

Abstract
Non‐heme (L)FeIII and (L)FeIII‐O‐FeIII(L) complexes (L=1,1‐di(pyridin‐2‐yl)‐N,N‐bis(pyridin‐2‐ylmethyl)ethan‐1‐amine) underwent reduction under irradiation to the FeII state with concomitant oxidation of methanol to methanal, without the need for a secondary photosensitizer. Spectroscopic and DFT studies support a mechanism in which irradiation results in charge‐transfer excitation of a FeIII−μ‐O−FeIII complex to generate [(L)FeIV=O]2+ (observed transiently during irradiation in acetonitrile), and an equivalent of (L)FeII. Under aerobic conditions, irradiation accelerates reoxidation from the FeII to the FeIII state with O2, thus closing the cycle of methanol oxidation to methanal.
Keywords: diiron complexes, iron, oxidation, photochemistry, reaction mechanisms
Photoredox catalysis has emerged as a versatile method to access highly reactive species in a selective and clean manner.1, 2 The redox‐active photosensitizers available include organic dyes,3 inorganic clusters,4 and transition‐metal complexes, such as [Ru(bpy)3]2+ and its derivatives,5, 6 whose redox potentials can be fine‐tuned by ligand modification.7, 8, 9 Photoredox catalysis can bypass reactive stoichiometric oxidants, such as H2O2 and ClO−, to generate high‐valent transition‐metal oxido species by electron‐transfer oxidation. Non‐heme iron complexes that are well‐known catalysts for a wide range of oxidation reactions have been combined with photoredox catalysts, such as [Ru(bpy)3]2+, for light‐driven oxidation reactions.9, 10, 11 In this multicatalyst strategy (Scheme 1 a), excitation of the photoredox sensitizer is followed by electron‐transfer oxidation of the catalyst to raise it to a higher oxidation state so that it can subsequently oxidize substrates. The photoredox sensitizer is reoxidized by an electron acceptor (EA); however, the use of atom‐economical terminal oxidants (e.g., O2) is a key challenge, and it would be preferable to use a single catalyst that is driven directly by light through the entire redox cycle. Furthermore, the generation of other species, such as singlet oxygen, by the organic and RuII/IrIII photosensitizers is difficult to avoid.12, 13, 14, 15, 16, 17
Scheme 1.
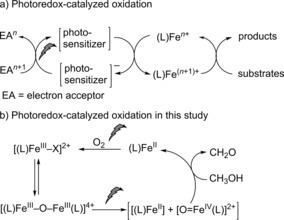
a) Multicatalyst strategy for photocatalytic reactions, and b) the single‐catalyst photocatalytic oxidation described herein. L=MeN4Py, X=OMe or Cl.
The photochemistry of iron complexes and especially the reduction of complexes from the FeIII to the FeII state when irradiated is well‐established,18 not least in the widely used chemical actinometer [FeIII(oxalato)3]3−[19] and other iron(III) carboxylato complexes.20 Photoreduction in such systems is irreversible and accompanied by ligand oxidation (e.g., CO2 formation from carboxylate ligands), and hence FeIII complexes are of limited use in the photocatalytic oxidation of organic substrates. Notable exceptions (see below) are to be found in the reports of Richman,21, 22 Karlin,23 and co‐workers on the photochemistry of μ‐oxido‐bridged diiron(III) complexes.
Previously, we reported that non‐heme FeII complexes (such as [(MeN4Py)FeII(CH3CN)]2+ 1, Figure 1) are photoinert in acetonitrile, but undergo light‐driven oxidation (from the FeII to the FeIII redox state) with O2 in solvents in which the CH3CN ligand is displaced by the solvent used.24 The photochemically driven oxidation of an FeII complex together with the earlier reports of photoreduction of FeIII complexes raises the possibility that a fully light driven photocatalytic oxidation cycle can be achieved without the need for a separate photosensitiser, dor example, [Ru(bpy)3]2+. However, simple non‐heme FeIII systems lack the distinct photophysics and chromophoric properties of the heme unit present in the systems of Richman,21, 22 Karlin,23 and co‐workers, and hence it would seem unlikely that a fully non‐heme FeIII complex would show similar photoreactivity.
