


Abstract
We systematically studied the association between somatic copy number aberration (SCNA), DNA methylation and gene expression using -omic data from The Cancer Genome Atlas (TCGA) on six cancer types: breast cancer, colon cancer, glioblastoma, leukemia, lower-grade glioma and prostate cancer. A major challenge for such integrated study is that the association between DNA methylation and gene expression is severely confounded by tumor purity and cell type composition, which are often unobserved and difficult to estimate. To overcome this challenge, we developed a method to remove confounding effects by calculating the principal components that span the space of the latent factors. Another intriguing findings of our study is that there could be both positive and negative associations between SCNA and DNA methylation, while the CpGs with negative/positive associations with SCNA are often located around CpG islands/ocean, respectively. A joint study of SCNA, DNA methylation, and gene expression suggest that SCNA often affect DNA methylation and gene expression independently.
INTRODUCTION
Cancer arises from the accumulation of somatic DNA aberrations and epigenetic modifications that alter transcription, protein products, and cell behavior. Thus, studies of molecular features such as gene expression or DNA methylation may inform the underlying mechanism of carcinogenesis and progression. The Cancer Genome Atlas (TCGA) project has collected multiple types of -omic data from >10 000 tumor samples (1), and this dataset allows systematic study of the genetic or epigenetic basis of cancer. Many previous TCGA studies focus on each types of -omic data in order to identify genetic loci/pathways with more somatic alterations/perturbations than expected by chance. Other studies have examined multiple types of -omic data to identify tumor subtypes or driver pathways. Several methods and results have been published for the integrated analysis of multiple types of -omic data (2,3). For example, Shen et al. (4) developed an integrative clustering approach (iCluster) and used it to identify molecular subtypes for breast, lung and colon cancers. Wang et al. (5) integrated different types of omic data using a hierarchical Bayesian model called ‘integrative Bayesian analysis of genomics data’ (iBAG). Zhu et al. (6) have used multiple types of omic data to predict survival time.
TCGA data also provide rare resources to systematically study the interplay between different types of -omic features, such as the association between gene expression trait and genetic variation, known as gene expression quantitative trait locus (eQTL) analysis (7). Analogous to eQTL, the genetic basis of DNA methylation has also been studied (8). In this paper, using the -omic data from TCGA, we systematically studied the association between somatic copy number aberration (SCNA), DNA methylation, and gene expression for six types of cancers: breast cancer (BRCA), colon cancer (COAD), acute myeloid leukemia (LAML), glioblastoma (GBM), lower-grade glioma (LGG) and prostate cancer (PRAD).
DNA methylation is one of the most widely studied epigenetic marks in cancer genome. Genome-wide hypo-methylation and hyper-methylation at some gene promoters have been observed in multiple types of cancer (9). Epigenetic marks such as chromatin organization or DNA methylation are often associated with the regulation of gene expression. Thus, studies on the association between DNA methylation and gene expression are critical to understanding the functional role of DNA methylation in cancer (10,11). It has been reported that DNA methylation at promoter regions is often negatively associated with gene expression while DNA methylation in gene bodies is often positively associated with gene expression (12). However, the functional role of DNA methylation (i.e. whether DNA methylation is a passive mark of transcription activity or an active regulator that modifies gene expression) is still being debated (13).
In this paper, we systematically studied the functional role of DNA methylation by jointly analyzing -omic data of SCNA, gene expression and DNA methylation. Toward this end, we developed a new method to accurately estimate the association between gene expression and DNA methylation by removing the confounding effect caused by tumor purity and cell type composition.
MATERIALS AND METHODS
For each type of cancer, we selected samples with associated clinical data plus all three types of -omic data, and we filtered out samples of rare tissue sites or plates (Supplementary Figure S1). For the breast cancer study, we also restricted our analysis to those samples with tumor purity, ploidy and subtype information. To avoid possible confounding due to race, we chose to study tumor samples from Caucasian samples, as inferred from genotype Principal component analysis (PCA) of both HapMap samples and all TCGA samples within a cancer type (Supplementary Figure S2). Then, we performed PCA again on the selected Caucasian samples and used the resulting top PCs as part of the demographical covariates.
We included the following 3 groups of covariates when we assessed associations: (i) batch effects, including tissue sites and plates (Supplementary Figure S3); (ii) demographical covariates such as age, gender, and genotype PCs to account for population stratification and (iii) cancer subtypes when such information was available. Specifically, for breast cancer, we employed the subtype inferred from gene expression data: Basal, Her2, LumA, LumB and Normal (14). For colon cancer, we partitioned the samples into two groups: hyper-mutated tumors and non-hyper-mutated tumors (15). The hyper-mutated tumors are defined as those with >1000 point mutation calls from exome-seq data. Subtype information for glioblastoma was taken from a recent publication (16) that classifies patients into 4 expression subtypes: classical, mesenchymal, neural and proneural, and additionally, a subgroup of G-CIMP based on methylation data. The subtype of lower-grade glioma was decided by IDH1/2 mutation and chromosome 1p/19q deletion (17). The subtypes of acute myeloid leukemia were defined by cytogenetic risk group (18). There were no well-defined subtypes for prostate cancer, and thus, we did not include subtypes in our analysis for prostate cancer.
