

Summary
Considerable progress has been made in understanding the influence of physical and genetic factors on the patterns of cell division in various model systems. However, how each of these factors directs changes in subcellular structures has remained unclear. Generic machineries for the execution of cell expansion and division have been characterized, but how these are influenced by genetic regulators and physical cell properties remains an open question. To a large degree, the complexity of growing post‐embryonic tissues and a lack of precise predictability have prevented the extraction of rigid correlations between subcellular structures and future orientation of cell division. The Arabidopsis embryo offers an exquisitely predictable and simple model for studying such correlations, but so far the tools and methodology for studying subcellular structures in the early embryo have been lacking. Here, we describe a set of markers to visualize a range of subcellular structures in the early Arabidopsis embryo. We have designed a series of fluorescent cellular reporters optimized for embryos, and demonstrate the effectiveness of using these ‘ACE’ reporters with simple three‐dimensional imaging procedures that preserve delicate cellular structures. We describe the ontogeny of subcellular structures in the early embryo and find that central/peripheral cell polarity is established much earlier than suspected. In addition, we show that the actin and microtubule cytoskeleton has distinct topologies in the embryo. These tools and methods will allow detailed analysis of the events of cellular reorganization that underlie morphogenesis in the Arabidopsis embryo.
Keywords: embryogenesis, Arabidopsis thaliana, cell biology, oriented cell division, imaging
Significance Statement
The early Arabidopsis embryo represents a predictable and compact model for studying the control of cellular reorganization during plant development. This paper describes a genetic toolkit and microscopy methodology to visualize subcellular structures in the Arabidopsis embryo.
Introduction
Morphogenesis in multicellular organisms depends on the decisions of individual cells about the direction in which growth or division occurs. Particularly in plants, where rigid cell walls prevent cell migration, pre‐mitotic control of cell division orientation makes a crucial contribution to organized three‐dimensional development (Sablowski, 2016). A key question in plant developmental biology, therefore, is what regulates the decision of individual cells about the direction in which to divide? It has been established that three major factors drive the orientation of cell division: (i) the initial geometry of a cell biases the preferred orientation of cell division (Besson and Dumais, 2011); (ii) tissue‐scale stresses influence cell division orientation (Landrein et al., 2015; Louveaux et al., 2016); and (iii) genetic factors can drive defined orientations of cells divisions such that cell division orientation is an output of developmental regulations (De Rybel et al., 2013; van Dop et al., 2015). Whatever triggers the oriented cell division, the input signal must be translated into cellular re‐organization to facilitate re‐orientation of division plane. A key challenge is therefore to identify the cellular components that are being modulated by regulatory input to effect cell division orientation.
From various different model systems it is clear that there is a core cell division machinery that executes pre‐mitotic orientation cues (Rasmussen et al., 2011; Buschmann and Zachgo, 2016). This core mechanism – the mitotic and cytokinetic machinery – forms a spindle perpendicular to the preprophase band to segregate chromosomes. Following chromosome segregation, the midline of the spindle is targeted by Golgi‐derived vesicles along microtubule and actin filaments, thus forming a phragmoplast to build a cell plate between the newly divided nuclei (Smertenko et al., 2017).
Thus, three key cytoskeletal indicators of oriented cell division are (i) the preprophase band, whose position and orientation forecast the phragmoplast (Pickett‐Heaps and Northcote, 1966a,b), (ii) cortical actin and actin cables anchoring the spindles during mitosis and later daughter nuclei during cytokinesis (Traas et al., 1987; Lloyd and Traas, 1988), and (iii) phragmoplast that determines the physical division plane (Gunning and Wick, 1985). These cytoskeletal structures follow both the microtubule and actin cytoskeleton that are important for determining the division plane (Rasmussen et al., 2013). Indeed there is ample evidence for the involvement of these structures in different contexts (Buschmann and Zachgo, 2016). How the cytoskeleton is manipulated to modify the orientation of division, however, is not well understood.
Essentially all organelles and cellular structures are coordinately orchestrated to allow cell division (Boruc and Van Damme, 2015; Smertenko et al., 2017). Thus, each of these, especially the vacuole (Cutler and Ehrhardt, 2002) and the nucleus (Murata and Wada, 1991), can either facilitate or constrain choices for cell division orientation. The roles that these structures play in oriented cell division have remained largely unexplored. In addition to subcellular structures that mediate the execution of cell division, the perception of cellular coordinates relative to the body–tissue axis could serve as a reference for protein polar transport–localization and the determination of cell division orientation. How polarity information is established and integrated into the division plane is also far from clear.
Analysis of the contributions of any cellular structure to oriented cell division requires that cellular morphology is relatively simple and/or predictable. Alternatively, structures should be followed over time in order to draw correlations between changes in these structures and future cell division orientation. Thus far, most analysis of oriented cell division has been performed in meristematic tissues of generally complex topologies with limited predictability (Sablowski, 2016). Here we use the early Arabidopsis embryo as a model for oriented cell division. During the first rounds of cell division, a highly regular pattern of cells emerges from which the precursors of all seedling tissues and organs are formed (Palovaara et al., 2016). From earlier 3D analysis, it has become clear that some cell divisions are symmetric, following only the geometry of the cell, while others are highly asymmetric, following tight genetic control to deviate from a default, symmetric division (Yoshida et al., 2014). In this system, the choice of cell division orientation is influenced by the plant hormone auxin, whose activity favors asymmetric division (Yoshida et al., 2014). While this is an excellent model system for studying symmetric and asymmetric division, as well as switches in cell division orientations, a key open question is which cellular structures are subject to auxin‐dependent regulation. However, thus far it has been impossible to address this question, as few if any tools were available to visualize subcellular structures in early embryos.
Here we have generated the tools required to answer this important question and to study cellular (re)organization in early Arabidopsis embryos.
Results
A set of fluorescent cellular markers for embryogenesis
To develop a panel of markers for subcellular structures in the Arabidopsis embryo we first selected a set of established reporters (Table S1 in the online Supporting Information) that includes: (i) uniformly distributed plasma membrane proteins for highlighting cell contours (Nelson et al., 2007; Geldner et al., 2009); (ii) plasmodesmata‐specific proteins for evaluating connectivity between cells (Simpson et al., 2009); (iii) organelle‐specific proteins labeling endosomes, the trans‐Golgi network or tonoplast (Geldner et al., 2009); (iv) nuclear pore markers (Tamura et al., 2010); (v) cytoskeletal markers labeling either F‐actin (Riedl et al., 2008; Era et al., 2009) or tubulin (Ueda et al., 1998). In addition to the dynamics of cellular components, in order to evaluate the establishment of polarity, which could provide spatial information to the cell, we also include (vi) a set of polar‐localized proteins labeling the apical (Truernit et al., 2012), basal (Dong et al., 2009), central (Takano et al., 2010) or peripheral (Takano et al., 2010) plasma membrane in post‐embryonic tissues. To achieve embryo‐specific expression of reporters, each was expressed from the embryo‐specific Arabidopsis WOX2 promoter (Breuninger et al., 2008) as well as from the meristematic Arabidopsis RPS5A promoter (Weijers et al., 2001). These markers were named ACE (for ‘Arabidopsis cellular markers for embryogenesis’; Table S1). Though ACE driven by pRPS5A (ACE‐R) were uniformly expressed in the complete embryo, the high activity of pRPS5A in the endosperm and maternal tissues made imaging difficult because fluorescence intensity was very high in cells surrounding embryos in embryo preparations (Figure 1a). In contrast, pWOX2‐driven ACE markers (ACE‐W) were specifically expressed in the pro‐embryo, two uppermost suspensor cells and the chalaza from the two‐cell to early globular stage, with a higher expression level than ACE‐R (Figure 1b–i). After the early globular stage, activity of pWOX2 started to decrease in the basal part of the embryo and two topmost cells in the suspensor (Figure 1h). These properties made ACE‐R more suitable for dissecting cell biology in the suspensor and the embryo proper after early globular stage and in seed development while ACE‐W were more suitable for dissecting cell biology in the early embryo proper and thus were used here unless mentioned otherwise.
Figure 1.
