

Abstract
Background:
Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) are common X-linked recessive neuromuscular disorders caused by mutations in dystrophin gene. Multiplex polymerase chain reaction (multiplex PCR) and multiplex ligation-dependent probe amplification (MLPA) are the most common methods for detecting dystrophin gene mutations. This study aimed to contrast the two methods and discern the genetic characterization of patients with DMD/BMD in Eastern China.
Methods:
We collected 121 probands, 64 mothers of probands, and 15 fetuses in our study. The dystrophin gene was detected by multiplex PCR primarily in 28 probands, and MLPA was used in multiplex PCR-negative cases subsequently. The dystrophin gene of the remaining 93 probands and 62 female potential carriers was tested by MLPA directly. In fetuses, multiplex PCR and MLPA were performed on 4 fetuses and 10 fetuses, respectively. In addition, sequencing was also performed in 4 probands with negative MLPA.
Results:
We found that 61.98% of the subjects had genetic mutations including deletions (50.41%) and duplications (11.57%). There were 43.75% of mothers as carriers of the mutation. In 15 fetuses, 2 out of 7 male fetuses were found to be unhealthy and 2 out of 8 female fetuses were found to be carriers. Exons 3–26 and 45–52 have the maximum frequency in mutation regions. In the frequency of exons individually, exon 47 and exon 50 were the most common in deleted regions and exons 5, 6, and 7 were found most frequently in duplicated regions.
Conclusions:
MLPA has better productivity and sensitivity than multiplex PCR. Prenatal diagnosis should be applied in DMD high-risk fetuses to reduce the disease incidence. Furthermore, it is the responsibility of physicians to inform female carriers the importance of prenatal diagnosis.
Keywords: Becker Muscular Dystrophy, Duchenne Muscular Dystrophy, Dystrophin, Multiplex Ligation-dependent Probe Amplification, Multiplex Polymerase Chain Reaction, Prenatal Diagnosis
摘要
背景:
杜氏肌营养不良症(DMD)和贝氏肌营养不良症(BMD)是抗肌萎缩蛋白基因突变导致的常见神经肌肉疾病,为X连锁隐性遗传。多重聚合酶链式反应技术(mPCR)和多重连接探针扩增技术(MLPA)是检测抗肌萎缩蛋白基因突变的常用方法。本研究旨在比较这两种方法以及总结中国东部地区DMD/BMD患者的基因突变特征。
方法:
本研究中,我们收集了先证者121例,先证者母亲64例以及胎儿15例。28例先证者抗肌萎缩蛋白基因使用mPCR检测,MLPA用于检测mPCR阴性病例,其余93例先证者和62例女性潜在携带者直接进行MLPA检测。在胎儿中,分别对4例胎儿和10例胎儿进行mPCR技术和MLPA检测。此外,四例MLPA阴性先证者通过测序方法进行检测。
结果:
我们发现61.98%的受试者有基因突变,包括缺失突变(50.41%)和重复突变(11.57%)。43.75%的母亲为携带者。在15例胎儿中,7例男性胎儿中有2例为患者,8例女性胎儿中有2例为携带者。外显子3-26和45-52在突变区域呈现出最高突变频率。在单个外显子突变频率统计中,在缺失突变中,外显子47和外显子50最为常见;而在重复突变中,外显子5,6,7最为常见。
结论:
MLPA在检测率和灵敏度上优于mPCR。产前诊断应该用于患病高危胎儿以降低疾病发病率。此外,医生有责任让女性携带者了解产前诊断的重要性。
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a common X-linked recessive neuromuscular disorder with a prevalence of 1/3500 among newborn boys of all races.[1] Clinically, DMD is characterized by rapidly progressive muscular weakness and degeneration, often associated with lumbar lordosis and calf hypertrophy. Patients with DMD are usually diagnosed before the age of 5 years and wheelchair-bound by the age of 12 years. They often die of respiratory or heart failure[2,3] in their late teens or early twenties. In contrast, Becker muscular dystrophy (BMD) is less severe allelic form of DMD with a prevalence of 1/18,000 among male live births.[4] Symptoms begin to manifest at about 11 years of age; progression is slower than DMD. The life expectancy of patients with BMD is longer than that of patients with DMD.[5,6]
DMD/BMD is caused by mutations in dystrophin gene, the biggest human gene known so far, occupying a genomic region of 2.4 Mb on Xp21.[6] The dystrophin gene contains 79 exons and encodes a 14.6 kb mRNA.[7] The dystrophin protein contains an N-terminal actin-binding domain and 24 spectrin-like repeat units interspersed by four hinge regions, followed by a cysteine-rich domain and a C-terminal domain.[1] The protein fulfills the link between cytoskeletal actin and the extracellular matrix, mainly expressing in the skeleton, muscles, and brain.[7] If the mutation maintains an open reading frame, a shortened but functional protein and a milder phenotype (BMD) are observed. If the mutation shifts the translational reading frame of the transcripts or creates a stop codon, a severe phenotype (DMD) is observed.[8]
