

Abstract
The objectives of this analysis were to develop a population pharmacokinetics (PK) model of durvalumab, an anti‐PD‐L1 antibody, and quantify the impact of baseline and time‐varying patient/disease characteristics on PK. Pooled data from two studies (1,409 patients providing 7,407 PK samples) were analyzed with nonlinear mixed effects modeling. Durvalumab PK was best described by a two‐compartment model with both linear and nonlinear clearances. Three candidate models were evaluated: a time‐invariant clearance (CL) model, an empirical time‐varying CL model, and a semimechanistic time‐varying CL model incorporating longitudinal covariates related to disease status (tumor shrinkage and albumin). The data supported a slight decrease in durvalumab clearance with time and suggested that it may be associated with a decrease in nonspecific protein catabolic rate among cancer patients who benefit from therapy. No covariates were clinically relevant, indicating no need for dose adjustment. Simulations indicated similar overall PK exposures following weight‐based and flat‐dosing regimens.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Durvalumab is a human monoclonal antibody that binds to PD‐L1 and blocks its interaction with PD‐1 and CD80. Durvalumab was granted accelerated approval for second‐line urothelial carcinoma, breakthrough designation for stage III non‐small cell lung cancer, and is investigated in a number of malignancies. The population pharmacokinetics (PK) of durvalumab has not yet been described in advanced solid tumors.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The analysis characterized the PK of durvalumab and quantified the determinants of durvalumab exposure in humans to better appraise the requirement for dose adjustment in special populations, and whether a flat‐dosing regimen would be comparable to the currently approved weight‐based dosing regimen of 10 mg/kg q2w i.v.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ This study proposes a novel semimechanistic PK model of durvalumab able to quantify the interplay between disease status and durvalumab exposure change over time at both the population and the individual level by incorporation of time‐varying pharmacodynamic biomarkers on linear clearance. The model also describes the nonlinear clearance at low doses and association with sPD‐L1 levels. As a direct application, PK model simulations support the potential switch from a weight‐based dosing to a flat‐dosing regimen.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
☑ The model we propose enables PK and pharmacodynamics (PD) to crosstalk according to a semimechanistic framework that is statistically superior to an empirical time‐varying description of clearance recently proposed for monoclonal antibodies in cancer patients. It elucidates the mechanistic role of disease status on durvalumab PK and permits quantification of the magnitude of change over time of exposure based on disease status and patient characteristics. These findings support the hypothesis that decreased durvalumab clearance with time may be associated with a decrease in nonspecific protein catabolic rate among cancer patients who benefit from therapy.
A number of antibody‐based anticancer therapies involving the PD‐1/PD‐L1 (programmed cell death‐1/programmed cell death ligand‐1) axis have emerged in recent years.1 PD‐L1, a ligand for PD‐1, is upregulated in cancer cells and supports their evasion from the immune system by inhibiting the action of tumor‐infiltrating T cells. Durvalumab (MEDI4736) is an anti‐PD‐L1 human immunoglobulin G1 (IgG1) kappa monoclonal antibody currently being evaluated in a number of malignancies. It blocks multiple interactions with PD‐L1, thus releasing immune activity by T cells against tumor cells. The human pharmacokinetics (PK) of these agents is of crucial interest for dose optimization. Several covariates for anti‐PD‐1/PD‐L1 antibodies have been reported based on population PK analyses. Nivolumab and pembrolizumab are anti‐PD‐1 antibodies currently approved for several cancer indications (including melanoma and non‐small cell lung cancer (NSCLC)) while atezolizumab, avelumab, and durvalumab are anti‐PD‐L1 antibodies (all received approval for urothelial carcinoma).
The population PK analysis of pembrolizumab2 in advanced solid tumors showed a typical IgG4 PK, with effects on exposure of body weight, sex, performance status, renal function, albumin, tumor type, and tumor size (all at baseline), and prior treatment with ipilimumab, an anti‐CTLA4 (cytotoxic T‐lymphocyte‐associated protein 4, another immune checkpoint) monoclonal antibody. Despite reaching statistical significance, none of these covariates, which included a number of disease‐related factors, had a significant impact on pembrolizumab exposure at 2 mg/kg q3w. Use of the approved dose of 2 mg/kg q3w was supported by the analysis, although a more recent study3 supported the use of a flat dose of 200 mg q3w.
The population PK of nivolumab in a pooled dataset including patients with advanced solid tumors was reported previously.4 Nivolumab, an IgG4 antibody, was found to have linear PK with time‐varying clearance, described empirically by a sigmoidal function4 decreasing over time with a 24.5% mean maximal reduction from baseline. Statistically significant exposure covariates included body weight, sex, performance status, albumin, race, renal function, and lactate dehydrogenase (LDH). Liu et al.5 found in a complementary analysis that the maximum decrease in nivolumab clearance was statistically associated with baseline disease status and hinted that posttreatment disease status may also play a role in nivolumab exposure. None of the covariates were found clinically relevant and the same conclusion was reached in the analysis described in the approval summary.6 While this analysis focused on the 3 mg/kg q2w (every 2 weeks) dosing regimen, a subsequent study supported the use of 240 mg q2w.7
The population PK of the IgG1 antibody atezolizumab (approved at 1,200 mg q3w) in hematologic and solid malignancies was described in a recent report.8 The PK was linear over a wide dose range. Population PK indicated several statistically significant covariates (body weight, sex, anti‐drug antibody (ADA), albumin, and tumor burden), none of which would require dose adjustment. Accumulation was well described, but a visual predictive check (VPC) indicated a small trend of increased exposure with time from cycle 3 onwards that was not fully captured by their linear clearance model predictions. Lastly, avelumab, an IgG1 antibody, reported similar PK properties as other antibodies targeting the PD‐1/PD‐L1 axis, with no clinically relevant covariates impacting avelumab exposure levels that would warrant dose adaptation.9
The objective of this work was to develop a population PK model of durvalumab, thus quantifying the effect of patient/disease characteristics on PK, including the explanatory value of time‐varying biomarkers on durvalumab clearance. Subsequently, the model was used to compare weight‐based vs. flat‐dosing regimens.
RESULTS
Data
A total of 1,409 patients provided data following durvalumab administration. Dose levels in Study 1108 (NCT01693562) ranged from 0.1–10 mg/kg q2w i.v. and from 15 mg/kg q3w i.v. to 20 mg/kg q4w i.v. ATLANTIC (NCT02087423) used a dose of 10 mg/kg q2w i.v. The study design details are provided in Supplementary Materials Table S1. Covariate summary statistics are provided in Table 1. The population included in this PK analysis was typical of an all‐comer cancer patient pool, with the majority of patients being male (56.7%), with median age 62, and median body weight at baseline of 69.8 kg. Around two‐thirds of the patients had a baseline Eastern Cooperative Oncology Group (ECOG) performance status score of 1. In the pan‐tumor pool used for this analysis, UC patients were 162 and lung cancer patients represented the biggest pool (n = 776).