Figure 1.
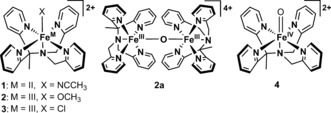
Structures of complexes 1–4 used in this study (see Figures S1 and S2 in the Supporting Information for the single‐crystal structure of 3 together with its solid‐state and calculated Raman spectra).
Herein, we show that a single iron‐based catalyst can promote catalytic oxidation reactions without the use of a secondary photosensitizer (Scheme 1 b). We report a light‐driven double photocycle capable of high‐turnover oxidation of methanol with O2 as the terminal oxidant. Photoreduction of the non‐heme iron(III) complexes to the FeII state occurs concomitant with the oxidation of methanol and is followed by light‐driven reoxidation of the iron(II) complex, with O2 as the terminal oxidant (Scheme 1 b). The whole cycle proceeds without significant ligand degradation.
Density functional (DFT) methods support the assignment of the μ‐oxido diiron(III) complex 2 a (Figure 1) as the photochemically reactive species with photoreduction proceeding via a [(L)FeIV=O]2+ intermediate analogous to that reported for the heme‐based systems.21, 22, 23 [(L)FeIV=O]2+ (4) is itself photoreactive, as we have shown recently.25 However, under certain conditions this species can also be observed during the irradiation of 2 a in acetonitrile. The formation of [(L)FeIV(O)]2+ (4) during irradiation opens the possibility for selective photocatalytic oxidation reactions.
Irradiation of the FeIII complexes [(L)FeIII(OCH3)]2+ (2) and [(L)FeIII(Cl)]2+ (3) in argon‐purged methanol at 365 nm resulted in a decrease in absorbance at 310 nm and concomitant increase in absorbance at 380 and 480 nm corresponding to the formation of FeII complexes (Figure 2; see also Figure S3 in the Supporting Information). Irradiation of 3 at 300 nm resulted in similar changes; however, there was a pronounced wavelength dependence of the photochemical quantum yield26, 27 (Φ 300 nm=0.31±0.01, Φ 365 or 355 nm=0.07±0.01). Irradiation at 490 nm did not affect the absorption spectrum (see Figure S4) even though this wavelength is in resonance with a weak absorption band. Changes in absorbance were not observed without irradiation (Figure 2; see also Figure S5). Essentially identical changes were observed upon irradiation of 2 a in methanol at 365 nm as with 2 and 3 (see Figure S6). The identical behavior of all three complexes in argon‐purged methanol reflects the rapid equilibration of 2 a and 3 with methanol to form predominantly 2, as confirmed by resonance Raman (λexc=355 nm; see Figure S7), EPR, and UV/Vis absorption spectroscopy (see the Supporting Information, Figures S8–S13, for further details).
Figure 2.
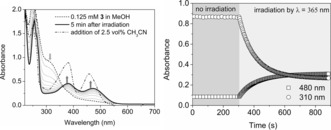
Left: UV/Vis absorption spectrum of 3 (0.125 mm, dashed line) in deoxygenated methanol, during (dotted lines) and after (thick solid line) irradiation at 365 nm, and after the subsequent addition of acetonitrile (2.5 vol %; black dash–dotted line). Right: Absorbance at 310 and 480 nm over time in the dark and under irradiation.
The addition of acetonitrile (to 2.5 vol%) after irradiation confirmed the integrity of the ligands by yielding the corresponding [(L)FeII(CH3CN)]2+ complex (1) quantitatively, as shown by comparison with the absorption spectrum of [(L)FeII(CH3CN)]2+ (1) in acetonitrile (Figure 2; see also Figure S14).24, 28 The concomitant formation of 0.5 equivalents of formaldehyde (see the Supporting Information) confirmed that methanol was the source of electrons for the reduction.