The majority of DMD cases are caused by gene deletion or duplication in single or multiple exons, which account for about 65% and 5–8%, respectively.[9] The remaining cases are attributable to point mutations, splicing mutation, and small deletions and insertions.[3,9] Previous reports indicate that approximately 70% of these lethal mutations are inherited from a carrier mother,[2] whereas 30% of the mutation are de novo.[10] To date, no effective therapy is widely available for DMD/BMD patients. Therefore, an effective and accessible method of prenatal diagnosis and genetic counseling is necessary to inhibit the birth of defective male.[11]
Many methodologies have been used to detect mutations in the DMD gene. The most basic and common method is the multiplex polymerase chain reaction (multiplex PCR) technique,[12,13] which offers a simple and rapid screening of the most common 18 exons in male patients.[14,15] Almost all DMD patients with deletion can be detected by multiplex PCR.[16] However, multiplex PCR cannot detect duplication and cannot be used as a test of female carriers.[13,17]
Multiplex ligation-dependent probe amplification (MLPA) has proved to be a reliable tool for the diagnosis of genetic diseases.[11] MLPA has been introduced and is now widely used to detect both deletions and duplications of the dystrophin gene. In contrast with multiplex PCR, MLPA can also be used for detecting female carriers in whom deletion is masked by the amplification of the normal X chromosome. As a sensible and available tool, MLPA can be used in prenatal diagnosis and genetic counseling.[13,14,17]
In this study, multiplex PCR, MLPA, and sequencing analysis were performed in 121 unrelated Chinese families with 200 individuals, including 121 probands, 64 mothers of probands, and 15 fetus samples to explore the characteristics of dystrophin gene, and illustrate the significance of prenatal diagnosis in female carriers.
METHODS
Ethical approval
This study was conducted in accordance with the procedure of disease in China and was approved by the Institutional Review Boards of the First Affiliated Hospital of Nanjing Medical University. All patients enrolled wrote informed consent before starting the study and have their medical data used for research purposes.
Subjects
A total of 121 unrelated Chinese families with 200 individuals were referred for gene analysis of the dystrophin gene between 2010 and 2014 in Nanjing, China. There were 121 probands, 64 mothers of probands, and 15 fetuses. Patients were mostly from Jiangsu Province, Anhui Province, Shandong Province, and Zhejiang Province. After providing a detailed history, each patient was examined by one of the investigators (Neurologists) and verified by a senior neurologist with particular emphasis on atrophy and hypertrophy of muscles and power of individual muscle and the presence of Gower's sign and valley sign. Diagnosis was based on clinical manifestation and symptoms, high serum creatine kinase levels, changed electromyography, and muscle biopsy. Electrocardiogram and echocardiography were done in some cases. Some patients were also subjected to psychometry and assessment of IQ using Binet–Kamath test of intelligence. Twenty-eight probands were detected by multiplex PCR primarily, and MLPA was used in multiplex PCR-negative cases subsequently. The remaining 93 probands and 62 female potential carriers were tested with MLPA directly. In fetuses, multiplex PCR technique and MLPA were performed on 4 fetuses and 10 fetuses, respectively. In addition, there were three probands, two female potential carriers, and one fetus with negative MLPA performed by sequencing.
