


Abstract
Organized in tandem repeat arrays in most eukaryotes and transcribed by RNA polymerase III, expression of 5S rRNA genes is under epigenetic control. To unveil mechanisms of transcriptional regulation, we obtained here in depth sequence information on 5S rRNA genes from the Arabidopsis thaliana genome and identified differential enrichment in epigenetic marks between the three 5S rDNA loci situated on chromosomes 3, 4 and 5. We reveal the chromosome 5 locus as the major source of an atypical, long 5S rRNA transcript characteristic of an open chromatin structure. 5S rRNA genes from this locus translocated in the Landsberg erecta ecotype as shown by linkage mapping and chromosome-specific FISH analysis. These variations in 5S rDNA locus organization cause changes in the spatial arrangement of chromosomes in the nucleus. Furthermore, 5S rRNA gene arrangements are highly dynamic with alterations in chromosomal positions through translocations in certain mutants of the RNA-directed DNA methylation pathway and important copy number variations among ecotypes. Finally, variations in 5S rRNA gene sequence, chromatin organization and transcripts indicate differential usage of 5S rDNA loci in distinct ecotypes. We suggest that both the usage of existing and new 5S rDNA loci resulting from translocations may impact neighboring chromatin organization.
INTRODUCTION
Ribosomal RNAs (rRNAs) are vital components of the translational machinery and constitute a large fraction of the total cellular RNA pool. 5S rRNA, the smallest RNA component of the ribosome, is encoded by 5S rRNA genes, which in most eukaryotic genomes are organized in multi-copy arrays. In Arabidopsis thaliana, fluorescence in situ hybridization (FISH) (1,2) and physical mapping (3) situated the 5S rRNA gene arrays in the pericentromeric regions of chromosome 3, 4 and 5. Despite the publication of the Arabidopsis Columbia-0 (Col-0) reference genome in 2000 (4), these chromosomal domains remain incompletely assembled. Furthermore, as typical for ribosomal DNA (rDNA), 5S rRNA gene copies within the array are nearly identical due to concerted evolution, a process that promotes homogeneity among the many repeat units (5–7). To date, specifics on the structure of the 5S rRNA gene arrays including arrangement, polymorphisms or variability within the A. thaliana species is still sparse, and available information is mostly derived from a small number of sequences (8–11).
A typical Arabidopsis 5S rRNA gene is 500 bp long, comprising a 120 bp transcribed sequence and a 380 bp spacer region. 5S rRNA transcription by RNA Polymerase III necessitates an internal promoter and a TATA-like motif located 28 bp upstream of the transcribed region, and terminates in a T-rich termination signal (12,13). 5S rRNA gene copies can contain polymorphisms in both transcribed sequences and spacer regions. Based on these polymorphisms, major 5S rRNA genes encoding the consensus 5S transcript are distinguished from minor 5S rRNA genes that carry one to several polymorphisms in the transcribed sequence (11). While in leaf tissue only major genes are expressed, sets of minor 5S rRNA genes are transcribed in specific tissues, such as seeds (14,15). Based on these observations, the 5S rRNA genes with polymorphisms are considered to be selectively repressed, while retaining the potential to be transcribed in certain cell types or developmental stages. Epigenetic mechanisms are involved in the selective silencing of these polymorphic 5S rRNA gene copies (14,16,17). Indeed, loss of the chromatin remodeling factor DECREASE IN DNA METHYLATION 1 (DDM1) alleviates the repression of minor 5S rRNA gene copies (14,18) similarly to mutants impaired in DNA methylation maintenance, histone de-acetylation or the RNA-directed DNA methylation (RdDM) pathway (19). The importance of epigenetic control of 5S rRNA gene transcription is also revealed by the expression of an atypical 5S rRNA transcript of 210 bases, which extends beyond the termination sequence into the intergenic spacer region, in different chromatin mutant contexts (18–20). Furthermore, specific modes of epigenetic regulation may operate at the different 5S rRNA loci, exemplified by the role of RNA polymerase V in controlling transcription and chromatin organization of the 5S rRNA gene copies situated on chromosome 4 (17). However, so far detailed information on potential differences in epigenetic marks between the three 5S rRNA gene loci and their role in the control of 5S rRNA gene transcription are missing.
Using Next Generation Sequencing (NGS) datasets, we show here that the Col-0 genome comprises over 2000 5S rRNA gene copies distributed in three major loci. While 5S rRNA gene sequences situated on chromosome 3 are the most polymorphic and enriched in repressive histone marks, gene copies of chromosome 5—and to a lesser extent of chromosome 4—are moderately enriched in transcriptionally permissive marks. In agreement, the locus on chromosome 5 is the major source of the atypical 5S-210 transcript, indicating a more open chromatin configuration. A similar in-depth analysis of a different ecotype, Landsberg erecta (Ler), revealed different enrichment in epigenetic marks and distinct prevalence of 5S-210 transcripts among 5S loci, indicating differential usage of 5S rRNA gene loci among ecotypes. In silico analysis of NGS datasets of a large number of ecotypes further revealed conservation of chromosome-specific transcription termination sequences in 5S rRNA genes coupled to important copy number variations, while 5S rDNA copy numbers are stably maintained during shorter time scales within the same ecotype. Finally, by combining in silico analysis, linkage mapping in a segregating population, chromosome-specific FISH and qPCR we characterized novel 5S rDNA insertions in the Ler genome and in several mutants in the RNA-directed DNA methylation (RdDM) pathway. We suggest that new 5S rDNA loci resulting from translocations impact chromatin organization by preferentially clustering with heterochromatic regions.
MATERIALS AND METHODS
Plant material
Arabidopsis ecotypes as well as mutant Arabidopsis lines were obtained from the Nottingham Arabidopsis Stock Center and/or were provided by other laboratories. Homozygous mutants ago4-2 (Col-0 background; (21)) and ago4-1 (N6364, Ler background) (22), dcl2-2 (SALK_123586, Col-0 background) and dcl3-1 (SALK_005512, Col-0 background) were confirmed by PCR-based genotyping. After 2 days of stratification at 4°C in the dark, plants were grown on soil in a growth chamber under 16-h light/8-h dark cycles at 22°C. For in vitro culture, seeds were sterilized and sown on germination medium containing 0.8% (w/v) agar, 1% (w/v) sucrose and Murashige & Skoog salts (M0255; Duchefa Biochemie, Netherlands). After 2 days of stratification at 4°C in the dark, plants were grown under 16-h light/8-h dark cycles at 23°C.
DNA extraction and qPCR conditions
The aerial parts of about five 3-week-old plants for each of the three biological replicates were shock frozen in liquid nitrogen, ground to powder and incubated at 65°C in extraction buffer (Tris pH 8 0.1 M, EDTA pH 8 50 mM, NaCl 500 mM, SDS 1.23%), potassium acetate was then added to a final concentration of 7.45 mM before centrifugation at 16 000g. DNA was precipitated in isopropanol. The pellet was resuspended into Tris–EDTA buffer and ethanol precipitated. The final DNA pellet was resuspended in water and stored at –20°C. DNA purity and quantity was assessed with a Nanodrop. 500 ng of purified DNA were used per 10 μl reaction in the Lightcycler 480 (Roche) with the LightCycler® 480 SYBR Green I Master mix (Roche). For each biological replicate, a mean of two technical replicates was assessed and qPCR data were analyzed using the ΔΔCt method. Primer sequences for chromosome specific primers are in Supplemental Table S6. The following conditions of amplifications were applied: 3 min at 95°C; 40 cycles of 15 s at 95°C, 15 s at 55°C and 20 s at 72°C except for the copies situated on chromosome 4 for which elongation was performed at 60°C. Copy numbers were standardized to two single copy genes (HXK1, At4g29130 and UEV1C, At2g36060). Real primer efficiencies of 5S rDNA were determined simultaneously on a serial DNA dilution.
ChIP analysis
Chromatin from 3-week-old plantlets grown on soil and harvested in triplicates was formaldehyde cross-linked for 20 min and chromatin immunoprecipitation carried out as previously described (23) with minor modifications: Chromatin was sheared using the Diagenode Bioruptor (10 cycles of 30 s ON and 1.5 min OFF) to fragments of ∼300 bp. Protein A-coupled Dynabeads (Invitrogen) were used to pre-clear sonicated chromatin for 3 h and for immuno-precipitation with anti-H3 (ab1791, Abcam), anti-H3K9me2 (ab1220, Abcam) and anti-H3K4me3 (04-745, Millipore) antibodies. Enrichment in histone marks was quantified using qPCR (Roche) as described above and normalized to H3 levels.
Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) was performed on somatic or meiotic nuclei at pachytene stage obtained from 10-day-old cotyledons or flower buds fixed in ethanol-acetic acid using directly labeled Locked Nucleic Acid probes (Exiqon): for global 5S ‘56FAM_CAAGCACGCTTAACTGCGGAGTTCTGAT’, specific for the 5S locus of chr4 ‘TEX615_ACCAAAAAAAAAAAAAAAAAAAAGAGGGATG’, of chr5 ‘56FAM_AAAGGTTAAACATAAAAGAGGGATG’, and specific for 180 bp centromeric repeats ‘TEX615_GTATGATTGAGTATAAGAACTTAAACCG’. Somatic nuclear spreads were obtained as previously described (24). Hybridization was performed for 1 hour at 55°C for the locus-specific probes and at 50°C for the global 5S and the 180 bp probes. Post-hybridization washes were carried out at 55°C twice in 2× SSC and once in 0.75× SSC. Slides were mounted in Vectashield containing DAPI (Vector laboratories). Imaging was performed with a Leica DM6000B with an ORCA-Flash4.0 V2 Digital CMOS camera C11440 (HAMAMATSU).
Next-generation sequencing of Col-0 DNA
DNA from aerial parts of 3-week-old Col-0 plants was extracted using the DNeasy plant Mini Kit (Qiagen) and sheared to a size of about 500 bp with a M220 Focused-ultrasonicator™ (Covaris). Library preparation was performed with the LTP Library Preparation Kit for Illumina® platforms (KAPA) followed by a size selection of around 500 bp using AMPure XP (Agencourt) and ligation of adapters (Pentabase). The KAPA library quantification kit for Illumina platforms was then used to quantify the library. Paired-end reads of 2 × 300 bp were obtained using the Illumina Mi-Seq with Mi-Seq Reagent Kit v3.
RNA extraction and RNA-Seq
Total RNA from 2-day-old seedlings were extracted with Tri-Reagent (Euromedex) according to manufacturer's instructions, then treated with RQ1 DNase I (Promega) and purified with phenol–chloroform extraction. For sequencing of 5S-210 cDNA, total RNA was reverse transcribed using random primers supplemented with primer RT-210 for RNA-Seq with M-MLV reverse transcriptase (Promega) followed by PCR amplification for 20 cycles with primers 5S-210 RNA-Seq-For and a mix of 5S-210 RNA-Seq-Rev1/2 using Phusion High-Fidelity DNA Polymerase (ThermoScientific). NEBNext Ultra DNA library Prep Kit for Illumina was used for library preparation with 15 cycles of amplification. Library quality control involved size determination by fragment analyzer run (Advanced Analytical, High sensitivity NGS Fragment analysis Kit) and qPCR for quantification (KAPA library quantification Kit for Illumina). RNA-Seq analysis was carried out in triplicates for Ler and Col-0 on an Illumina Mi-Seq using paired end 75 sequencing with V3 chemistry. Libraries for Illumina sequencing of 5S-120 transcripts were prepared from total RNA by sequence specific reverse transcription followed by a template switch and subsequent PCR amplification. One microgram of total RNA was reverse transcribed using a 5S rRNA specific primer and Superscript II reverse transcriptase. Ten cycles of amplification by PCR were performed for library preparation. Library quality control involved size determination by fragment analyzer run (Advanced Analytical, High sensitivity NGS Fragment analysis Kit) and qPCR for quantification (KAPA library quantification Kit for Illumina). RNA-Seq analysis was carried out in triplicates for Ler and Col-0 on the Illumina Hi-Seq2500 using paired end 125 bp sequencing.
Bioinformatics analyses
Copy number estimation
To estimate 5S rRNA gene copy number through next-generation-sequencing (NGS), we divided the average coverage along the 120 bp transcribed sequence of the 5S rRNA by the average coverage along 16 reference genes (At1g13320, At1g58050, At1g59830, At2g28390, At2g32170, At3g01150, At3g53090, At4g26410, At4g27960, At4g33380, At4g34270, At4g38070, At5g08290, At5g15710, At5g46630 and At5g55840 (25)), in addition to the genes At2g36060 and At4g29130 used for qPCR normalization in this study. For each individual dataset analyzed in this study we mapped 50 bp single-end (SE) reads separately to a single reference consisting of the 120 bp transcribed sequence of the 5S rRNA gene and to the Arabidopsis TAIR10 reference genome with BWA-MEM (v0.7.8) (26; Li, arXiv:1303.3997v2). We retrieved per-base read depth of the 5S rRNA gene and the 18 single copy genes described above with the function Depthofcoverage from GATK (v3.5) (27). Since read lengths of the 1135 ecotypes from The 1001 Genomes Consortium are very heterogeneous (ranges from 30 bp to 143 bp; (28) we report 5S rRNA gene copy number for a subset of ecotypes sequenced in a single study (29). We trimmed the reads to 50 bp in length with trimmomatic (30).
Extraction of T-stretch signatures
To determine the sequence of the different T-stretch signatures downstream of the 120 bp transcribed sequence without an a priori, we generated an in-house pipeline (https://gist.github.com/laurianesimon/0ae2dd7b8c34c23cdacec217aeaab79c). This pipeline maps 50 bp reads using BWA-ALN with low stringency settings (allowing four mismatches in the last 15 nucleotides) against a single reference consisting of the consensus 120 bp transcribed sequence of the 5S rRNA gene prolonged in 3′ by 30 Ns and extracts the downstream sequence. The T-stretch sequence for each read was then affected manually to its locus of origin based on sequence information from chromosome-specific YACs (11). For reads longer than 50 pb, reads were trimmed. The same pipeline was applied to determine the T-stretches in 5S-210 RNA-Seq datasets.
Identification of chromosome-specific polymorphisms
To determine the polymorphisms in the 5S rRNA genes in a chromosome specific manner, we developed a pipeline that isolates flashed reads from the Mi-Seq dataset using grep on an exhaustive list of the identified T-stretches (https://gist.github.com/laurianesimon/a9fc44aa83305c576e914710cae75f87). The isolated reads were then mapped using BWA-MEM to multiple references of 160 bp in length, which comprise the 120 bp transcribed sequence of the 5S rRNA gene as well as 20 nucleotides upstream and downstream; the latter including the specific T-stretches. Only reads covering the whole 120 bp transcribed sequence were retained to determine the polymorphisms in the transcribed sequence. The observed polymorphisms for each reference were extracted and quantified. The same procedure was carried out to determine the polymorphisms along the complete 5S rRNA gene using the whole 5S rRNA gene as reference.
Mapping of polymorphisms in the MAGIC population
To detect polymorphisms along the 5S rRNA gene in the MAGIC population, we first mapped the reads with BWA-MEM as described above to a 300 bp consensus reference from chromosome 5 that spans from 100 bp upstream to 80 bp downstream of the transcribed sequence of the 5S rRNA gene. Subsequently, to obtain the proportion of each polymorphism in the MAGIC population we employed a pipeline described elsewhere (31).
Quantification of enrichment in epigenetic marks
To analyze the enrichment of different epigenetic marks, available raw ChIP-seq data (Supplemental Table S3) have been re-analyzed: For the immunoprecipitation (IP) we extracted the number of reads mapped by BWA ALN to ACT2, HXK1, Ta3 and the 120 bp transcribed sequence of the 5S rRNA gene divided by the total number of reads of the ChIP-Seq dataset. For normalization by the length of the gene of interest or the 5S copy number, this value was divided by the mean read number from three Col-0 input DNA datasets (32,33), as most of the ChIP-seq datasets were not provided with an internal input DNA control. To determine the enrichment at each 5S locus, specific T-stretch signatures were extracted from each ChIP-Seq dataset as above and normalized to the proportion of 5S reads from a particular chromosome in the input datasets.
RESULTS
5S rRNA gene copy number and organization in the Col-0 genome
We first examined by BLASTn analysis the available sequence information on 5S rRNA genes in the Col-0 TAIR10 reference genome (4) (Supplemental Figure S1A). The reference genome comprises about 370 5S rDNA copies, which is substantially less than the ∼1000 copies estimated in a previous study (10). Only few copies were scored on chromosome 4 (Supplemental Figure S1A, Table S1), in spite of this locus being previously reported as one of the main 5S rDNA loci by FISH (2). This underlines the difficulties to assemble repeated sequences with low sequence diversity and suggests that the majority of 5S rRNA genes are missing from the reference sequence. Conversely, genome assembly favors polymorphic sequences as 93.8% of the 5S rDNA sequences are polymorphic with respect to the consensus of the 120 bp transcribed sequence. To determine more precisely 5S rRNA gene copy number in the Col-0 genome, we used high coverage NGS data. We generated an Illumina Mi-Seq dataset with 300 bp paired-end reads and fragments of roughly 500 bp in size (74.5% reads >QC 30) and analyzed two publicly available Illumina Hi-Seq datasets (Supplemental Table S2) for Col-0 (28,34,35). Read mapping to the consensus of the 120 bp transcribed sequence of the 5S rRNA gene (Figure 1A, Supplemental Figure S1B) revealed ∼2000 5S rRNA gene copies per haploid genome, which were confirmed by qPCR (Figure 1B).
Figure 1.