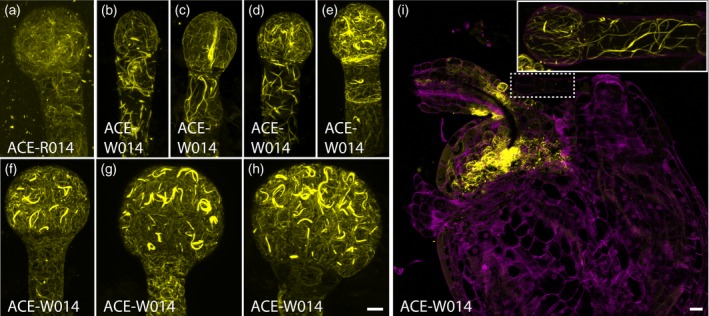
Expression of Arabidopsis cellular markers for embryogenesis (ACE) driven by pRPS5A (ACE‐R) and by pWOX2 (ACE‐W) during embryogenesis
(a) Maximum intensity projection of actin filaments labeled by ACE‐R14 (Lifeact:tdTomato) in the 16‐cell embryo. (b)–(h) Maximum intensity projections of actin filaments labeled by ACE‐W14 (Lifeact:tdTomato) in 1‐cell (b), 2‐cell (c), 4‐cell (d), 8‐cell (e), 16‐cell (f), early‐globular (g), and late‐globular (h) embryos. (i) Overview of ACE‐W14 (Lifeact:tdTomato) expression in the seed. Different acquisition settings were used to accommodate high expression in chalaza. Inset: maximum intensity projection of the 2‐cell embryo in the main panel marked by a dashed box with the acquisition setting used for embryos. Scale bar for (a)–(h) = 5 μm. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Optimizing preservation of delicate cellular structures
Using common microscopy procedures (Llavata‐Peris et al., 2013; Musielak et al., 2015) we examined the expression of ACE‐W lines in the embryos. We found that particularly lines that mark delicate cellular structures did not show the expected patterns. Microtubule and actin architectures were either fragmentary or completely depolymerized, with reporter protein only in the cytosol (Figure 2a, b). Intact microtubule and actin architectures could be observed in the corresponding ACE‐R root apical meristem (Figure S1), suggesting that the markers’ capacities to label each component were not compromised. Given that a structured cytoskeleton must exist in embryos, we conclude that a different procedure is required to preserve such structures.
Figure 2.

Effects of mounting media on cytoskeletal integrity in embryos
(a), (b) Maximum intensity projections of microtubule (a; ACE‐W15; mGFP:AtTUA6) and actin (b; ACE‐W14; Lifeact:tdTomato) markers in 8‐cell embryos in 5% glycerol and 4% paraformaldehyde in 1 × PBS solution. (c), (d) Depth color coding key of all panels illustrated by rotating embryos in (a) and (b), respectively, by 90°. The look‐up table shows color values corresponding to the depth of the image in the z‐dimension. (e)–(l) Maximum projections of microtubule (e–h; ACE‐W15; mGFP:AtTUA6) and actin (i–l; ACE‐W14; Lifeact:tdTomato) markers in 8‐cell embryos imaged in embryo general mounting solution (EGM) (e, i), embryo microtubule mounting solution (EMTM) (f, j), embryo microtubule counterstaining solution (EMTC) (g), embryo general counterstaining solution (EGC) (k) or EMTM followed by the addition of EMTC (h, l).
All images from the same markers were acquired with the same acquisition settings. Scale bar = 5 μm. [Colour figure can be viewed at http://wileyonlinelibrary.com]
To preserve microtubule architectures for qualitative and quantitative analysis we tested various mounting media (Table S2) and various sample preparation procedures. Intact microtubule architectures in live protoplasts and late embryos had been achieved with 0.55 m mannitol (Shi et al., 2011) and 10% glucose (Wang and Huang, 2014), respectively. However, although embryos extracted with the established procedure (Llavata‐Peris et al., 2013; Musielak et al., 2015) with 5% glycerol in 1 × PBS solution replaced with either 0.55 m mannitol or 10% glucose showed no sign of plasmolysis nor swelling with preserved spindles and preprophase band, intact cortical microtubule arrays were rarely found. In the rare cases that the cortical microtubule arrays remained intact, they showed no clear sign of deterioration after 60 min of exposure in 0.55 m mannitol and 10% glucose (Figure 2e). Therefore, since both 0.55 m mannitol and 10% glucose provided conditions to preserve microtubule architectures in the embryo for at least 60 min, microtubule depolymerization must have occurred before exposure of the embryos to the mounting medium, probably due to the pressure applied during the extraction of embryos from seeds. To stabilize microtubules, microtubule stabilizing buffer (MTSB) (Soltys and Borisy, 1985) and 10 μm microtubule stabilizer paclitaxel (Taxol) (Baskin et al., 1994) were included, creating embryo microtubule mounting (EMTM) solution. While Taxol treatment could alter microtubule architecture and cause the swelling of cells after prolonged (2–24 h) treatment (Baskin et al., 1994; Mathur and Chua, 2000; Bannigan et al., 2006), no significant differences in microtubule orientation or microtubule anisotropy were detected between root epidermal cells incubated with or without Taxol, even after 90 min of treatment (Figure S2, Table S3). Seeds incubated in EMTM solution for 15 min before embryo extraction preserved the microtubule architectures homogeneously labelled by TUA6‐GFP in over 90% (n = 74/81) of extracted embryos for at least 60 min after extraction (Figure 2f). However, while this procedure allowed visualization of microtubules, the topology of the network relative to cell shape could not be inferred without counterstaining the plasma membrane or the cell wall. Thus, we included 0.1% of the cell wall stain Renaissance SR2200 in EMTM (named the embryo microtubule counterstaining solution, EMTC). This led to depolymerization of cortical microtubules (Figure 2g) and suggests that Renaissance SR2200 may compromise Taxol‐dependent microtubule stabilization. To circumvent the negative effect of SR2200, seeds were first incubated in EMTM for 15 min to stabilize microtubules, and an equal volume of EMTC was then added immediately before embryo extraction. With this modification, microtubule architectures were preserved (Figure 2h).
Actin architectures, on the other hand, could be preserved in all tested mounting media except for 5% glycerol in 1 × PBS solution with actin filaments and cables homogenously labelled by Lifeact (Figure 2i–k). Since Taxol specifically targets tubulin (Manfredi et al., 1982), we thus tested omitting MTSB and Taxol in EMTM and EMTC [named embryo general mounting solution (EGM) and embryo general counterstaining solution (EGC), respectively] and found no clear negative effect on the integrity of actin architecture (Figure 2l).
Having established optimized imaging media and methods, we next imaged a panel of ACE‐W for subcellular structures (Figure S3). Reporters marking plasma membrane domains, plasmodesmata, organelles or nuclei were imaged in EGC. Plasma membrane‐localized protein PIP2, NPSN12, BOR1 and NIP5;1 evenly labelled their corresponding domains, although PIP2, NPSN12 and NIP5;1 were also found intracellularly (Figures 3a–c and S3). Markers derived from Rab protein families labelled various stages of endosomes that appeared as punctate, dot‐like structures in the cytoplasm (Figure 3d) with minor variation in fluorescence intensity between each endosome labeled by RabF2b and RabC1 when compared with those labeled by VTI12 (Figure S3). Golgi bodies labelled by GOT1p and SYP32 also appeared as punctate, dot‐like structures in the cytoplasm but with uniform fluorescence intensity among all labeled Golgi bodies (Figures 3e and S3). Tonoplast‐specific VAMP711 evenly labelled the tonoplast marking the contour of vacuoles (Figures 3f and S3). NUP54 and NUP75 labelled the nuclear pore complexes as dots embedded in the nuclear envelope and in the cytoplasm (Figures 3g and S3). Finally, plasmodesmata callose‐binding PDCB1 labelled the plasmodesmata as dots on the interfaces between cells (Figures 3h, i and S3). All markers labelled patterns consistent with their designated cellular components, and in line with morphologies described in other organs (Ueda et al., 1998; Takano et al., 2005, 2006, 2010; Hunter et al., 2007; Nelson et al., 2007; Era et al., 2009; Geldner et al., 2009; Simpson et al., 2009; Tamura et al., 2010; van der Honing et al., 2011; Benitez‐Alfonso et al., 2013; Ivanov and Harrison, 2014; Wang et al., 2017).
Figure 3.