Table 1.
Summary of baseline covariates and post‐baseline ADA status
| Categorical covariate | Patients, n (%) |
|---|---|
| ADA statusa | |
| Missing | 210 (14.9) |
| Negative post‐baseline | 1,155 (82.0) |
| Positive post‐baseline | 44 (3.1) |
| Race | |
| American Indian or Alaska native (=1) | 0 (0.0) |
| Asian (=2) | 270 (19.2) |
| Black or African American (=3) | 44 (3.1) |
| Native Hawaiian or other Pacific islander (=4) | 5 (0.4) |
| White (=5) | 1,000 (71.0) |
| Other or nonspecified or missing (=6) | 88 (6.2) |
| Multi‐race (=7) | 2 (0.1) |
| Gender | |
| Male | 799 (56.7) |
| Female | 610 (43.3) |
| ECOG performance status | |
| Missing | 5 (0.4) |
| 0 | 480 (34.1) |
| 1 | 921 (65.4) |
| 2 | 3 (0.2) |
| Tumor type | |
| Advanced cutaneous melanoma | 22 (1.6) |
| Bladder cancer (urothelial carcinoma) | 162 (11.5) |
| Colorectal cancer | 2 (0.1) |
| Gastroesophageal cancer | 54 (3.8) |
| Glioblastoma | 20 (1.4) |
| Hepatocellular carcinoma | 40 (2.8) |
| HPV positive cancer | 22 (1.6) |
| MSI‐high cancer | 62 (4.4) |
| Nasopharyngeal carcinoma | 10 (0.7) |
| Nonsquamous NSCLC | 520 (36.9) |
| Squamous NSCLC | 235 (16.7) |
| Ovarian cancer | 46 (3.3) |
| Pancreatic adenocarcinoma | 36 (2.6) |
| Renal cell cancer | 2 (0.1) |
| Squamous cell carcinoma of head and neck | 62 (4.4) |
| Small cell lung cancer | 21 (1.5) |
| Soft tissue sarcoma | 20 (1.4) |
| Triple negative breast cancer | 41 (2.9) |
| Uveal melanoma | 24 (1.7) |
| NSCLC (nonspecified histology) | 14 (1.0) |
| Advanced malignant melanoma | 8 (0.6) |
| Continuous covariate (units) | n | Mean (SD) | Median | Range | n (%) missing | Normal range (LLN–ULN) | n (%) patients with values ≥ ULN or ≤ LLN |
|---|---|---|---|---|---|---|---|
| Age (years) | 1,409 | 60.9 (11.6) | 62.0 | 19.0–96.0 | 0 (0.0) | NA | NA |
| Weight (kg) | 1,409 | 71.7 (17.1) | 69.8 | 34.0–149.1 | 0 (0.0) | NA | NA |
| Bilirubin (mg/dL) | 1,394 | 0.5 (0.3) | 0.5 | 0.1–3.9 | 15 (1.1) | 0.3–1.9 | 9 (0.6) |
| AST (IU/L) | 1,389 | 27.2 (21.6) | 21.0 | 0.0–322.0 | 20 (1.4) | 10.0–34.0 | 247 (17.5) |
| ALT (IU/L) | 1,394 | 23.5 (18.8) | 18.0 | 0.0–245.0 | 15 (1.1) | 7.0–56.0 | 75 (5.3) |
| Albumin (g/L)b | 1,386 | 37.3 (5.3) | 38.0 | 20.0–57.1 | 23 (1.6) | 35.0–55.0 | 446 (31.7) |
| Cr (mg/dL) | 1,393 | 0.86 (0.3) | 0.8 | 0.3–2.2 | 16 (1.1) | 0.6–1.2 | 162 (8.7) |
| CRCL (mL/min) | 1,393 | 90.6 (32.0) | 85.7 | 26.6–270.5 | 16 (1.1) | 90.0–130.0 | 774 (54.9) |
| LDH (IU/L)b | 1,363 | 356.6 (395.0) | 241.0 | 18–5707 | 47 (3.3) | 105.0–303.0 | 493 (35.0) |
| sPD‐L1 (pg/mL) | 1,259 | 139.0 (115.6) | 124.8 | 67.1–3471 | 150 (10.6) | NA | NA |
| NLRb | 1,322 | 6.07 (6.0) | 4.31 | 0.45–89.3 | 87 (6.2) | 0.78–3.53 | 812 (61.4) |
| Tumor size (mm)b | 797 | 84.8 (52.6) | 74.8 | 10–366 | 612 (43.4) | NA | NA |
ADA status is defined based on postbaseline ADA positive or negative criteria (binomial covariate). Hence, it was not handled as a time‐varying covariate.
Covariates with time‐varying data also available in the analysis dataset (see Figure 2 for time‐course distribution of values).
ADA, anti‐drug antibody status; ALT, alanine transaminase; AST, aspartate transaminase; Cr, serum creatinine; CRCL, creatinine clearance; ECOG, Eastern Cooperative Oncology Group; HPV, human papillomavirus; LDH, lactate dehydrogenase; LLN, lower limit of normal; MSI, microsatellite instability; NA, not applicable; NLR, neutrophil‐to‐lymphocyte ratio; SD, standard deviation; sPD‐L1, soluble PD‐L1 level at baseline; ULN, upper limit of normal.
Most cancer patients were white (71.0%), with a sizeable pool of Asians (19.2%) and 3.1% Black or African American patients. Postbaseline ADA status was chosen in the analysis as the most relevant immunogenicity variable to evaluate the impact of ADA on durvalumab PK.
Primary population PK modeling
A two‐compartment PK model including both linear and nonlinear (Michaelis‐Menten) clearance adequately described the data (Supplementary Materials Figure S1). Durvalumab exhibited nonlinear PK with saturable target‐mediated clearance at doses <3 mg/kg and linearity was approached at doses ≥3 mg/kg. The best stochastic model included a random variable on clearance (CL), central and peripheral volume (V1 and V2, respectively), with banded‐structure block correlations. The residual error was best described by a combined error model and was moderate in size. Model parameter estimates for the time‐invariant CL model can be found in Table 2. Population PK analysis based on stepwise covariate modeling (SCM) identified some statistically significant covariates. Higher baseline albumin and low creatinine clearance (CRCL) levels were associated with reduced CL. Patients with a baseline ECOG score of 0 had lower CL than patients with higher scores. Patients with samples positive for postbaseline ADA status had higher CL than patients with negative ADA status. An increase in tumor size at baseline was associated with higher CL estimates. Patients with higher body weight (WT) were associated with higher CL and V1, whereas females had lower CL, V1, and V2 than males. A high sPD‐L1 level at baseline was associated with an increased Vmax, hence faster nonlinear clearance of durvalumab. All other covariate PK relationships were not statistically significant.