Characterization of 5S rRNA genes and their genomic distribution in Col-0. (A) Scheme of 5S rRNA genes, comprising the 120 bp transcribed sequence (gray arrow) containing the internal promoter sequence (box A, I, C), the 380 bp long intergenic region and the Thymine-rich termination sequence (T-stretch). TSS and TTS indicate transcription start and termination sequence respectively. Positions of primers (pink arrowheads) used to determine 5S rRNA gene copy number by qPCR in (B) are indicated. (B) Mean 5S rRNA gene copy number in Col-0 determined by in silico analysis (dark gray) of the Mi-Seq dataset and two publicly available Hi-Seq datasets from the 1001 genome project and the mutation accumulation (MA) lines (28,34,35) after normalization to 18 single copy genes, and by qPCR (light grey). For the latter, mean values of 5S rDNA copy numbers from three biological replicates of Col-0 plants normalized to two single copy genes (HXK1, At4g29130 and UEV1C, At2g36060) are shown. Error bars correspond to SEM for three biological replicates. (C) Schematic representation of the predominant 5S rDNA loci in the pericentromeric regions (white) of chromosome 3 (orange), 4 (red) and 5 (green) in the Col-0 ecotype. (D) (Top) Major T-stretch signatures of each 5S rDNA locus in the Col-0 genome derived from the Mi-Seq data set. (Bottom) Relative frequencies of the predominant T-stretch signatures (colored fractions) and the T-stretches with single nucleotide polymorphisms (uncolored fractions) assigned to the three different loci by their characteristic nucleotide combinations (details in Supplemental Figure S1C) are shown in the pie chart. (E) DNA FISH on meiotic bivalents with LNA-DNA mixmer probes designed to specifically recognize the major T-stretch signatures (described in (D)) of chromosome 4 (red) and chromosome 5 (green), DNA is counterstained with DAPI (blue). The scale bar presents 10 μM. (F) Percentage of 5S rRNA gene copies carrying the T-stretch signatures characteristic of 5S rRNA genes from chromosomes 3, 4 and 5 in the Col-0 genome.
Sanger sequencing of YAC-DNA previously showed the existence of particular T-rich termination sequences, termed T-stretch signatures, at the three main 5S rDNA loci situated on chromosomes 3, 4 and 5 (Figure 1C) (11). To obtain a high-resolution view of these T-stretch signatures in the Col-0 genome we extracted all sequences 3′ of the 120 bp transcribed sequence from the Mi-Seq dataset using an in house Perl script. T-stretch sequences could be identified as belonging to chromosome 3 by the specific stretch characterized by CGG(Nx)CTC, to chromosome 4 by the uninterrupted T-stretch or to chromosome 5 by the motif ATG(Nx)AACC. Besides the major DNA-signatures (Figure 1D), we revealed recurrent single nucleotide polymorphisms (SNPs) in the T-stretches, particularly for the polymorphic 5S rRNA genes assigned to chromosome 3 (Supplemental Figure S1C). To confirm that the different T-stretches are indeed specific to a particular chromosome, we then confronted the identified T-stretch sequences to sequencing datasets of six YACs (36) that comprise 5S rDNA sequences assigned to the genetic map (11). Indeed, T-stretches are specific, as we never find T-stretch signatures assigned to a specific chromosome in the 5S loci from other chromosomes (Supplemental Figure S1D). To corroborate that 5S rRNA genes with the same T-stretch signature are present at the same genomic position, we performed FISH with LNA-DNA mixmer FISH probes designed specifically to recognize the principal T-stretch signatures assigned to chromosomes 4 and 5 (37). The LNA-DNA mixmer probes label either the 5S rDNA locus of chromosome 4 or the one of chromosome 5 (Figure 1E) strongly suggesting that each locus contains predominantly 5S rRNA genes with a specific T-stretch signature. We then established the percentage of reads that were assigned to a specific chromosome. Contrary to the information from the reference genome (4), for which the majority of 5S rDNA copies were mapped at the pericentromeric regions of chromosome 3 (290 genes, 78%) and 5 (61 genes, 16%, Supplemental Figure S1A), our results reveal that most 5S rRNA gene copies carry the chromosome 5 signature (55.6%), followed by those with the signature of chromosome 4 (29.6%) and then of chromosome 3 (14.8%, Figure 1F). This is in agreement with FISH experiments establishing the locus of chromosome 5 as the largest locus (1,2).
Taken together, in Col-0, ∼2000 5S rRNA gene copies contribute to the pericentromeric repetitive DNA content, about twice the amount as previously described. 5S rRNA gene copies with the same T-stretch signature cluster within the same locus strongly suggesting gene amplification and homogenization of 5S rRNA gene copies within a given locus.
Identification of locus-specific polymorphisms
To obtain a better view on the 5S rRNA gene content of each locus, we partitioned the reads in three groups according to their chromosomal origin inferred from T-stretch signatures. For each group, we mapped all reads that covered the complete 120 bp of the transcribed sequence against a 5S rRNA gene reference, and determined the percentage of copies with 0 (major 5S rRNA gene), 1, 2 and more than 2 SNPs (minor 5S rRNA gene) (Figure 2A). Nearly all 5S rRNA gene copies assigned to chromosome 3 carry more than two SNPs. 5S rRNA genes from chromosomes 4 and 5 are less polymorphic: 43.4% and 33.8%, respectively, of the transcribed sequences are identical to the consensus 5S rRNA transcript. 5S rRNA genes are highly methylated (38) and spontaneous deamination of methylated cytosines leads to thymine substitutions. We therefore scored G to A and C to T transitions in the transcribed sequence, which make up for 53.9%, 40.1% and 27.5% of the SNPs for chromosomes 3, 4 and 5 respectively compared to 18.61% as expected by chance, suggesting that deamination is a major source of the observed SNPs. Closer investigation of the different SNPs revealed some that are preferentially found on chromosome 5, such as 53 T-C, while SNPs 30 G-A and 99 G-A are present in over 50% and 99% of the 5S rRNA genes on chromosome 3, respectively (Figure 2B). Similar profiles were detected when polymorphisms were determined in parts of 5S rRNA gene loci comprised in the analyzed YACs (Supplementary Figure S2A) with over 99% of the 5S rRNA genes from chromosome 3 carrying the 99 G-A polymorphism. To determine whether the identified SNPs in the transcribed portion of the 5S rRNA gene are characteristic for the chromosome 3 locus in other ecotypes as well, we made use of the Multiparent Advanced Generation Inter-Cross (MAGIC) population; a set of recombinant inbred lines derived from intercrossing 19 geographically and genetically diverse ecotypes (39). In the MAGIC population, 5S rDNA loci and marker SNPs in the flanking regions often remain in linkage making it possible to treat polymorphisms in the 5S rRNA genes as quantitative traits. We identified a single quantitative trait locus (QTL) at the pericentromeric region of chromosome 3 for polymorphisms 30 G-A, 41 G-A, 96 C-A and 99 G-A, which derives exclusively from founder ecotypes Bur-0, Can-0, Col-0 and Edi-0 (Figure 2C, Supplemental Figure S2B). Consistent with our linkage mapping results, these are the only four founder lines of this population for which we detected, in the NGS sequencing, chromosome 3 specific T-stretch signatures. This demonstrates that these specific signatures are characteristic for the 5S rRNA genes of chromosome 3 in other ecotypes as well.
Figure 2.
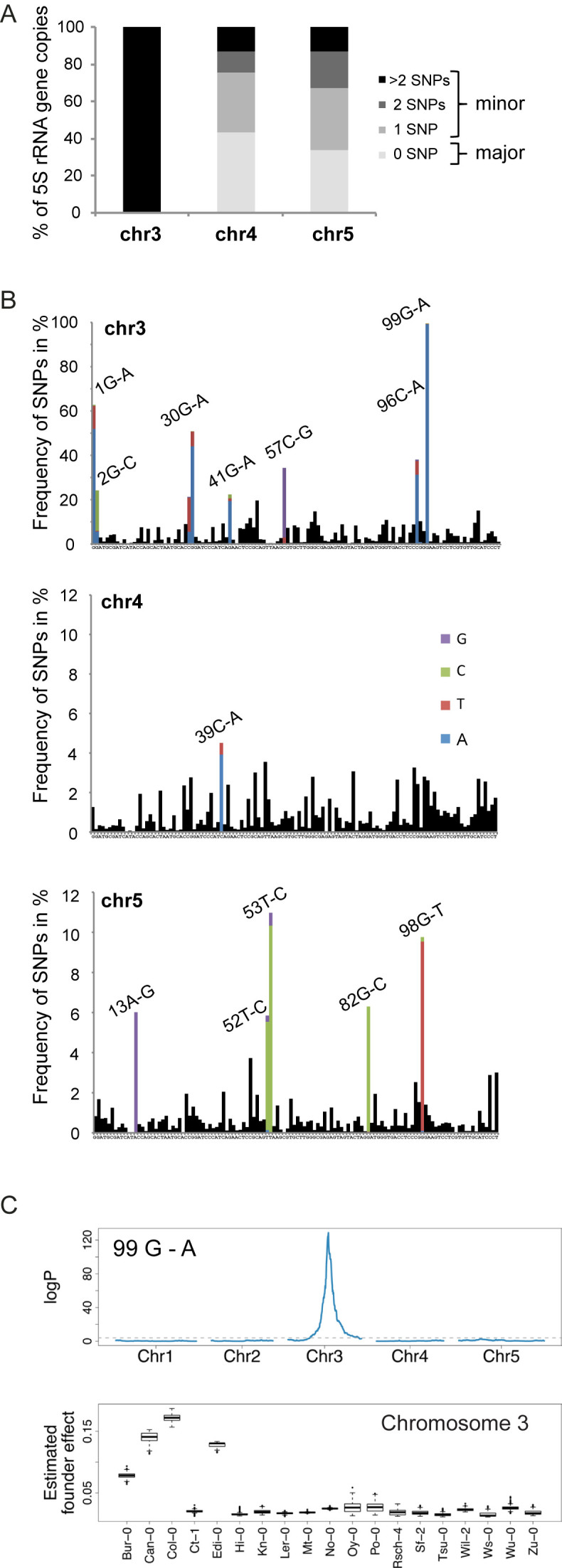
Chromosome specific single nucleotide polymorphisms. (A) Percentage of 5S rRNA gene copies assigned to chromosomes 3, 4 and 5 with 0 (major 5S rRNA genes), 1, 2 and more than 2 polymorphisms (minor 5S rRNA genes) in the transcribed sequence relative to the 5S rRNA consensus sequence. (B) Frequency of single nucleotide polymorphisms (SNPs) along the 120 bp transcribed sequence determined from the Mi-Seq NGS dataset. For the most frequent SNPs, the exchanged nucleotide at the given position is indicated in color. The consensus sequence is indicated below each graph. (C) (Top) Linkage mapping of the abundance of the G to A single nucleotide polymorphism at position 99 estimated by NGS in 393 individuals of the MAGIC population. (Bottom) Boxplot of the estimated founder ecotype effect by multiple imputation using R/happy at the major quantitative trait locus from the top panel in chromosome 3.