Multiplex polymerase chain reaction
Eighteen pairs of multiplex PCR primers covering exons in the deletion hotspots were divided into two groups to amplify: one encompasses exons 4, 8, 12, 17, 19, 44, 45, 48, and 51 and the other encompasses promoter and exons 3, 6, 13, 43, 47, 50, 52, and 60. The procedures were referred to previous researches.[12,16] DNA was amplified in a final volume of 50 ml using 250–500 ng genomic DNA, 10 buffer, 0.2 mmol of each primer, dNTP (200 mmol each), and 1 U Taq DNA polymerase. PCR cycling was performed under the following conditions: initial denaturation at 95°C for 6 min, 94°C for 1 min, 56.8°C for 1 min, 72.6°C for 1 min for a total of 30 cycles, and 72°C for 10 min. The products were visualized by electrophoresis on a 1.5% agarose gel. The PCR products were separated by capillary electrophoresis according to the size of fragments, and the measured data of separated DNA fragments were analyzed by Genemarker software (Softgenetic LLC, State college, PA, USA).
Multiplex ligation-dependent probe amplification
Genomic DNA was extracted from peripheral blood samples taken from the individuals. Two SALSA MLPA probe mix sets – P034 and P035 (MRC-Holland, The Netherlands) – were used for detecting all the exons of the DMD gene. Procedures followed the manufacturer's instruction and previous research;[15] 5 μl of the working DNA was used for each sample. All reactions were carried out on a standard thermal cycle. The carboxyfluorescein-labeled MLPA PCR products were separated by capillary electrophoresis on the ABI 3730xl DNA Analyzer (Applied Biosystems, USA). The sizes of the exon-specific amplified fragments were identified according to their migration relative to the GeneScan Rox-500 size standard using Gene Mapper version 4.0 software (Applied Biosystems, USA). Relative amounts of PCR products were determined using Coffalyser software (MRC-Holland, The Netherlands) provided online by the manufacturer (www.mlpa.com).
Sequencing analysis
A direct sequencing using forward and reverse primers complementary to screen all 79 exons and exon-intron junctions with the ABI Prism 3100 Genetic Analyzer (Applied Biosystems, USA) was performed on MLPA-negative results. Sequencing analysis was detected in the Beijing Genomics Institute (BGI, China).
RESULTS
Detection of Duchenne muscular dystrophy gene mutations in 121 probands
Fourteen probands were identified as having gene deletions, accounting for 50.00% (14/28) of patients analyzed by multiplex PCR. In 14 samples with negative multiplex PCR assay, 3 showed positive results: using MLPA, they were found to have gene duplications [Table 1]. In 93 probands subjected to MLPA directly, the rearrangements consisted of 47 deletions and 11 duplications [Table 2], accounting for 62.37% (58/93) of patients tested with MLPA. Combining the results, the rearrangements consisted of 61 large deletions and 14 large duplications spanning one or more exons, representing 50.41% (61/121) and 11.57% (14/121) of all mutations identified in this study, respectively [Figure 1]. There were 59 DMD and 2 BMD probands with deletions and 13 DMD and 1 BMD probands with duplications. Three BMD patients had positive results, in which two out of three had the mutation of del 45, 47, 48, 50, 51, 52 through the method of multiplex PCR, and the rest had the mutation of dup 2 through the method of MLPA. To detect the DMD gene definitively, 4 probands with negative MLPA accepted gene sequencing analysis in this study. There were one case with small deletion in intron 17, one case with substitution mutation in intron 65, and one case with nonsense point mutation in exon 8.
Table 1.
MPLA results of 14 patients with negative multiplex PCR
| Patient ID | Multiplex PCR | MLPA |
|---|---|---|
| M009 | Negative | Dup 68–79 |
| M021 | Negative | Dup 42–43 |
| M031 | Negative | Dup 3–7 |
| The remaining 11 patients | Negative | Negative |
Dup: Duplicated exons; Multiplex PCR: Multiplex polymerase chain reaction; MLPA: Multiple ligation-dependent probe amplification.
Table 2.