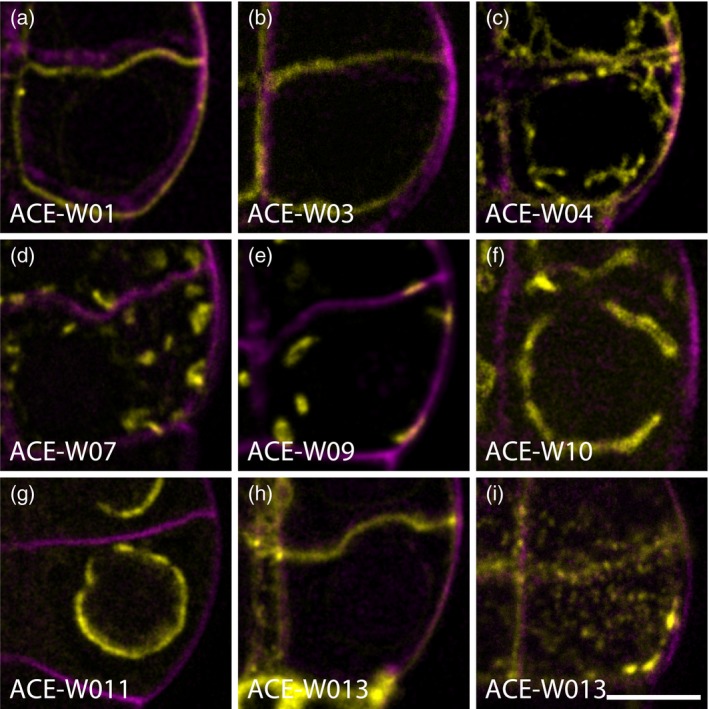
Arabidopsis cellular markers for embryogenesis driven by pWOX2 (ACE‐W) markers label cellular compartments in embryos.
Single optical sections of plasma membrane (a; ACE‐W01; AtPIP2A:GFP), inner membrane (b; ACE‐W03; BOR1:mCitrine), outer membrane (c; ACE‐W04; mCherry:NIP5;1), trans‐Golgi network and early endosomes (d; ACE‐W07; eYFP:VTI12), Golgi complex (e; ACE‐W09; eYFP:GOT1p), tonoplast and vacuole (f; ACE‐W10; eYFP:VAMP711), nuclear pore complex (g; ACE‐W11; AtNUP54:GFP) and plasmodesmata (h, i; ACE‐W13; mCherry:AtPDCB1) markers. Note that all markers are imaged in the center of one of the lower‐tier cells in an 8‐cell embryo, except panel (i), which is imaged at the upper cell surface. Scale bar for all panels = 5 μm. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Thus, with this panel of markers, and an optimized imaging procedure, we can now visualize both robust and fragile subcellular structures in the early Arabidopsis embryo.
Early establishment of central/peripheral polarity
To answer the question of when polarity axes are established and implemented in each cell during embryogenesis, we studied four polar‐localized proteins, OPS (Truernit et al., 2012), BASL (Dong et al., 2009), BOR1 (Takano et al., 2010) and NIP5;1 (Takano et al., 2010), that labelled the apical, basal, central and central side of the cells relative to the body axis, respectively. While ACE lines harboring BOR1 (ACE‐W03) and NIP5;1 (ACE‐W04) were obtained, and showed clear fluorescence signals (Figure 4a–c, g–i), no transgenic plants were obtained from reporters containing OPS nor BASL despite multiple attempts.
Figure 4.
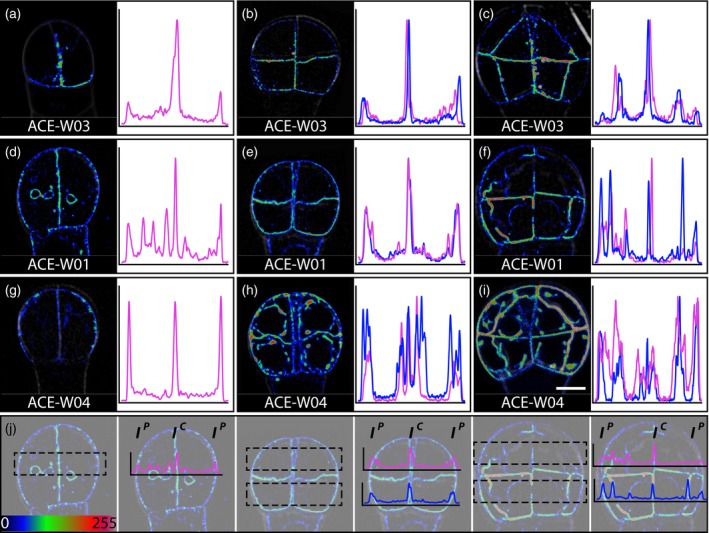
Early establishment of inner–outer cell polarity in embryos.
Single optical sections through 2‐cell (a, d, g), 8‐cell (b, e, h) and 16‐cell (c, f, i) embryos expressing the inner membrane marker ACE‐W03 (a–c; BOR1:mCitrine), general plasma membrane marker ACE‐W01 (d–f; AtPIP2A:GFP) or outer membrane marker ACE‐W04 (g–i; mCherry:NIP5;1). Images are shown in an intensity color scale according to the look‐up table in the bottom left. In each panel, the fluorescence intensity profile is shown in a graph on the right side. Positions of each region for intensity profiles are indicated in panel (j) underneath each row of embryos. Fluorescence intensity profiles of upper (magenta) and lower (blue) tier cells were generated by averaging the fluorescence intensity of each pixel with the same x coordinate in the regions of interest (ROI) shown as dashed boxes. The fluorescence intensity ratios are ratios between I C and I P. Images from the same markers were acquired with the same acquisition setting and are in same scale. Scale bar = 5 μm. [Colour figure can be viewed at http://wileyonlinelibrary.com]
We hypothesized that polarity was established at the latest before the 16‐cell stage, when protoderm is separated from inner cells in a genetically regulated asymmetric cell division (Yoshida et al., 2014). It was likely that polarity perception would be involved in positioning the cell division plane parallel to the embryo surface.
The BOR1 marker was found most abundantly on plasma membrane facing toward the central axis and the suspensor with the lower fluorescence intensity in the peripheral plasma membrane at the 4‐ and 8‐cell stages (Figure 4a, b). Fluorescence became exclusive to the plasma membrane facing toward the central axis and the suspensor in 16‐cell embryos (Figure 4c). However, the higher BOR1 levels found in plasma membrane facing toward the central axis and towards the suspensor could be due to the sum of fluorescence intensity from two neighboring plasma membranes compared with the single peripheral plasma membrane. To non‐invasively verify the polar localization of BOR1 in the early embryo, we compared the fluorescence intensity profiles with that of the homogeneous plasma membrane proteins PIP2 (Nelson et al., 2007) (ACE‐W01) and NPSN12 (Geldner et al., 2009) (ACE‐W02). A 5‐μm wide cross section across the embryo was selected for fluorescence intensity profiles, and the central/peripheral fluorescence intensity ratios were calculated (Figure 4j). If there was any preferential localization, the fluorescence intensity profile of the given protein would be different from the fluorescence intensity profiles of homogeneous plasma membrane proteins. We first compared fluorescence profiles and central/peripheral fluorescence intensity ratios between PIP2 and NPSN12 to validate this approach. From the 4‐ to 16‐cell stages fluorescence profiles of PIP2 and NPSN12 all had peaks at positions corresponding to the plasma membrane with comparable height and sometimes even a lower peak at the central plasma membrane (Figure 4d–f). There was no significant difference in their central/peripheral fluorescence intensity ratios (Table S4). When compared with the fluorescence intensity profiles of PIP2, BOR1 had a distinct peak at the center and lower peaks at the flanks of the fluorescence intensity profile in 4‐ and 8‐cell embryos, while no peaks remain in the flanks with a distinct peak in the center and middle of the fluorescence intensity profile for the 16‐cell embryo (Figure 4a–f). The differences in the fluorescence intensity profiles were further supported by the significantly higher central/peripheral fluorescence intensity ratios of BOR1 compared with those of PIP2 and NPSN12 from 4‐ to 16‐cell embryos (Table S4). We thus concluded that BOR1 was localized at the pole on the central faces of plasma membranes from at least as early as the 4‐cell embryo.
We next analyzed NIP5;1 which had previously been shown to mark outer membranes (Takano et al., 2010). Unlike the consistent polar plasma membrane‐localized BOR1, NIP5;1 was not only found on the peripheral plasma membrane but also accumulated in intracellular compartments in 4‐ to 16‐cell embryos (Figure 4g–i). The fluorescence intensity profiles of NIP5;1 in most 4‐ and 8‐cell embryos had three peaks with comparable height, with additional peaks when the sampling area crossed the intracellular compartment with accumulated NIP5;1. The fluorescence intensity profiles of NIP5;1 in most 4‐ and 8‐cell embryos were similar to those of PIP2 (Figure 4g, h). In 16‐cell embryos, the peripheral localization of NIP5;1 became more consistent and distinct (Figure 4i). The fluorescence intensity profiles of NIP5;1 had a central peak lower than the flanking peaks (Figure 4i) and its central/peripheral fluorescence intensity ratios, though having a lower average, were not significantly different from those of PIP2 and NPSN12 until 16‐cell and 8‐cell embryos, respectively (Table S4). These results suggested that while NIP5;1 could be localized polewards to the peripheral plasma membrane from the 4‐cell embryo its peripheral localization only became robust in the 16‐cell embryo.
