In contrast, the presence of the enzyme in the multilayered surface coating resulted in a negligible proliferation of cells. These results illustrate that localized generation of NO is a highly effective method to control the proliferation of adhering myoblasts.
Figure 7.

Cell number and viability for myoblasts cultured on the PSS/PAH multilayered polyelectrolyte films over 24, 48, and 72 h. Multilayered films were equipped with the β-Gal enzyme with the feed protein content of 0, 0.2, 2, and 20 mg/L. Cell culture was performed in the presence of 100 μM NONOate. Results are presented as means ± SD for at least three independent experiments.
Figure 8.

Fluorescence microscopy images of myoblast cells proliferating on the multilayered polyelectrolyte coatings in the presence of 100 μM NONOate over 72 h of cell culture either with or without β-Gal incorporated into the polymer film. Scale bar: 50 μm.
We next aimed to investigate if the localized synthesis of NO via enzymatic conversion of prodrugs achieves the highly sought-after site-specific effect of drug delivery. The main aspect contributing to this highly advantageous prospect is the short lifetime of the generated NO in serum. Site-specific drug delivery was investigated using multiwell coculture slides (Figure 9A). Each of the two major wells is separated into nine minor wells, allowing individual cell cultures to be established in each minor well. This allowed us to assemble individually designed multilayered polyelectrolyte coatings such that only the nominated minor wells contained the immobilized enzyme (denoted with “E”). Upon cell attachment, cell culture medium was added to cover the entire major well such that the nutrients, the prodrugs, and the newly synthesized drugs are shared among the minor wells. Myoblasts were cultured in the presence of 100 μM NONOate for 48 h with the replenishment of the media and the prodrug at 24 h. The resulting cultures were imaged using fluorescence microscopy (Figure 9B), and the cell density was quantified through a direct cell count (Figure 9C). The microscopy images demonstrate a clear negative correlation between the presence of the enzyme in the underlying surface coating and the resulting cell density in the minor well—attributable to the localized enzymatic production of NO. This conclusion is supported by the cell count that illustrates a statistically significant, almost 10-fold decrease in the number of cells in the minor wells with the immobilized enzyme. Despite the cell culture medium being shared among the nine minor wells, the therapeutic effect due to the synthesized NO is only observed locally in the well where NO is produced. This provides a direct evidence of the site-specific nature of this mode of delivery of NO.
Figure 9.

(A) Schematic representation of coculture μ-slides indicating the multilayered-coated wells. (B) Fluorescence microscopy imaging of myoblast cells. Selected wells were coated with biocatalytic multilayers with 20 mg/L β-Gal for local delivery of NO. Cells were incubated for 48 h in the presence of 100 μM NONOate, replenished at 24 h. Scale bar: 100 μm. (C) Averaged cell count of coated vs noncoated wells. Results are presented as mean ± SD for at least three independent experiments. ***P < 0.001.
Encouraged by the successful design and implementation of the localized synthesis of NO on model substrates, biocatalytic coatings were engineered on the surface of metallic wires produced industrially for the manufacturing of cardiovascular stents and other implantable biomaterials. Three corrosion-resistant alloys commonly used in clinical practice (316L, 35N LT, and L605) were used as substrates to assemble the polyelectrolyte multilayered coatings containing β-Gal enzyme. The wire samples were identical dimensionally, such that individual specimens had closely matched surface area. The wires with deposited multilayered coatings were first used to evaluate the resulting biocatalytic output, that is, ascertain enzymatic catalysis mediated by the coated wires. We note that in our preliminary experiments, we observed that the assembly conditions used for planar substrates did not afford sufficient buildup of the material on the wires (as evidenced by the low enzymatic output of the modified wire). We therefore optimized the assembly conditions to maximize the deposited quantities for the polymers and the protein for details, see Materials and Methods). The resulting biocatalytic coatings on the three alloys were nearly identical in their performance in converting the fluorogenic substrate into its fluorescent product (Figure 10). However, to much surprise, the alloy composition had a significant impact on the rate of deactivation of the enzyme within the coating. Thus, 316L alloy appears to deactivate the enzyme quickly, and at day seven measurement, the enzymatic activity of the biocatalytic coating was hardly detectable. Although the origin of this deactivation is not fully clear, a possible cause of this may be that the 316L alloy contains copper; the latter is a known inhibitor of β-Gal.46 In contrast, when deposited on the wires based on 35N LT and L605 alloys, the biocatalytic coatings were rather stable in terms of their enzymatic performance, revealing a half-life of the enzyme of ca. 7 days. Following 2 weeks of incubation in a physiological buffer at 37 °C, these implantable biomaterials revealed at least 20% of the initial enzymatic activity. We are now working toward optimization of the enzyme-containing coatings to extend the lifetime of the enzyme.