We then extended our analysis to the complete 5S rRNA gene and established a consensus 5S rRNA gene reference sequence per chromosome (Supplemental Figure S2C). We noted, and confirmed in the mapping population (Supplemental Figure S2D), that 57% of 5S rRNA genes from chromosome 4, but only 0.06% from chromosome 5, carry the mutation –26 T-G in the TATA-box, a mutation that has been shown to reduce transcription efficiency by 50% in vitro (12). This mutation is not only present in genes with polymorphisms, but also in 52.7% of the major 5S rRNA genes without polymorphism in the transcribed region situated on chromosome 4, suggesting that in Col-0 this locus may be less transcriptionally active.
5S rRNA gene loci are differentially enriched in certain histone marks and histone variants
To investigate whether the distinct sequence features at the three 5S rDNA loci are reflected at the level of their chromatin organization, we exploited available ChIP-seq datasets (Supplemental Table S3) to quantify nucleosome occupancy, enrichment in histone modifications and the presence of certain histone variants at the 5S rRNA genes (Figure 3A and B, Supplemental Figure S3A and B). First, enrichment at all 5S rRNA genes was compared to two active genes with different expression levels (Supplemental Figure S3C) as well as the silent Ta3 retrotransposon, situated in the pericentromeric region of chromosome 1. In contrast to active genes and similarly to Ta3, 5S rRNA genes globally show elevated nucleosome occupancy and are enriched in H3K9me2 and the histone variant H2A.W.6, typical markers of heterochromatin (32,33,40). Furthermore, they are depleted of H3.3 and H3K36me3, marks indicative of transcription (Figure 3A). We then determined the differential enrichment at the three 5S rRNA gene loci based on their specific T-stretch signatures. The locus on chromosome 3 that carries predominantly polymorphic copies is densely packed in nucleosomes, which are depleted of H3.3 and H3K36me3 and enriched in H2A.W.6 and H3K9me2 (Figure 3B). A closer inspection of the distribution of the different histone marks associated with transcription revealed a moderate enrichment of the 5S rRNA genes from chromosomes 5 and—to a lesser extent—4 in H3.3, H3K36me3 and H3K4me3 relative to the gene copies from chromosome 3 (Figure 3, Supplemental Figure S3B). This observation, coupled with the prevalence of the –26 T-G mutation in the promoter region of 5S rRNA genes of chromosome 4 prompted us to investigate whether the locus on chromosome 5 could be preferentially expressed. We therefore sequenced the pool of 5S-120 transcripts in 2-day-old plantlets, known to be enriched in polymorphic transcripts (14). Indeed, 12.2% of transcripts at this developmental stage contained SNPs (Figure 3C). When we ascribed the observed polymorphisms to the three main loci, we noted that the most frequent SNPs in the 5S-120 RNA pool are not necessarily the most abundant in the genome, and that they could not be unambiguously assigned to a specific chromosome (Supplemental Table S4). By taking advantage of the diagnostic T-stretch region, we therefore sequenced with an alternative approach the 5S-210 transcripts that extend into the intergenic region and comprise the T-stretch. The large majority of 5S-210 transcripts contained T-stretches with chromosome 5 specific signatures (Figure 3D). Since 5S-210 transcripts are suggested to represent read-through transcripts by RNA polymerase III and to be a marker for a more permissive chromatin organization (18,20), this indicates that 5S rDNA copies from the chromosome 5 locus are the main contributors to the 5S rRNA pool.
Figure 3.

Epigenetic marks and expression of 5S rRNA genes. (A and B) Nucleosome occupancy estimated by H3-ChIP and enrichment in histone post-translational modifications (H3K9me2 and H3K36me3) as well as histone variants H2A.W.6 and H3.3 at (A) the transcriptionally active genes HEXOKINASE1 (HXK1, At4g29130) and ACTIN2 (ACT2, At3g18780), the retrotransposon Ta3 and the 120 bp transcribed sequence of a 5S rRNA gene or (B) at the 3 different 5S rDNA loci. The y-axis corresponds to the number of reads in the ChIP-seq datasets normalized to the number of reads in input datasets. (C) Mean percentage of 5S rRNA transcripts with 0 (major 5S rRNA genes) or 1, 2 and more than 2 single nucleotide polymorphisms (minor 5S rRNA genes) determined in RNA-seq datasets from 3 biological replicates of 2-day-old Col-0 seedlings. (D) Left: Schematic representation of the 5S-120 and the 5S-210 transcripts. Right: Mean percentage of 5S-210 reads with the chromosome 4 or chromosome 5 specific T-stretch signatures determined in RNA-seq datasets prepared from 2-day old seedlings in three biological replicates. For all panels the error bar corresponds to SEM.
Taken as a whole, occurrence of single nucleotide polymorphisms, including the –26 T-G mutation in the TATA-box on chromosome 4, chromatin modifications and RNA-Seq datasets, suggest that the locus in chromosome 5 has a more open chromatin configuration and is the major source of 5S rRNA in Col-0.
5S rRNA gene copy number varies in different ecotypes
The availability of large sets of Arabidopsis ecotypes that are completely sequenced using Illumina short read sequencing (28) allowed us to investigate whether 5S rRNA gene copy number or T-stretch signatures vary between different populations. Due to the heterogeneity of Illumina platforms, read-lengths and insert sizes used to sequence the 1001 genomes, we conducted our analyses in a subset of ecotypes sequenced for a single study (29). We identified ecotypes with as few as 800 and to over 4800 5S rRNA gene copies, illustrating that 5S rRNA gene copy number varies considerably between Arabidopsis ecotypes (Figure 4A) without significantly affecting 5S rRNA transcript levels (Supplemental Figure S4A). To investigate whether the copy number changes between different ecotypes are caused by loss or gain of certain rRNA gene sequences, we extracted the T-stretch signatures and assigned them to the different loci for selected ecotypes with few (∼800), average (∼2000) and high (∼4500) copy numbers. T-stretch signatures identified in Col-0 are conserved in different ecotypes, but only few of them comprise a 5S rRNA gene locus on chromosome 3, as reflected in the MAGIC population (Supplemental Figure S2B). 5S rRNA genes with signatures from both chromosome 4 and chromosome 5 have undergone copy number variations, those with signatures from chromosome 5 being the most variable (Figure 4B). To examine the variation in a shorter time-scale in a single ecotype, we took advantage of the Col-0 Mutation Accumulation (MA) lines that have been maintained by single-seed descent for 30 generations in the absence of selection (34). We found a rather constant 5S rRNA gene copy number in this population (coefficient of variation = 0.043, Supplemental Figure S4B), as well as constant relative size of the three different 5S rDNA loci (Supplemental Figure S4C). This is in sharp contrast to the variation in 45S rRNA gene copy number reported in the same MA population (41) (coefficient of variation = 0.24). In summary, 5S rRNA genes carrying characteristic signatures from chromosomes 4 and 5 are conserved in most Arabidopsis populations, and show high level of variation in global copy number.
Figure 4.