In conclusion, BOR1 was robustly centrally localized from the 4‐cell embryo onwards while NIP5;1, although peripheral localization was observed from 4‐cell embryos, was only robustly peripherally localized in the upper tier of embryos from 16‐cell embryos. These results suggest that the ability to distinguish between central and peripheral plasma membrane and to deploy specific proteins to their corresponding locations is already established in the 4‐cell embryo.
Crowded embryo cells show no preferential organelle position
To determine if there was any preferential localization or morphological change of the endomembrane system during early embryogenesis, we examined the distribution and morphology of the Golgi complex, various stages of endosomes and vacuoles from the 4‐cell to 16‐cell stage.
Endosomes marked by ACE‐W07 and Golgi complexes marked by ACE‐W09 were both loosely distributed close to the plasma membrane in 4‐ and 8‐cell embryos, and their density in each cell seemed to increase in 16‐cell embryos (Figure 5a, b). This increased density suggested an increased secretion and endocytosis activity to accommodate the increase in embryo volume from the 16‐cell stage onward (Yoshida et al., 2014). However, no distinguishable local aggregation of Golgi complexes and endosomes was observed from 4‐ to 16‐cell embryos (Figure 5a, b).
Figure 5.

Endomembrane morphologies in early embryos.
(a), (b) Single optical sections (top rows) and depth‐coded maximum intensity projections (bottom rows) of endosomes and the trans‐Golgi network (a; ACE‐W07; eYFP:VTI12) and Golgi complex (b; ACE‐W09; eYFP:GOT1p) in 4‐cell (left row), 8‐cell (middle row) and 16‐cell (right row) embryos. (c)–(h) Maximum intensity projections of depth‐coded stacks of vacuoles labeled by ACE‐W10 (eYFP:VAMP711) in 4‐cell (c, e, g; left rows), 8‐cell (c, e, g; middle rows) and 16‐cell (c, e, g; right rows) embryos. Panels (d), (f), (h) represent zoomed in images of single cells representing corresponding vacuole morphologies in (c), (e) and (g), respectively, and are shown as single optical sections (left row) or depth‐coded maximum intensity projections (right row). Distinct vacuole morphologies are Type 1 (c, d), Type 2 (e, f) and Type 3 (g, h). (i)–(k) Maximum intensity projection of depth‐coded stacks of nuclear envelopes labeled by the nuclear pore complex marker ACE‐W11 (AtNUP54:GFP) in 4‐cell (left row in i, j), 8‐cell (middle row in i, j) and 16‐cell (right row in i, j) embryos. Panel (i) represents embryos with smooth nuclear envelopes and panel (j) shows embryos with wavy nuclear envelopes. (k) Maximum projection of a depth‐coded stack of an 8‐cell embryo with both smooth (arrows) and wavy (thick arrow) nuclear envelopes, and nuclear pore complexes accumulating in the spindle during mitosis (arrowhead). All images of the same marker were acquired with identical acquisition settings. Scale bars = 5 μm. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Vacuoles marked by tonoplast specific ACE‐W10, on the other hand, showed a wide range of morphologies and distributions during early embryogenesis. Three types of vacuole morphologies were found in 4‐ to 16‐cell embryos. Type 1 represented large foil‐like vacuoles with distinguishable transvacuolar strands, but without distinct bulbs (Figure 5c, d). This type of vacuole tightly enclosed a spherical space in the cell, presumably the nucleus (Figure 5d). Type 2 vacuoles were also large but with several distinguishable bulbs emanating from the spherical space, which the vacuoles loosely enclosed (Figure 5e, f). Type 3, unlike the former two, was composed of vacuoles with many small bulbs seemingly filling the cell (Figure 5 g, h). All types of vacuole morphologies were observed from the 4‐ to 16‐cell stages and could vary between cells in each embryo (Figure 5c, e, g). Though all cells in most embryos have the same vacuole morphology, combinations of different vacuole morphologies could occasionally be found (Figure 5g). Similar changes of vacuole morphology were also observed in earlier findings in shoot meristem cells (Seguí‐Simarro and Staehelin, 2006), suggesting that embryos might share vacuole dynamics with meristematic cells.
Nuclear envelopes marked by ACE‐W11 revealed that nuclear envelope morphology also changed as cellular development progressed in 4‐ to 16‐cell embryos. Two types of nuclear envelope morphologies were found in both embryo proper and suspensor at all stages examined. The first type had a smooth surface with NUP54 evenly distributed and enclosing a spherical space (Figure 5i), while the second type showed a wavy surface (Figure 5j). Both types could be found in the same embryo (Figure 5k). To confirm the identity/nature of the spherical space surrounded by the vacuole and determine if the movement of nuclei was involved in the asymmetric cell division between 8‐ and 16‐cell embryos, we measured the diameter of the nuclei in 8‐cell embryos to determine if it was consistent with the diameter of the spherical space encompassed by the vacuoles. The average diameter of the nuclei in an 8‐cell embryo was 5.6 ± 0.7 μm (n = 131 nuclei) at the 8‐cell stage. Therefore, with the relatively large nuclei in the embryo cells and the similarity in both shape and positioning with the spherical space surrounded by the vacuole, it was likely that the spherical space encompassed by the vacuole could only be the nucleus but also suggested very limited room for movement of the nucleus without deforming it.
In conclusion, each cell in 4‐ to 16‐cell embryos was filled with non‐polar localized endosome and Golgi close to the plasma membrane, with vacuoles, either tightly or loosely enclosing the nucleus, which showed no clear movement during symmetric and asymmetric division between 4‐ and 8‐cell embryos and between 8‐ and 16‐cell embryos, respectively. The lack of polar aggregation of endosomes and Golgi complexes or preferential positioning of vacuoles suggested that there might not be directional cell growth or expansion at early embryogenesis. While the lack of nuclear movement of nuclei suggested that such movement might not be required for asymmetric cell division between 8‐ and 16‐cell embryos.
Cytoskeletal organization during embryogenesis
To understand how the cytoskeleton is rearranged to accommodate the constant change of division plane during early embryogenesis, we examined actin and microtubule architecture labelled by Lifeact (ACE‐W14) and TUA6 (ACE‐W15) in 4‐cell to early globular embryos.
We found that actin architectures in early embryos were very different from earlier studies in post‐embryonic meristem and differentiated cells (Ketelaar et al., 2004; Sheahan et al., 2004; Wang et al., 2004, 2008; Voigt et al., 2005; Rahman et al., 2007; Era et al., 2009; Dyachok et al., 2014). In mitotic cells in the root apical meristem, actin is found mainly as a fine and dense meshwork without F‐actin bundles (Sheahan et al., 2004; Voigt et al., 2005). In the embryo, however, cells had thick F‐actin bundles forming a dense meshwork throughout the cytoplasm without prominent orientation (Figure 6a–e). From early‐globular embryos, the F‐actin meshwork seemed more organized than in younger embryos (Figure 6e). Notably, F‐actin bundles stretched around the nuclei at later stages (Figure 6c). In addition, from the 8‐cell embryo onwards, a thick arched F‐actin bundle could be found in each embryo proper cell (Figure 6c–e). These arch structures were associated with the nuclei, although not always at the equator of the nucleus, and orientations were not correlated to the orientations of previous or following cell divisions (Figure 6c–e). In suspensor cells, the noticeable difference is the absence of the peri‐nuclear arch and the cortical actin filaments when compared with those in embryo proper cells (Figure 6a–e). In addition, the F‐actin bundle meshworks in the suspensor cells were looser than in embryo proper cells and also showed no prominent orientation (Figure 6a–e).
Figure 6.