The genotype of 121 probands with deletions and duplications
| Deletion detected by multiplex PCR | Deletion detected by MLPA | Duplication | |
|---|---|---|---|
| Del 3, 4, 6, 8 | Del 2–17 | Del 45–50 | Dup 2 |
| Del 8, 12, 13, 17, 19 | Del 3–7 | Del 45–52* | Dup 2–7 |
| Del 17, 19, 43 | Del 5–7 | Del 45–53 | Dup 3–13 |
| Del 45* | Del 8–19 | Del 45–76 | Dup 3–34 |
| Del 45, 47* | Del 8–44 | Del 46–47* | Dup 3–7* |
| Del 45, 47, 48, 50, 51, 52* | Del 10–11 | Del 46–48* | Dup 5–11 |
| Del 47, 48 | Del 10–30 | Del 46–50 | Dup 8–9 |
| Del 47, 48, 50, 51, 52 | Del 14–19 | Del 46–55 | Dup 12 |
| Del 48, 50, 51, 52 | Del 18–26 | Del 48–50† | Dup 42–43 |
| Del 51 | Del 18–29 | Del 48–52 | Dup 48–52 |
| Del 60 | Del 18–37 | Del 49–50* | Dup 62 |
| Del 19 | Del 49–52* | Dup 64–67 | |
| Del 22–27 | Del 50 | Dup 68–79 | |
| Del 22–44 | Del 51* | ||
| Del 43 | Del 51–54 | ||
| Del 44 | Del 52 | ||
| Del 45 | Del 52–54 | ||
| Del 45–47* | Del 53–55 | ||
| Del 45–48 | Del 55 | ||
The information was listed from smallest to largest depending on the number of first exon. *Repetition of twice; †Repetition of three times. Del: Deleted exons; Dup: Duplicated exons; Multiplex PCR: Multiplex polymerase chain reaction; MLPA: Multiple ligation-dependent probe amplification.
Figure 1.
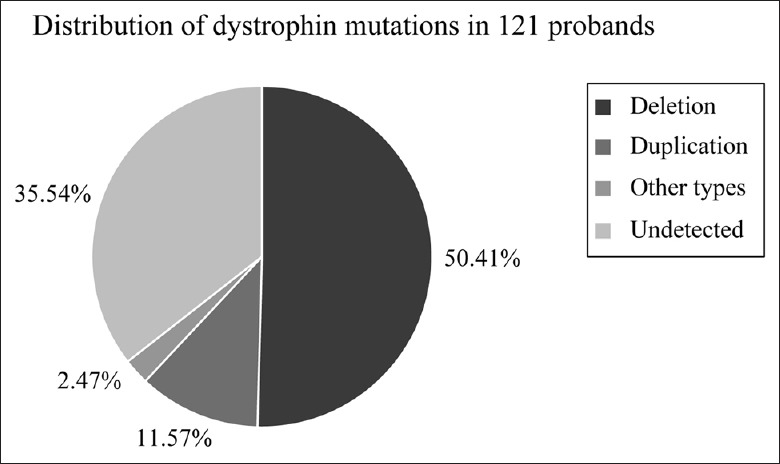
Distribution of dystrophin mutations in 121 probands. Although applying multiplex PCR, MLPA, and sequencing, we found 61 cases of deletion (50.41%), 14 cases of duplication (11.57%), 3 cases of other type mutations, 1 case of small deletion (0.83%), 1 case of nonsense mutation (0.83%), and 1 case of substitution mutation (0.83%). In addition, there were 43 undetected cases by these three methods in this study. PCR: Polymerase chain reaction; MLPA: Multiplex ligation-dependent probe amplification.
Small fragment deletions and duplications are predominant in the Duchenne muscular dystrophy gene
Amid 121 probands with exonic deletion, single-exon deletions made the largest proportion, accounting for 21.31% (13/61). Our study also showed that 18.03% (11/61) of the deletions involved 3-exon deletions; 13.11% (8/61) had 2-exon deletions; 9.83% (6/61) had each 4-exon deletions and 5-exon deletions; 8.20% (5/61) had 6-exon deletions; and 3.27% (2/61) had each 8-exon deletions and 9-exon deletions. Among 14 patients with exonic duplication, single-exon duplication is most common, accounting for 21.43% (3/14). 14.29% (2/14) exhibited 2-exon duplication. Combining all data, the deletions and duplications with nine exons or smaller account for 86.89% (53/61) and 78.57% (11/14), respectively.