Table 2.
Parameter estimates of three candidate PK models of durvalumab (a = final model) (with 95% confidence interval derived from nonparametric bootstrapping)
| Parameter | Time‐invariant CL model [95% CI] | Empirical time‐varying CL model [95% CI] | Semimechanistic time‐varying CL modela [95% CI] |
|---|---|---|---|
| CL (L/day) | 0.232 [0.221, 0.240] | 0.249 [0.237, 0.273] | 0.232 [0.224, 0.238] |
| V1 (L) | 3.51 [3.44, 3.58] | 3.50 [3.43, 3.56] | 3.51 [3.44, 3.59] |
| V2 (L) | 3.56 [3.36, 3.78] | 3.20 [2.80, 3.41] | 3.45 [3.26, 3.66] |
| Intercompartmental clearance Q (L/day) | 0.477 [0.403, 0.565] | 0.511 [0.43, 0.61] | 0.476 [0.406, 0.556] |
| Michaelis‐Menten constant Km (mg/L) | 0.608 [0.117, 1.71] | 0.452 [0.0408, 1.47] | 0.344 [0.0317, 1.32] |
| Maximum elimination rate Vmax (mg/day) | 0.961 [0.59, 1.53] | 0.744 [0.434, 1.17] | 0.824 [0.544, 1.25] |
| Tmax (unitless) | — | −0.185 [−0.344,−0.101] | — |
| TC50 (days) | — | 173.1 [74.2, 395] | — |
| Lambda (unitless) | — | 1.817 [1.22, 4.22] | — |
| Correlation CL.V1 | 0.269 [0.203, 0.312] | 0.271 [0.251, 0.342] | 0.279 [0.211, 0.321] |
| Correlation V1.V2 | 0.600 [0.565, 0.630] | 0.627 [0.518, 0.614] | 0.560 [0.512, 0.587] |
| Covariate 1: ALB on CL | −0.0272 [−0.0306, −0.0157] | −0.0241 [−0.0307,−0.0222] | −0.0350 [−0.0383, −0.0317]a |
| Covariate 2: WT on CL (power) | 0.400 [0.247, 0.497] | 0.369 [0.295, 0.481] | 0.389 [0.299, 0.477] |
| Covariate 3: ADA on CL | 0.256 [0.0890, 0.438] | 0.308 [0.139, 0.490] | 0.234 [0.0905, 0.401] |
| Covariate 4: CRCL on CL (linear) | 0.00128 [0.000637, 0.00208] | 0.00135 [0.000601, 0.00211] | 0.00149 [0.000834, 0.00218] |
| Covariate 5: ECOG (0 score) on CL | −0.0802 [−0.117, −0.0451] | −0.0763 [−0.106, −0.0366] | −0.0630 [−0.0935, −0.0288] |
| Covariate 6: tumor size on CL | 0.00169 [0.00113, 0.00237] | 0.00168 [0.00102, 0.00214] | 0.00178 [0.00131, 0.00223]a |
| Covariate 7: SEX (female) on CL | −0.129 [−0.165,−0.0875] | −0.127 [−0.173, −0.0942] | −0.143 [−0.177, −0.107] |
| Covariate 8: sPD‐L1 on Vmax (power) | 0.00397 [0.00209, 0.0126] | 0.00500 [0.00272, 0.0149] | 0.00336 [0.00145, 0.0134] |
| Covariate 9: SEX (female) on V1 | −0.166 [−0.189,−0.137] | −0.166 [−0.191,−0.136] | −0.165 [−0.192, −0.136] |
| Covariate 10: WT on V1 (power) | 0.406 [0.335,0.470] | 0.405 [0.345, 0.479] | 0.406 [0.337, 0.474] |
| Covariate 11: SEX (female) on V2 | −0.236 [−0.302,−0.175] | −0.240 [−0.290,−0.154] | −0.205 [−0.261, −0.149] |
| Between‐subject variability CL ω (CV%) | 29.3% [27.7, 32.5] | 28.50% [25.8, 30.2] | 27.0% [25.1, 28.7] |
| Between‐subject variability V1 ω (CV%) | 20.9% [18.9, 22.9] | 21.30% [18.9, 22.7] | 20.9% [18.9, 22.8] |
| Between‐subject variability V2 ω (CV%) | 38.4% [23.0, 44.2] | 37.60% [29.4, 43.1] | 33.6% [28.1, 39.3] |
| Between‐subject variability Tmax ω (SD) | — | 0.234 [0.132, 0.357] | — |
| Proportional residual error σ (CV%) | 21.70% [20.9, 22.6] | 21.40% [20.5, 22.2] | 21.3% [20.5, 22.1] |
| Additive error standard deviation σ (μg/mL) | 0.351 [0.0984, 0.474] | 0.371 [0.122, 0.506] | 0.301 [0.0954, 0.522] |
| Amount of successful bootstraps out of 1000 replicates | 608 | 495 | 694 |
ADA, anti‐drug antibody status; ALB, serum albumin expressed in g/L; CI, confidence interval; CL, clearance; CRCL, creatinine clearance expressed in ml/min; CV%, coefficient of variation in percent; ECOG, Eastern Cooperative Oncology Group performance status; LDH, lactate dehydrogenase; PK, pharmacokinetic; SD, standard deviation; SEX is 1 for female; sPD‐L1, soluble PD‐L1 level expressed in pg/mL; tumor size is expressed in mm; V1, central volume; V2, peripheral volume; WT, body weight expressed in Kg; ω, standard deviation of omega; σ, standard deviation of sigma.
Tumor size and albumin for the semimechanistic time varying CL model were based on samples taken during the trials where the last observation is carried forward for each PK sample. For the other two models tumor size and albumin at baseline were used.
η‐shrinkage for the time‐invariant CL model were 16.8%, 21.5%, 33.3% for CL, V1, and V2, respectively. ε‐shrinkage estimate was 13.9%. η‐shrinkage for the empirical time‐varying CL model were 17.6%, 20.9%, 37.3%, and 67.0% for CL, V1, V2, and Tmax, respectively. ɛ‐shrinkage estimate was 14.4%. η‐shrinkage for the semimechanistic time‐varying CL model were 17.5%, 20.6%, and 36.2% for CL, V1, and V2, respectively. ε‐shrinkage estimate was 13.7%.
Model validation by VPC showed good predictive performance and provided an adequate description of the entire patient pool (data not shown). Terminal half‐life (t1/2) was estimated to be ∼21 days.