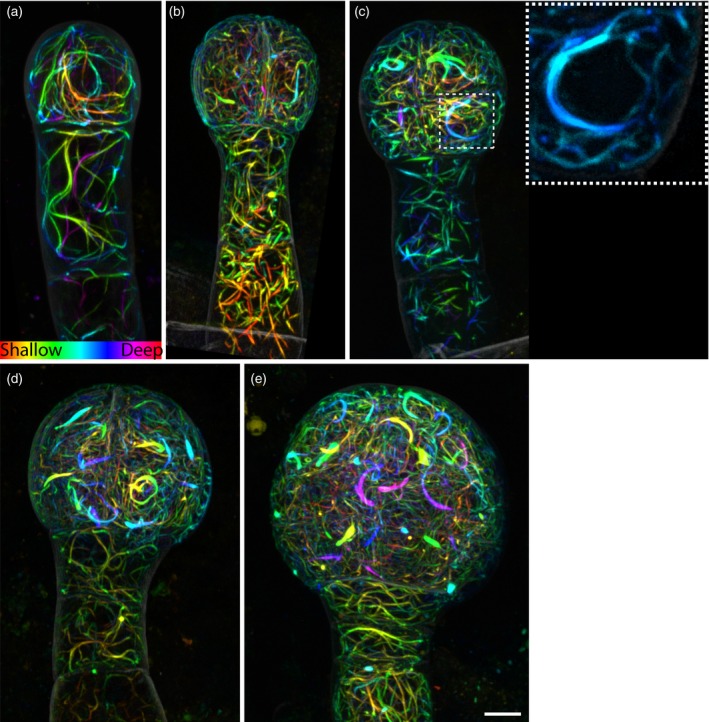
Actin topology in early embryos.
Maximum intensity projections of depth‐coded stacks of actin labeling (ACE‐W14; Lifeact:tdTomato) in 2‐cell (a), 4‐cell (b), 8‐cell (c), 16‐cell (d) and early globular (e) embryos. The inset in panel (c) shows a peri‐nuclear actin arch.
Images were acquired with the same acquisition settings and are in the same scale. Scale bar = 5 μm. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Microtubules in early embryos were straight cables (Figure 7a). Microtubule structures including cortical microtubule arrays, cytoplasmic microtubule bundles, pre‐prophase bands, spindles and phragmoplast were present with mitotic‐ and cytokinesis‐related structures with their orientations consistent with expected cell division orientation (Figure 7b). Due to technical limitations only cortical microtubule arrays on the periclinal faces could be examined (see Discussion), and pro‐embryo cells and suspensor cells showed obvious differences in their cortical microtubule arrangements. In pro‐embryo cells, the cortical microtubule arrays were dense and seemed to lack prominent orientation during most of the interphase, although occasionally, presumably at early G1 or/and late G2 phase, they became more organized (Figure 7c–f). In suspensor cells, cortical microtubule arrays were less dense and seemed more organized with their orientation being perpendicular to the embryo axis (Figure 7c–e). With the uniform periclinal cortical microtubule array and the single‐cell‐layer nature of suspensor cells, the periclinal cortical microtubule array of suspensor cells could be readily analysed with FibrilTool (Boudaoud et al., 2014); the results supported the dominant orientation (Figure 7g–i).
Figure 7.
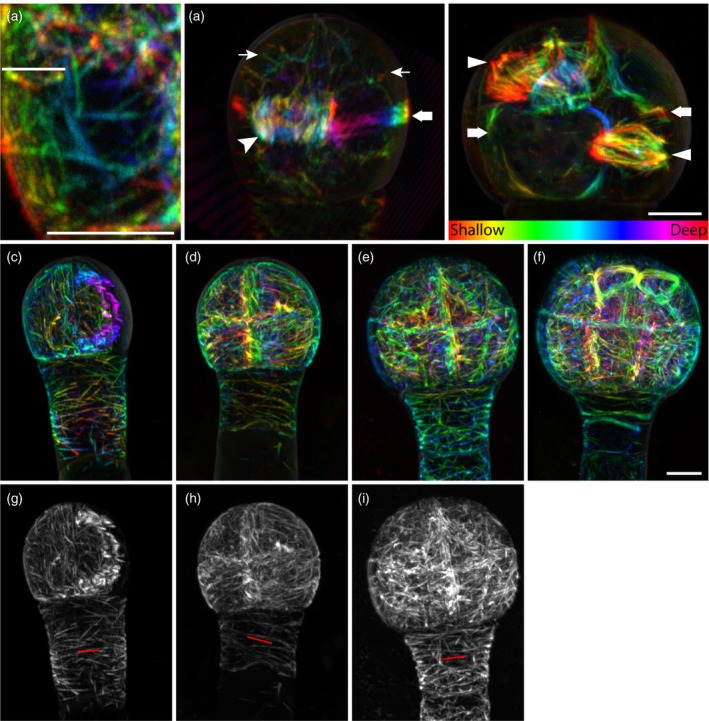
Microtubule topologies in early embryos.
Labeling of microtubules with the ACE‐W15 (mGFP:AtTUA6) marker reveals filaments (a), as well as cytoplasmic signals (thin arrows in the left image of panel b), phragmoplast (arrowhead in the left image of panel b), preprophase band (thick arrows in both left and right images in panel b) and spindles (triangles in the right image in panel b) in early embryos. (c)–(i) Microtubules in 4‐cell (c, g), 8‐cell (d, h), 16‐cell (e, i) and early globular (f) embryos shown either as depth‐coded stacks (c–f) or maximum intensity projections (g–i). The orientation of cortical microtubules in the suspensor is marked by red lines in (g)–(i). Images were acquired with same acquisition settings and are in the same scale, except for (a) and (b). Scale bar = 5 μm. [Colour figure can be viewed at http://wileyonlinelibrary.com]
In conclusion, all mitotic and cytokinetic cytoskeletal structures were found in early embryos and their orientations followed the expected cell division orientations. This suggests that both F‐actin meshwork and microtubules in embryo cells behave as described in late and post‐embryonic cells.
Discussion
In this study we have generated a comprehensive set of cellular reporters driven by RPS5A and WOX2 promoters that were specifically designed for imaging cellular reorganization in early Arabidopsis embryos. With these cell type‐specific promoters, the expression level of the reporter genes could be maximized while minimizing the background signal from surrounding cells and preventing the morphological defects commonly found when using constitutive and ubiquitous reporters (Abe and Hashimoto, 2005; Dyachok et al., 2014). This feature allowed us to capture clear cellular structures to dissect the cellular organization and regulation in embryos. The combination of embryo‐specific ACEs and our optimized imaging procedure allowed the preservation of delicate cellular structures and detailed documentation of their three‐dimensional architectures, and thus overcomes the major obstructions involved in dissecting cellular reorganization in embryos.
Using this panel of markers we addressed a number of questions. First, we found that central–peripheral cellular polarity is established extremely early in the embryo. This was evident from the polar localization of BOR1 and, later, NIP5;1 proteins. Both are polarity probes that were mis‐expressed in early embryos and still targeted to expected inner or outer domains based on imaging in multi‐layered post‐embryonic tissues (Takano et al., 2010). This suggests that a polar transport and/or sorting mechanism is already established in early embryos, but also that the central–peripheral faces of early embryo cells are molecularly distinguished as early as from the second, and perhaps even the first, cell division. Importantly, establishment of the outer–inner polarity of a cell does not depend on the presence of multiple cell layers. In addition, a large portion of embryonic cells is occupied by the nucleus, vacuoles and other organelles along with the dense F‐actin bundle meshwork. This suggests that cytoplasmic volume is very limited, and therefore intracellular trafficking and active cytoplasmic streaming may be relatively efficient in early embryonic cells. These properties of early embryonic cells make it unlikely that gradients of small‐molecule diffusible morphogens in each cell serve as a guide for the establishment of polarity. Therefore, polarity establishment in the early embryo might be caused by other factors, such as a combination of the molecular properties of cell walls and genetic regulation.
Besides limiting the small molecule gradients within the cells, the small sizes of embryo proper cells with their relatively large nuclei also restrict nuclear movement. In an 8‐cell embryo, with a diameter of 5.6 μm, each nucleus could only move within a sphere with a diameter 6.4 μm, or 0.4 μm in each direction when the nucleus is in the center of this sphere, without deforming the nucleus as we observed. This suggests that nuclear movement might not occur nor be required for asymmetric cell division between 8‐ and 16‐cell embryos to establish the central and peripheral cell layers for proper pattern formation. The position of the nucleus has been shown to be correlated with the division plane in asymmetric divisions (Robinson et al., 2011; Kimata et al., 2016), where the division plane goes through the center of the previous position of the nucleus. However, both symmetric and asymmetric divisions in early embryo cells can be generated following this principle without moving the nucleus.
