

Abstract
Brown adipose tissue (BAT) has received enormous scientific and lay attention in the recent past as its thermogenic, energy‐consuming capacities represent prime candidates for therapeutic interventions toward obesity, glucose intolerance, and diabetes even in humans. The overall positive effects of BAT activation and recruitment on systemic energy homeostasis have been largely attributed to the inherent ability of brown adipocytes to combust fatty acid and glucose energy substrates through mitochondrial uncoupling, driven by the unique expression of uncoupling protein 1 (UCP1). Two recent reports by Boutant et al and Mahdaviani et al now identify the GTPase mitofusin (Mfn) 2 as a key determinant of BAT thermogenic function that is largely independent of its previously described role in mitochondrial fusion [1,2].
Subject Categories: Membrane & Intracellular Transport, Metabolism
The current obesity epidemic has focused a great deal of attention on mechanisms controlling energy balance. While energy uptake is affected by diet and nutrient absorption, on the other side of the equation, energy expenditure is determined by basal metabolism, physical activity, and adaptive heat production (thermogenesis). In this context, BAT, which dissipates chemical energy via non‐shivering thermogenesis by combusting fatty acid and glucose energy substrates, emerged as an important player for thermogenesis‐mediated energy expenditure. Indeed, as the amount of BAT declines with increasing body mass index and years of age and as women have a greater BAT mass and glucose uptake activity of BAT, a role for BAT in adult human metabolism has been suggested 3. Its thermogenic property relies predominantly on the action of UCP1, which is activated by fatty acids and subsequently uncouples the mitochondrial electron transport chain activity from ATP formation. However, regulation of UCP1 does not seem to fully explain thermogenic activity, and indeed, UCP1‐independent non‐shivering thermogenesis via futile cycles has been described in muscle and adipose tissue 4. Strikingly, recent studies revealed the vast capacity of BAT for triglyceride clearance and glucose disposal 5. This let us and others conclude that BAT activation/recruitment has beneficial effects on systemic metabolism through its thermogenic potential.
Figure 1. Metabolic programming in the presence and absence of Mfn2 in brown adipocytes.
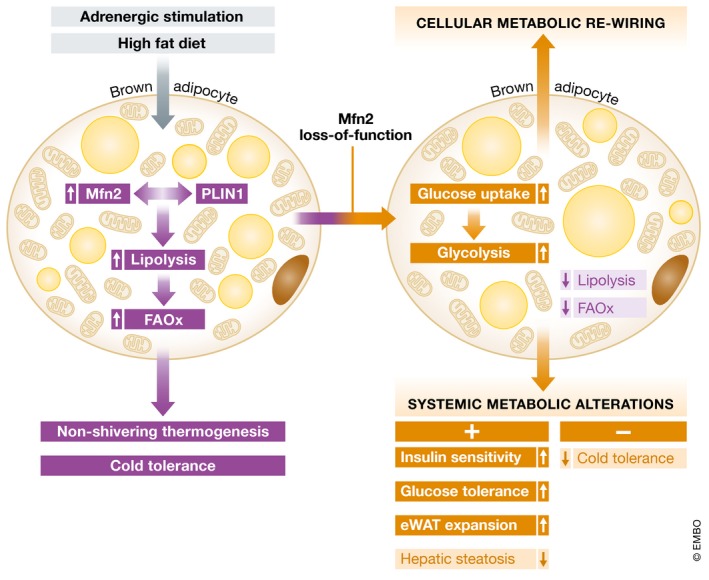
(Left) Mfn via its interaction with perilipin (PLIN) 1 ensures proper lipolysis and fatty acid oxidation (FAOX) in response to adrenergic or dietary stimuli, thereby ensuring cold‐/diet‐induced thermogenesis and cold tolerance in mice. (Right) Mfn2 loss of function (LOF) leads to intra‐adipose metabolic re‐wiring as indicated by a diminished lipolytic response but enhanced glucose uptake and glycolytic breakdown. As a result, systemic glucose tolerance, insulin sensitivity and hepatic steatosis are improved while cold tolerance is diminished.
Mitochondria play an essential role in brown adipocyte activity, and mitochondrial fission and fusion are critical in maintaining functional mitochondria. While fusion helps mitigate stress of partially damaged mitochondria, fission creates new mitochondria but can also lead to apoptosis during high levels of cellular stress.
While Mfn2 had been implicated in the fusion process of the outer mitochondrial membrane already 6, the two new studies by Boutant et al and Mahdaviani et al now report a new role for Mfn2 in BAT functionality using adipose tissue and BAT‐specific knockout mice, respectively 1, 2. Mfn2 is highly expressed in BAT and abrogation of Mfn2 in adipose tissue and/or BAT leads to thermogenic dysfunction, characterized by defects in lipid mobilization and fatty acid oxidation, overall leading to reduced (UCP1‐dependent) respiratory capacity. Interestingly, Mfn2 seems to fulfill its pro‐thermogenic action independent from its role in mitochondrial fission/fusion events. Indeed, Mfn2 was found to be responsible for a functional lipid droplet–mitochondria interaction through direct interaction with the adipose‐specific lipid droplet‐associated protein perilipin 1. The Mfn2‐perilipin 1 interaction was critical for an appropriate lipolytic response and the subsequent induction of mitochondrial respiration 2.