The dependence of the photochemistry on wavelength (ee above) indicates that not all of the species (2, 2 a, etc.) present in solution are photoactive (see below). Although the expected S= FeIII (X‐band) EPR signals of 2 were observed at 77 K (see Figure S8), quantification indicates that in deoxygenated methanol, only 40 % of the FeIII is present as a mononuclear S= FeIII−OCH3 complex. The remaining 60 % is EPR‐silent, possibly present in the FeIII−O−FeIII form, for example, 2 a, or as mononuclear complexes with coordination modes that lead to fast electron‐spin relaxation (and hence EPR silence as observed for 3 in acetonitrile; see the Supporting Information). Hence the UV/Vis absorption spectrum of 2 (and 3) in deoxygenated methanol and in acetonitrile is a weighted sum of the spectra of [(L)FeIII−OCH3]2+ (2) (or [(L)FeIII−Cl]2+, 3; see Figures S8 and S23), [(L)FeIII−μ‐O−FeIII(L)]4+ (2 a), and other related species.29
The addition of NaOAc (50 equiv) to 2 in argon‐purged methanol resulted in a slight but immediate change in its UV/Vis absorption spectrum (Figure 3; for 3, see Figure S15), but thereafter no further thermally induced changes were observed. The rate of photoreduction was, however, increased fourfold (Figure 3). Again subsequent addition of acetonitrile (see above, Figure S16) resulted in the quantitative formation of [(L)FeII(CH3CN)]2+ (1), thus confirming the integrity of the ligand (L).
Figure 3.
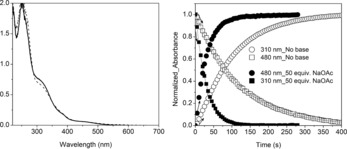
Left: UV/Vis absorption spectrum of 2 in methanol (solid line) and after the addition of NaOAc (50 equiv; dashed line). Right: Comparison of normalized absorbance at 310 and 480 nm over time under irradiation (λ exc=365 nm) with (closed circles and squares) and without (open circles and squares) NaOAc (6.25 mm).
CH3CN did not significantly displace CH3O−, μ‐O2− (see below), or Cl− in the ferric state, as confirmed, for example, by the EPR spectrum of 2, which shows the characteristic low‐spin S= signal (g=2.28, 2.12, 1.96) for FeIII−OCH3 (see Figure S17; see the Supporting Information for further discussion). Nevertheless, photoreduction of 2, 2 a, and 3 was also observed in acetonitrile; however, in contrast to methanol, the initial form of the FeIII complex used played an important role in the observed photochemistry (see below). Furthermore, adventitious water could displace CH3O−, μ‐O2−, or Cl− to form [(L)FeIII−OH]2+, as manifested in weaker signals, g=2.36, 2.16, and 1.94 (see Figure S17).
The photoreduction of 2 in acetonitrile was orders of magnitude slower than in methanol (Figure 4), with a k obs value (from fitting of the change in the absorbance at 310 nm as an exponential decay) of 0.15 s−1 in methanol and 0.0066 s−1 in acetonitrile (with the same incident light flux). The addition of H2O (2 vol% ; see Figure S18) or triflic acid (1.0, 5.0, or 50 equiv; see Figure S19) to 2 in acetonitrile resulted in a substantial decrease in the rate of photoreduction.
Figure 4.
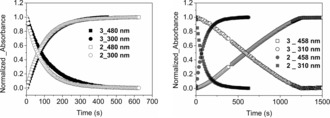
Absorbance of 2 and 3 (0.125 mm) in argon‐purged methanol (left; at 300 and 480 nm) and acetonitrile (right; at 310 and 458 nm) during irradiation (λ exc=365 nm). The initial absorbance at 300/310 and final absorbance at 458/480 nm were used for normalization.
Irradiation of 3 at 365 nm in acetonitrile resulted in an almost linear decrease and increase in absorbance at 310 and 480 nm, respectively, due to formation of 1, and was again much slower than observed in methanol (Figure 4). The lower rate is due to the stronger binding of the chlorido ligand of 3 (see the Supporting Information for a discussion) and hence a reduced extent of exchange with adventitious water to form aqua and dinuclear complexes, such as 2 a. This conclusion was confirmed by the addition of chloride to 2 in acetonitrile, which resulted in a lower rate of reduction. The observed rate is dependent on irradiation power, thus confirming photokinetic control (see Figure S20), and the linear decay indicates that the photoreactive species maintains a steady‐state concentration throughout most of the reaction.