The SCNA data were level 3 data from the TCGA data portal after segmentation of probe-level data. More specifically, the probe-level SCNA data of each individual were obtained from Affymetrix 6.0 array using log ratio of the intensity of the tumor sample versus the intensity of the paired-normal sample. Such measurement of SCNA is often referred to as LRR (log R ratio), which quantifies underlying copy number changes, i.e. higher values indicate copy number gain and lower values indicate copy number loss. Such LRR values were segmented by circular binary segmentation (CBS) and saved as level 3 data in TCGA data portal. We used gene-level SCNA data by taking the segmental mean of the corresponding gene (Supplementary Figures S4 and S5).
The DNA methylation data were analyzed for each CpG separately (Supplementary Figure S6). The gene expression data were summarized by read count per gene and per sample, normalized by sample-specific read-depth (Supplementary Figure S7). The gene expression and DNA methylation data were further transformed using normal quantile transformation for each gene and each CpG separately. This transformation is commonly used in large-scale eQTL studies to obtain more robust results when there is non-linear relations or outliers (19). Overall the methylation-expression association strength estimated before and after normal quantile transformation are similar (Supplementary Figure S10), suggesting that linear relation is reasonable for most methylation-expression associations. We have illustrated such linear relations for a few CpG-gene pairs (Supplementary Figure S16). Previous studies have reported highly non-linear relation between methylation and gene expression when examining all the genes within one individual (20,21). In contrast, our focus is the association of one gene and one CpG across individuals. The across individual associations may not have the same property as across gene associations (22). We used MatrixEQTL (23) for all computation to assess pairwise associations of SCNA, DNA methylation, and gene expression.
In order to assess methylation-expression (ME) associations while accounting for the effects of tumor purity and cell type composition, we proposed to identify the subspace spanned by tumor purity and cell type composition using top PCs from PCA of significantly associated distant ME pairs. Specifically, we first performed the ME association analysis for each ME pair, while accounting for the effects of all other covariates such as batch effect, demographical variables, cancer subtypes, as well as the SCNA of the corresponding gene. Then, we selected those significantly associated ME pairs that are not located on the same chromosomes. Suppose that K such ME pairs are selected. Denote the methylation and expression data of each pair as Mk and Ek, respectively. Note that both Mk and Ek are vectors of length n, where n is the sample size. We regressed Mk and Ek against all other covariates and obtained residuals. We then standardized the residuals to have mean 0 and standard deviation 1 and denoted the resulting data by 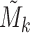 and
and 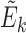 . The association between Mk and Ek could be positive or negative. βk is the regression coefficient of regressing Ek versus Mk, and thus sign(βk) > 0 means gene expression increases as methylation increases. We took the average of
. The association between Mk and Ek could be positive or negative. βk is the regression coefficient of regressing Ek versus Mk, and thus sign(βk) > 0 means gene expression increases as methylation increases. We took the average of 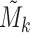 and
and 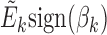 , denoted by
, denoted by 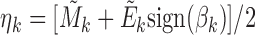 , which is a vector of length n. Let Γ = (η1, η2, ..., ηK)T, which is a matrix of size K × n. We performed PCA on Γ and then used the resulting PCs (referred to as ME PCs) as covariates in ME association testing. It is reasonable to take average of
, which is a vector of length n. Let Γ = (η1, η2, ..., ηK)T, which is a matrix of size K × n. We performed PCA on Γ and then used the resulting PCs (referred to as ME PCs) as covariates in ME association testing. It is reasonable to take average of 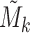 and
and 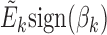 because we want to identify the signals shared between
because we want to identify the signals shared between 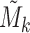 and
and 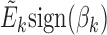 , and they are on the same scale after standardization.
, and they are on the same scale after standardization.
The above procedure implicitly assumes the vast majority of the distant ME associations are false positives due to tumor purity or cell type composition. If this conjecture is true, then a few top PCs can explain most variation in the data. We have confirmed this in our data analysis. For example, for breast cancer, the top 7 PCs explains >92% of the variation of >136 000 distant ME pairs (Supplementary Figure S13). This situation fits to the ‘spiked eigenvalue model’ for high-dimensional data where a few population eigenvalues are substantially larger than the rest. We refer the readers to (24,25) for more details of the statistical property of eigen-values and eigen-vectors in such settings.