5S rRNA gene copy number variation in different Arabidopsis thaliana ecotypes. (A) Total 5S rRNA gene copy number distribution in different Arabidopsis ecotypes as determined by in silico analysis of NGS datasets with 50 bp-long reads (see Materials and Methods). (B) Number of 5S rRNA gene copies with the respective T-stretches of chromosome 3, 4 and 5 in 9 different ecotypes: 3 with few (∼800 copies, red, Ga-0, Westkar-4, Sei-0), 3 with medium (∼2,000 copies, green, Benk-1, Dra-0, Db-1) and 3 with high (∼4500 copies, blue, Ang-0, Pi-0, Van-0) total 5S rDNA copy numbers.
The additional 5S rRNA gene locus on chromosome 3 in Landsberg erecta (Ler) plants carries the chromosome 5-specific T-stretch signature
To further investigate the variations in 5S rRNA gene loci organization, we analyzed in more detail the Landsberg erecta (Ler) ecotype. Ler has been selected after irradiation from the original Landsberg line (42,43), and it is the only other ecotype for which a fully independently assembled genome is available (43). Interestingly, Ler plants comprise a 5S rRNA gene locus on the long arm of chromosome 3, but the origin of this locus remains unknown. An analysis of publicly available Ler Illumina NGS datasets (44) showed neither T-stretch signatures nor polymorphisms characteristic for Col-0 chromosome 3 (Supplemental Figure S2A). This result confirms previous FISH studies that failed to reveal a 5S rDNA locus in the pericentromeric regions of chromosome 3 in Landsberg (La-0) and Ler (2,43), and suggests that the 5S rRNA gene locus on chromosome 3 in Ler does not have the same origin as in Col-0. We further noticed some variations in the number of thymines in the T-stretches of chromosome 4 and 5, as well as overrepresentation of specific SNPs in the T-stretch such as 123 T-C and 142 T-G in about 30% of the 5S rRNA genes (Supplemental Figure S5A). As Ler is one of the founders of the MAGIC population, we used this population to analyze abundant Ler polymorphisms. 123 T-C ( Figure 5A) in the T-stretch as well as 56 G-A in the transcribed sequence (Supplemental Figure S5B) map to a major QTL in the pericentromeric region of chromosome 5. Remarkably, both polymorphisms also display a minor QTL that maps to the longer arm of chromosome 3, suggesting that the new locus originated from chromosome 5. Furthermore, mapping located this locus to position 6.24 Mb on chromosome 3 according to the Col-0 TAIR10 reference. Further in silico analysis revealed more 5S rDNA copies in Ler compared to La-0 and a higher number of 5S rRNA genes with the chromosome 5 specific T-stretch, which could be explained by the additional locus on chromosome 3 (Figure 5B). Finally, the detection of 5S rDNA loci with locus-specific LNA-DNA mixmer probes clearly identified a second locus with the chromosome 5-specific signature in Ler (Figure 5C). Taken together, analysis of the MAGIC population combined with locus specific FISH reveals that the 5S rRNA genes situated on the longer arm of chromosome 3 in Ler derive from the chromosome 5 locus.
Figure 5.

Variations of 5S rDNA copy number and genomic position. (A) (Top) Linkage mapping of the abundance of the T to C single nucleotide polymorphism at position 123 estimated by NGS in 393 individuals of the MAGIC population. (Bottom) Boxplots of the estimated founder ecotype effect by multiple imputation using R/happy at the major quantitative trait locus from the top panel in chromosome 5 and chromosome 3. (B) Number of total 5S rRNA genes and genes with chromosome 4 or 5 specific T-stretch signatures as determined by in silico analysis of La-0 and Ler Illumina sequencing datasets. (C) FISH using LNA-DNA mixmer probes specific for chromosome 4 (red) and chromosome 5 (green) on pachytene spreads of Col-0 and Ler. DNA is counterstained by DAPI (in grey). Arrow indicates weak cross hybridization of the chromosome 4 probe to the 5S rRNA gene copies from chromosome 3. Arrowhead indicates the 5S locus on the long arm of bivalent 3 in Ler. The scale bar presents 5 μM. (D) FISH with 5S (red) and 45S (green) rDNA probes on metaphase I bivalents of Col-0, Ler and the ago4 mutant alleles, ago4-2 (Col-0 background) and ago4-1 (Ler background). The scale bar presents 5 μM. (E) FISH on ago4-2 and ago4-1 pachytene spreads with LNA-DNA mixmer probes as in (C). Arrow indicates weak cross hybridization of the chromosome 4 probe with the 5S rRNA gene copies of chromosome 3 in ago4-2. Arrowhead designates the additional locus (green) on the long arm of bivalent 3 in ago4-2. The locus on the long arm of bivalent 3 in Ler is lost in ago4-1. The scale bar presents 5 μM. (F) Relative 5S rDNA copy number for each locus in Col-0, ago4-2, Ler and ago4-1 determined by qPCR in three biological replicates. Copy numbers in Col-0 are set to 1 for each chromosome. *P < 0.05, ANOVA.
5S rDNA loci organization is altered in certain mutants of the RdDM pathway
The RdDM pathway is involved in 5S rRNA gene methylation (19,20,45,46). DICER-LIKE (DCL) and ARGONAUTE 4 (AGO4) are key components of this pathway, and loss of AGO4 induces hypomethylation of 5S rDNA as well as expression of minor 5S rRNA genes (19). We therefore investigated whether loss of these key regulators of epigenetic control at 5S rRNA genes could impact 5S rDNA stability. Interestingly, metaphase I spreads of ago4-2, dcl2-2 and dcl3-1 mutants in the Col-0 background revealed small additional 5S rDNA loci, while in the ago4-1 mutant allele in the Ler background the 5S rDNA locus on the shorter arm of chromosome 3 is absent (Figure 5D, Supplemental Figure S5C). To identify 5S rRNA genes involved in these rearrangements, we performed FISH with LNA-DNA mixmer probes on pachytene spreads. We confirmed the loss of the 5S rRNA genes from the longer arm of chromosome 3 in ago4-1 and found that genes with the chromosome 5-specific T-stretch had translocated to chromosome 3 in ago4-2 mutants (Figure 5E). The results from qPCR using primer pairs positioned on the T-stretch signatures that amplify specifically the 5S rRNA gene copies from chromosome 3, 4 or 5 (Supplemental Figure S5D-G) confirmed the loss of chromosome 5-specific 5S rRNA gene copies in ago4-1 (Figure 5F). Taken together, 5S rRNA genes with the chromosome 5-specific T-stretches are dynamically reorganized in argonaute 4 mutants suggesting that frequent translocations may preferentially initiate from the chromosome 5 locus.
Differential enrichment in epigenetic marks at 5S rDNA loci and impact of altered localization of 5S rRNA gene loci on nuclear organization in Ler
Given that 5S rDNA loci are differentially positioned in Col-0 and Ler plants, we investigated whether chromosomal position impacts 5S rDNA chromatin marks. We carried out ChIP-qPCR in Col-0 and Ler plants and determined the enrichment in H3K4me3, a post-translational modification associated with transcription, and the repressive mark H3K9me2 at all 5S rRNA gene copies and at those with chromosome 4 or 5-specific signatures (Figure 6A, Supplemental Figure S6A). Surprisingly, although parts of the 5S rRNA genes that carry the chromosome 5 specific signature are located on the euchromatic arm, no loss and even slight enrichment in H3K9me2 were observed. In contrast, the copies situated on chromosome 4 show a transcriptionally more favorable environment in Ler plants with increased enrichment in H3K4me3 and reduced H3K9me2 levels. These differences in enrichment in epigenetic marks at the 5S rRNA genes situated on chromosome 4 may imply differential usage of the 5S rRNA gene pool in Ler plants compared to Col-0. We therefore sequenced 5S-120 and 5S-210 transcripts of 2-day-old plantlets from Ler under identical growth conditions as in Col-0. More 5S-120 transcripts with polymorphisms are present in the Ler RNA-pool (Figure 6B). While most of the SNPs are common to both ecotypes (Supplemental Figure S6B), polymorphisms are differentially enriched in the RNA pools suggesting that globally distinct sets of genes are transcribed in the two ecotypes (Supplemental Table S5). Furthermore, in the pool of 5S-210 transcripts, the percentage of transcripts with uninterrupted T-stretches characteristic for chromosome 4 is higher in Ler (19.1% versus 9.3%, P < 0.05, Figure 6C), which argues for a more open chromatin state at the 5S rDNA locus from chromosome 4 in Ler. Moreover, analysis of the Ler NGS datasets revealed no evidence for –26 T-G mutations in the TATA-box (Supplemental Figure S2D), while in Col-0 they were frequent in 5S rRNA genes of chromosome 4.
Figure 6.