Figure 10.

(A) Scanning electron microscopy images of the 35N LT and L605 wires in pristine form (bare) and after the assembly of a biocatalytic coating; scale bars: 100 μm. (B) Enzymatic activity of the multilayered polyelectrolyte coatings containing β-Gal and assembled on the corrosion-resistant alloys (35N LT, L605, 316L) during incubation in PBS at 37 °C over 28 days. Statistical evaluation was performed to compare the enzymatic activity of the coated wires with the background fluorescence of a metal without the enzyme, calculated via a two-way ANOVA, followed by Tukey’s multiple comparison test.
The two alloys that supported the enzymatic activity over extended times were then used in an ex vivo physiological activity validation study. Specifically, we used a wire myograph technique to quantify the vasodilation in rat mesenteric arteries. Vasodilation is among the most important and the most well-characterized physiological effects of NO.47 Of high importance, specific inhibitors of NO signaling pathways such as ODQ and IbTX allowed us to confirm that the experimentally observed effects are indeed due to the released NO and not due to the off-site effects (vasodilation because of the tissue cell death).
The samples of wires based on 35N LT and L605 were coated with the biocatalytic multilayered polyelectrolyte film and inserted into the artery mounted in the myograph and covered with a physiological saline solution. Vasoconstriction was achieved through the addition of NE, following which increasing concentrations of the NONOate were added to the bath. As expected, NONOate revealed only a minor vasodilation activity, likely because of the spontaneous, nonenzymatic degradation of the prodrug (Figure 11, top row). Pristine metallic wires mediated no prodrug conversion, as evidenced by a negligible decrease in the vascular contraction force at all doses of the NONOate. In contrast, addition of NONOate to the biocatalytic wires resulted in a pronounced, concentration-dependent decrease in contraction, illustrating a physiological effect of the NO produced via SMEPT. The NONOate vasodilation was abolished in the presence of inhibitors of NO-induced signaling pathways, soluble guanylate cyclase, and large conductance calcium-activated K channels by ODQ and IbTX. These findings confirm the specificity of the physiological response because of the release of NO. The dose response curves for the NONOate (Figure 11, bottom) reveal that vasodilation was registered at NONOate concentrations significantly (100- to 1000-fold) lower than the toxicity discussed above toward cell proliferation and using the 35N LT wires. Thus, statistically significant relaxation was achieved at nanomolar concentrations of the prodrug. This experiment also explicitly illustrates the highly advantageous opportunity associated with SMEPT in that the adjustment of the physiological effect in a mammalian tissue was achieved using the same implantable biomaterial via the choice of the concentration of NONOate. In other words, the same biomaterial can be instructed externally such as to achieve personalized, fine-tuned therapeutic or physiological response—an opportunity not available with the stents and the overall majority of implants on the market today.
Figure 11.

Ex vivo wire myograph quantification of the contraction force exerted ex vivo by the rat mesenteric arteries (A,B) and calculated degree of vasorelaxation (C,D) in the presence of NONOate (0.5 nM to 15 μM) and the wires based on 35N LT and L605 alloys coated with the biocatalytic multilayered polyelectrolyte coatings (denoted as wire + Enz + NONOate). Control experiments include administering the NONOate in the absence of wires (denoted as NONOate), using the wires and multilayered coatings with no incorporated enzyme (denoted wire + NONOate), and using the samples identical to the experimental group and also containing specific inhibitors of the NO-mediated signaling pathways (denoted as wire + Enz + NONOate + ODQ/IbTX). Data are presented as mean ± SEM, n = 5 or greater. Statistics is shown for comparing the effects mediated by the biocatalytic coatings with those mediated by the NONOate (¤), the coatings with no enzyme (#), and the biocatalytic coatings in the presence of inhibitors (*) and calculated via a two-way ANOVA followed by Tukey’s multiple comparison test.