Intriguingly, despite thermogenic and fatty acid oxidation defects, mice deficient in adipose tissue Mfn2 were resistant against diet‐induced obesity phenotypes, including improvements in glucose tolerance and insulin sensitivity as compared with wild‐type littermates. This seemingly paradoxical finding could be explained by a cell‐intrinsic metabolic re‐wiring within the brown adipocytes as both studies report on a highly induced glycolytic flux in Mfn2‐deficient cells, responding in a compensatory manner to impairments in fatty acid usage. As a consequence, Mfn2 deficiency significantly triggered BAT‐dependent glucose clearance and ameliorated hepatic steatosis, thereby overall improving metabolic dysfunction upon dietary challenges 2. Of note, in the BAT‐specific Mfn2 knockout animals, clear sex‐specific differences were observed as demonstrated by a loss of body fat and lean mass in female and male animals, respectively 1.
These two studies now place Mfn2 into a broader metabolic context. While whole‐body Mfn2 knock out is embryonically lethal, tissue‐specific knockout mice have been implicated in metabolic control already. Indeed, ablation of Mfn2 specifically in the liver promotes glucose intolerance and enhanced hepatic gluconeogenesis 7, while Mfn2 deficiency in hypothalamic POMC neurons leads to decreased energy expenditure and hyperphagia, resulting in an obese phenotype 8. In combination with the two current reports by Boutant et al and Mahdaviani et al, the combined data suggest Mfn2 to play a rather protective role against non‐favorable metabolic conditions across a number of distinct organ compartments 1, 2. In this scenario, the reported elevated levels of Mfn2 in BAT of high fat diet‐fed mice 1 could be interpreted as a rather counter‐regulatory consequence toward excessive caloric intake. It will be interesting to determine whether Mfn2‐dependent metabolic control requires mitochondrial fusion events in distinct tissues or can recruit alternative mechanisms, for example, lipid droplet remodeling, in a cell‐type specific manner. In any case, the regulation of mitochondrial integrity/functionality seems to be centerpiece for the impact of Mfn2 on metabolic control. However, a number of discrepancies between the two recent papers will require attention by investigators in order to evaluate the potential of Mfn2 to serve as a checkpoint in mitochondrial‐centered therapeutic avenues in metabolic diseases: (i) The general adipose tissue deficiency of Mfn2 is characterized by improvements in liver function and a blunting effect of thermoneutrality on improvements in insulin sensitivity while BAT‐specific Mfn2 deletion showed no alterations in liver parameters and thermoneutrality even further amplified the insulin‐sensitizing effects of Mfn2 deficiency. Of note, the presence or absence of adipose Mfn2 effects on liver parameters correlated with existing or not existing inductions of circulating FGF21 levels, respectively, indicating that Mfn2 may differentially control FGF21 expression in white vs. brown adipocytes. (ii) The study by Mahdaviani et al provided significant differences between male and female mice that have not yet been explored beyond a descriptive stage but clearly provide intriguing and relevant starting points for future studies 1. (iii) Both studies do not fully address the (patho)physiological conditions upon which Mfn2 levels are significantly altered to mimic the knock out (or overexpression) experimental conditions. This holds particularly true for the relevance of the reported findings for the human system.
Despite these important but still unresolved issues, the studies by Boutant et al and Mahdaviani et al help us to make a number of general conclusions for future BAT research 1, 2. It becomes obvious that an enhancement of mitochondrial fission is probably not sufficient to promote uncoupling potential, despite earlier findings that mitochondrial fission is needed to increase brown adipocyte energy dissipation 9. Moreover, these reports on Mfn2 adipose tissue function further underline the notion that systemic insulin sensitivity can be unchained from thermogenic capacity which aligns well with previous studies describing preventive effects of genetic manipulations on the development of insulin resistance despite a decreased thermogenic capacity 10. Overall, it is tempting to speculate that cellular metabolic re‐wiring in BAT can be principally used to disentangle organismic adaptations to either cold or nutrient exposure which may become relevant in the search for new therapeutic options in the fight against obesity and associated complications.
See also: https://doi.org/10.15252/embr.201643827 (July 2017) and https://doi.org/10.15252/embj.201694914 (June 2017)
References
- 1. Mahdaviani K, Benador I, Su S et al (2017) EMBO Rep 18: 1123–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boutant M, Kulkarni SS, Joffraud M et al (2017) EMBO J 36: 1543–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Algire C, Medrikova D, Herzig S (2013) Biochim Biophys Acta 1831: 896–904 [DOI] [PubMed] [Google Scholar]
- 4. Bertholet AM, Kazak L, Chouchani ET et al (2017) Cell Metab 25: 811–822 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nedergaard J, Bengtsson T, Cannon B (2011) Cell Metab 13: 238–240 [DOI] [PubMed] [Google Scholar]
- 6. Bach D, Pich S, Soriano FX et al (2003) J Biol Chem 278: 17190–17197 [DOI] [PubMed] [Google Scholar]
- 7. Sebastian D, Hernandez‐Alvarez MI, Segales J et al (2012) Proc Natl Acad Sci USA 109: 5523–5528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schneeberger M, Dietrich MO, Sebastian D et al (2013) Cell 155: 172–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wikstrom JD, Mahdaviani K, Liesa M et al (2014) EMBO J 33: 418–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schoiswohl G, Stefanovic‐Racic M, Menke MN et al (2015) Endocrinology 156: 3610–3624 [DOI] [PMC free article] [PubMed] [Google Scholar]