Exons 3–26 and 45–52 have the maximum frequency in mutation regions
Analyzing all deletions, we found that both exons 45 and 51 were the most common single-delete exons in 13 single-exon delete ones. Among the 48 cases with multiple exons, deletion of three exons in 48–50 was the most common (6.25%). Of all detected exons, exons 47 and exon 50 were the most common deleted ones, followed by exon 48 and then exon 51 and exons 45, 49, and 52. No deletion was found in exon 1 and exons 77–79. Among all duplicated exons, exons 5, 6, and 7 had the highest numbers. Counting the frequency of deletions and duplications together, the common regions in this study are exons 3–26 and 45–52 in this study [Figure 2].
Figure 2.

It is a rearrangement of dystrophin gene. Frequency of dystrophin gene rearrangement showed that exons 47 and 50 were most common in all exonic deletions and exons 5, 6, and 7 were the most common in all exonic duplications.
Detection of Duchenne muscular dystrophy gene mutations in female potential carriers
MLPA analysis was performed on 64 mothers of DMD boys to detect whether or not they were carriers. Among the 64 females, 26 carriers were detected by MLPA, including 20 with deletions and 6 with duplications. Moreover, two mothers were identified as carriers using sequencing. Analyzing information from the 64 females and their unhealthy boys, we could see that 43.75% (28/64) of mothers were carriers. In other words, 43.75% of pathogenic mutations were inherited from a carrier mother and 56.25% of the mutations are surfaced spontaneously.
Detection of Duchenne muscular dystrophy gene mutations in fetuses
Multiplex PCR technique and MLPA were performed on 14 fetuses with high-risk prevalence of DMD. In multiplex PCR group, one male fetus was identified as a patient with gene exonic deletion. In MLPA group, one female fetus was confirmed to be a carrier of gene exonic deletion and duplication each and carried the same maternal chromatids with probands. We also observed that one case was determined to be defective with the mutation in intron 65 by sequencing. Therefore, 2 out of 7 male fetuses were found to be unhealthy and 2 out of 8 female fetuses were found to be carriers [Table 3].
Table 3.
Prenatal diagnosis and pregnant suggestion of 15 fetuses
| Patient ID of proband | Genotype of proband | Carrier status of mother | Prenatal diagnosis of fetus | Gender of fetus | Continued gestation |
|---|---|---|---|---|---|
| M010 | Del 45, 47, 48, 50, 51, 52 | Yes | Normal | Male | Yes |
| M011 | Del 8, 12, 13, 17, 19 | Yes | Del 8, 12, 13, 17, 19 | Male | No |
| M020 | Intron 65 mutation | Yes | Normal | Female | Yes |
| M020 | Intron 65 mutation | Yes | Intron 65 mutation | Male | No |
| M023 | Del 45 | Yes | Normal | Female | Yes |
| M029 | Del 48, 50, 51, 52 | No | Normal | Female | Yes |
| M033 | Del 45, 47, 48, 50, 51, 52 | Unknown | Normal | Male | Yes |
| M037 | Del 51 | Yes | Del 51 | Female | Yes |
| M053 | Del 52 | No | Normal | Female | Yes |
| M062 | Del 22–44 | No | Normal | Male | Yes |
| M062 | Del 22–44 | No | Normal | Male | Yes |
| M067 | Del 22–27 | Yes | Normal | Male | Yes |
| M070 | Del 46–48 | Yes | Normal | Female | Yes |
| M075 | Dup 62 | No | Normal | Female | Yes |
| M079 | Dup 12 | Yes | Dup 12 | Female | Yes |
Del: Deleted exons; Dup: Duplicated exons.