Then a related question is how to decide the number of ME PCs to use. These ME PCs are calculated based on distant ME associations and some of those distant associations may be true biological signals. Therefor if we include too many ME PCs in the analysis, there is risk to remove such true distant ME associations. We believe that the choice on the number of ME PCs does not impose serious limitations on our approach because those true distant ME association signals are usually weak and sparse, and thus may not lead to ME PCs with relatively large eigen-values. From our experience, there are a few helpful guidelines. First, one may choose the cutoff of the number of top ME PCs where there is apparent drop on the size of eigen-values. Second, one can choose the smallest number of top ME PCs such that after adjusting for those ME PCs, most methylation-expression associations are local. Additionally, we may exclude those ME PCs that are associated with the methylation of CpGs within a short genomic region, because such ME PCs may be due to eQTM (expression quantitative trait methylation) hotspots where the methylation of a short genomic region is associated with the expression of many genes.
RESULTS
We will mainly report the results from the breast cancer data. The results from other types of cancer are similar and are reported in the Supplementary Materials.
SCNA versus gene expression or DNA methylation
The vast majority of the SCNA-expression and SCNA-methylation associations are local, meaning that the SCNA of a gene affects gene expression or DNA methylation in nearby genomic regions (Figure 1A and B, Supplementary Figures S21–S25). As expected, SCNAs are positively associated with gene expression. We are not aware of any previous systematic studies on the association between SCNA and DNA methylation. We found that such associations can be positive or negative. When they have negative associations (i.e. DNA methylation decreases with copy number gain and DNA methylation increases with copy number loss), the corresponding CpGs are often located at CpG islands (Figure 1C and D, Supplementary Figures S26–S30). In contrast, positive associations between SCNA and DNA methylation are often observed on those CpG’s located at CpG ocean.
Figure 1.

Associations between SCNA and gene expression or DNA methylation. (A) Associations between SCNA and DNA methylation, after accounting for the effects of batches (sites and plates), age, three genotype PCs and breast cancer subtypes. Each point in this plot indicates the association between the SCNA of one gene (x-axis) and the DNA methylation of one probe (y-axis). The color of each point indicates the range of the corresponding –log10(P-value), as shown in the legend at the top of the panel. The associations with P-values larger than 10–20 are not shown. (B) Associations between SCNA and gene expression, after accounting for the effects of batches (sites and plates), age, three genotype PCs and breast cancer subtypes. (C) The distribution of the regression coefficients of methylation Quantitate trait Copy Number (mQCN), i.e. the regression coefficients of SCNA within the model: DNA methylation = SCNA + other covariates. (D) ‘mQCN+’ and ‘mQCN–’ indicate the DNA methylation probes that have only positive or negative associations, respectively, with SCNA. This plot shows the proportion of mQCN+/mQCN– probes located at different regions with respect to CpG islands. CpGs were annotated based on annotation file provided by Illumina: CpG shore are defined as 2,000 bp either side of an island with north/south shore means 5′ or 3′ relative to the associated gene. North and south shelves are 2000 bp flanking the CpG shores. The other CpGs are referred to as ‘none’, or sometime ‘CpG ocean’ or ‘open sea’.
We accounted for both technical and demographic/clinical covariates in our analyses. The technical batch effects (e.g. tissue collection sites, plates for sample handling) often have a very strong influence on gene expression or DNA methylation (Supplementary Figure S3). We also included cancer subtypes in our analysis (see Materials and Methods section for the definition of cancer subtypes). Failure to account for cancer subtypes may lead to misleading results. For example, in breast cancer studies, without conditioning on the subtypes of breast cancer, one may identify associations due to subtype-specific SCNA, DNA methylation and gene expression (Supplementary Figures S8 and S9), including some confusing results, such as negative associations between SCNA and gene expression. Another potential confounding factor is tumor purity. Adding tumor purity as an additional covariate does not remove much of the SCNA-methylation or SCNA-expression associations. For example, for breast cancer and at p-value cutoff 1e–30, we identified around 328,000 SCNA-methylation associations without purity as a covariate. After including purity as a covariate, we can recover around 88% or 98% of these 328 000 SCNA-methylation associations at P-value cutoff 1e–30 and 1e–28, respectively.
After accounting for all the known batch effects and tumor subtypes, we still observe a hotspot of SCNA vs. DNA methylation associations in breast cancer data, where DNA methylation of hundreds of CpGs from multiple chromosomes are associated with SCNA of multiple genes on chr16 (Figure 1A). This is likely due to copy number changes of CTCF. CTCF is a well-known transcription factor that plays important roles in epigenetic regulation. CTCF is among those genes in chr16 whose copy number changes are associated with many CpGs, and the gene expression of CTCF is strongly associated with its SCNA (P-value 4.6 × 10−50). At P-value threshold 10−20, there are 134 CpGs that are not located in chr16 and their methylation levels are associated with at least one SCNA on chr16. Among them, 120 (97%) are located in CTCF binding sites, while we expect 5% overlap by chance. CTCF binding sites information were obtained from CTCFBSDB 2.0 (26), see Supplementary Materials Section B.1.7 for more details. The methylation levels of these 134 CpGs are all negatively associated with CTCF copy number, which implies that when CTCF amplifies, the CTCF protein is more likely to bind those binding sites, and lead to reduction of methylation in these regions.