Our finding that the mitotic and cytokinetic microtubule architectures aligned with the expected division orientation in the early embryo suggested that microtubule dynamics may be similar to those in post‐embryonic cells. Given that early embryo cells can easily be segmented in three dimensions (Yoshida et al., 2014), and that cell division patterns in Arabidopsis embryos are highly predictable, this now opens the possibility of determining contributions of genetically regulated cytoskeletal remodeling to the control of cell division. However, there are limitations to imaging microtubules in the embryo that can not be easily overcome. Firstly, all outer membranes are curved, and thus cytoskeletal topology cannot easily be extracted. Secondly, high‐resolution imaging is limited to those membranes that lie in the x–y plane because resolution along the z‐axis is much lower using confocal microscopy, and embryos cannot easily be tilted during observation. Therefore, instead of treating each cell as a plane and processing images through maximum projection, three‐dimensional reconstruction and analysis will be the next technical challenge for any quantitative analysis of cytoskeletal organization during embryogenesis.
Beyond these examples, we expect that many outstanding questions related to the cell biology of tissue patterning can now be addressed using this set of marker lines.
Experimental procedures
Plasmid construction and plant material
All constructs, reporter cassettes and their corresponding DNA templates as well as all oligonucleotides used in this study are listed in Table S5. pGIIB/pWOX2::LIC:tNOS was generated through excising the pRPS5A cassette in pPLV28 (de Rybel et al., 2011) with Acc65I digestion and ligated with an Acc65I‐digested pWOX2 cassette. The pWOX2::LIC cassette in pGIIB/pWOX2::LIC:tNOS was then excised with Acc65I and NotI double digestion and ligated with pGIIN (a kind gift from T Laux, University Freiburg, Germany) linearized with the same restriction digestion to generate pGIIN/pWOX2::LIC:tNOS. All reporting cassettes except Lifeact were introduced into HpaI linearized pPLV28 and pGIIN/pWOX2::LIC:tNOS through ligation‐independent cloning (Aslanidis and de Jong, 1990) to generate ACE reporter constructs with the corresponding promoter. pGIIB/pRPS5A::Lifeact:tdTomato:tNOS was generated by introducing the oligonucleotide dimer Lifeact into pGIIB/pRPS5A::LIC:tdTomato:tNOS through ligation‐independent cloning. pGIIB/pRPS5A::LIC:tdTomato:tNOS was generated by ligating BamHI‐linearized pPLV28 with tdTomato excised from pPLV23 (de Rybel et al., 2011). pGIIN/pWOX2::Lifeact:tdTomato:tNOS was generated by introducing Lifeact:tdTomato amplified from pGIIB/pRPS5A::LIC:tdTomato:tNOS into HpaI‐linearized pGIIN/pWOX2::LIC:tNOS via ligation‐independent cloning.
All ACE reporting constructs were introduced into A. thaliana ecotype Col‐Utrecht with the mpB4149 mutation (Willemsen et al., 2003) heterozygous in the background, through floral dip (Clough and Bent, 1998). T1 transgenic plants of ACE‐R markers were selected with DL‐phosphinotricin (Sigma, http://www.sigmaaldrich.com/), and T1 transgenic plants of ACE‐W markers were selected with Norflurazon (Supelco, https://www.sigmaaldrich.com/catalog/product/supelco/ps1044?lang=en%26region=NL) (Misawa et al., 1993). T3 homozygotes without the mpB4149 allele were used in this study.
Seeds harboring p35S:: GFP: TUA6 were sterilized by incubating in five times dilution of household bleach containing approximately 5% sodium hypochlorite with demineralized water for 10 min followed by washing five times with sterilized demineralized water. The sterilized seeds were plated on 1/2 MS0 medium plates containing 0.8% agar. After stratification at 4°C for 2 days, the plates were transferred to a phytochamber (22°C, 16 h light and 8 h dark). After 6 days of growth in the phytochamber the seedlings were used to test the short‐term effect of Taxol on microtubule organization.
Microscopy and image analysis
Embryo samples were prepared as described in Figure S4 with mounting and counterstaining solutions listed in Table S2. Fluorescence intensity profiles used for verifying polar localization of BOR1 and NIP5;1 were generated through the ‘Analyze/plot profile’ function in Fiji (Schindelin et al., 2012). A 5‐μm wide band perpendicular to the suspensor axis that crossed a region directly above or under the cell wall, which separates the upper and lower tiers of embryo proper, was defined as the region of interest (ROI) for quantification. The ROIs and results were saved, and the fluorescence intensity profiles were exported and plotted in MS Excel™. Plasma membrane signals of each face in each fluorescence intensity profile were defined by examining the corresponding confocal image and identifying the coordinate ranges of each face in the ROI; the maximum intensity in each coordinate range was then extracted using MS Excel™ to calculate the central‐peripheral fluorescence intensity ratio.
Root treatments with paclitaxel (Sigma) were conducted through live imaging and the samples were prepared as described in Figure S5, and microtubule orientation and anisotropy were analyzed as described in Boudaoud et al. (2014).
Images were acquired in 8‐bit format using a Leica TCS SP5II confocal laser scanning microscope with a 63 × NA = 1.20 water‐immersion objective with pinhole set to 1.0 Airy unit. mGFP and mCitrine were excited by an argon‐ion laser and tdTomato and SCRI Renaissance Stain 2200 (SR2200) (Renaissance Chemicals, http://www.renchem.co.uk/) were excited using a diode laser, and their emissions were detected sequentially with a Leica HyD in photon counting mode. Excitation and detection of fluorophores were configured as follows: mGFP was excited at 488 nm and detected at 498–528 nm; Venus was excited at 514 nm and detected at 524–554 nm; tdTomato was excited at 561 nm and detected at 571–630 nm; Renaissance 2200 was excited at 405 nm and detected at 430–470 nm.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Figure S1. Microtubule and actin structures in root tip labeled by ACE‐R15 and ACE‐R14.
Figure S2. Effect of Taxol on microtubules in roots.
Figure S3. Overview of Arabidopsis cellular markers for embryogenesis (ACEs).
Figure S4. Sample preparation for imaging cellular structures.
Figure S5. Seedling sample preparation for short‐term chemical treatment and live imaging.
Table S1. Catalog of Arabidopsis cellular markers for embryogenesis (ACE) lines.
Table S2. Mounting solutions used in this study.
Table S3. Effects of 90 min treatment with 10 μm Taxol on cortical microtubule dynamics in root epidermal cells.
Table S4. Central/peripheral fluorescence intensity ratio (I C/I P) of ACE‐W01, ACE‐W02, ACE‐03 and ACE‐W04 in early embryos.
Table S5. Primers used in this study.
Acknowledgements
We thank Thomas Laux (Universität Freiburg) for the pGII/N plasmid backbone, Niko Geldner (University of Lausanne) for the mCherry‐NIP5;1 and BOR1‐Cit reporting cassettes, and Tijs Ketelaar (Wageningen University) for the GFP‐TUA6 reporting cassette and seeds of the p35S:: GFP: TUA6 reporter line. This research was supported by the European Research Council (CELLPATTERN; contract no. 281573).
Linked article: This paper is the subject of a Research Highlight article. To view this Research Highlight article visit https://doi.org/10.1111/tpj.13879.