DISCUSSION
DMD and BMD are common X-linked neuromuscular disorders with high prevalence rate and low diagnostic rate. Therefore, a number of patients suffer greatly due to delayed diagnosis. In our study, we used multiplex PCR and MLPA methods in 200 people to identify whether or not they exhibited dystrophin gene mutations. We already knew that multiplex PCR is a relatively simple and rapid method. Our results indicated that 50.00% of the 28 male probands carried gene deletions detected by multiplex PCR. Hence, because the dystrophin gene is X-linked, deletions in male patients can be easily identified by multiplex PCR.[18] However, multiplex PCR is neither suitable for detecting gene duplication nor useful for detecting mutations in female carriers.[19]
MLPA is a new and useful technique for the quantification of gene abnormalities, not only for deletions but also for duplications and female carriers in a wide range of conditions.[20] It is based on comparative quantitation of hybridized probes that are amplified by PCR with a single pair of universal primers. The number of ligated oligonucleotide products is proportional to the amount of original target DNA. The PCR fragments of different lengths can be separated and quantitated in an automated capillary DNA sequencer. Therefore, it allows interrogation of gene dosage at multiple target loci in a single reaction.[14] In our study, 50.54% of our probands carried deletions and 11.83% carried duplications, which increased the sensitivity of detecting DNA rearrangement by MLPA up to 62.37%. In addition, 3 of 14 multiplex PCR-negative probands got positive results with MLPA. Therefore, considering the productivity and sensitivity of detection, MLPA is superior to multiplex PCR[19] and MLPA should be considered for the initial test in the detection of exon deletions and duplications of the dystrophin gene.[11,15,20] Our observation also showed that sequencing could be performed on the patients with negative MLPA, similar to other studies.[21]
In 64 mothers of DMD boys, 43.75% carried the same mutations as their defective sons whereas 56.25% probands showed no mutation. These results suggested that 43.75% probands were inherited from a carrier mother and 56.25% of the mutations are de novo. This percentage is remarkably higher than in previous studies, which showed that 30% of the mutations occur naturally. That some real female carriers of probands were not enrolled in the study or some other potential causes may have induced the higher percentage of de novo.[22] Further investigation and data are necessary to provide more evidence. However, from the 43.75% female carriers, we strongly suggest that effective and available methods should be used to detect the dystrophin gene of the carrier females, in order to reduce the incidence of disease.
Among the 15 fetuses, 2 out of 7 male fetuses were found to be unhealthy and 2 out of 8 female fetuses were found to be carriers. Thus, we suggested that the 2 pathogenic male fetuses had to achieve termination of pregnancy. On the contrary, the 2 carrier fetuses and remaining normal fetuses could continue to grow in the uterus. The subsequent follow-up visit showed that the actual situation was consistent with the results of prenatal diagnosis. Compared with multiplex PCR, MLPA has a higher accuracy in detecting male fetuses and break the limits of testing carriers and fetuses. Besides, MLPA is an available method which can be used for prenatal diagnosis when the deletions or duplications are not detected by multiplex PCR.[23] Certainly, sequencing can be used to detect small gene mutations in MLPA-negative individuals. It is essential to perform antenatal diagnosis on the carriers of childbearing age by MLPA primarily. In addition, clinical experiences reveal that the pregnant women will benefit greatly if the fetuses with hereditary disease can be prediagnosed.
Exons 45–54 and exons 3–22 are the most commonly deleted regions.[21] Exons 2–20 and 44–53 were previously reported as hot-spot regions in the dystrophin gene. In this study, we have analyzed the distribution of each exon regarding their frequency of deletions or duplications. Counting the frequency of deletions and duplications, we found in this study that hot-spot regions were exons 3–26 and 45–52. Here, after analysis of the frequency of each individual exon, exon 47 and exon 50 were the most common deleted exons and exons 5, 6, and 7 were the most common duplicated exons. Exon skipping is an available therapy for DMD that can transform DMD into BMD, which is based on the recovery of the reading frame induced by alternative splicing of antisense oligonucleotides.[24] Combing these data, researchers can create exon skipping drugs depending on the high incidence exons.
In China, although physicians know that DMD is a hereditary neuromuscular disease, most of DMD patients do not receive timely diagnosis, appropriate treatment, or useful suggestions.[19] If there are some genetic diseases such as DMD, physicians should suggest the patients to take a precise molecular test[18] for disease diagnosis and genetic consulting. For the molecular test, MLPA should be the first choice, and sequencing can be a supplement. In addition, the muscle biopsy should be conducted to verify previous diagnosis and provide sequence analysis for dystrophin mRNA.[25] In the diagnosis of DMD/BMD, physicians combine clinical signs with auxiliary examinations. Above all, molecular analysis of dystrophin gene is indispensable.