The 1H NMR spectrum of 2 a in CD3CN (see Figure S21) is similar to that reported for its N4Py analogue29 and shows moderate paramagnetic line broadening and shift, which is consistent with strong antiferromagnetic coupling of the FeIII centers, and also further confirmed by the absence of signals in its EPR spectrum at 77 K (see Figure S22). The UV/Vis absorption spectrum of 2 a in anhydrous acetonitrile shows the strong absorption at 312 nm (see Figure S23), which has been assigned as an oxo → Fe charge‐transfer band,30 with symmetric and asymmetric bands of a near‐linear Fe−O−Fe core31 at 407 and 810 cm−1, respectively, observed in its resonance Raman (λ exc=355 nm) spectrum (see Figure S24). The data confirm that the complex retains its dinuclear structure in anhydrous acetonitrile, in contrast to the equilibration with mononuclear complexes observed in methanol (see above).
Irradiation of 2 a in anhydrous acetonitrile resulted in an increase in the absorbance at 458 nm due to formation of the FeII complex (1). At higher concentrations, that is, 0.5 mm, an absorption band at 686 nm, characteristic of [(L)FeIV=O]2+ (4), appeared also (Figures 5; see also Figure S25). The addition of excess H2O to 2 a in acetonitrile had a minor effect on the resonance Raman and EPR spectra (see Figure S22 and S24, respectively), thus indicating that the dinuclear structure is largely retained, but accelerated the rate and extent of the increase in absorbance at 686 nm (Figure 5; see also Figure S26). The subsequent decrease in absorbance at 686 nm after 300 s is due to the photochemical reduction of [(L)FeIV=O]2+ formed.25 The absence of [(L)FeIV=O]2+ under irradiation of 2 a at lower concentrations in acetonitrile (see Figure S25) or in methanol (see Figure S6) is expected considering its low molar absorptivity (400 m −1 cm−1) and its own photoreactivity.25 At higher concentrations of 2 a in acetonitrile, at which the absorbance at 365 nm is above 2, the inner‐filter effect allows only partial penetration of light into the solution and the buildup of a significant steady‐state concentration of 4 within the bulk.
Figure 5.
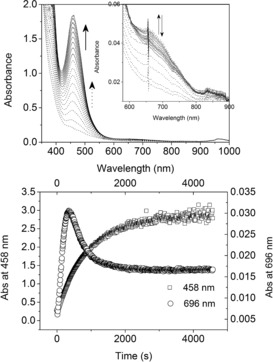
Top: UV/Vis absorption spectrum of 2 a (0.5 mm) in acetonitrile with H2O (10 vol%) during the first 1000 s of irradiation (365 nm). Bottom: Absorbance at 458 (left y‐axis) and 686 nm (right y‐axis) over time during irradiation.
Overall, the non‐heme iron(III) complexes 2, 2 a, and 3 equilibrate rapidly with argon‐purged methanol and show identical photochemical reduction to the FeII oxidation state without ligand degradation. Both EPR spectroscopy and the wavelength dependence of Φ indicate that there are several species present in solution, not all of which are photochemically reactive. In non‐heme systems, the equilibrium between mononuclear and μ‐oxido‐bridged dinuclear FeIII complexes with pentadentate ligands (N4Py, P2DA, 6‐OC6H4‐TPA, etc.),29, 32, 33 has been shown earlier to be rapid. Addition of base (NaOAc) and proton sources (H2O or TfOH) shifts the equilibrium towards complexes, such as mononuclear FeIII−OH and FeIII−OH2 and dinuclear FeIII−O−FeIII complexes. In the present reaction, conditions which favor dimer formation (base addition) are accompanied by an increase in the rate of photoreduction, while an added proton source or added chloride favor the formation of mononuclear FeIII complexes and retard photoreduction. A possible mechanism for the photoreduction is shown in Scheme 2.
Scheme 2.