Gene expression versus DNA methylation
We assessed methylation-expression (ME) associations after accounting for technical and demographic/clinical covariates, as well as SCNA. An initial assessment identified a huge number of significant ME associations, and most of them were distant associations (Figure 2A). A majority, but not all of the distant ME associations can be removed by conditioning on SCNA-based estimates of tumor purity (Supplementary Figure S10) (27,28). This suggests that tumor purity is a confounding factor. This observation is consistent with recent findings that tumor purity affects both gene expression and DNA methylation in tumor samples (29,30).
Figure 2.

Associations between DNA methylation and gene expression. (A) Associations between DNA methylation and gene expression. Each point in this plot indicates the association between the DNA methylation of one probe (x-axis) and the expression of one gene (y-axis), after accounting for the effects of batches (sites and plates), age, three genotype PCs, SCNA of the corresponding gene and breast cancer subtypes. The color of each point indicates the range of the corresponding –log10(P-value), as shown in the legend at the top of the panel. (B) Association between DNA methylation and gene expression, after including all covariates used in (A) plus seven ME PCs. (C) The proportion of ‘hot’ (‘cold’) CpGs that are located at specific regions with respect to CpG Island. (D) The genomic location of gene expression quantitative trait methylation probes (eQTM) with respect to the location of its associated genes. [–1,0] is the region 1 kb upstream of transcription starting site (TSS), and [0,1] corresponds to gene body. All eQTMs are divided into two classes: those that are positively or negatively associated with gene expression, denoted by eQTM+ and eQTM–, respectively.
We developed a new computational approach that allows us to remove ME associations due to latent confounding factors. Specifically, we performed a joint PCA of methylation and expression data for significantly associated ME pairs (see Materials and Methods section for details) and added the resulting PCs (referred to as ME PCs) as covariates when assessing ME associations. This approach removed most distant ME associations even at more liberal p-value cutoffs (Figure 2(B), Supplementary Table S4, Supplementary Figures S11 and S12).
We conjecture that ME PCs capture cell type composition information within tumor tissue. The most dramatic cell type difference is between tumor and non-tumor cells, and indeed some of these ME PCs have strong associations with tumor purity, which was estimated by SCNA data (Supplementary Figures S13 and S14). However, there are also multiple types of non-tumor cells that may contribute to ME associations. This explains why we need multiple PCs to remove suspicious ME associations. To validate this conjecture, we examined the associations between the top seven ME PCs and cell type-specific gene expression, which were calculated as the median expression of cell-type-specific genes. We identified such cell-type-specific genes from the list of 812 immune metagenes (each immune metagene is associated with one of 31 cell types) reported by Angelova et al. (31), and they identified such immune metagenes by differential expression analysis of purified immune cells. Each PC is associated with a variety of cell types (Figure 3, Supplementary Figures S42–S46). Together these seven ME PCs explain a substantial amount of variation in cell type-specific genes. For example, for breast cancer, they explain more than 60% of variation for 12 of 31 cell types, and >40% of variation in 21 of 31 cell types (Supplementary Figures S47–S52). It is important to note that these ME PCs jointly span a space of underlying cell type composition, and a specific PC may not correspond to a distinct subset of cell types.
Figure 3.

Relations between cell type prevalence and ME PCs. The median expression of the genes annotated to each cell type was used as a surrogate for cell type prevalence, and it was regressed against top seven ME PCs. This Figure illustrates the proportion of variance explained by each of the top seven PCs. Values >0.5 are truncated for visualization contrast.
Having demonstrated that ME PCs capture cell type composition information, including tumor purity, we conclude that by including ME PCs in our analysis, we are able to account for such confounding effects without actually estimating these quantities. Without accounting for such confounding factors, most ME associations are false positives in the sense that they are due to differential expression or methylation across cell types and do not imply any functional associations. Even if we only focus on those gene and CpG pairs within 1 Mb, >90% of significant ME associations are false positives (Supplementary Figure S15).
Next, we sought to characterize those genes and CpGs that have major contributions to the ME PCs. We can identify such genes by those ‘hot’ genes that are associated with the methylation of a large number of CpGs. These hot genes are expressed in different types of immune cells, and their expression are negatively associated with tumor purity (Supplementary Tables S5, S10–S14, Supplementary Figure S17), suggesting that these genes have relatively low expression in tumor and thus can be used to quantify the amount of infiltrating immune cells. The results remain similar if we identify hot genes based on effect size (regression coefficients) rather than P-value (Supplementary Figure S18, Supplementary Table S6). The hot CpGs are not strongly associated with any location annotations that we have tested, though it does have higher likelihood of being located at CpG shores or CpG ocean (i.e. regions farther away from CpG islands) (Figure 2C, Supplementary Figures S36–S40), which is consistent with previous findings about cancer-specific methylation in CpG shores (16).