We have applied multiplex PCR, MLPA, and sequencing to detect large rearrangements in gene and concluded some characteristics of deletions and duplications among male patients, female potential carriers, and suspected fetuses. Considering the productivity and sensitivity of detection, MLPA is superior to multiplex PCR. Sequencing analysis can detect the small mutation in dystrophin gene, making up for deficiency of MLPA. In this study, MLPA analysis plays an important role in detecting rearrangements and prenatal diagnosis. It is a responsibility for physicians to inform female carriers the importance of prenatal diagnosis with appropriate methods.
Financial support and sponsorship
This study was supported by grants from the National Natural Science Foundation of China (No. 81671117), the Jiangsu Province Natural Science Foundation (No. BK20141439), and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (No. JXC10231802).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Yi Cui
REFERENCES
- 1.Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the leiden duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006;34:135–44. doi: 10.1002/mus.20586. doi:10.1002/mus.20586. [DOI] [PubMed] [Google Scholar]
- 2.Zhang T, Liu S, Wei T, Yong J, Mao Y, Lu X, et al. Development of a comprehensive real-time PCR assay for dystrophin gene analysis and prenatal diagnosis of Chinese families. Clin Chim Acta. 2013;424:33–8. doi: 10.1016/j.cca.2013.05.006. doi:10.1016/cca.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 3.Hegde MR, Chin EL, Mulle JG, Okou DT, Warren ST, Zwick ME, et al. Microarray-based mutation detection in the dystrophin gene. Hum Mutat. 2008;29:1091–9. doi: 10.1002/humu.20831. doi:10.1002/humu.20831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bushby KM, Thambyayah M, Gardner-Medwin D. Prevalence and incidence of Becker muscular dystrophy. Lancet. 1991;337:1022–4. doi: 10.1016/0140-6736(91)92671-n. doi:10.1016/0140-6736(91)92671-N. [DOI] [PubMed] [Google Scholar]
- 5.van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;357:2677–86. doi: 10.1056/NEJMoa073108. doi:10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- 6.Bakker E. DNA-based techniques for detection of carriers of duchenne and becker muscular dystrophy. Muscular Dystrophy. 2001;43:111–35. doi:10.1385/1-59259-138-8:111. [Google Scholar]
- 7.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–28. doi: 10.1016/0092-8674(87)90579-4. doi: 10.1016/0092.8674(87)90579.4. [DOI] [PubMed] [Google Scholar]
- 8.Bellayou H, Hamzi K, Rafai MA, Karkouri M, Slassi I, Azeddoug H, et al. Duchenne and Becker muscular dystrophy: Contribution of a molecular and immunohistochemical analysis in diagnosis in morocco. J Biomed Biotechnol. 2009;2009:325210. doi: 10.1155/2009/325210. doi:10.1155/2009/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lalic T, Vossen RH, Coffa J, Schouten JP, Guc-Scekic M, Radivojevic D, et al. Deletion and duplication screening in the DMD gene using MLPA. Eur J Hum Genet. 2005;13:1231–4. doi: 10.1038/sj.ejhg.5201465. doi:10.1038/sj.ejhg.5201465. [DOI] [PubMed] [Google Scholar]
- 10.Alcántara MA, García-Cavazos R, Hernández-U E, González-del Angel A, Carnevale A, Orozco L, et al. Carrier detection and prenatal molecular diagnosis in a Duchenne muscular dystrophy family without any affected relative available. Ann Genet. 2001;44:149–53. doi: 10.1016/s0003-3995(01)01084-x. doi:10.1016/S0003-3995(01)01084-X. [DOI] [PubMed] [Google Scholar]
- 11.Gatta V, Scarciolla O, Gaspari AR, Palka C, De Angelis MV, Di Muzio A, et al. Identification of deletions and duplications of the DMD gene in affected males and carrier females by multiple ligation probe amplification (MLPA) Hum Genet. 2005;117:92–8. doi: 10.1007/s00439-005-1270-7. doi:10.1007/s00439-005-1270-7. [DOI] [PubMed] [Google Scholar]