Conclusions
In this work, we engineered an enzyme–prodrug therapy onto the surface of metallic wires based on the alloys commonly used in clinical practice. The resulting coatings performed localized bioconversion of prodrugs and produced the physiological messenger molecule NO with a flux similar to the level produced by the healthy human endothelium. We successfully demonstrated physiological responses to the locally produced NO, in an ex vivo wire myograph model. The NO-mediated vasorelaxation was instructed by the concentration of the administered NONOate, thus illustrating that the physiological response itself is not engineered into the implantable biomaterial. Rather, it is the capacity to respond that was successfully incorporated and preserved after biomaterial processing and delivery to tissue. This design paradigm can be used toward personalized treatments using therapeutic implants.
Acknowledgments
The authors wish to acknowledge Dr. Aslan Hüsnü and Rikke Meyer (Aarhus University) for AFM imaging and Essi Taipaleenmäki and Dr. Brigitte Städler (Aarhus University) for SEM imaging of the biocatalytic coatings. The authors wish to acknowledge the financial support from the European Research Council Consolidator grant (A.N.Z., ERC-2013-CoG 617336 BTVI), the ERC Seventh Framework Programme Consolidator grant “Naturale CG” under grant agreement no. 616417 (M.M.S.), a Wellcome Trust Senior Investigator Award (M.M.S., 098411/Z/12/Z), and the Novo Nordisk Foundation (US, NNF16OC0023284).
Author Present Address
∇ School of Chemical and Biomolecular Engineering, The University of Sydney, Sydney, NSW 2006, Australia.
Author Contributions
¶ A.K.W. and B.F. contributed equally.
The authors declare no competing financial interest.
References
- Bredt D. S.; Snyder S. H. Nitric Oxide: A Physiologic Messenger Molecule. Annu. Rev. Biochem. 1994, 63, 175–195. 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]
- Garthwaite J.; Boulton C. L. Nitric Oxide Signaling in the Central Nervous System. Annu. Rev. Physiol. 1995, 57, 683–706. 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- Änggård E. Nitric oxide: mediator, murderer, and medicine. Lancet 1994, 343, 1199–1206. 10.1016/s0140-6736(94)92405-8. [DOI] [PubMed] [Google Scholar]
- Carpenter A. W.; Schoenfisch M. H. Nitric oxide release: Part II. Therapeutic applications. Chem. Soc. Rev. 2012, 41, 3742–3752. 10.1039/c2cs15273h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking J.; Xie Q.-w.; Nathan C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997, 15, 323–350. 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- Felley-Bosco E. Role of nitric oxide in genotoxicity: Implication for carcinogenesis. Cancer Metastasis Rev. 1998, 17, 25–37. 10.1023/a:1005948420548. [DOI] [PubMed] [Google Scholar]
- Akaike T.; Maeda H. Nitric oxide and virus infection. Immunology 2000, 101, 300–308. 10.1046/j.1365-2567.2000.00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mel A.; Murad F.; Seifalian A. M. Nitric Oxide: A Guardian for Vascular Grafts?. Chem. Rev. 2011, 111, 5742–5767. 10.1021/cr200008n. [DOI] [PubMed] [Google Scholar]