Time‐varying CL post‐hoc analysis
First, an adaptation of an empirical time‐changing sigmoidal function4 was implemented to investigate whether durvalumab clearance was time‐dependent (Figure 1 left hand side). Model parameter estimates for this empirical time‐varying CL model can be found in Table 2. A mean of 16.9% (1‐exp(Tmax)) decrease in clearance from baseline described the data adequately. However, the empirical nature of this model and its lack of stability warranted investigating alternatives, including longitudinal biomarkers able to explain the clearance time‐dependency in a more mechanistic manner. By visual inspection, longitudinal data of LDH, neutrophil‐to‐lymphocyte ratio (NLR), albumin (ALB), and tumor size all showed a trend over time (Figure 2). The incorporation of time‐dependent changes in albumin and tumor size levels on durvalumab CL improved the statistical fit (ΔOFV of –392 and –94, respectively, compared to the model incorporating baseline covariates) and yielded a semimechanistic time‐varying CL model (see Table 2 and Figure 1right hand side). This model provided good stability and explained 15% of the interindividual variability on clearance (11% being attributable to ALB and 4% to tumor size) (see Supplementary Materials Figures S2 and S3).
Figure 1.
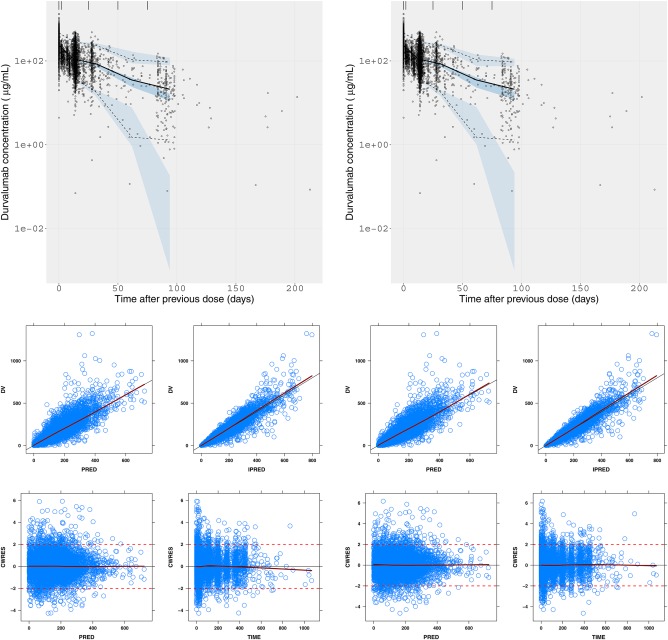
Left panels: Empirical time‐varying CL model. Right panels: Semimechanistic time‐varying CL model, where, Top: VPC (10 mg/kg q2w i.v.); Bottom: Goodness‐of‐fit (all dose levels). Dark blue: smoother line. Red dotted line: indicators of –2 and 2 conditional weighted residuals. Black lines: line of identity. CWRES, conditional weighted residues; DV, data value; IPRED, individual prediction; i.v., intravenous; PRED, population predicted; q2w, every 2 weeks; VPC, visual predictive check. [Color figure can be viewed at http://www.cpt-journal.com]
Figure 2.
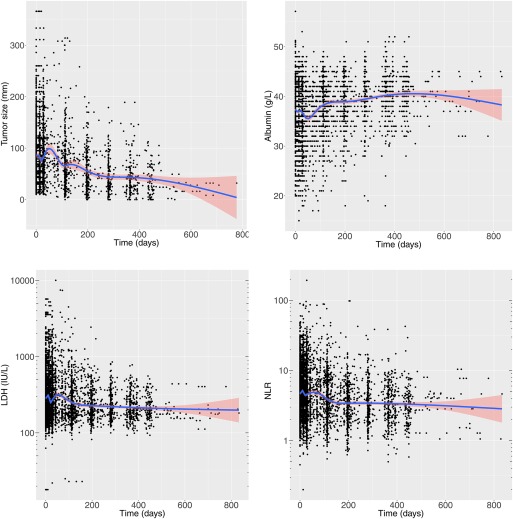
Changes in tumor size, serum albumin, LDH, and NLR over time in the analysis dataset upon which the semimechanistic time‐varying CL model was built. An LOCF imputation technique was used for interpolation during the merging of PK data and time‐varying covariate data. Blue lines represent loess smoother and pink area is the 95% confidence interval of this regression. LDH, lactate dehydrogenase; LOCF, last observation carried forward; NLR, neutrophil‐to‐lymphocyte ratio. [Color figure can be viewed at http://www.cpt-journal.com]
Typical durvalumab clearance in the semimechanistic time‐varying CL model was 0.232 L/day, V1 was 3.51 L, V2 was 3.45 L, and the Michaelis‐Menten constant (Km) was 0.344 mg/L, with moderate interindividual variability in clearance (27.0% coefficient of variation (CV)), V1 (20.9% CV), and V2 (33.6% CV) (Table 2). Km was poorly estimated, likely due to the lack of data at low doses (3.78% of data is at doses ≤3 mg/kg) at which PD‐L1 alters the kinetics of durvalumab.
Time‐changing LDH and NLR did not improve the model further and were not incorporated in the final model. Note that tumor size at baseline measurable based on a blinded independent central review was missing in a large portion of patients (43.4%), suggesting that its true explanatory value on the CL time‐course was not fully quantifiable from this dataset. The VPC of the empirical time‐varying CL model and the semimechanistic time‐varying CL model performed similarly (Figure 1); however, the semimechanistic model performed better based on stability, parsimony, and potential mechanistic value for extrapolation (Table 3, Supplementary Materials Figure S3).
Table 3.
Comparisons of the empirical time‐varying CL model performance with the semimechanistic time‐varying CL model
| Model | Semimechanistic time‐varying CL model | Empirical time‐varying CL model |
|---|---|---|
| Statistical criteria | OFV=60386 (ΔOFV=‐368) | OFV=60754 (reference) |
| Parsimony | 0 d.o.f.a | +4 d.o.f.a |
| Model stability | Successful minimization and covariance step | Run terminated due to rounding errors and aborted covariance step |
| Mechanistic explanatory value | Change in clearance explained by changes in disease state | Change in clearance explained by an empirical formula |
| Application for PK prediction | Model can predict PK based on individual and population albumin concentrations and tumor size changes | Model has limited predictive value since its parameters only fit changes in clearance observed in trials |
| Application for PD prediction | Model can be linked to a PK/PD model with changes in PK informing PD, and also mechanistically account for changes in PD informing PK | Model can be linked to a PK/PD model, but changes in disease state will not automatically impact PK behavior |
d.o.f., degree of freedom relative to the time‐invariant CL model presented in Table 2; CL, clearance; OFV, objective function value; PD, pharmacodynamics.