By accounting for those ME PCs, we have a more accurate picture of the association between gene expression and DNA methylation. Across different cancer types, we found that gene expression is often negatively associated with DNA methylation around Transcription Starting Sites (TSSs), but positively associated with DNA methylation in gene bodies (Figure 2D, Supplementary Figures S33–S37), which is consistent with previous findings (13).
SCNA, DNA methylation, and gene expression
We further study the association of any two types of -omic features given the third one. Since we have accounted for SCNA in ME association studies, here we focus on the conditional associations of SCNA and DNA methylation given gene expression (CM given E) and CE given M. Among those cases with significant CE associations, given M, CE associations have little changes (Supplementary Figures S53–S58), suggesting DNA methylation does not have strong mediation role on the relations between SCNA and gene expression.
Next we focus on those cases with significant CM associations to study the relations of these three variables. We perform a gene-centric analysis. For each gene, we consider its SCNA, gene expression, and local CpGs (within 200 kp of this gene) of which the DNA methylation is significantly associated with the SCNA of this gene, and we ignore those genes without any significant local CM associations (Figure 4A).
Figure 4.

Study the associations of SCNA (C), DNA methylation (M), and gene expression (E). (A) A gene-centric organization of the three types of data. Here, we focus on those cases with significant CM associations and the CpG is with 200 kb of the gene. (B) We compare the likelihood of three models and for each cancer type, and identify the number of CM pairs where one of the three models has significantly higher likelihood, or that the likelihoods of the top two models are not significantly different.
For each triplet of C, M and E, we compare three possible models, causal, reactive, and conditional independence (Figure 4B) using likelihood ratio test for non-nested models (32). The difference of these models can be quantified by likelihood function because they entail different conditional dependence assumption. For example, causal model implies gene expression is independent of SCNA given DNA methylation, i.e., the likelihood can be written as L(C)L(M|C)L(E|M). In contrast, the likelihood for reactive and conditional independence models can be written as L(C)L(E|C)L(M|E) and L(C)L(E|C)L(M|C). Standard likelihood model can compare two nested models. However, in this case, the three models are not nested with each other. Therefore we employ a likelihood ratio test for non-nested model. Under the null hypothesis, the two non-nested models have equal distance to the underlying unknown true model. We conclude one model fit the data better than the other two models if the likelihood ratio test p-value for the best versus the second best model is smaller than 0.01. We found that the independence model fit the data better in most cases, and this conclusion remains the same regardless the choice of p-value cutoff (Figure 4(B)). To further illuminate this conclusion, we also perform three sets of unconditional/conditional regressions for CE, CM and ME associations. For example, for each CE pair, we asses its association strength before and after adding DNA methylation into the regression model, while the other covariates, such as batch effects, demographical variables (age, gender, genotype PC), tumor subtypes, and ME PCs are always included in the model. Similar, we compare the CM versus CM | E associations, and ME versus ME | C associations (Supplementary Figures S53-S58).
When we cannot distinguish top two models, i.e. the likelihood ratio test conclude that these two models have similar distances to the underlying true model (Figure 4B), the underlying model is likely a complete model where any two of the three omic features are connected. In other words, this is the situation where both DNA methylation and SCNA make their own contributions to explain the variation in gene expression. For example, PVT1 is a known oncogene and is amplified in a subset of tumor samples. Copy number amplification of PVT1 is associated with higher gene expression. Given such copy number changes, we still observe association between DNA methylation in the promoter of PVT1 and its gene expression (Figure 5).
Figure 5.

An example where gene expression are associated with both SCNA and DNA methylation. (A) Scatter plots of SCNA of PVT1, the methylation of a CpG at its promoter, and its expression. The values of SCNA and methylation are partitioned into three categories based on 25th and 75th percentiles. (B) Distribution of gene expression with respect to SCNA and DNA methylation categories. Gene expression increases as SCNA increases and methylation decreases.
DISCUSSION
We have made two major contributions in this paper. One is to report that the underlying cell type composition (e.g. tumor cells versus different types of normal cells including infiltrating immune cells) is a crucial confounding factor when studying the associations between DNA methylation and gene expression of tumor samples. We have developed a computational method to remove such confounding factors while bypassing the challenging task of inferring cell type composition. Our method is different from surrogate variable analysis (33) or supervised normalization of microarrays (34). They study the association between one type of omic data (e.g. gene expression) and some covariates of interest, and identify latent confounding factors by PCA of the omic data while carefully handling the effects of known covariates. Our work is different because we exploit the correlation of two types of omic data to identify the underlying latent factors. Our second major contribution is to report that DNA methylation may be either positively or negatively associated with somatic copy number aberrations.