Differences in nuclear organization and epigenetic marks at 5S rDNA loci in Col-0 and Ler. (A) Differential enrichment in H3K4me3 and H3K9me2 normalized to H3 levels in Col-0 and Ler plants at all 5S rRNA genes (left) and specifically at copies carrying the chromosome 4 (middle) or chromosome 5-specific signature (right). Error bars correspond to SEM from three biological replicates. *P < 0.05, Wilcoxon test. (B) Percentage of 5S-120 reads with polymorphisms in RNA-seq datasets of three biological replicates of Col-0 and Ler 2-day old plantlets. *P < 0.05, t-test. (C) Percentage of 5S-210 reads with chromosome 4 or chromosome 5 specific T-stretches determined from an RNA-seq dataset comprising three biological replicates of 2-day old Ler plantlets. (D) Representative nuclei of 10-day-old cotyledons stained by FISH with 5S rDNA probes (green) showing 5S rDNA loci associated or not with a chromocenter revealed by a probe against the centromeric 180 bp repeats (red). In Col-0, all 5S signals are associated with chromocenters. Representative nuclei for Ler showing six or four 5S rDNA signals associated with chromocenters or six 5S rDNA signals out of which two are located distant from chromocenters (indicated by arrowheads). (E) Percentage of cotyledon interphase nuclei presenting 3, 4, 5, 6 or 7 5S rDNA signals per nucleus as revealed by a FISH probe detecting all 5S rRNA genes (300 nuclei per genotype, n = 3 experiments; Error bars correspond to SEM). ****P < 0.0001, ANOVA.
Finally, we investigated whether the altered position of 5S rRNA gene loci affects nuclear organization in interphase. FISH with a 5S rDNA probe detecting all 5S rRNA gene loci and the centromeric 180 bp probe to mark chromocenters revealed that ∼80% of Col-0 nuclei show six independent 5S rDNA FISH signals partly co-localizing with chromocenters (Figure 6D and E). In contrast, in Ler, ∼30% and ∼20% of interphase nuclei show only four or five signals, respectively, revealing frequent co-localization of the 5S rDNA loci on the arm of chromosome 3 with themselves and/or with the loci present in the pericentromeric regions of chromosomes 4 and 5. Indeed, despite the fact that this locus is physically located far from the pericentromeric region, in 79 ± 5.5% of Ler nuclei all 5S rDNA signals co-localize with chromocenters. To establish whether the 5S loci on the chromosome arm cluster preferentially with a specific chromocenter, we scored the co-localization events between chromosomes 4 and 5 specific hybridization signals at the same chromocenters using locus-specific LNA-DNA mixmer probes. Only in about 10% of Ler nuclei loci from chromosome 4 and from chromosome 5 can be found at the same chromocenter (Supplementary Figure S6C and D).
In summary, the 5S rRNA genes with chromosome 5 specific T-stretch signatures in Ler show –despite localization in both the pericentromeric region of chromosome 5 and the chromosome arm of chromosome 3– a similar chromatin state as in Col-0. Furthermore, the novel locus on chromosome 3 preferentially associates with the chromocenter of chromosome 5 in interphase nuclei. Finally, both 5S-210 transcript sequencing data and ChIP-qPCR indicate that 5S rRNA genes from chromosome 4 may have a greater contribution to the 5S rRNA pool in Ler, suggesting the existence of various gene dosage mechanisms operating between the two major 5S rDNA loci.
DISCUSSION
5S ribosomal RNA is of critical importance for protein synthesis and contributes substantially to the total cellular RNA pool. Present in excess in the genome, only a subset of 5S rRNA genes is actively transcribed at a given time, prompting interesting questions on the mode of gene choice and regulation. Here, using DNA, RNA and ChIP-sequencing datasets we reveal variation in 5S rRNA gene sequences as well as differential chromatin profiles and 5S rRNA transcripts between loci and ecotypes, suggesting preferential expression of specific sets of genes among ecotypes.
Based on our exhaustive characterization of T-stretch signatures in different whole genome datasets and in subsets of 5S rDNA loci cloned in YACs combined with the detection of specific T-stretches by FISH, we conclude that 5S rRNA genes with the same T-stretch characteristics cluster within the same locus. While we cannot exclude, without a complete assembly of the 5S rDNA loci, that few highly polymorphic 5S rRNA gene copies have been missed in the analysis or were falsely assigned, such observation is in agreement with homogenization of rRNA genes within a given locus (47). The characteristic nucleotides for the different T-stretch signatures are also conserved between ecotypes, but differences in the number of thymines in the T-stretch, likely due to slippage during replication, are frequent. While all analyzed ecotypes carry 5S rDNA loci with signatures characteristic of those on chromosomes 4 and 5, only a subset (8/34 ecotypes analyzed in this study) comprises those from the pericentromeric locus on chromosome 3. Therefore, this locus may have been gained or lost during evolution without affecting the cellular 5S rRNA production. In addition to differences concerning the chromosome 3 locus, analysis of a subset of individuals from the 1001 genome population also revealed substantial 5S rRNA gene copy number variation among ecotypes. However, over shorter time scales, 5S rRNA gene copy numbers are less dynamic than 45S rRNA genes (41) as the Col-0 MA lines propagated for 30 generations show little variation in absolute 5S rDNA copy numbers. Furthermore, in agreement with previous studies of chromatin mutants that loose 45S but not 5S rRNA genes (48,49), we find no clear evidence of concerted copy number variation of 5S and 45S rDNA, questioning the existence of a common mode of copy number regulation in plants, in contrast to mammals (50).
In addition to copy numbers, genomic locations of 5S rRNA gene loci are variable as well. Using independent approaches including linkage mapping in the MAGIC population, in silico analysis of available Illumina sequencing datasets, qPCR analysis and FISH studies we characterized a novel locus on the long arm of chromosome 3 in Ler as originating from chromosome 5. Mapping narrowed down the position of this locus to ∼6.24 Mb, close to a large-scale inversion of 170 kb recently found in the Ler genome (43). Irradiation of the original Landsberg line that resulted in L. erecta (51) might therefore have induced an inversion on chromosome 3 as well as translocation of part of the 5S rDNA locus of chromosome 5. Furthermore, differences in 5S rDNA locus positions between WT and ago4 or dcl mutants illustrate the mobility of 5S rRNA gene copies. Together, these observations support the notion that 5S rRNA genes show frequent reorganization and may therefore be only under low selective pressure during evolution (52). The contribution of chromosome 5 specific copies to most of these rearrangements, indicates a role for the particular chromatin state at these 5S rRNA genes (see below) or a preferential implication of this locus in interchromosomal interacting domains that may explain preferential translocations (53).
Rearrangements of 5S rDNA loci may have a substantial impact on genome function, either by disrupting genomic sequences, locally changing chromatin status or altering 3D organization of chromatin within the nucleus. In agreement with the recently suggested autonomous property of chromatin regions to associate with domains of similar chromatin state in mammals (54) and the observed preferential clustering of repetitive elements in interphase in Arabidopsis (55), the 5S rDNA locus on the long arm of chromosome 3 in Ler frequently co-localizes with chromocenters that comprise 5S rDNA loci. We can speculate that these inter-chromosomal interactions influence genome expression (56–58). Indeed, in humans, 5S rDNA contacts a wide range of different chromosomal regions including genes in nuclear space (59) and in mouse, a 5S rDNA transgene mediates nucleolar association of its linked genomic region, demonstrating that 5S rDNA sequences can contribute to nuclear positioning (60).
Despite copy number variations, rRNA genes are assumed to be maintained in excess over the amount required for organism survival (61) and consequently a subset of genes undergoes silencing through epigenetic mechanisms. Based on the mostly repressive chromatin marks observed at 5S rRNA genes, we suggest that only a small number of 5S rRNA genes –or a specific cluster– is expressed at a given time, similar to the 45S rRNA genes, for which a range of 10 to 27% are estimated to be transcriptionally active (62,63). Indeed, we find similar relative 5S rRNA levels whether an ecotype comprises ∼800 or ∼4500 5S rRNA gene copies. The ‘extra’ copies could represent a functionally diverse 5S rRNA gene reservoir that is expressed only under certain conditions that require the production of high amounts of rRNA (13–15) or play a role in genome stability as suggested for 45S rDNA (64,65). Notwithstanding the global enrichment in repressive marks, differences can be detected among the three 5S rDNA loci and transcriptionally permissive marks are slightly more enriched at the chromosome 5-specific gene copies. In this regard, it is interesting to note that most of the 5S-210 transcripts originated from chromosome 5, and that this locus does not contain a TATA-box mutation in contrast to the genes from chromosome 4. Together, this implies that this locus predominantly contributes to the 5S rRNA pool in Col-0. In yeast, transcription of rRNA genes favors recombination resulting in rRNA copy number variation (66,67). This is particular interesting in light of our observation that copy numbers of 5S rRNA genes with the chromosome 5-specific signature are highly variable among ecotypes and that they engage in translocations in ago4 mutants, in which chromatin organization is affected (16).
In comparison to Col-0, the 5S rRNA genes of chromosome 4 in Ler show a more permissive chromatin environment and relatively more 5S-210 transcripts originate from this locus. This hints to a combination of sequence variation and epigenetic regulation in the transcriptional control of 5S rDNA loci. While the polymorphisms in the RNA-seq dataset did not allow us to unequivocally assign the different reads to a specific 5S rDNA locus, the prevalence of the different SNPs nevertheless indicates the expression of distinct sets of genes in different ecotypes, similar to the variation in 45S rDNA cluster-usage (31). Expression of these different polymorphic transcripts that have been shown to be incorporated into the ribosome (11) may contribute to the functional diversity of ribosomes (68).