- Jen M. C.; Serrano M. C.; van Lith R.; Ameer G. A. Polymer-Based Nitric Oxide Therapies: Recent Insights for Biomedical Applications. Adv. Funct. Mater. 2012, 22, 239–260. 10.1002/adfm.201101707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio D. A.; Schoenfisch M. H. Nitric oxide release: Part I. Macromolecular scaffolds. Chem. Soc. Rev. 2012, 41, 3731–3741. 10.1039/c2cs15272j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejerskov B.; Olesen M. T. J.; Zelikin A. N. Substrate mediated enzyme prodrug therapy. Adv. Drug Delivery Rev. 2017, 118, 24–34. 10.1016/j.addr.2017.04.013. [DOI] [PubMed] [Google Scholar]
- Fejerskov B.; Zelikin A. N. Substrate Mediated Enzyme Prodrug Therapy. PLoS One 2012, 7, e49619 10.1371/journal.pone.0049619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes A. C.; Zelikin A. N. Enzyme Prodrug Therapy Engineered into Biomaterials. Adv. Funct. Mater. 2014, 24, 5202–5210. 10.1002/adfm.201304312. [DOI] [Google Scholar]
- Andreasen S. Ø.; Fejerskov B.; Zelikin A. N. Biocatalytic polymer thin films: optimization of the multilayered architecture towards in situ synthesis of anti-proliferative drugs. Nanoscale 2014, 6, 4131–4140. 10.1039/c3nr05999e. [DOI] [PubMed] [Google Scholar]
- Fejerskov B.; Jensen N. B. S.; Teo B. M.; Städler B.; Zelikin A. N. Biocatalytic Polymer Coatings: On-Demand Drug Synthesis and Localized Therapeutic Effect under Dynamic Cell Culture Conditions. Small 2014, 10, 1314–1324. 10.1002/smll.201303101. [DOI] [PubMed] [Google Scholar]
- Cha W.; Meyerhoff M. E. Catalytic generation of nitric oxide from S-nitrosothiols using immobilized organoselenium species. Biomaterials 2007, 28, 19–27. 10.1016/j.biomaterials.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Fan Y.; Pan X.; Wang K.; Wu S.; Han H.; Yang P.; Luo R.; Wang H.; Huang N.; Tan W.; Weng Y. Influence of chirality on catalytic generation of nitric oxide and platelet behavior on selenocystine immobilized TiO2 films. Colloids Surf., B 2016, 145, 122–129. 10.1016/j.colsurfb.2016.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng Y.; Song Q.; Zhou Y.; Zhang L.; Wang J.; Chen J.; Leng Y.; Li S.; Huang N. Immobilization of selenocystamine on TiO2 surfaces for in situ catalytic generation of nitric oxide and potential application in intravascular stents. Biomaterials 2011, 32, 1253–1263. 10.1016/j.biomaterials.2010.10.039. [DOI] [PubMed] [Google Scholar]
- Hwang S.; Meyerhoff M. E. Organoditelluride-tethered polymers that spontaneously generate nitric oxide when in contact with fresh blood. J. Mater. Chem. 2008, 18, 1784–1791. 10.1039/b715523a. [DOI] [Google Scholar]
- Yang Z.; Yang Y.; Xiong K.; Li X.; Qi P.; Tu Q.; Jing F.; Weng Y.; Wang J.; Huang N. Nitric oxide producing coating mimicking endothelium function for multifunctional vascular stents. Biomaterials 2015, 63, 80–92. 10.1016/j.biomaterials.2015.06.016. [DOI] [PubMed] [Google Scholar]
- Walther R.; Rautio J.; Zelikin A. N. Prodrugs in medicinal chemistry and enzyme prodrug therapies. Adv. Drug Delivery Rev. 2017, 118, 65–77. 10.1016/j.addr.2017.06.013. [DOI] [PubMed] [Google Scholar]
- Chandrawati R.; Chang J. Y. H.; Reina-Torres E.; Jumeaux C.; Sherwood J. M.; Stamer W. D.; Zelikin A. N.; Overby D. R.; Stevens M. M. Localized and Controlled Delivery of Nitric Oxide to the Conventional Outflow Pathway via Enzyme Biocatalysis: Toward Therapy for Glaucoma. Adv. Mater. 2017, 29, 1604932. 10.1002/adma.201604932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Lu Y.; Qin K.; Wu Y.; Tian Y.; Wang J.; Zhang J.; Hou J.; Cui Y.; Wang K.; Shen J.; Xu Q.; Kong D.; Zhao Q. Enzyme-functionalized vascular grafts catalyze in-situ release of nitric oxide from exogenous NO prodrug. J. Controlled Release 2015, 210, 179–188. 10.1016/j.jconrel.2015.05.283. [DOI] [PubMed] [Google Scholar]