Clinical relevance of covariates
None of the covariates that were statistically significant (P < 0.001) predictors of durvalumab PK were found to be clinically relevant as judged by the magnitude of covariate effect on durvalumab PK or exposure parameters not exceeding 30%, a threshold set a priori (Figure 3). Since the variability of observed values at baseline did not increase with time (see Figure 2), baseline evaluation of covariates as displayed in Figure 3 was sufficient to appraise their clinical relevance. Low albumin levels gave rise to a 22% reduction in AUCss for the 10th percentile level (30 g/L) compared to a typical patient. This level is below the upper limit of normal and is representative of a patient with hypoalbuminemia. This reduction in durvalumab exposure would not translate into a substantial reduction in the target suppression coverage since the trough concentration would still result in >99% suppression of the target in the serum throughout the dosing interval. Hence, albumin effect on durvalumab serum levels does not warrant dose adaptation. Tumor size at baseline is predicted to result in an exposure drop of –13% in AUCss at the distribution higher end (90th percentile = 158 mm) compared to a typical patient (median target lesion size at baseline of 74.8 mm). Conversely, an increase in AUCss by + 10% is predicted by the model at the other extreme of the tumor size distribution (10th percentile = 26.4 mm) compared to a typical patient. A combined effect of increased CL and V1 with increasing body weight did not translate into more than a 30% difference in AUCss with –17% respectively, for the 10th percentile patient (WT = 51.4 kg) and +20% for the 90th percentile patient (WT = 93.7 kg) compared to a typical patient (WT = 69.8 kg). This increase in exposure for high body weight patients is mainly linked to the weight‐based dosing scheme of durvalumab used in simulations. Females had + 17% higher AUCss, due to the impact of sex on CL, V1, and V2, which are reduced for women. CRCL levels at the 10th and 90th percentiles had a marginal impact on exposure (+5% and –7% impact on AUCss for the 10th and 90th percentiles, respectively). Patients with ECOG score of 0 showed a + 7% increase in AUCss compared to a typical patient. Finally, patients whose samples tested positive for ADA had lower exposure levels of durvalumab but the reduction of –19% in AUCss did not reach clinical relevance.
Figure 3.
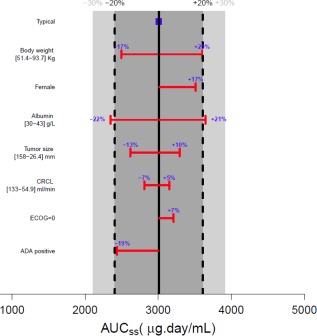
Effect of baseline covariates on exposure parameter AUCss. Simulations obtained using Berkeley Madonna software based on the final PK model (semimechanistic time‐varying CL model) estimates of NONMEM for each covariate at baseline separately. The time‐varying nature of covariate (tumor size and albumin) was not accounted for in this evaluation, provided that the variability at baseline did not increase with time. Solid black vertical line and blue square show steady‐state exposure level of durvalumab for a typical patient (male, without positive ADA, with baseline values as follows: ECOG performance status of 1 or higher, body weight of 69.8 kg, serum albumin level of 38 g/L, target lesion tumor size of 74.8 mm, and CRCL estimate of 85.65 mL/min). Light gray area shows 30% change from the typical patient; dark gray delineated by dotted black lines shows 20% change. Red horizontal bar represents the covariate being evaluated with values of the 10th and 90th percentiles of the covariate distribution displayed for continuous covariate in square brackets. The length of each bar describes the impact of that particular covariate on durvalumab exposure metric, with the percent change of exposure from the typical value being displayed (bold blue); ADA, anti‐drug antibody; AUCss, area under the curve steady state (derived from analytical solution Dose/CLss, with CLss taken on Day 365); CRCL, creatinine clearance; ECOG, Eastern Cooperative Oncology Group performance status. [Color figure can be viewed at http://www.cpt-journal.com]
Other covariates, such as age and race, were not identified as influencing durvalumab PK. Similar findings as described here for AUCss were seen for other PK metrics (Cmax,ss and Cmin,ss). Based on these cumulative results, no dose adjustment is required for special populations.
Comparison of weight‐based vs. flat‐dosing regimens
The effect of weight‐based and flat‐dosing regimens were evaluated using simulations based on the final population PK model (semimechanistic time‐varying CL). Two i.v. flat‐dosing regimens were evaluated against 10 mg/kg q2w i.v.: 750 mg q2w and 1,500 mg q4w. Simulation results presented in Figure 4 indicated that both regimens yield a similar median steady‐state exposure and associated variability, with no increased incidence of extreme concentration values for a flat‐dosing regimen compared to an equivalent weight‐based dosing regimen (see Supplementary Materials Figure S4). This result supports a potential switch to a flat‐dosing regimen of 750 mg q2w i.v. or an equivalent, but less frequent, flat‐dosing regimen of 1,500 mg q4w i.v.
Figure 4.
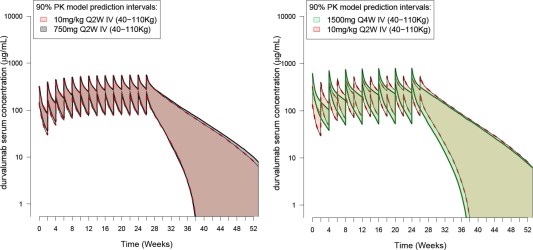
Simulated PK profiles of durvalumab following weight‐based dosing regimens (10 mg/kg q2w i.v.) compared with flat‐dosing. (a) 750 mg q2w i.v.; (b) 1,500 mg q4w i.v. The area (pink, gray, and green) represents the 90% prediction interval from the semimechanistic time‐varying CL model according to three different dosing schemes; they are delimited by the 5th and 95th percentiles of the simulated PK data obtained from a pool of n = 1,000 virtual patients. Only the body weight covariate effect was investigated (no time‐varying covariate were used for simulations). [Color figure can be viewed at http://www.cpt-journal.com]
DISCUSSION
A semimechanistic time‐varying CL model, featuring post‐hoc inclusion of albumin and tumor size time courses, was proposed to explain the change in clearance of durvalumab over time. A two‐compartment PK model including both linear and nonlinear clearance adequately described PK data for all dosing regimens. Typically, durvalumab clearance was 0.232 L/day, V1 was 3.51 L, V2 was 3.45 L, and Km was 0.344 mg/L with moderate interindividual variability in clearance (27.0% coefficient of variation (CV)), V1 (20.9% CV), and V2 (33.6% CV). The estimated t1/2 was about 21 days. The PK model identified 10 mg/kg i.v. q2w as the dose to maintain exposure levels above 50 μg/mL throughout the dosing interval, with >90% of patients expected to reach almost complete saturation of both soluble and membrane‐bound PD‐L1 in serum (>99% target suppression, based on mean Km value).
Although population PK analysis identified statistically significant covariates (body weight, sex, postbaseline ADA, CRCL, ECOG performance status, sPD‐L1 levels, tumor size, and ALB), none were found to be clinically relevant (impact on PK parameters <30%). Age, race, tumor type, LDH, NLR, renal function (mild to moderate), and hepatic function (mild) had no impact on PK.