Following our association study, immediate questions are the mechanisms or causal relations for those ME (methylation-expression) or CM (copy number-methylation) associations. Ultimately, these questions need to be answered by experiments with carefully designed interventions on the molecular system. However, our computational analysis can provide some insights. Previous studies have shown that hyper-methylation on gene promoters often maintain, rather than initialize transcriptional repression (13). For example, Lock et al. (35) showed that DNA methylation occurs after inactivation of gene expression, and Verma et al. (36) demonstrated that promoter hypermethylation does not lead to decrease of gene expression. Our results on the joint analysis of SCNA (C), DNA methylation (M), and gene expression (E) are consistent with the ‘maintainer’ role of DNA methylation. If DNA methylation actively modifies gene expression, given that we start with triplets with C-M associations, the model of C, M, and E should be a causal model of C → M → E or a full model with connections C–M, and M–E and C–E. However, our results show that in most cases, the most likely model is the conditional independence model in which the variation of DNA methylation is not associated with changes in gene expression. If methylation only plays a ‘maintainer’ role, it remains a question what is the factor that causes gene expression changes, and a potential route to answer this question is to explore other epigenetic marks such as chromatin modification or transcription factor binding (37).
It is an intriguing question why SCNA is associated with DNA methylation changes. If the probability that a CpG is methylated is cis-regulated, e.g. it is controlled by surrounding DNA sequence of the same allele, then DNA methylation should remain the same regardless of copy number changes. Therefore the fact that we observe strong SCNA-methylation associations suggests that DNA methylation level is regulated in trans upon SCNA events. The genome-wide changes of methylation pattern upon SCNA events is unlikely due to somatic mutations of particular proteins or pathways, e.g. the association between DNA methylation and copy number changes of CTCF observed in breast cancer patients. Instead, it is more likely to be initiated and maintained by a generic machinery.
To understand the underlying mechanism of CM associations, we summarize the locations of the genes with significant local CM associations (within 2 kb) as well as the magnitude of methylation changes, and note three observations. First, there is significant overlap of CM associations across cancer types. For example, 7056 and 829 genes are involved in local CM associations in BRCA and COAD patients at P-value cutoffs 10−20 and 10−10, respectively. Different p-value cutoffs are used to account for sample size difference. About 88.7% of the 829 genes identified from COAD are also identified in BRCA (P-value 9.7 × 10−243). Part of such overlap can be explained by the similarity of SCNA events across cancer types (Supplementary Figures S59–S69). Another observation is that genes with positive and negative CM associations are often located next to each other, or one gene’s expression may be positively and negatively associated with the methylation of different CpGs (Supplementary Figures S65–S69). Third, the magnitude of DNA methylation changes with respect to copy number changes is relatively small. For example, as shown in Supplementary Figure S70, the average DNA methylation of one CpG changes from 0.06 to 0.02 as SCNA measurement varies from 0 to 2.0.
Based on these three observations, it is attempting to conjecture that the CM associations may be due to redistribution of the methylase complex in a relatively short region of the genome, and this redistribution is not a cancer type-specific process. For example, when there is copy number amplification, we observe that DNA methylation decreases around CpG islands and increases in CpG ocean. This may be due to redistribution of DNA methylation from CpG islands to nearby CpG poor regions. When there is copy number deletion, we observe that DNA methylation increases around CpG islands and decreases in CpG ocean, and this may be due to redistribution of DNA methylation from CpG ocean to nearby CpG islands. It has been reported that oxidative damage can induce such redistribution of DNA methylation (38). Low level increase of methylation at CpG islands has been associated with aging and cancer, and it may be due to redistribution of the methylase complex (39). The magnitude of aging-associated methylation changes is similar to what we have observed in CM associations in tumor samples (40).
Such conjecture of ‘redistribution of DNA methylation’ upon copy number changes can be validated using longitudinal copy number and DNA methylation measurements along the timeline of tumor initiation and progression. Without such luxury resource, we use adjacent normal tissue as an proxy of pre-tumor sample. We focus on colon cancer due to the availability of adjacent normal samples in TCGA data and the fact that tumor cells and adjacent normal cells in colon cancer are likely derived from the same set of stem cells (41). We downloaded and processed SCNA data for 90 adjacent normal samples and we found that there are no copy number aberrations in these adjacent normal samples (Supplementary Section B.4). Adjacent DNA methylation data were available for 38 samples. The DNA methylation of the CpGs involved in CM associations is more similar between tumor and adjacent normal samples than the other CpGs (Supplementary Figure S72). This result indicates that those CpGs involved in CM associations are not those that are differentially methylated between tumor and normal samples. Focusing on those CpGs involved in CM associations, we found that DNA methylation is more similar between tumor and normal samples if we only considered the tumor samples with copy number close to two (Supplementary Figure S72). Therefore, this reinforces the conjecture that DNA methylation is perturbed upon copy number changes.