DATA AVAILABILITY
The bioinformatic pipelines to extract specific T-stretch signatures is available on (https://gist.github.com/laurianesimon/0ae2dd7b8c34c23cdacec217aeaab79c) and (https://gist.github.com/laurianesimon/a9fc44aa83305c576e914710cae75f87).
The Col-0 DNA Mi-Seq data, the 5S-210 RNA-Seq data, the 5S-120 RNA-Seq data as well as the sequencing data from the six YACs have been deposited under PRJNA369183, PRJNA369190, PRJNA378941 and PRJNA378941 respectively.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to J-P Pichon (Biogemma) for valuable advice in library preparation and for performing the high-throughput sequencing of Col-0 DNA. We thank C. Poncet (GENTYANE sequencing facility) for advice and help with DNA shearing using the Covaris technology and P. Vera for having kindly donated ago4-2 seeds. We thank P. Arnaud, M. Benhamed, Y. Bidet, F. Choulet, F. Pontvianne, J. Saez-Vasquez, S. Tutois and in particular S. Tourmente for insightful discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Region Auvergne; Agence National de Recherche [‘Dynam’Het’ ANR-11 JSV2 009 01, ‘SINUDYN’ ANR-12 ISV6 0001 to AVP]; European Union's Horizon 2020 research and innovation program [Marie Sklodowska-Curie grant agreement no. 752846 to FAR]; Centre National de la Recherche Scientifique, the Institut National de la Santé et de la Recherche Médicale and the University Clermont Auvergne. Funding for open access charge: Agence National de Recherche [ANR-12 ISV6 0001].
Conflict of interest statement. None declared.
REFERENCES
- 1. Murata M., Heslop-Harrison J.S., Motoyoshi F.. Physical mapping of the 5S ribosomal RNA genes in Arabidopsis thaliana by multi-color fluorescence in situ hybridization with cosmid clones. Plant J. 1997; 12:31–37. [DOI] [PubMed] [Google Scholar]
- 2. Fransz P., Armstrong S., Alonso-Blanco C., Fischer T.C., Torres-Ruiz R.A., Jones G.. Cytogenetics for the model system Arabidopsis thaliana. Plant J. 1998; 13:867–876. [DOI] [PubMed] [Google Scholar]
- 3. Tutois S., Cloix C., Cuvillier C., Espagnol M.C., Lafleuriel J., Picard G., Tourmente S.. Structural analysis and physical mapping of a pericentromeric region of chromosome 5 of Arabidopsis thaliana. Chromosome Res. 1999; 7:143–156. [DOI] [PubMed] [Google Scholar]
- 4. Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000; 408:796–815. [DOI] [PubMed] [Google Scholar]
- 5. Brown D.D., Wensink P.C., Jordan E.. A comparison of the ribosomal DNA’s of Xenopus laevis and Xenopus mulleri: the evolution of tandem genes. J. Mol. Biol. 1972; 63:57–73. [DOI] [PubMed] [Google Scholar]
- 6. Dover G. Molecular drive: a cohesive mode of species evolution. Nature. 1982; 299:111–117. [DOI] [PubMed] [Google Scholar]
- 7. Nei M., Rooney A.P.. Concerted and birth-and-death evolution of multigene families. Annu. Rev. Genet. 2005; 39:121–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tutois S., Cloix C., Mathieu O., Cuvillier C., Tourmente S.. Analysis of 5S rDNA Loci among Arabidopsis Ecotypes and subspecies. Genome Lett. 2002; 1:115–122. [Google Scholar]
- 9. Cloix C., Tutois S., Mathieu O., Cuvillier C., Espagnol M.C., Picard G., Tourmente S.. Analysis of 5S rDNA arrays in Arabidopsis thaliana: physical mapping and chromosome-specific polymorphisms. Genome Res. 2000; 10:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Campell B.R., Song Y., Posch T.E., Cullis C.A., Town C.D.. Sequence and organization of 5 S ribosomal RNA-encoding genes of Arabidopsis thaliana. Gene. 1992; 112:225–228. [DOI] [PubMed] [Google Scholar]
- 11. Cloix C., Tutois S., Yukawa Y., Mathieu O., Cuvillier C., Espagnol M.C., Picard G., Tourmente S.. Analysis of the 5S RNA pool in Arabidopsis thaliana: RNAs are heterogeneous and only two of the genomic 5S loci produce mature 5S RNA. Genome Res. 2002; 12:132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cloix C., Yukawa Y., Tutois S., Sugiura M., Tourmente S.. In vitro analysis of the sequences required for transcription of the Arabidopsis thaliana 5S rRNA genes. Plant J. 2003; 35:251–261. [DOI] [PubMed] [Google Scholar]
- 13. Layat E., Sáez-Vásquez J., Tourmente S.. Regulation of Pol I-transcribed 45S rDNA and Pol III-transcribed 5S rDNA in Arabidopsis. Plant Cell Physiol. 2012; 53:267–276. [DOI] [PubMed] [Google Scholar]
- 14. Mathieu O., Jasencakova Z., Vaillant I., Gendrel A.V., Colot V., Schubert I., Tourmente S.. Changes in 5S rDNA chromatin organization and transcription during heterochromatin establishment in Arabidopsis. Plant Cell. 2003; 15:2929–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Layat E., Cotterell S., Vaillant I., Yukawa Y., Tutois S., Tourmente S.. Transcript levels, alternative splicing and proteolytic cleavage of TFIIIA control 5S rRNA accumulation during Arabidopsis thaliana development. Plant J. 2012; 71:35–44. [DOI] [PubMed] [Google Scholar]
- 16. Vaillant I., Tutois S., Jasencakova Z., Douet J., Schubert I., Tourmente S.. Hypomethylation and hypermethylation of the tandem repetitive 5S rRNA genes in Arabidopsis. Plant J. 2008; 54:299–309. [DOI] [PubMed] [Google Scholar]
- 17. Douet J., Tutois S., Tourmente S.. A Pol V-mediated silencing, independent of RNA-directed DNA methylation, applies to 5S rDNA. PLoS Genet. 2009; 5:e1000690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vaillant I., Schubert I., Tourmente S., Mathieu O.. MOM1 mediates DNA-methylation-independent silencing of repetitive sequences in Arabidopsis. EMBO Rep. 2006; 7:1273–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vaillant I., Tutois S., Cuvillier C., Schubert I., Tourmente S.. Regulation of Arabidopsis thaliana 5S rRNA Genes. Plant Cell Physiol. 2007; 48:745–752. [DOI] [PubMed] [Google Scholar]
- 20. Blevins T., Pontes O., Pikaard C.S., Meins F.. Heterochromatic siRNAs and DDM1 independently silence aberrant 5S rDNA transcripts in Arabidopsis. PLoS One. 2009; 4:e5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Agorio A., Vera P.. ARGONAUTE4 is required for resistance to Pseudomonas syringae in Arabidopsis. Plant Cell. 2007; 19:3778–3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zilberman D., Cao X., Jacobsen S.E.. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science. 2003; 299:716–719. [DOI] [PubMed] [Google Scholar]
- 23. Bowler C., Benvenuto G., Laflamme P., Molino D., Probst A.V., Tariq M., Paszkowski J.. Chromatin techniques for plant cells. Plant J. 2004; 39:776–789. [DOI] [PubMed] [Google Scholar]
- 24. Probst A.V., Fransz P.F., Paszkowski J., Mittelsten Scheid O.. Two means of transcriptional reactivation within heterochromatin. Plant J. 2003; 33:743–749. [DOI] [PubMed] [Google Scholar]
- 25. Czechowski T., Stitt M., Altmann T., Udvardi M.K., Scheible W.R.. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005; 139:5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J. et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013; 11:11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. The 1001 Genomes Consortium, X. 1, 135 genomes reveal the global pattern of polymorphism in arabidopsis thaliana. Cell. 2016; 166:481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schmitz R.J., Schultz M.D., Urich M.A., Nery J.R., Pelizzola M., Libiger O., Alix A., McCosh R.B., Chen H., Schork N.J. et al. Patterns of population epigenomic diversity. Nature. 2013; 495:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rabanal F.A., Mandáková T., Soto-Jiménez L.M., Greenhalgh R., Parrott D.L., Lutzmayer S., Steffen J.G., Nizhynska V., Mott R., Lysak M.A. et al. Epistatic and allelic interactions control expression of ribosomal RNA gene clusters in Arabidopsis thaliana. Genome Biol. 2017; 18:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stroud H., Otero S., Desvoyes B., Ramírez-Parra E., Jacobsen S.E., Gutierrez C.. Genome-wide analysis of histone H3.1 and H3.3 variants in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:5370–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yelagandula R., Stroud H., Holec S., Zhou K., Feng S., Zhong X., Muthurajan U.M., Nie X., Kawashima T., Groth M. et al. The histone variant H2A.W defines heterochromatin and promotes chromatin condensation in arabidopsis. Cell. 2014; 158:98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shaw R.G., Byers D.L., Darmo E.. Spontaneous mutational effects on reproductive traits of Arabidopsis thaliana. Genetics. 2000; 155:369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hagmann J., Becker C., Müller J., Stegle O., Meyer R.C., Wang G., Schneeberger K., Fitz J., Altmann T., Bergelson J. et al. Century-scale methylome stability in a recently diverged arabidopsis thaliana Lineage. PLoS Genet. 2015; 11:e1004920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Creusot F., Fouilloux E., Dron M., Lafleuriel J., Picard G., Billault A., Le Paslier D., Cohen D., Chabouté M.E., Durr A. et al. The CIC library: a large insert YAC library for genome mapping in Arabidopsis thaliana. Plant J. 1995; 8:763–770. [DOI] [PubMed] [Google Scholar]