References
- Abe, T. and Hashimoto, T. (2005) Altered microtubule dynamics by expression of modified α‐tubulin protein causes right‐handed helical growth in transgenic Arabidopsis plants. Plant J. 43, 191–204. [DOI] [PubMed] [Google Scholar]
- Aslanidis, C. and de Jong, P.J. (1990) Ligation‐independent cloning of PCR products (LIC‐PCR). Nucl. Acids Res. 18, 6069–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannigan, A. , Wiedemeier, A.M.D. , Williamson, R.E. , Overall, R.L. and Baskin, T.I. (2006) Cortical microtubule arrays lose uniform alignment between cells and are oryzalin resistant in the arabidopsis mutant, radially swollen 6. Plant Cell Physiol. 47, 949–958. [DOI] [PubMed] [Google Scholar]
- Baskin, T.I. , Wilson, J.E. , Cork, A. and Williamson, R.E. (1994) Morphology and microtubule organization in arabidopsis roots exposed to oryzalin or taxol. Plant Cell Physiol. 35, 935–942. [PubMed] [Google Scholar]
- Benitez‐Alfonso, Y. , Faulkner, C. , Pendle, A. , Miyashima, S. , Helariutta, Y. and Maule, A. (2013) Symplastic intercellular connectivity regulates lateral root patterning. Dev. Cell, 26, 136–147. [DOI] [PubMed] [Google Scholar]
- Besson, S. and Dumais, J. (2011) Universal rule for the symmetric division of plant cells. Proc. Natl Acad. Sci. USA, 108, 6294–6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boruc, J. and Van Damme, D. (2015) Endomembrane trafficking overarching cell plate formation. Curr. Opin. Plant Biol. 28, 92–98. [DOI] [PubMed] [Google Scholar]
- Boudaoud, A. , Burian, A. , Borowska‐Wykrȩt, D. , Uyttewaal, M. , Wrzalik, R. , Kwiatkowska, D. and Hamant, O. (2014) FibrilTool, an ImageJ plug‐in to quantify fibrillar structures in raw microscopy images. Nat. Protoc. 9, 457–463. [DOI] [PubMed] [Google Scholar]
- Breuninger, H. , Rikirsch, E. , Hermann, M. , Ueda, M. and Laux, T. (2008) Differential expression of WOX genes mediates apical‐basal axis formation in the arabidopsis embryo. Dev. Cell, 14, 867–876. [DOI] [PubMed] [Google Scholar]
- Buschmann, H. and Zachgo, S. (2016) The evolution of cell division: from streptophyte algae to land plants. Trends Plant Sci. 21, 872–883. [DOI] [PubMed] [Google Scholar]
- Clough, S.J. and Bent, A.F. (1998) Floral dip: a simplified method for Agrobacterium‐mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743. [DOI] [PubMed] [Google Scholar]
- Cutler, S.R. and Ehrhardt, D.W. (2002) Polarized cytokinesis in vacuolate cells of Arabidopsis. Proc. Natl Acad. Sci. USA, 99, 2812–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rybel, B. , Möller, B. , Yoshida, S. , Grabowicz, I. , Barbier de Reuille, P. , Boeren, S. , Smith, R.S. , Borst, J.W. and Weijers, D. (2013) A bHLH complex controls embryonic vascular tissue establishment and indeterminate growth in Arabidopsis. Dev. Cell, 24, 426–437. [DOI] [PubMed] [Google Scholar]
- Dong, J. , MacAlister, C.A. and Bergmann, D.C. (2009) BASL controls asymmetric cell division in arabidopsis. Cell, 137, 1320–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dop, M. , Liao, C.Y. and Weijers, D. (2015) Control of oriented cell division in the Arabidopsis embryo. Curr. Opin. Plant Biol. 23, 25–30. [DOI] [PubMed] [Google Scholar]
- Dyachok, J. , Sparks, J.A. , Liao, F. , Wang, Y.S. and Blancaflor, E.B. (2014) Fluorescent protein‐based reporters of the actin cytoskeleton in living plant cells: fluorophore variant, actin binding domain, and promoter considerations. Cytoskeleton, 71, 311–327. [DOI] [PubMed] [Google Scholar]
- Era, A. , Tominaga, M. , Ebine, K. , Awai, C. , Saito, C. , Ishizaki, K. , Yamato, K.T. , Kohchi, T. , Nakano, A. and Ueda, T. (2009) Application of lifeact reveals F‐actin dynamics in arabidopsis thaliana and the liverwort, marchantia polymorpha. Plant Cell Physiol. 50, 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geldner, N. , Dénervaud‐Tendon, V. , Hyman, D.L. , Mayer, U. , Stierhof, Y.D. and Chory, J. (2009) Rapid, combinatorial analysis of membrane compartments in intact plants with a multicolor marker set. Plant J. 59, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunning, B.E. and Wick, S.M. (1985) Preprophase bands, phragmoplasts, and spatial control of cytokinesis. J. Cell Sci. Suppl. 2, 157–179. [DOI] [PubMed] [Google Scholar]
- van der Honing, H.S. , van Bezouwen, L.S. , Emons, A.M.C. and Ketelaar, T. (2011) High expression of Lifeact in Arabidopsis thaliana reduces dynamic reorganization of actin filaments but does not affect plant development. Cytoskeleton, 68, 578–587. [DOI] [PubMed] [Google Scholar]
- Hunter, P.R. , Craddock, C.P. , Di Benedetto, S. , Roberts, L.M. and Frigerio, L. (2007) Fluorescent reporter proteins for the tonoplast and the vacuolar lumen identify a single vacuolar compartment in arabidopsis cells. Plant Physiol. 145, 1371–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov, S. and Harrison, M.J. (2014) A set of fluorescent protein‐based markers expressed from constitutive and arbuscular mycorrhiza‐inducible promoters to label organelles, membranes and cytoskeletal elements in Medicago truncatula. Plant J. 80, 1151–1163. [DOI] [PubMed] [Google Scholar]
- Ketelaar, T. , Allwood, E.G. , Anthony, R. , Voigt, B. , Menzel, D. and Hussey, P.J. (2004) The actin‐interacting protein AIP1 is essential for actin organization and plant development. Curr. Biol. 14, 145–149. [DOI] [PubMed] [Google Scholar]
- Kimata, Y. , Higaki, T. , Kawashima, T. , Kurihara, D. , Sato, Y. , Yamada, T. , Hasezawa, S. , Berger, F. , Higashiyama, T. and Ueda, M. (2016) Cytoskeleton dynamics control the first asymmetric cell division in Arabidopsis zygote. Proc. Natl Acad. Sci. USA, 113, 14157–14162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrein, B. , Kiss, A. , Sassi, M. et al. (2015) Mechanical stress contributes to the expression of the STM homeobox gene in Arabidopsis shoot meristems. eLife, 4, e07811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llavata‐Peris, C. , Lokerse, A. , Möller, B. , De Rybel, B. and Weijers, D. (2013)Imaging of phenotypes, gene expression, and protein localization during embryonic root formation in arabidopsis. Methods Mol. Biol. 959, 137–148. [DOI] [PubMed] [Google Scholar]
- Lloyd, C.W. and Traas, J.A. (1988) The role of F‐actin in determining the division plane of carrot suspension cells. Drug studies. Development, 102, 211–221. [Google Scholar]
- Louveaux, M. , Julien, J.D. , Mirabet, V. , Boudaoud, A. and Hamant, O. (2016) Cell division plane orientation based on tensile stress in Arabidopsis thaliana. Proc. Natl Acad. Sci. USA, 113, E4294–E4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredi, J.J. , Parness, J. and Horwitz, S.B. (1982) Taxol binds to cellular microtubules. J. Cell Biol. 94, 688–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur, J. and Chua, N.H. (2000) Microtubule stabilization leads to growth reorientation in Arabidopsis trichomes. Plant Cell, 12, 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misawa, N. , Yamano, S. , Linden, H. , De Felipe, M.R. , Lucas, M. , Ikenaga, H. and Sandmann, G. (1993) Functional expression of the Erwinia uredovora carotenoid biosynthesis gene crtl in transgenic plants showing an increase of β‐carotene biosynthesis activity and resistance to the bleaching herbicide norflurazon. Plant J. 4, 833–840. [DOI] [PubMed] [Google Scholar]
- Murata, T. and Wada, M. (1991) Effects of centrifugation on preprophase‐band formation in Adiantum protonemata. Planta, 183, 391–398. [DOI] [PubMed] [Google Scholar]
- Musielak, T.J. , Schenkel, L. , Kolb, M. , Henschen, A. and Bayer, M. (2015) A simple and versatile cell wall staining protocol to study plant reproduction. Plant Reproduction, 28, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, B.K. , Cai, X. and Nebenführ, A. (2007) A multicolored set of in vivo organelle markers for co‐localization studies in Arabidopsis and other plants. Plant J. 51, 1126–1136. [DOI] [PubMed] [Google Scholar]
- Palovaara, J. , De Zeeuw, T. and Weijers, D. (2016) Tissue and organ initiation in the plant embryo: a first time for everything. Annu. Rev. Cell Dev. Biol. 32, 47–75. [DOI] [PubMed] [Google Scholar]
- Pickett‐Heaps, J.D. and Northcote, D.H. (1966a) Cell division in the formation of the stomatal complex of the young leaves of wheat. J. Cell Sci. 1, 121–128. [DOI] [PubMed] [Google Scholar]