In addition to SCNA, it is also interesting to study the associations between gene expression/DNA methylation and somatic point mutations such as single nucleotide variants (SNVs) or indels. Systematic study of somatic point mutation associations, which is beyond the scope of this paper, faces the challenges of imperfect somatic mutation calling accuracy as well as low recurrence rates of somatic mutations. Here we show an interesting example. Somatic mutations in IDH1/IDH2 are strongly associated with the DNA methylation of many CpGs in lower grade glioma (LGG) (42). In our analysis of TCGA LGG samples, we considered IDH1/IDH2 mutation as part of the tumor subtype, and thus it does not affect our results. However, if we study the associations between IDH mutation, DNA methylation, and gene expression, we observed that DNA methylation often mediates the association between IDH mutation and gene expression. In contrast, the association between IDH mutation and DNA methylation remains similar after conditioning on gene expression (Supplementary Figure S76).
Our method to estimate the associations between gene expression and DNA methylation allows us to bypass the challenging task of estimating tumor purity and cell type composition. However, these are still very important problems that warrant further studies. Currently, tumor purity is often inferred by SCNA data (27), and cell type composition may be decomposed using gene expression data, while ignoring tumor purity (43). Our results suggest that jointly studying multiple types of -omic data to infer both tumor purity and cell type composition is a promising approach.
Supplementary Material
ACKNOWLEDGEMENTS
W.S., D.Y.L., M.C. designed the study. W.S., D.Y.L. with input from C.M.P. and D.N.H. wrote the manuscript. W.S., P.B., C.J., P.L., V.Z. conducted analysis. All authors read and approved the final manuscript. Jin Wang, a graduate student in UNC’s Department of Biostatistics, helped with part of the R codes for genotype calling. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [GM105785, P01 CA142538, U10CA181009]. Funding for open access charge: NIGMS [GM105785].
Conflict of interest statement. C.M.P. is an equity stock holder, consultant and Board of Director Member, of BioClassifier LLC. C.M.P. is also listed an inventor on patents for the Breast Cancer PAM50 assay.
REFERENCES
- 1. Hoadley K.A., Yau C., Wolf D.M., Cherniack A.D., Tamborero D., Ng S., Leiserson M.D., Niu B., McLellan M.D., Uzunangelov V. et al. . Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014; 158:929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kristensen V.N., Lingjærde O.C., Russnes H.G., Vollan H. K.M., Frigessi A., Børresen-Dale A.-L.. Principles and methods of integrative genomic analyses in cancer. Nat. Rev. Cancer. 2014; 14:299–313. [DOI] [PubMed] [Google Scholar]
- 3. Richardson S., Tseng G.C., Sun W.. Statistical methods in integrative genomics. Annu. Rev. Stat. Appl. 2016; 3:181–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shen R., Olshen A.B., Ladanyi M.. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. 2009; 25:2906–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang W., Baladandayuthapani V., Morris J.S., Broom B.M., Manyam G., Do K.-A.. iBAG: integrative Bayesian analysis of high-dimensional multiplatform genomics data. Bioinformatics. 2013; 29:149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhu B., Song N., Shen R., Arora A., Machiela M.J., Song L., Landi M.T., Ghosh D., Chatterjee N., Baladandayuthapani V. et al. . Integrating clinical and multiple omics data for prognostic assessment across human cancers. Scientific Rep. 2017; 7:16954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kendziorski C., Chen M., Yuan M., Lan H., Attie A.. Statistical methods for expression quantitative trait loci (eQTL) mapping. Biometrics. 2006; 62:19–27. [DOI] [PubMed] [Google Scholar]
- 8. Zhang H., Wang F., Kranzler H.R., Yang C., Xu H., Wang Z., Zhao H., Gelernter J.. Identification of methylation quantitative trait loci (mQTLs) influencing promoter DNA methylation of alcohol dependence risk genes. Hum. Genet. 2014; 133:1093–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Irizarry R.A., Ladd-Acosta C., Wen B., Wu Z., Montano C., Onyango P., Cui H., Gabo K., Rongione M., Webster M. et al. . The human colon cancer methylome shows similar hypo-and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009; 41:178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fleischer T., Frigessi A., Johnson K.C., Edvardsen H., Touleimat N., Klajic J., Riis M.L., Haakensen V.D., Wärnberg F., Naume B. et al. . Genome-wide DNA methylation profiles in progression to in situ and invasive carcinoma of the breast with impact on gene transcription and prognosis. Genome Biol. 2014; 15:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gevaert O., Tibshirani R., Plevritis S.K.. Pancancer analysis of DNA methylation-driven genes using MethylMix. Genome Biol. 2015; 16:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shen H., Laird P.W.. Interplay between the cancer genome and epigenome. Cell. 2013; 153:38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jones P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012; 13:484–492. [DOI] [PubMed] [Google Scholar]