- Jewell C. M.; Zhang J.; Fredin N. J.; Wolff M. R.; Hacker T. A.; Lynn D. M. Release of Plasmid DNA from Intravascular Stents Coated with Ultrathin Multilayered Polyelectrolyte Films. Biomacromolecules 2006, 7, 2483–2491. 10.1021/bm0604808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry B.; Winnik F. M.; Merhi Y.; Tabrizian M. Nanocoatings onto arteries via layer-by-layer deposition: Toward the in vivo repair of damaged blood vessels. J. Am. Chem. Soc. 2003, 125, 7494–7495. 10.1021/ja034321x. [DOI] [PubMed] [Google Scholar]
- Yang J.; Welby J. L.; Meyerhoff M. E. Generic nitric oxide (NO) generating surface by immobilizing organoselenium species via Layer-by-Layer assembly. Langmuir 2008, 24, 10265–10272. 10.1021/la801466e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelikin A. N. Drug Releasing Polymer Thin Films: New Era of Surface-Mediated Drug Delivery. ACS Nano 2010, 4, 2494–2509. 10.1021/nn100634r. [DOI] [PubMed] [Google Scholar]
- Schaffer J. E.; Nauman E. A.; Stanciu L. A. Cold-Drawn Bioabsorbable Ferrous and Ferrous Composite Wires: An Evaluation of Mechanical Strength and Fatigue Durability. Metall. Mater. Trans. B 2012, 43, 984–994. 10.1007/s11663-012-9661-3. [DOI] [Google Scholar]
- Appadoo V.; Carter M. C. D.; Lynn D. M. Controlling the surface-mediated release of DNA using “mixed multilayers”. Bioeng. Transl. Med. 2016, 1, 181–192. 10.1002/btm2.10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen U.; Wadsworth R. M.; Buus N. H.; Mulvany M. J. In vitro simultaneous measurements of relaxation and nitric oxide concentration in rat superior mesenteric artery. J. Physiol. 1999, 516, 271–282. 10.1111/j.1469-7793.1999.271aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruths J.; Essler F.; Decher G.; Riegler H. Polyelectrolytes I: Polyanion/Polycation Multilayers at the Air/Monolayer/Water Interface as Elements for Quantitative Polymer Adsorption Studies and Preparation of Hetero-superlattices on Solid Surfaces. Langmuir 2000, 16, 8871–8878. 10.1021/la000257a. [DOI] [Google Scholar]
- Gong H.; Garcia-Turiel J.; Vasilev K.; Vinogradova O. I. Interaction and Adhesion Properties of Polyelectrolyte Multilayers. Langmuir 2005, 21, 7545–7550. 10.1021/la051045m. [DOI] [PubMed] [Google Scholar]
- Picart C.; Mutterer J.; Richert L.; Luo Y.; Prestwich G. D.; Schaaf P.; Voegel J.-C.; Lavalle P. Molecular basis for the explanation of the exponential growth of polyelectrolyte multilayers. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 12531–12535. 10.1073/pnas.202486099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn J. D.; Yang S. Y.; Hiller J.; Hochbaum A. I.; Rubner M. F. Rational Design of Cytophilic and Cytophobic Polyelectrolyte Multilayer Thin Films. Biomacromolecules 2003, 4, 96–106. 10.1021/bm0256101. [DOI] [PubMed] [Google Scholar]
- Salloum D. S.; Schlenoff J. B. Protein Adsorption Modalities on Polyelectrolyte Multilayers. Biomacromolecules 2004, 5, 1089–1096. 10.1021/bm034522t. [DOI] [PubMed] [Google Scholar]