The empirical time‐varying CL model approach supported a slight decrease in clearance of 16.9%. To investigate the possible origin of this phenomenon, longitudinal time‐varying covariates indicative of potential changes in the patients' health status were selected. Durvalumab CL decreased with increasing albumin level, and with decreased tumor burden. Hypoalbuminemia is a well‐known marker of cachexia, inflammatory conditions, and increased catabolic activity,10 and the impact of albumin on PK of monoclonal antibodies has been previously reported for infliximab,11 bevacizumab,12 ustekinumab,13 and pertuzumab.14 However, these interactions were only identified based on baseline albumin measures. The observed albumin increase posttreatment in cancer patients responding to therapy may be associated with an improvement in cancer inflammation and cachexia. Recently, experimental evidence has been reported that tumor cells can directly catabolize albumin and other extracellular proteins by micropinocytosis, and potentially contribute to hypoalbuminemia in cancer patients.15 Because albumin and IgG share the same FcRn salvaging pathway, hypoalbuminemia could reflect a higher protein catabolic rate in cancer patients that also affects the clearance of durvalumab and other IgG monoclonal antibodies. Interestingly, the time‐varying clearance was associated with the linear clearance of durvalumab, while the nonlinear clearance was associated only with baseline sPD‐L1 levels. These results suggest that the decrease in clearance with time in patients with tumor shrinkage is not caused by a decrease in target‐mediated clearance by tumor cells, but instead by a nonspecific decrease in IgG catabolism. Besides, the positive association found by covariate analysis between the sPD‐L1 level at baseline and Vmax implies that faster elimination of durvalumab at low doses is likely linked to the faster turnover of PD‐L1 compared to the rate of elimination of durvalumab through the reticuloendothelial system and neonatal FcRn recycling process.
The magnitude of individual change in CL being associated with response status of patients was previously hypothesized by Wang et al.16 Although more than 40% of the patients had missing tumor size information in our covariate analysis, tumor shrinkage was associated with decreased durvalumab CL, resulting in higher exposure for responders. As such, the recovery of albumin levels and reduction in tumor burden in durvalumab‐treated patients may be linked with disease status and protein catabolic rate. This suggests an interaction of disease status and durvalumab PK that is embedded in the semimechanistic time‐varying CL model presented here.
All models, including the time‐invariant CL model, empirical time‐varying CL model, and semimechanistic time‐varying CL model, were compared and our analysis favored the more mechanistic model when considering statistical fit, stability, and predictive value (Table 3). The model also provides a broader understanding of the mechanism of action of durvalumab, cross‐linking the PK and the disease status (tumor burden, cancer inflammation, and cachexia). Quantifying the association between PK and pharmacodynamics (or clinical endpoints) is critical to prevent potential confounding of exposure–response analyses in cancer treatment. Overall survival or progression‐free survival stratification by exposure (or any other biomarker) in Kaplan–Meier univariate analysis could suggest an exposure–response that is only apparent, due to the high correlation of PK with disease status. This is further complicated by the fact that disease status is evolving with time, and patients benefiting from therapy have a higher steady‐state exposure due to their decreased catabolic status and/or improved cachexia. This can provide a false impression of predictive or prognostic value of some biomarkers, or that higher doses may provide more efficacy, as exemplified by a trastuzumab case study in metastatic gastric cancer.17, 18 Without a clear understanding of the degree of correlation between all these variables, incorrect conclusions could be easily drawn when analyzing data in only one dimension without accounting for potential interplays between PK and disease variables. The pitfalls of exposure–response without accounting for time‐varying exposure in oncology are further discussed elsewhere.10, 16
A similar magnitude of change in clearance over time was previously reported for nivolumab4 and did not reach clinical significance. Liu et al. reported the impact of baseline albumin, baseline tumor size, and best overall response on CL of nivolumab.5 The results from our population PK analysis allow description of the time‐varying CL of durvalumab with a more parsimonious model that accounts for patient characteristics at baseline and during the course of treatment.
Moreover, the strength of population PK modeling to rationalize dose selection and to corroborate the requirement of dose adaptation was exemplified. These results indicate that the change in CL over time was not clinically relevant and there is no need for dose adjustment based on baseline patient characteristics. One limitation of this approach is that the clinical relevance of the covariate effect on exposure levels is evaluated in isolation rather than in a multivariate manner. However, the intent is to provide clinicians and health practitioners a simple guide to quantify the relative effect of each covariate on exposure levels. In clinical practice, a summation of the covariate effects when a patient presents a combination of multiple risk factors could be considered to evaluate the optimal dosing regimen of a given therapeutic. Lastly, population PK modeling of durvalumab supports the potential switch to a flat‐dosing regimen of 750 mg q2w i.v. or a regimen of 1,500 mg q4w i.v. The flat dose regimen of durvalumab of 1,500 mg q4w i.v. is currently pursued in multiple confirmatory trials across several indications.
METHODS
Study design and patient population
This analysis is based on the pooling of two clinical studies investigating durvalumab in solid tumors (see Supplementary Materials Table S1 and Table 1):
CD‐ON‐MEDI4736‐1108 (NCT01693562; referred to as Study 1108): A phase Ib/II study to evaluate the safety, tolerability, and pharmacokinetics of MEDI4736 in subjects with advanced solid tumors.
ATLANTIC or D4191C00003 (NCT02087423): A phase II, noncomparative, open‐label, multicenter, international study of MEDI4736, in patients with locally advanced or metastatic non‐small cell lung cancer NSCLC (Stage IIIB–IV) who have received at least two prior systemic treatment regimens including one platinum‐based chemotherapy regimen.
Both studies were conducted in compliance with the Declaration of Helsinki and the Guidelines for Good Clinical Practice. Written informed consent was obtained from all patients.
Primary population PK modeling analysis
A nonlinear mixed‐effects modeling approach was used in NONMEM software (v. 7.3, ICON Development Solutions, Ellicott City, MD; 200619) to analyze durvalumab PK data, using a first‐order conditional estimation method with interaction maximum likelihood estimation method. Data management and graphical exploration were conducted in R (v. 3.3.1 or higher, Vienna, Austria).20 Model parameters' uncertainty was computed using the default option for the NONMEM $COV record or by bootstrap estimation.21 A series of structural models were evaluated based on objective function values (OFV), precision, plausibility of parameter estimates, and goodness‐of‐fit plots, including simulation‐based visual predictive checks (VPC).22, 23 Parameter distribution assumptions are discussed in the Supplementary Materials.