- 14. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Berman B.P., Weisenberger D.J., Aman J.F., Hinoue T., Ramjan Z., Liu Y., Noushmehr H., Lange C.P., van Dijk C.M., Tollenaar R.A. et al. . Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2012; 44:40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brat D.J., Verhaak R.G., Aldape K.D., Yung W.A., Salama S.R., Cooper L.A., Rheinbay E., Miller C.R., Vitucci M., Morozova O. et al. . Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med. 2015; 372:2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cancer Genome Atlas Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013; 368:2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wright F.A., Sullivan P.F., Brooks A.I., Zou F., Sun W., Xia K., Madar V., Jansen R., Chung W., Zhou Y.-H. et al. . Heritability and genomics of gene expression in peripheral blood. Nat. Genet. 2014; 46:430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lou S., Lee H., Qin H., Li J., Gao Z., Liu X., Chan L.L., Lam V. K.L., So W., Wang Y. et al. . Whole-genome bisulfite sequencing of multiple individuals reveals complementary roles of promoter and gene body methylation in transcriptional regulation. Genome Biol. 2014; 15:408–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jjingo D., Conley A., Yi S., Lunyak V., Jordan I.. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012; 3:462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gutierrezarcelus M., Lappalainen T., Montgomery S.B., Buil A., Ongen H., Yurovsky A., Bryois J., Giger T., Romano L., Planchon A. et al. . Passive and active DNA methylation and the interplay with genetic variation in gene regulation. eLife. 2013; 2:e00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shabalin A.A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012; 28:1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnstone I.M. On the distribution of the largest eigenvalue in principal components analysis. Ann. Stat. 2001; 29:295–327. [Google Scholar]
- 25. Lee S., Zou F., Wright F.A.. Convergence of sample eigenvalues, eigenvectors, and principal component scores for ultra-high dimensional data. Biometrika. 2014; 101:484–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ziebarth J.D., Bhattacharya A., Cui Y.. CTCFBSDB 2.0: a database for CTCF-binding sites and genome organization. Nucleic Acids Res. 2012; 41:D188–D194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carter S.L., Cibulskis K., Helman E., McKenna A., Shen H., Zack T., Laird P.W., Onofrio R.C., Winckler W., Weir B.A. et al. . Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012; 30:413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zack T.I., Schumacher S.E., Carter S.L., Cherniack A.D., Saksena G., Tabak B., Lawrence M.S., Zhang C.-Z., Wala J., Mermel C.H. et al. . Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013; 45:1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoshihara K., Shahmoradgoli M., Martínez E., Vegesna R., Kim H., Torres-Garcia W., Treviño V., Shen H., Laird P.W., Levine D.A. et al. . Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013; 4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang N., Wu H., Zhang W., Wang J., Wu H., Zheng X.. Predicting tumor purity from methylation microarray data. Bioinformatics (Oxford, England). 2015; 31:3401. [DOI] [PubMed] [Google Scholar]
- 31. Angelova M., Charoentong P., Hackl H., Fischer M., Snajder R., Krogsdam A., Waldner M., Bindea G., Mlecnik B., Galon J. et al. . Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2014; 16:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun W., Yu T., Li K.-C.. Detection of eQTL modules mediated by activity levels of transcription factors. Bioinformatics. 2007; 23:2290–2297. [DOI] [PubMed] [Google Scholar]
- 33. Leek J.T., Storey J.D.. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007; 3:e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mecham B.H., Nelson P.S., Storey J.D.. Supervised normalization of microarrays. Bioinformatics. 2010; 26:1308–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lock L.F., Takagi N., Martin G.R.. Methylation of the Hprt gene on the inactive X occurs after chromosome inactivation. Cell. 1987; 48:39–46. [DOI] [PubMed] [Google Scholar]
- 36. Verma N., Pan H., Doré L.C., Shukla A., Li Q.V., Pelham-Webb B., Teijeiro V., González F., Krivtsov A., Chang C.-J. et al. . TET proteins safeguard bivalent promoters from de novo methylation in human embryonic stem cells. Nat. Genet. 2018; 50:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ji H., Jiang H., Ma W., Johnson D.S., Myers R.M., Wong W.H.. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat. Biotechnol. 2008; 26:1293–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O’Hagan H.M., Wang W., Sen S., Shields C.D., Lee S.S., Zhang Y.W., Clements E.G., Cai Y., Van Neste L., Easwaran H. et al. . Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer cell. 2011; 20:606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Teschendorff A.E., Menon U., Gentry-Maharaj A., Ramus S.J., Weisenberger D.J., Shen H., Campan M., Noushmehr H., Bell C.G., Maxwell A.P. et al. . Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010; 20:440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Klutstein M., Nejman D., Greenfield R., Cedar H.. DNA methylation in cancer and aging. Cancer Res. 2016; 76:3446–3450. [DOI] [PubMed] [Google Scholar]
- 41. Vermeulen L., Snippert H.J.. Stem cell dynamics in homeostasis and cancer of the intestine. Nat. Rev. Cancer. 2014; 14:468–480. [DOI] [PubMed] [Google Scholar]
- 42. Turcan S., Rohle D., Goenka A., Walsh L.A., Fang F., Yilmaz E., Campos C., Fabius A.W., Lu C., Ward P.S. et al. . IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012; 483:479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A.. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015; 12:453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.