- Dimitrova M.; Affolter C.; Meyer F.; Nguyen I.; Richard D. G.; Schuster C.; Bartenschlager R.; Voegel J.-C.; Ogier J.; Baumert T. F. Sustained delivery of siRNAs targeting viral infection by cell-degradable multilayered polyelectrolyte films. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 16320–16325. 10.1073/pnas.0800156105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerdjoudj H.; Boura C.; Marchal L.; Dumas D.; Schaff P.; Voegel J. C.; Stoltz J. F.; Menu P. Decellularized umbilical artery treated with thin polyelectrolyte multilayer films: potential use in vascular engineering. Biomed. Mater. Eng. 2006, 16, S123–S129. [PubMed] [Google Scholar]
- Feldötö Z.; Lundin M.; Braesch-Andersen S.; Blomberg E. Adsorption of IgG on/in a PAH/PSS multilayer film: Layer structure and cell response. J. Colloid Interface Sci. 2011, 354, 31–37. 10.1016/j.jcis.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Cortez C.; Quinn J. F.; Hao X.; Gudipati C. S.; Stenzel M. H.; Davis T. P.; Caruso F. Multilayer Buildup and Biofouling Characteristics of PSS-b-PEG Containing Films. Langmuir 2010, 26, 9720–9727. 10.1021/la100430g. [DOI] [PubMed] [Google Scholar]
- Smith R. N.; McCormick M.; Barrett C. J.; Reven L.; Spiess H. W. NMR Studies of PAH/PSS Polyelectrolyte Multilayers Adsorbed onto Silica. Macromolecules 2004, 37, 4830–4838. 10.1021/ma049797v. [DOI] [Google Scholar]
- Duong H. T. T.; Kamarudin Z. M.; Erlich R. B.; Li Y.; Jones M. W.; Kavallaris M.; Boyer C.; Davis T. P. Intracellular nitric oxide delivery from stable NO-polymeric nanoparticle carriers. Chem. Commun. 2013, 49, 4190–4192. 10.1039/c2cc37181b. [DOI] [PubMed] [Google Scholar]
- Radomski M. W.; Palmer R. M. J.; Moncada S. The role of nitric oxide and cGMP in platelet adhesion to vascular endothelium. Biochem. Biophys. Res. Commun. 1987, 148, 1482–1489. 10.1016/s0006-291x(87)80299-1. [DOI] [PubMed] [Google Scholar]
- Vaughn M. W.; Kuo L.; Liao J. C. Estimation of nitric oxide production and reaction rates in tissue by use of a mathematical model. Am. J. Physiol. 1998, 274, H2163–H2176. 10.1152/ajpheart.1998.274.6.h2163. [DOI] [PubMed] [Google Scholar]
- de Winter R. J.; Katagiri Y.; Asano T.; Milewski K. P.; Lurz P.; Buszman P.; Jessurun G. A. J.; Koch K. T.; Troquay R. P. T.; Hamer B. J. B.; Ophuis T. O.; Wöhrle J.; Wyderka R.; Cayla G.; Hofma S. H.; Levesque S.; Żurakowski A.; Fischer D.; Kośmider M.; Goube P.; Arkenbout E. K.; Noutsias M.; Ferrari M. W.; Onuma Y.; Wijns W.; Serruys P. W. A sirolimus-eluting bioabsorbable polymer-coated stent (MiStent) versus an everolimus-eluting durable polymer stent (Xience) after percutaneous coronary intervention (DESSOLVE III): a randomised, single-blind, multicentre, non-inferiority, phase 3 trial. Lancet 2018, 391, 431–440. 10.1016/s0140-6736(17)33103-3. [DOI] [PubMed] [Google Scholar]
- Guh J.-H.; Hwang T.-L.; Ko F.-N.; Chueh S.-C.; Lai M.-K.; Teng C.-M. Antiproliferative Effect in Human Prostatic Smooth Muscle Cells by Nitric Oxide Donor. Mol. Pharmacol. 1998, 53, 467–474. 10.1124/mol.53.3.467. [DOI] [PubMed] [Google Scholar]
- Guven R. G.; Kaplan A.; Guven K.; Matpan F.; Dogru M. Effects of various inhibitors on β-galactosidase purified from the thermoacidophilic Alicyclobacillus acidocaldarius subsp. Rittmannii isolated from Antarctica. Biotechnol. Bioprocess Eng. 2011, 16, 114–119. 10.1007/s12257-010-0070-7. [DOI] [Google Scholar]
- Moncada S.; Palmer R. M. J.; Higgs E. A. Nitric-Oxide: Physiology, Pathophysiology, and Pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [PubMed] [Google Scholar]