- Pickett‐Heaps, J.D. and Northcote, D.H. (1966b) Organization of microtubules and endoplasmic reticulum during mitosis and cytokinesis in wheat meristems. J. Cell Sci. 1, 109–120. [DOI] [PubMed] [Google Scholar]
- Rahman, A. , Bannigan, A. , Sulaman, W. , Pechter, P. , Blancaflor, E.B. and Baskin, T.I. (2007) Auxin, actin and growth of the Arabidopsis thaliana primary root. Plant J. 50, 514–528. [DOI] [PubMed] [Google Scholar]
- Rasmussen, C.G. , Humphries, J.A. and Smith, L.G. (2011) Determination of symmetric and asymmetric division planes in plant cells. Annu. Rev. Plant Biol. 62, 387–409. [DOI] [PubMed] [Google Scholar]
- Rasmussen, C.G. , Wright, A.J. and Müller, S. (2013) The role of the cytoskeleton and associated proteins in determination of the plant cell division plane. Plant J. 75, 258–269. [DOI] [PubMed] [Google Scholar]
- Riedl, J. , Crevenna, A.H. , Kessenbrock, K. et al. (2008) Lifeact: a versatile marker to visualize F‐actin. Nat. Methods, 5, 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, S. , De Reuille, P.B. , Chan, J. , Bergmann, D. , Prusinkiewicz, P. and Coen, E. (2011) Generation of spatial patterns through cell polarity switching. Science, 333, 1436–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rybel, B.D. , van Den Berg, W. , Lokerse, A. , Liao, C.Y. , Van Mourik, H. , Möller, B. , Peris, C.L. and Weijers, D. (2011) A versatile set of ligation‐independent cloning vectors for functional studies in plants. Plant Physiol. 156, 1292–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sablowski, R. (2016) Coordination of plant cell growth and division: collective control or mutual agreement? Curr. Opin. Plant Biol. 34, 54–60. [DOI] [PubMed] [Google Scholar]
- Schindelin, J. , Arganda‐Carreras, I. , Frise, E. et al. (2012) Fiji: an open‐source platform for biological‐image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seguí‐Simarro, J.M. and Staehelin, L.A. (2006) Cell cycle‐dependent changes in Golgi stacks, vacuoles, clathrin‐coated vesicles and multivesicular bodies in meristematic cells of Arabidopsis thaliana: a quantitative and spatial analysis. Planta, 223, 223–236. [DOI] [PubMed] [Google Scholar]
- Sheahan, M.B. , Staiger, C.J. , Rose, R.J. and McCurdy, D.W. (2004) A green fluorescent protein fusion to actin‐binding domain 2 of arabidopsis fimbrin highlights new features of a dynamic actin cytoskeleton in live plant cells. Plant Physiol. 136, 3968–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, L. , Wang, B. , Gong, W. , Zhang, Y. , Zhu, L. and Yang, X. (2011) Actin filaments and microtubules of Arabidopsis suspension cells show different responses to changing turgor pressure. Biochem. Biophys. Res. Commun. 405, 632–637. [DOI] [PubMed] [Google Scholar]
- Simpson, C. , Thomas, C. , Findlay, K. , Bayer, E. and Maule, A.J. (2009) An arabidopsis GPI‐anchor plasmodesmal neck protein with callose binding activity and potential to regulate cell‐to‐cell trafficking. Plant Cell, 21, 581–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smertenko, A. , Assaad, F. , Baluška, F. et al. (2017) Plant cytokinesis: terminology for structures and processes. Trends Cell Biol. 27, 885–894. [DOI] [PubMed] [Google Scholar]
- Soltys, B.I. and Borisy, G.G. (1985) Polymerization of tubulin in vivo: direct evidence for assembly onto microtubule ends and from centrosomes. J. Cell Biol. 100, 1682–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano, J. , Miwa, K. , Yuan, L. , Von Wirén, N. and Fujiwara, T. (2005) Endocytosis and degradation of BOR1, a boron transporter of Arabidopsis thaliana, regulated by boron availability. Proc. Natl Acad. Sci. USA, 102, 12276–12281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano, J. , Wada, M. , Ludewig, U. , Schaaf, G. , Von Wirén, N. and Fujiwara, T. (2006) The Arabidopsis major intrinsic protein NIP5;1 is essential for efficient boron uptake and plant development under boron limitation. Plant Cell, 18, 1498–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano, J. , Tanaka, M. , Toyoda, A. , Miwa, K. , Kasai, K. , Fuji, K. , Onouchi, H. , Naito, S. and Fujiwara, T. (2010) Polar localization and degradation of Arabidopsis boron transporters through distinct trafficking pathways. Proc. Natl Acad. Sci. USA, 107, 5220–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Fukao, Y. , Iwamoto, M. , Haraguchi, T. and Hara‐Nishimura, I. (2010) Identification and characterization of nuclear pore complex components in Arabidopsis thaliana. Plant Cell, 22, 4084–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traas, J.A. , Doonan, J.H. , Rawlins, D.J. , Shaw, P.J. , Watts, J. and Lloyd, C.W. (1987) An actin network is present in the cytoplasm throughout the cell cycle of carrot cells and associates with the dividing nucleus. J. Cell Biol. 105, 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truernit, E. , Bauby, H. , Belcram, K. , Barthélémy, J. and Palauqui, J.C. (2012) OCTOPUS, a polarly localised membrane‐associated protein, regulates phloem differentiation entry in Arabidopsis thaliana. Development, 139, 1306–1315. [DOI] [PubMed] [Google Scholar]
- Ueda, K. , Matsuyama, T. and Hashimoto, T. (1998) Visualization of microtubules in living cells of transgenic Arabidopsis thaliana. Protoplasma, 206, 201–206. [Google Scholar]
- Voigt, B. , Timmers, A.C.J. , Šamaj, J. , Müller, J. , Baluška, F. and Menzel, D. (2005) GFP‐FABD2 fusion construct allows in vivo visualization of the dynamic actin cytoskeleton in all cells of Arabidopsis seedlings. Eur. J. Cell Biol. 84, 595–608. [DOI] [PubMed] [Google Scholar]
- Wang, Q. and Huang, S. (2014) Visualization of microtubule organization and dynamics in living Arabidopsis embryonic cells. Mol Plant, 7, 1397–1401. [DOI] [PubMed] [Google Scholar]
- Wang, Y.S. , Motes, C.M. , Mohamalawari, D.R. and Blancaflor, E.B. (2004) Green fluorescent protein fusions to Arabidopsis Fimbrin 1 for spatio‐temporal imaging of F‐actin dynamics in roots. Cell Motil. Cytoskeleton, 59, 79–93. [DOI] [PubMed] [Google Scholar]
- Wang, Y.S. , Yoo, C.M. and Blancaflor, E.B. (2008) Improved imaging of actin filaments in transgenic Arabidopsis plants expressing a green fluorescent protein fusion to the C‐ and N‐termini of the fimbrin actin‐binding domain 2. New Phytol. 177, 525–536. [DOI] [PubMed] [Google Scholar]
- Wang, H.J. , Hsu, Y.W. , Guo, C.L. , Jane, W.N. , Wang, H. , Jiang, L. and Jauh, G.Y. (2017) VPS36‐dependent multivesicular bodies are critical for plasmamembrane protein turnover and vacuolar biogenesis. Plant Physiol. 173, 566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weijers, D. , Franke‐van Dijk, M. , Vencken, R.J. , Quint, A. , Hooykaas, P. and Offringa, R. (2001) An Arabidopsis minute‐like phenotype caused by a semi‐dominant mutation in a RIBOSOMAL PROTEIN S5 gene. Development, 128, 4289–4299. [DOI] [PubMed] [Google Scholar]
- Willemsen, V. , Friml, J. , Grebe, M. , Van Den Toorn, A. , Palme, K. and Scheres, B. (2003) Cell polarity and PIN protein positioning in Arabidopsis require STEROL METHYLTRANSFERASE1 function. Plant Cell, 15, 612–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, S. , BarbierdeReuille, P. , Lane, B. , Bassel, G.W. , Prusinkiewicz, P. , Smith, R.S. and Weijers, D. (2014) Genetic control of plant development by overriding a geometric division rule. Dev. Cell, 29, 75–87. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Microtubule and actin structures in root tip labeled by ACE‐R15 and ACE‐R14.
Figure S2. Effect of Taxol on microtubules in roots.
Figure S3. Overview of Arabidopsis cellular markers for embryogenesis (ACEs).
Figure S4. Sample preparation for imaging cellular structures.
Figure S5. Seedling sample preparation for short‐term chemical treatment and live imaging.
Table S1. Catalog of Arabidopsis cellular markers for embryogenesis (ACE) lines.
Table S2. Mounting solutions used in this study.
Table S3. Effects of 90 min treatment with 10 μm Taxol on cortical microtubule dynamics in root epidermal cells.
Table S4. Central/peripheral fluorescence intensity ratio (I C/I P) of ACE‐W01, ACE‐W02, ACE‐03 and ACE‐W04 in early embryos.
Table S5. Primers used in this study.