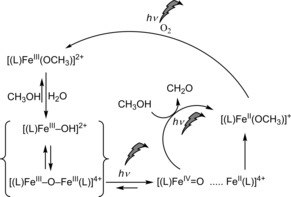
Overall scheme for the catalytic oxidation of methanol under irradiation.
Photoinduced heterolysis was reported first by Richman and co‐workers. In the case of μ‐oxido‐bridged diiron(III) porphyrin complexes, visible irradiation resulted in the reduction of both FeIII centers to the FeII redox state via an intermediate FeIV/FeII species34 in the presence of oxidizable substrates;21, 22 reoxidation of the dinuclear FeII complex was not spontaneous, thus limiting the potential for catalytic turnover. In the absence of substrates with weak C−H bonds, the quantum yield for the reduction was negligible due to rapid recombination of the FeIV=O/FeII centers to the FeIII−O−FeIII state. Karlin and co‐workers23 have shown that photocatalytic oxidation and aromatic dehalogenation are possible with turnover by using a nonsymmetric dinuclear FeIII complex based on a non‐heme FeIII unit and an FeIII porphyrin, which were bridged by both a μ‐oxido unit and a covalent link between the heme and non‐heme ligands. As in the double iron(III) porphyrin systems,34 an intermediate FeIV=O/FeII species was observed by flash photolysis. The FeIV=O/FeII species was sufficiently long‐lived to react with organic substrates with relatively strong C−H bonds, and the FeIII−μ‐O−FeIII complex was recovered subsequently by aerobic oxidation. The formation of tetranuclear complexes bearing an inert non‐heme FeIII−μ‐O−FeIII unit was observed especially in dechlorination reactions.
For heme cofacial porphyrin μ‐oxido‐bridged diiron(III) complexes, irradiation into the oxido → FeIII charge‐transfer band35 results in photoinduced disproportionation to FeII and FeIV=O monomers.21, 22, 34 In the present non‐heme system, an analogous model would see an FeIV=O species formed upon excitation of 2 a in methanol or acetonitrile, which can recombine with the FeII fragment to reform 2 a or react with methanol to form methanal and a second equivalent of an FeII complex. The electronic nature of the photoreaction and the thermodynamic energies of possible dissociation products were explored by DFT methods (see the Supporting Information). In brief, the electronic structure of the μ‐oxido‐bridged dinuclear complex 2 a and all accessible spin states revealed an antiferromagnetically coupled ground state (see Table S4 in the Supporting Information), in accordance with the experimental data.29 The excited states of 2 a are predicted to result in Fe−O bond elongation owing to the charge‐transfer character of the spin‐allowed transitions to low‐lying excited states. For possible dissociation products formed following photoexcitation, that is, {(L)FeIII−O+(L)FeIII} and {(L)FeIV=O+(L)FeII}, a triplet ground state for (L)FeIV=O and quartet ground state for (L)FeIII−O is indicated, whereas for (L)FeIII and (L)FeII low‐spin ground states were found both with and without coordinated CH3CN (see Tables S5–S10). The electronic and Gibbs free energies indicate that both dissociation pathways are stabilized through solvent coordination; however, the (L)FeIV=O+(L)FeII charge‐transfer path is substantially more favorable. Importantly, when coordination of CH3CN is included explicitly, both 2 a and (1+FeIV=O) are similar in energy (see Tables S11–S14).
The oxidation of [(MeN4Py)FeII(CH3CN)]2+ (1) in methanol to its FeIII state (i.e., 2) with O2 as the terminal oxidant was reported by our group earlier with visible and UV light.24 In the present study, we have shown that the iron(III) complexes of the ligand N4Py undergo reduction upon irradiation in methanol. This observation prompted us to explore whether both reactions could proceed under the same conditions simultaneously and thereby enable the catalytic use of O2 as a terminal oxidant. Irradiation of [(MeN4Py)FeII(CH3CN)]2+ (1) at 365 nm in methanol at room temperature under aerobic conditions resulted in a steady increase in the amount of formaldehyde formed over time (Scheme 2 and Figure 6) with a relatively minor decrease in visible absorbance (33 % after irradiation for 3 h; see Figure S29). Over 50 turnovers were observed with respect to 1, thus confirming that the process is catalytic.