- 12.Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res. 1988;16:11141–56. doi: 10.1093/nar/16.23.11141. doi:10.1093/nar/16.23.11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itto AB, Hamzi K, Bellayou H, Itri M, Slassi I, Nadifi S, et al. Evolution of molecular diagnosis of Duchenne muscular dystrophy. J Mol Neurosci. 2013;50:314–6. doi: 10.1007/s12031-013-9971-1. doi:10.1007/s12031-013-9971-1. [DOI] [PubMed] [Google Scholar]
- 14.Hwa HL, Chang YY, Chen CH, Kao YS, Jong YJ, Chao MC, et al. Multiplex ligation-dependent probe amplification identification of deletions and duplications of the Duchenne muscular dystrophy gene in Taiwanese subjects. J Formos Med Assoc. 2007;106:339–46. doi: 10.1016/S0929-6646(09)60318-1. doi:10.1016/S0929-6646(09)60318-1. [DOI] [PubMed] [Google Scholar]
- 15.Janssen B, Hartmann C, Scholz V, Jauch A, Zschocke J. MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: Potential and pitfalls. Neurogenetics. 2005;6:29–35. doi: 10.1007/s10048-004-0204-1. doi:10.1007/s10048-004-0204-1. [DOI] [PubMed] [Google Scholar]
- 16.Sbiti A, El Kerch F, Sefiani A. Analysis of dystrophin gene deletions by multiplex PCR in Moroccan patients. J Biomed Biotechnol. 2002;2:158–60. doi: 10.1155/S1110724302205069. doi:10.1155/S1110724302205069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Q, Li-Ling J, Lin C, Wu Y, Sun K, Ma H, et al. Characteristics of dystrophin gene mutations among Chinese patients as revealed by multiplex ligation-dependent probe amplification. Genet Test Mol Biomarkers. 2009;13:23–30. doi: 10.1089/gtmb.2008.0059. doi:10.1089/gtmb.2008.0059. [DOI] [PubMed] [Google Scholar]
- 18.Sansović I, Barišić I, Dumić K. Improved detection of deletions and duplications in the DMD gene using the multiplex ligation-dependent probe amplification (MLPA) method. Biochem Genet. 2013;51:189–201. doi: 10.1007/s10528-012-9554-9. doi:10.1007/s10528-012-9554-9. [DOI] [PubMed] [Google Scholar]
- 19.Lai KK, Lo IF, Tong TM, Cheng LY, Lam ST. Detecting exon deletions and duplications of the DMD gene using multiplex ligation-dependent probe amplification (MLPA) Clin Biochem. 2006;39:367–72. doi: 10.1016/j.clinbiochem.2005.11.019. doi:10.1016/j.clinbiochem.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Wang Z, Yan M, Huang S, Chen TJ, Zhong N, et al. Similarity of DMD gene deletion and duplication in the Chinese patients compared to global populations. Behav Brain Funct. 2008;4:20. doi: 10.1186/1744-9081-4-20. doi:10.1186/1744-9081-4- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J, Li SY, Li YQ, Cao JQ, Feng SW, Wang YY, et al. MLPA-based genotype-phenotype analysis in 1053 Chinese patients with DMD/BMD. BMC Med Genet. 2013;14:29. doi: 10.1186/1471-2350-14-29. doi:10.1186/1471-2350-14- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ji X, Zhang J, Xu Y, Long F, Sun W, Liu X, et al. MLPA application in clinical diagnosis of DMD/BMD in shanghai. J Clin Lab Anal. 2015;29:405–11. doi: 10.1002/jcla.21787. doi:10.1002/jcla.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verma PK, Dalal A, Mittal B, Phadke SR. Utility of MLPA in mutation analysis and carrier detection for Duchenne muscular dystrophy. Indian J Hum Genet. 2012;18:91–4. doi: 10.4103/0971-6866.96667. doi:10.4103/0971-6866.96667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim KR, Maruyama R, Yokota T. Eteplirsen in the treatment of duchenne muscular dystrophy. Drug Des Devel Ther. 2017;11:533–45. doi: 10.2147/DDDT.S97635. doi:10.2147/DDDT.S97635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takeshima Y, Yagi M, Okizuka Y, Awano H, Zhang Z, Yamauchi Y, et al. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J Hum Genet. 2010;55:379–88. doi: 10.1038/jhg.2010.49. doi:10.1038/jhg.2010.49. [DOI] [PubMed] [Google Scholar]