Baseline covariate analysis was automated in Perl‐speaks‐Nonmem (PsN) v. 4.0 or higher24, 25, 26, 27 following the stepwise covariate model (SCM) building technique.28, 29 The SCM included two steps—a forward selection and a backward elimination phase (with statistical criteria prespecified respectively at a type‐I error rate of P < 0.01 and P < 0.001). The P‐values were derived from the change in the OFV provided by NONMEM based on the likelihood ratio test (LRT) for nested models. Covariates were first investigated graphically and summarized numerically. If initial graphical exploration pointed to a relationship between a covariate and η, suggesting an influence on the interindividual variability (ω2) of durvalumab PK, the covariate‐PK relationship was then assessed in the nonlinear mixed‐effects model framework. The covariates to be evaluated were selected based on scientific interest, mechanistic plausibility, and prior knowledge and the implementation is described in the Supplementary Materials.
The primary population PK analysis evaluated the impact of baseline covariates assuming a time‐invariant clearance (the time‐invariant CL model). For baseline covariate with missing information, no data imputation was performed. The primary analysis evaluated the impact of covariates related to demographics (age, sex, body weight, race); disease status (performance status, tumor type, NLR, and tumor size defined as the sum of the longest diameter of target lesions); liver function (aspartate transaminase, alanine transaminase, total bilirubin, serum albumin); kidney function (CRCL, serum creatinine); metabolic marker (LDH); level of sPD‐L1; and postbaseline ADA. The significance of a covariate effect was then evaluated by its clinical or physiological relevance. Based on the final model parameter estimates, a final filter was applied to appraise the clinical relevance of the covariate‐PK relationship found to be statistically significant by means of deterministic simulations in the software Berkeley Madonna (v. 8.3.18, Berkeley, CA).30 If the magnitude of covariate effect was less than 30% of the parameter estimates (or key predicted PK exposure metrics, such as area under the concentration–time curve at steady state (AUCss), and steady state minimum and maximum concentrations of durvalumab (Cmin,ss, and Cmax,ss)) for a typical patient, the covariates were not considered clinically relevant.
Post‐hoc analysis of time‐varying CL
The primary analysis model (time‐invariant CL model) was complemented with a post‐hoc analysis accommodating a time‐varying clearance component. Two competing time‐varying durvalumab CL models were evaluated: an empirical time‐varying CL model, and a semimechanistic time‐varying CL model. First, an empirical model already published for other PD‐1/PDL‐1 mAbs4, 5 was implemented to explain the time‐changing clearance of durvalumab. The empirical time‐varying CL model employed a sigmoid time‐course to describe longitudinal changes in clearance values, without links to biologically relevant explanatory variables, as described below:
In this sigmoidal saturation model, CL is the systemic clearance, TVCLcov is the typical value of clearance for a given covariate, ηCL is the interindividual clearance variability random effect, t is time after first dose, Tmax is the logarithm maximum fractional change in clearance over the trial time course, and TC50 is the time at which the change is half of its maximum, while Lambda is an exponential shape parameter.
A semimechanistic time‐varying CL model was implemented to incorporate longitudinal time‐varying covariates to explain changes in durvalumab CL over time. Four time‐varying covariates, including tumor size, ALB, LDH, and NLR, were investigated in this approach. The last‐observation‐carry‐forward (LOCF) technique was employed to interpolate the missing covariate values in the PK dataset. NLR was evaluated as a potential covariate on PK of durvalumab, as this metric was shown to have either prognostic or predictive value for immune checkpoint inhibitors,31, 32, 33 but may be correlated with other variables such as PK or other disease indices. Time‐dependent profiles related to tumor burden (target lesion size and LDH), and cancer, inflammation, cachexia, and protein catabolic rate (NLR and albumin) were tested for inclusion in the population PK model. They either replaced the baseline covariate or were newly added to the existing covariate relationship. The rationale is that accounting for longitudinal changes in these covariates would explain the variation in time‐dependent changes in CL in a semimechanistic manner, thus facilitating biologically plausible extrapolation of results.
A comparison of three approaches (time‐invariant CL model, empirical time‐varying CL model, and semimechanistic time‐varying CL model) was conducted by means of statistical comparison of overall fit (LRTs), parameter estimates and precision, mechanistic plausibility, parsimony, and predictive performance (VPCs).
The methodology for assessing the weight‐based vs. fixed dosing simulations and a brief description of assay methodology can be found in the Supplementary Materials.
CONFLICT OF INTEREST
P.G. Baverel, V.F.S. Dubois, C. Jin, Y. Zheng, X. Song, A.K. Gupta, P. Vicini, L. Roskos, R. Narwal are employees of MedImmune, a wholly owned subsidiary of AstraZeneca. P. Mukhopadhyay and P.A. Dennis are employees of AstraZeneca. X. Jin and Y. Ben are former employees of MedImmune and AstraZeneca, respectively.
FUNDING
The ATLANTIC study was sponsored by AstraZeneca and Study 1108 was sponsored by MedImmune. This analysis was funded by MedImmune.
AUTHOR CONTRIBUTIONS
P.G.B., V.F.S.D., P.V., L.R., and R.N. wrote the article; P.G.B., C.J., A.K.G., L.R., and R.N. designed the research; P.G.B., V.F.S.D., C.J., X.S., X.J., P.M., P.A.D., Y.B., L.R., and R.N. performed the research; P.G.B., V.F.S.D., C.J., Y.Z., X.S., P.V., L.R., and R.N. analyzed the data.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The first two authors contributed equally to this work. The contents of this article were partially presented at the ASCO Annual Meeting 2017, held on April 1–5, 2017 (Paul G. Baverel, Vincent F.S. Dubois, Chao Yu Jin, Xuyang Song, Xiaoping Jin, Pralay Mukhopadhyay, Ashok Kumar Gupta, Phillip A. Dennis, Yong Ben, Lorin Roskos, Rajesh Narwal) and published in abstract form in the conference proceedings in the Journal of Clinical Oncology 35, no. 15 (Suppl) 2566 (May 2017).
References
- 1. Balar, A.V. & Weber, S.J. PD‐1 and PD‐L1 antibodies in cancer: current status and future directions. Cancer Immunol. Immunother. 66, 551–564 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahamadi, M. et al Model‐based characterization of the pharmacokinetics of pembrolizumab: a humanized anti ‐ PD‐1 monoclonal antibody in advanced solid tumors. CPT Pharmacometrics Syst. Pharmacol. 6, 49–57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Freshwater, T. et al Evaluation of dosing strategy for pembrolizumab for oncology indications. J. Immunother. Cancer 5, 43 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bajaj, G. et al Model‐based population pharmacokinetic analysis of nivolumab in patients with solid tumors. CPT Pharmacometrics Syst. Pharmacol. 6, 58–66 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu, C. et al Association of time‐varying clearance of nivolumab with disease dynamics and its implications on exposure response analysis. Clin. Pharmacol. Ther. 101, 657–666 (2017). [DOI] [PubMed] [Google Scholar]
- 6. Hazarika, M. et al U. S. FDA approval summary: Nivolumab for treatment of unresectable or metastatic melanoma following progression on ipilimumab. Clin. Cancer Res. 23, 3484–3489 (2017). [DOI] [PubMed] [Google Scholar]
- 7. Zhao, X. et al Assessment of nivolumab benefit‐risk profile of a 240‐mg flat dose relative to a 3‐mg/kg dosing regimen in patients with advanced tumors. Ann. Oncol. 28, 2002–2008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stroh, M. et al Clinical pharmacokinetics and pharmacodynamics of atezolizumab in metastatic urothelial carcinoma. Clin. Pharmacol. Ther. 102, 305–312 (2017). [DOI] [PubMed] [Google Scholar]
- 9. Heery, C.R. et al Pharmacokinetic profile and receptor occupancy of avelumab (MSB0010718C), an anti‐PD‐L1 monoclonal antibody, in a phase I, open‐label, dose escalation trial in patients with advanced solid tumors. J. Clin. Oncol. 33(15_suppl), 3055 (2015). 26304891 [Google Scholar]
- 10. Ryman, J.T. & Meibohm, B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst. Pharmacol. 6, 576–588 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fasanmade, A.A. , Adedokun, O.J. , Olson, A. , Strauss, R. & Davis, H.M. Serum albumin concentration: a predictive factor of infliximab pharmacokinetics and clinical response in patients with ulcerative colitis. Int. J. Clin. Pharmacol. Ther. 48, 297–308 (2010). [DOI] [PubMed] [Google Scholar]
- 12. Lu, J.‐F. et al Clinical pharmacokinetics of bevacizumab in patients with solid tumors. Cancer Chemother. Pharmacol. 62, 779–786 (2008). [DOI] [PubMed] [Google Scholar]
- 13. Zhu, Y. et al Population pharmacokinetic modeling of ustekinumab, a human monoclonal antibody targeting IL‐12/23p40, in patients with moderate to severe plaque psoriasis. J. Clin. Pharmacol. 49, 162–175 (2009). [DOI] [PubMed] [Google Scholar]
- 14. Garg, A. et al Population pharmacokinetic and covariate analysis of pertuzumab, a HER2‐targeted monoclonal antibody, and evaluation of a fixed, non‐weight‐based dose in patients with a variety of solid tumors. Cancer Chemother. Pharmacol. 74, 819–829 (2014). [DOI] [PubMed] [Google Scholar]
- 15. Davidson, S.M. et al Direct evidence for cancer‐cell‐autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med. 23, 235–241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang, Y. et al Toward greater insights on pharmacokinetics and exposure‐response relationships for therapeutic biologics in oncology drug development. Clin. Pharmacol. Ther. 101, 582–584 (2017). [DOI] [PubMed] [Google Scholar]
- 17. Yang, J. et al The combination of exposure‐response and case‐control analyses in regulatory decision making. J. Clin. Pharmacol. 53, 160–166 (2013). [DOI] [PubMed] [Google Scholar]
- 18. Shah, M.A. et al HELOISE: Phase IIIb randomized multicenter study comparing standard‐of‐care and higher‐dose trastuzumab regimens combined with chemotherapy as first‐line therapy in patients with human epidermal growth factor receptor 2‐positive metastatic gastric or gast. J. Clin. Oncol. 35, 2558–2567 (2017). [DOI] [PubMed] [Google Scholar]
- 19. Beal, S.L. , Sheiner, L.B. , Boeckmann, A.J. & Bauer, R.J. NONMEM Users Guides. Ellicott City, MD: Icon Development Solutions; 1989. [Google Scholar]
- 20. R Development Core Team . R: A Language and Environment for Statistical Computing. Vol. 3.3.1 (R Foundation for Statistical Computing, Vienna, Austria, 2013). [Google Scholar]
- 21. Efron, B. Bootstrap methods: another look at the jackknife. Ann. Math. Stat. 7, 1–26 (1979). [Google Scholar]
- 22. Karlsson, M.O. & Holford, N. Model evaluation. Visual predictive checks. In Popul. Approach Gr. Eur. 17, Abstr. 1434 (Marseille, France, 2008). <http://www.page-meeting.org/?abstract=1434>.
- 23. Keizer R, Pastoor D, Savic R. New open source R libraries for simulation & visualization: PKPDsim and vpc In Popul. Approach Gr. Eur. 24, Abstr. 3636 (Hersonissos, Crete, Grece, 2015). <http://www.page-meeting.org/?abstract=3636>. [Google Scholar]
- 24. Lindbom, L. , Ribbing, J. & Jonsson, E.N. Perl‐speaks‐NONMEM (PsN) — A Perl module for NONMEM related programming. Comput. Methods Programs Biomed. 75, 85–94 (2004). [DOI] [PubMed] [Google Scholar]
- 25. Lindbom, L. , Pihlgren, P. & Jonsson, N. PsN‐Toolkit — a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput. Methods Programs Biomed. 79, 241–257 (2005). [DOI] [PubMed] [Google Scholar]
- 26. Harling, K. , Ueckert, S. , Hooker, A.C. , Jonsson, E.N. & Karlsson, M.O. Xpose and Perl speaks NONMEM (PsN) In Popul. Approach Gr. Eur. 19, Abstr. 1842 (Berlin, Germany, 2010). <http://www.page-meeting.org/?abstract=1842>. [Google Scholar]
- 27. Keizer, R.J. , Karlsson, M.O. & Hooker, A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst. Pharmacol. 2, e50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jonsson, E.N. & Karlsson, M.O. Automated covariate model building within NONMEM. Pharm. Res. 15, 1463–1468 (1998). [DOI] [PubMed] [Google Scholar]
- 29. Khandelwal, A. , Harling, K. , Jonsson, E.N. , Hooker, A.C. & Karlsson, M.O. A fast method for testing covariates in population PK/PD models. AAPS J. 13, 464–472 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Macey, R. , Oster, G. & Zahnley, T. Berkeley Madonna User's Guide (Berkeley, CA, University of California, 2000). [Google Scholar]
- 31. Templeton, A.J. et al Prognostic role of neutrophil‐to‐lymphocyte ratio in solid tumors: a systematic review and meta‐analysis. J. Natl. Cancer Inst. 106, 1–11 (2014). [DOI] [PubMed] [Google Scholar]
- 32. Bagley, S.J. et al Pretreatment neutrophil‐to‐lymphocyte ratio as a marker of outcomes in nivolumab‐treated patients with advanced non‐small‐cell lung cancer. Lung Cancer 106, 1–7 (2017). [DOI] [PubMed] [Google Scholar]
- 33. Ferrucci, P.F. et al Baseline neutrophils and derived neutrophil‐to‐lymphocyte ratio: prognostic relevance in metastatic melanoma patients receiving ipilimumab. Ann. Oncol. 27, 732–738 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information