Figure 6.
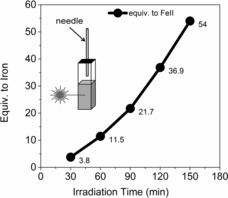
Formaldehyde formation over time under irradiation under aerated conditions with 1 (0.125 m) in methanol.
In summary, the photoreduction of non‐heme FeIII complexes proceeds via an intermediate formed from the mononuclear complexes 2 and 3 or the μ‐oxido‐bridged diiron(III) complex 2 a. DFT calculations indicate that photoexcitation of 2 a would result in the population of antibonding orbitals and drive heterolytic cleavage to form a five‐coordinate FeII species and an FeIV=O species in an excited electronic state (HS) rather than in its intermediate‐spin (IS) ground state. Recombination to reform 2 a competes with solvent coordination (e.g., in acetonitrile to form 1) and oxidation of solvent (e.g., methanol to methanal) by the FeIV=O species formed. This mechanism is analogous to those proposed for the heme FeIII systems reported earlier. Importantly, we show that the present system can use light to achieve a full catalytic cycle in methanol without the need for a secondary photosensitizer. In the presence of O2, the FeII species formed undergoes light‐driven oxidation by O2 to close a full photocatalytic cycle with a single catalyst, and oxidation of methanol with O2 occurs with high turnover numbers. The present system opens opportunities for selective photocatalytic reactions with a single catalyst.
In memory of John J. McGarvey
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The COST association action CM1305 ECOSTBio (STSM grant 38503), the European Research Council (ERC 279549, WRB), Labex ARCANE (ANR‐11‐LABX‐003), the Serbian Ministry of Science (OI172035), and the Chinese Scholarship Council (CSC) are acknowledged for financial support. We thank the Center for Information Technology of the University of Groningen for their support and for providing access to the Peregrine high‐performance computing cluster. We thank Prof. Edwin Otten for X‐ray structural analysis of 3, and Dr. Carole Duboc and Dr. Sandeep Padamati for recording X‐band EPR spectra of 3 at 4 K.
J. Chen, S. Stepanovic, A. Draksharapu, M. Gruden, W. R. Browne, Angew. Chem. Int. Ed. 2018, 57, 3207.
Contributor Information
Dr. Apparao Draksharapu, Email: adraksha@umn.edu.
Prof. Dr. Maja Gruden, Email: gmaja@chem.bg.ac.rs.
Prof. Dr. Wesley R. Browne, Email: w.r.browne@rug.nl.
References
- 1. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yoon T. P., Ischay M. A., Du J., Nat. Chem. 2010, 2, 527–532. [DOI] [PubMed] [Google Scholar]
- 3. Ooyama Y., Harima Y., Eur. J. Org. Chem. 2009, 2903–2934. [Google Scholar]
- 4. Hoffmann M. R., Martin S., Choi W., Bahnemann D. W., Chem. Rev. 1995, 95, 69–96. [Google Scholar]
- 5. Steel P. J., Constable E. C., J. Chem. Soc. Dalton Trans. 1990, 1389–1396. [Google Scholar]
- 6. Roundhill D. M., Photochemistry and Photophysics of Metal Complexes, Springer US, New York, 1994. [Google Scholar]
- 7. Hoffmann N., ChemSusChem 2012, 5, 352–371. [DOI] [PubMed] [Google Scholar]
- 8. Adamson A. W., Waltz W. L., Zinato E., Watts D. W., Fleischauer P. D., Lindholm R. D., Chem. Rev. 1968, 68, 541–585. [Google Scholar]
- 9. Perutz R. N., Procacci B., Chem. Rev. 2016, 116, 8506–8544. [DOI] [PubMed] [Google Scholar]