- 37. Simon L., Probst A.V.. Bemer M, Baroux C. High-Affinity LNA–DNA mixmer probes for detection of chromosome-Specific polymorphisms of 5S rDNA repeats in arabidopsis thaliana. Plant Chromatin Dynamics. 2018; 1675:NY: Methods in Molecular Biology Humana Press; 481–491. [DOI] [PubMed] [Google Scholar]
- 38. Mathieu O., Yukawa Y., Sugiura M., Picard G., Tourmente S.. 5S rRNA genes expression is not inhibited by DNA methylation in Arabidopsis. Plant J. 2002; 29:313–323. [DOI] [PubMed] [Google Scholar]
- 39. Kover P.X., Valdar W., Trakalo J., Scarcelli N., Ehrenreich I.M., Purugganan M.D., Durrant C., Mott R.. A multiparent advanced generation inter-cross to fine-map quantitative traits in Arabidopsis thaliana. PLoS Genet. 2009; 5:e1000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chodavarapu R.K., Feng S., Bernatavichute Y.V., Chen P.Y., Stroud H., Yu Y., Hetzel J.A., Kuo F., Kim J., Cokus S.J. et al. Relationship between nucleosome positioning and DNA methylation. Nature. 2010; 466:388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rabanal F.A., Nizhynska V., Mandáková T., Novikova P.Y., Lysak M.A., Mott R., Nordborg M.. Unstable Inheritance of 45S rRNA Genes in Arabidopsis thaliana. G3 (Bethesda). 2017; 7:1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Abraham M.C., Metheetrairut C., Irish V.F.. Natural variation identifies multiple loci controlling petal shape and size in arabidopsis thaliana. PLoS One. 2013; 8:e56743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zapata L., Ding J., Willing E., Hartwig B., Bezdan D., Jiao W., Patel V.. Chromosome-level assembly of Arabidopsis thaliana Ler reveals the extent of translocation and inversion polymorphisms. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E4052–E4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Abe M., Kaya H., Watanabe-Taneda A., Shibuta M., Yamaguchi A., Sakamoto T., Kurata T., Ausín I., Araki T., Alonso-Blanco C.. FE, a phloem-specific Myb-related protein, promotes flowering through transcriptional activation of flowering locus T and flowering locus t interacting protein 1. Plant J. 2015; 83:1059–1068. [DOI] [PubMed] [Google Scholar]
- 45. He X.J., Hsu Y.F., Pontes O., Zhu J., Lu J., Bressan R.A., Pikaard C., Wang C.S., Zhu J.K.. NRPD4, a protein related to the RPB4 subunit of RNA polymerase II, is a component of RNA polymerases IV and v and is required for RNA-directed DNA methylation. Genes Dev. 2009; 23:318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gao Z., Liu H.-L., Daxinger L., Pontes O., He X., Qian W., Lin H., Xie M., Lorkovic Z.J., Zhang S. et al. An RNA polymerase II- and AGO4-associated protein acts in RNA-directed DNA methylation. Nature. 2010; 465:106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sastri D.C., Hilu K., Appels R., Lagudah E.S., Playford J., Baum B.R.. An overview of evolution in plant 5S DNA. Plant Syst. Evol. 1992; 183:169–181. [Google Scholar]
- 48. Pontvianne F., Blevins T., Chandrasekhara C., Feng W., Stroud H., Jacobsen S.E., Michaels S.D., Pikaard C.S.. Histone methyltransferases regulating rRNA gene dose and dosage control in Arabidopsis. Genes Dev. 2012; 26:945–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mozgová I., Mokros P., Fajkus J.. Dysfunction of chromatin assembly factor 1 induces shortening of telomeres and loss of 45S rDNA in Arabidopsis thaliana. Plant Cell. 2010; 22:2768–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gibbons J.G., Branco A.T., Godinho S.A., Yu S., Lemos B.. Concerted copy number variation balances ribosomal DNA dosage in human and mouse genomes. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:2485–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rédei G.P. Supervital mutants of Arabidopsis. Genetics. 1962; 47:443–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Garcia S., Kovařík A.. Dancing together and separate again: gymnosperms exhibit frequent changes of fundamental 5S and 35S rRNA gene (rDNA) organisation. Heredity (Edinb). 2013; 111:23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Roix J.J., McQueen P.G., Munson P.J., Parada L.A., Misteli T.. Spatial proximity of translocation-prone gene loci in human lymphomas. Nat. Genet. 2003; 34:287–291. [DOI] [PubMed] [Google Scholar]
- 54. Van De Werken H.J.G., Haan J.C., Feodorova Y., Bijos D., Weuts A., Theunis K., Holwerda S.J.B., Meuleman W., Pagie L., Thanisch K. et al. Small chromosomal regions position themselves autonomously according to their chromatin class. Genome Res. 2017; 27:922–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pecinka A., Kato N., Meister A., Probst A.V., Schubert I., Lam E.. Tandem repetitive transgenes and fluorescent chromatin tags alter local interphase chromosome arrangement in Arabidopsis thaliana. J. Cell Sci. 2005; 118:3751–3758. [DOI] [PubMed] [Google Scholar]
- 56. Feng S., Cokus S.J., Schubert V., Zhai J., Pellegrini M., Jacobsen S.E.. Genome-wide Hi-C analyses in wild-type and mutants reveal high-resolution chromatin interactions in Arabidopsis. Mol. Cell. 2014; 55:694–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Grob S., Schmid M.W., Grossniklaus U.. Hi-C analysis in Arabidopsis identifies the KNOT, a structure with similarities to the flamenco locus of Drosophila. Mol. Cell. 2014; 55:678–693. [DOI] [PubMed] [Google Scholar]
- 58. Fransz P., de Jong J.H., Lysak M., Castiglione M.R., Schubert I.. Interphase chromosomes in Arabidopsis are organized as well defined chromocenters from which euchromatin loops emanate. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:14584–14589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yu S., Lemos B.. A portrait of ribosomal DNA contacts with Hi-C reveals 5S and 45S rDNA anchoring points in the folded human genome. Genome Biol. Evol. 2016; 8:evw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fedoriw A.M., Starmer J., Yee D., Magnuson T.. Nucleolar association and transcriptional inhibition through 5S rDNA in mammals. PLoS Genet. 2012; 8:e1002468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Eickbush T.H., Eickbush D.G.. Finely orchestrated movements: evolution of the ribosomal RNA genes. Genetics. 2007; 175:477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pontes O., Lawrence R.J., Neves N., Silva M., Lee J.-H., Chen Z.J., Viegas W., Pikaard C.S.. Natural variation in nucleolar dominance reveals the relationship between nucleolus organizer chromatin topology and rRNA gene transcription in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:11418–11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Grummt I., Pikaard C.S.. Epigenetic silencing of RNA polymerase I transcription. Nat. Rev. Mol. Cell Biol. 2003; 4:641–649. [DOI] [PubMed] [Google Scholar]
- 64. Dvořáčková M., Fojtová M., Fajkus J.. Chromatin dynamics of plant telomeres and ribosomal genes. Plant J. 2015; 83:18–37. [DOI] [PubMed] [Google Scholar]
- 65. Kobayashi T. Regulation of ribosomal RNA gene copy number and its role in modulating genome integrity and evolutionary adaptability in yeast. Cell. Mol. Life Sci. 2011; 68:1395–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Takeuchi Y., Horiuchi T., Kobayashi T.. Transcription-dependent recombination and the role of fork collision in yeast rDNA. Genes Dev. 2003; 17:1497–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kobayashi T., Ganley A.R.. Recombination regulation by transcription-Induced cohesin dissociation in rDNA repeats. Science. 2005; 309:1581–1584. [DOI] [PubMed] [Google Scholar]
- 68. Xue S., Barna M.. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat. Rev. Mol. Cell Biol. 2012; 13:355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The bioinformatic pipelines to extract specific T-stretch signatures is available on (https://gist.github.com/laurianesimon/0ae2dd7b8c34c23cdacec217aeaab79c) and (https://gist.github.com/laurianesimon/a9fc44aa83305c576e914710cae75f87).
The Col-0 DNA Mi-Seq data, the 5S-210 RNA-Seq data, the 5S-120 RNA-Seq data as well as the sequencing data from the six YACs have been deposited under PRJNA369183, PRJNA369190, PRJNA378941 and PRJNA378941 respectively.