- 10. Company A. et al., J. Am. Chem. Soc. 2014, 136, 4624–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kotani H., Suenobu T., Lee Y.-M., Nam W., Fukuzumi S., J. Am. Chem. Soc. 2011, 133, 3249–3251. [DOI] [PubMed] [Google Scholar]
- 12. DeRosa M. C., Crutchley R. J., Coord. Chem. Rev. 2002, 233–234, 351–371. [Google Scholar]
- 13. Ashen-Garry D., Selke M., Photochem. Photobiol. 2014, 90, 257–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abdel-Shafi A. A., Worrall D. R., Ershov A. Y., Dalton Trans. 2004, 30–36. [DOI] [PubMed] [Google Scholar]
- 15. Hergueta-Bravo A., Jiménez-Hernández M. E., Montero F., Oliveros E., Orellana G., J. Phys. Chem. B 2002, 106, 4010–4017. [Google Scholar]
- 16. Whitten D. G., Acc. Chem. Res. 1980, 13, 83–90. [Google Scholar]
- 17. Demas J. N., Harris E. W., McBride R. P., J. Am. Chem. Soc. 1977, 99, 3547–3551. [Google Scholar]
- 18. Šima J., Makáňová J., Coord. Chem. Rev. 1997, 160, 161–189. [Google Scholar]
- 19. Hatchard C. G., Parker C. A., Proc. R. Soc. London Ser. A 1956, 235, 518–536. [Google Scholar]
- 20. Abrahamson H. B., Rezvani A. B., Brushmiller J. G., Inorg. Chim. Acta 1994, 226, 117–127. [Google Scholar]
- 21. Richman R. M., Peterson M. W., J. Am. Chem. Soc. 1982, 104, 5795–5796. [Google Scholar]
- 22. Peterson M. W., Rivers D. S., Richman R. M., J. Am. Chem. Soc. 1985, 107, 2907–2915. [Google Scholar]
- 23. Wasser I. M., Fry H. C., Hoertz P. G., Meyer G. J., Karlin K. D., Inorg. Chem. 2004, 43, 8272–8281. [DOI] [PubMed] [Google Scholar]
- 24. Draksharapu A., Li Q., Roelfes G., Browne W. R., Dalton Trans. 2012, 41, 13180–13190. [DOI] [PubMed] [Google Scholar]
- 25. Chen J., Draksharapu A., Harvey E., Rasheed W., Que L., Browne W. R., Chem. Commun. 2017, 53, 12357–12360. [DOI] [PubMed] [Google Scholar]
- 26. Maafi M., Maafi W., Int. J. Photoenergy 2015, 454895–454812. [Google Scholar]
- 27. Maafi W., Maafi M., Int. J. Pharm. 2013, 456, 153–164. [DOI] [PubMed] [Google Scholar]
- 28. Draksharapu A., Li Q., Logtenberg H., van den Berg T. A., Meetsma A., Killeen J. S., Feringa B. L., Hage R., Roelfes G., Browne W. R., Inorg. Chem. 2012, 51, 900–913. [DOI] [PubMed] [Google Scholar]
- 29. Roelfes G., Lubben M., Chen K., Ho R. Y. N., Meetsma A., Genseberger S., Hermant R. M., Hage R., Mandai S. K., V. G. Young, Jr. , Zang Y., Kooijman H., Spek A. L., L. Que, Jr. , Feringa B. L., Inorg. Chem. 1999, 38, 1929–1936. [DOI] [PubMed] [Google Scholar]
- 30. Kurtz D. M., Chem. Rev. 1990, 90, 585–606. [Google Scholar]
- 31. Sanders-Loehr J., Wheeler W. D., Shiemke A. K., Averill B. A., Loehr T. M., J. Am. Chem. Soc. 1989, 111, 8084–8093. [Google Scholar]
- 32. Lange S. J., Miyake H., Que L., J. Am. Chem. Soc. 1999, 121, 6330–6331. [Google Scholar]
- 33. McDonald A. R., Guo Y., Vu V. V., Bominaar E. L., Münck E., Que L., Chem. Sci. 2012, 3, 1680–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hodgkiss J. M., Chang C. J., Pistorio B. J., Nocera D. G., Inorg. Chem. 2003, 42, 8270–8277. [DOI] [PubMed] [Google Scholar]
- 35. Czernuszewicz R. S., Macor K. A., Li X. Y., Kincaid J. R., Spiro T. G., J. Am. Chem. Soc. 1989, 111, 3860–3869. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary