

Abstract
The kidneys, after the bone marrow and liver, are third in terms of the amounts of haem synthesized daily. Haem is incorporated into haemoproteins that are critical to renal physiology. In turn, disturbances in haem metabolism interfere with renal physiology and are tightly interrelated with kidney diseases. Acute intermittent porphyria causes kidney injury, whereas medical situations associated with end-stage renal disease, such as porphyrin accumulation, iron overload and hepatitis C, participate in the inhibition of uroporphyrinogen decarboxylase and predispose the individual to porphyria cutanea tarda. Even if some of these interactions have been known for a long time, the clinical situations associated with these interrelations have strikingly evolved over time with the advent of new therapeutic strategies for dialysis therapy and a better understanding of the pathophysiological mechanisms of porphyria-associated kidney disease. Physicians should be aware of these interactions. The aim of this review is to summarize the complex interactions between kidney physiology and pathology in the settings of porphyria and to emphasize their often-underestimated importance.
Keywords: acute intermittent porphyria, acute kidney injury, chronic kidney disease, porphyria cutanea tarda, porphyria
Outline
Knowledge has recently increased regarding relationships between the kidneys and porphyrias. The important relationship between peptide transporter 2 (PEPT2) variants and the development of renal disease in acute porphyrias is an example of how advances in the field can support new pathophysiological concepts, allow for the discovery of biomarkers of disease evolution and identify therapeutic targets [1]. The interactions between porphyria and renal diseases have been known for some time, and the aim of this review is to provide a comprehensive picture of the relationship between haem biosynthesis pathways, porphyria, renal homeostasis and kidney diseases. A description of the biochemical basis of the haem synthesis pathway, its perturbations and how it participates in renal physiology will be performed. Renal diseases associated with porphyrias will be addressed in describing the clinical and experimental basis of porphyria cutanea tarda (PCT) associated with end-stage renal disease (ESRD) and kidney diseases associated with acute intermittent porphyria (AIP). The available data on the management of kidney transplantation in individuals with acute porphyria will be examined. Finally, in addition to kidney diseases, acute porphyrias also affect water homeostasis by inducing inappropriate antidiuretic hormone secretion.
Background
The haem biosynthetic pathway
Haem participates in the formation of haemoproteins, such as haemoglobin, myoglobin, cytochromes, lipoxygenases, cyclooxygenases and peroxidases [2]. The binding of Fe2+ in the centre of the porphyrin ring with oxygen (O2), nitric oxide (NO) and carbon monoxide (CO) allows the physiological functions of these proteins, such as O2 transport and oxidative reactions. The synthesis of haem begins in the mitochondria with the conjugation of succinyl coenzyme A to the amino acid glycine by aminolevulinic acid synthase (ALAS), leading to the production of the porphyrin precursor ALA [2]. In erythroid precursors, ALA is produced by ALAS2, an isoform whose expression is positively regulated by iron, whereas in the liver and kidneys, ALA is produced by ALAS1, an isoform under the negative control of haem [3].
Porphobilinogen (PBG) results from the dehydration of ALA by ALA-dehydratase (Figure 1). PBG is a pyrrole-containing intermediate, and the third enzyme of the pathway, hydroxymethylbilane synthase (HMBS, also known as PBG deaminase), catalyzes the condensation of four PBG molecules into the linear hydroxymethylbilane. The cyclization of hydroxymethylbilane by uroporphyrinogen III synthase generates a large macrocyclic structure called uroporphyrinogen III. Next, successive steps of oxidative decarboxylation produce coproporphyrinogen III, protoporphyrinogen III, protoporphyrin IX and finally haem, after the addition of Fe2+ by ferrochelatase, the last enzyme of the pathway. These last steps are performed in the mitochondria [2]. In all, 80% of the haem is produced in the bone marrow (for haemoglobin production), 15% in the liver (for the production of haemoproteins, such as cytochromes) and 5% in the kidneys [4].
Fig. 1.

Representation of the haem biosynthesis pathway. The enzymatic activities that occur in the mitochondria are shown in red and the enzymatic activities that occur in the cytoplasm are shown in blue. Alteration of the activity of each of the eight enzymes causes a specific porphyria.
Disorders of haem biosynthesis
Porphyria encompasses a group of eight metabolic disorders of the haem biosynthesis pathway [5, 6], and knowledge of these biochemical sequential reactions is important for understanding some critical aspects of the pathophysiology and the diagnosis of porphyria. Because porphyrias are rare diseases and the symptoms of porphyrias are non-specific, porphyrias remain a relatively unknown group of diseases for many physicians. However, specific and simple tests allow for a diagnosis in symptomatic patients, which is a key step before the introduction of a specific therapy [4].
Each porphyria is caused by the abnormal function of an enzymatic step, resulting in an accumulation of porphyrins and porphyrin precursors. The symptoms and biological diagnosis of porphyrias are related to the systemic accumulation of compounds upstream of the deficient enzyme. Most porphyrias are transmitted in an autosomal dominant manner, allowing for residual enzymatic activity, which can vary by tissue based on environmental and endogenous factors, such as drugs, alcohol, hormonal status, inflammation or virus infection [6, 7]. The interaction of genetic features with additional susceptibility factors results in a highly multifactorial disease. Notably, PCT is a multifactorial, iron-related disorder and most cases do not have UROD mutations.
Acute porphyrias are a group of diseases that include AIP, variegate porphyria and hereditary coproporphyria. Acute porphyrias are characterized by the occurrence of neurovisceral attacks (abdominal pain, nausea, vomiting, constipation and neuropsychiatric symptoms) accompanied by the excretion of large amounts of the porphyrin precursors ALA and PBG in the urine. ALA is a highly hydrophilic molecule that can cross barriers, such as the blood–brain barrier, and is freely excreted in the urine. ALA is taken up through the di- and tri-peptide transporters PEPT1 and PEPT2 [8] and some sodium-dependant neutral amino acid transporters. [9, 10]. ALA is a cytotoxic molecule that promotes oxidative stress and is a potent vasoconstrictor that can promote injury in target organs. Thus ALA is considered to be directly responsible for the symptoms of acute porphyria [11].
Cutaneous porphyrias compose the second clinical group of porphyrias and have two main phenotypes: bullous porphyria (sporadic and familial PCT) and acute painful photosensitive porphyria (erythropoietic protoporphyria and X-linked erythropoietic protoporphyria). PCT is characterized by bullous and blister lesions on areas of exposed skin and is associated with skin fragility [12]. Notably, a photocutaneous disease similar to PCT can occur in individuals with variegate porphyria and hereditary coproporphyria. The tetrapyrrole rings of porphyrins are highly photoreactive; they absorb light energy and convey it to the surrounding tissues to mediate lipid, nucleic acid and peptide oxidation. The principal site of photoinjury appears to be the blood vessels of the papillary dermis [5]. Porphyrins generate oxidative free radicals when exposed to ultraviolet light, resulting in photosensitivity and skin lesions. The diagnosis of PCT is determined by urine or plasma porphyrin profiles with a predominance of uroporphyrin and heptacarboxyporphyrin. In PCT, levels of ALA and PBG are normal or only minimally elevated.
Other very rare porphyrias, such as congenital erythropoietic porphyria, hepatoerythropoietic porphyria and rare recessive acute hepatic porphyrias, such as 5-aminolevulinic acid dehydratase porphyria, will not be addressed in this review.
Kidneys and haem biosynthesis
Haem biosynthesis, metabolism and catabolism in the kidney
Haem synthesis in the kidney mainly occurs in the proximal tubules, and the renal cortex possesses a greater haem-synthesizing capacity than the medulla; this distribution of haem-synthesizing ability closely overlaps with that of the cytochrome P450 system [13]. The proximal tubules are highly active metabolically and haem is required for many functions that depend on a variety of haemoproteins, such as P450 cytochromes, which are involved in the oxidative metabolism of xenobiotics (CYP3A5) or endogenous molecules (CYP27B1 for 25-hydroxyvitamin D 1α-hydroxylation; CYP4F2/4A11 for eicosanoids metabolism); NO synthase, guanylate cyclase and prostaglandin synthase, which influence vascular tone; and catalase and peroxidases, which have antioxidant activities.
The regulation of haem biosynthesis in kidney cells differs from that observed in the liver with respect to the regulation of the expression and activity of ALAS1, which is less responsive to porphyrinogenic molecules in the kidney [13]. For example, the biological effects of repressor compounds such as glucose and haem reflect a substantially greater ratio of non-regulated haem biosynthetic activity to overall haem biosynthetic activity in renal cells than in hepatocytes [13]. The fact that the renal haem-synthesizing machinery is refractory to the inhibitory effects of substances that are excreted by the kidney allows for a steady supply of haem to be used for incorporating into haem proteins, including those that are involved in the cytochrome P450 system [13, 14]. However, the biological mechanisms that support differences of ALAS1 activity between liver and kidney are unknown.
Pathological processes also may contribute to increased levels of free haem in the kidney, ultimately leading to renal damage. For example, ischaemic and nephrotoxic insults may destabilize intracellular haem proteins normally present in the kidney, thereby incurring increased intracellular levels of haem, leading to oxidative stress and tissue injury [14, 15]. Increased renal content of haem may also occur as haem proteins that are extrinsic to the kidney, such as myoglobin from injured muscle or haemoglobin from lysed erythrocytes, are delivered and incorporated by the kidney. After oxidation and destabilization of these haem proteins, haem is ultimately freed and thus accumulates within cells. The catabolism of haem is effected by haem oxygenase, which catalyzes the conversion of haem to biliverdin, which in turn is metabolized into bilirubin. Experimental evidence supports haem oxygenase as a protectant against haem-induced renal injury. Interestingly, the induction of haem oxygenase is accentuated by pathophysiologically relevant stimuli such as hypoxia [14, 15].
Perturbations of renal haem biosynthesis
The activity of enzymes of the haem biosynthetic pathway can be selectively inhibited, leading to increased production of porphyrins and porphyrin precursors in the urine. Studies on the mechanisms of trace metal–induced renal injury support the view that the kidney can play an important role in the aetiology of excess urinary porphyrins that are excreted as a result of a disordered porphyrin metabolism. Studies on lead [16], arsenic [17] and mercury [18] compounds have shown that prolonged exposure to low levels of these metals in rats and mice elicits a pronounced increase in the rate of coproporphyrin excretion as a result of the inhibition of coproporphyrinogen oxidase activity [13]. In addition, lead inhibits ALA dehydratase activity, leading to increased excretion of ALA in the urine, which constitutes a biomarker of the disease, and it is likely that ALA dehydratase inhibition and ALA accumulation is causative of the symptoms of the disease [19]. Together, these observations suggest that porphyrinogenic metals interfere with and inhibit the activity of coproporphyrinogen oxidase and ALA dehydratase in the kidney, leading to the production of excess porphyrins and porphyrin precursors in the urine. Disturbances of haem biosynthesis in the kidney also occur during genetic diseases. In hereditary tyrosinaemia type I, succinylacetone accumulates in the urine, blood and liver, where it inhibits ALA dehydratase to give rise to symptoms that are very similar to those of acute porphyrias [20].
ESRD predisposes to porphyria
Evidence from experimental and clinical studies indicates that injured kidneys may also play an important role in the aetiology and manifestations of human porphyrias. Several abnormalities of porphyrin metabolism leading to PCT have been described in patients who reached ESRD (Figure 2A). The prevalence of acquired PCT in haemodialysis ranges from 5 to 18% [21, 22], although systematic studies have not been performed. Notably, the levels of uroporphyrins in unaffected ESRD patients can be similar to or exceed (due to the lack of renal clearance) the levels found in patients who have PCT and normal kidney function, suggesting that additional precipitating factors likely occur [23].
Fig. 2.

Summary of the interactions between kidney disease and porphyrias. (A) ESRD is associated with metabolic disturbances that lead to a deficiency of the enzymatic activity of uroporphorinogen decarboxylase: iron overload (due to multiple transfusions), chronic hepatitis (due to the transfusion of contaminated blood packs), aluminium intoxication (used as a phosphorus chelator) and uraemic toxins (accumulated upon ESRD) reduce the enzymatic activity of uroporphorinogen decarboxylase. In addition, reduced clearance of porphyrins leads to the systemic accumulation of uroporphyrinogen III and symptoms of PCT. (B) The porphyrin precursors ALA and PBG are produced in excess in the steady state of the disease and during attacks in patients with AIP. ALA and PBG promote vasoconstriction and cytotoxicity, leading to hypertension, kidney tissue injury and squaring. Repeated AIP attacks promote repeated episodes of acute kidney injury, ultimately leading to irreversible CKD.
During ESRD, uraemic toxins with potent biological activities accumulate in the body of patients and interfere with the biosynthetic pathway of haem. The activity of uroporphyrinogen decarboxylase (UROD) is reduced during ESRD [24–26], leading to the accumulation of water-soluble uroporphyrins in the plasma, liver and skin. In addition, patients with ESRD have an impaired ability to excrete porphyrins. For example, uroporphyrins that are highly bound to high-molecular-weight proteins are not well dialysed by low-flux haemodialysis and therefore are not effectively eliminated (21, 27–29).
Iron also plays a key role in the development of the symptoms of acquired PCT by inhibiting uroporphyrinogen decarboxylase. During a time when transfusion was the sole therapeutic strategy for anaemia associated with ESRD, the need for frequent red blood cell transfusions was associated with iron overload and a high rate of chronic viral hepatitis infections. In the liver, iron and increased oxidative stress lead to the formation of inhibitors of uroporphyrinogen decarboxylase [30]. In addition, hepatitis C infections are thought to precipitate the dysfunction of uroporphyrinogen decarboxylase. Chronic hepatitis C infection decreases hepcidin production by hepatocytes, which leads to increased iron absorption from the gut and ultimately to iron overload [31]. In addition, although it is very uncommon today, aluminum intoxication from the use of aluminum-containing phosphorus chelators has been suspected to play a role in PCT, as high serum aluminium concentrations in patients with ESRD are associated with an accumulation of uroporphyrins and coproporphyrins [32] (Figure 2A). However, the way they affect enzymes in the haem biosynthetic pathway to promote prophyrins accumulation remains an unresolved issue.
PCT is currently much less common (at least in France), although there is no rigorous documentation of such a change in disease prevalence [21]. Indeed, PCT was a common disorder in dialysis patients in the pre-erythropoietin era when transfusion-related iron overload and hepatitis infections were common [33, 34]. The use of recombinant erythropoietin has been a therapeutic revolution that reduced the use of blood transfusions and thereby iron overload and hepatitis C infections. In addition, whereas standard haemodialysis did not remove uroporphyrins, the use of high-flux membranes dramatically increased plasma porphyrins clearance [21, 28, 29]. Kidney transplantation has also been reported to improve refractory PCT in patients under dialysis therapy [35, 36].
Pseudoporphyria, a non-porphyric disorder with similar skin lesions and histopathology but normal porphyrin levels, can occur in patients with impaired renal function [37]. Whereas the pathophysiological pathways leading to pseudoporphyria are unknown, the disease manifestations appear to develop due to the association of photosensitizing medications such as naproxen, furosemide, tetracyclines and amiodarone, with excessive ultraviolet exposure and the presence of uraemic toxins.
AIP promotes kidney injury
Whereas the prevalence of PCT associated with ESRD significantly declined due to better management of patients under chronic dialysis therapy, several series and experimental studies since the 1990s have reported an increased frequency of CKD among patients with AIP and highlighted the deleterious impact of porphyrin precursors on renal homeostasis (Figure 2B). Porphyria-associated kidney disease (PAKD) occurs in >50% of the patients with symptomatic AIP and 60% of patients with PAKD have hypertension [38–40]. Even if hypertension participates in CKD, AIP is an independent factor that promotes CKD [38]. Supporting this, the specific role of ALA and PBG in the chronic renal structural deterioration that accompanies AIP has recently emerged [38].
AIP promotes arteriolar injury
In a large cohort of patients with PAKD, histological findings of kidney biopsies revealed that diffuse glomerulosclerosis and chronic interstitial changes were almost universal (albeit non-specific) and were associated with additional ischaemic lesions, suggesting that AIP can target renal arterioles and cause kidney injury [38] (Figure 2B). The vascular toxicity of porphyrin precursors is highly probable since ALA promotes vasoconstriction experimentally [41, 42]. Abdominal symptoms are thought to be, at least in part, related to gut ischaemia [43]. The brain autopsies of AIP patients who died during crisis indicate the existence of multiple small infarcts [44, 45], and AIP attacks can cause posterior reversible encephalopathy [46, 47]. Consistent with a role of arteriolar damage in the pathogenesis of PAKD, the findings of kidney biopsies highlight the importance of the arteriolopathy associated with AIP: in half of the cases, severe vascular lesions comprising fibrous intimal hyperplasia associated with focal cortical atrophy, a finding reminiscent of the primary antiphospholipid syndrome–associated renal vasculopathy, were observed [38, 48]. From these features, a first pathophysiological characteristic can be drawn linking the vasoactive properties of ALA to acute arteriolar vasospasm that occurs during AIP crisis and leading to acute kidney injury. In addition, there are histological arguments indicating that renal arterioles can be chronically injured (maybe as a result of repeated acute injuries), leading to a chronic vasculopathy with a narrowed lumen and tissue ischaemia that contributes to CKD.
AIP promotes tubular injury
In addition to arteriolar lesions, kidney biopsies in PAKD patients consistently revealed a chronic tubulointerstitial disease characterized by interstitial fibrosis and tubular atrophy without specific glomerular lesions [38, 49]. Experimental data support a model wherein tubules are separate targets of porphyrin precursors and ALA promotes phenotypic changes of epithelial cells reminiscent of epithelial-to-mesenchymal transition and tubular cell death [38]. The precise mechanism by which porphyrin precursors are cytotoxic remains to be established, but it could involve endoplasmic reticulum stress [38]. Even if there is no histological documentation of kidney damage during AIP attacks, tubular injury could occur during crisis, when the concentration of porphyrin precursors in the urine peaks, promoting tubular cell death followed by healing. In the pathophysiological model we propose, renal ischaemia generated by both acute vasoconstriction and hypovolaemia (that results from emesis) could promote acute ischaemic tubular injury. The succession of attacks and injury/reparation cycles could progressively generate chronic lesions, such as tubular atrophy, interstitial fibrosis and arteriosclerosis. In addition, a chronic toxic effect of slightly elevated concentrations of ALA and PBG could lead to a slowly evolving structural deterioration. This hypothesis is supported by the fact that the concentrations of porphyrin precursors in urine samples taken outside of AIP attacks are basally higher for patients with a lower estimated glomerular filtration rate than for patients without kidney dysfunction [38]. In line with the tubular toxicity of ALA, lead poisoning, during which ALA is overexcreted in urine, is also associated with chronic tubulointerstitial disease [16]. ALA likely contributes to tubular injury in this medical situation, although this hypothesis must be supported by experimental data.
Supporting the participation of ALA in tubular injury, human PEPT2 significantly impacts the natural history of PAKD. ALA, which is highly hydrophilic, is freely filtered in the urine and reabsorbed in the S2 and S3 segments of the proximal tubule by PEPT2. A variant of the gene that encodes PEPT2, termed PEPT2*2, reduces the affinity of the transporter for ALA and is associated with reduced severity and slower progression of PAKD [1] [Figure 3]. Notably, in the cohort that we have followed, all of the patients who suffered from hepatocarcinoma had CKD, which suggests a link between the processes and that a shared mechanism that drives PAKD and hepatocarcinoma (such as chronic exposition to ALA) is likely involved but remains to be identified (unpublished data).
Fig 3.
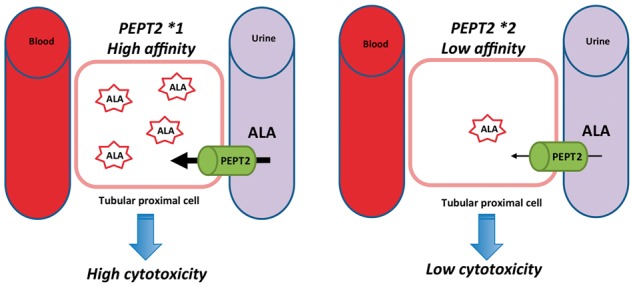
Model proposal for the role of PEPT2 in ALA tubular toxicity. The dipeptide transporter PEPT2 is expressed by the apical membrane of proximal tubular kidney cells. The PEPT*1 variant has a greater affinity for ALA than does PEPT2*2, and as a consequence, ALA could be less reabsorbed from the urine in the tubular cells that express PEPT2*2 than in the cells that express PEPT2*1, leading to a reduced cytotoxicity of ALA.
Kidney transplantation and AIP
PAKD is not a severe disease in most cases since it has a relatively low rate of decline of the glomerular filtration rate over time (1 mL/min/year, which is slightly higher than the physiological rate) and only a small proportion of patients apparently reach ESRD. However, and even if there is no well-described cohort of AIP patients under haemodialysis prospectively followed up, ESRD can be a devastating complication for AIP patients with a chronic active disease, leading to peripheral neuropathy and secondary cutaneous lesions. In such cases, the clinical course could not be reversed by medical treatment [50]. As a consequence, kidney transplantation can be considered as a therapeutic option. Numerous case reports and small series have reported that patients showed no evidence of activation of porphyria following transplantation, indicating that AIP is not a contraindication to kidney transplantation [51]. Importantly, AIP patients appear to safely tolerate several of the immunosuppressive medications currently used to prevent transplant rejection, including tacrolimus, mycophenolate mofetil and sirolimus [52, 53]. Due to the lack of well-described cohorts of AIP patients with a kidney transplant, critical issues cannot be resolved, including possible modifications of the activity of the disease after kidney transplantation.
AIP promotes inappropriate antidiuretic hormone (ADH) secretion
Finally, the syndrome of inappropriate ADH secretion is also attributable to AIP attacks [54–56]. AIP attacks are often associated with hyponatraemia characterized by the inappropriate release of ADH by the hypothalamus (independent of changes in plasma volume or osmolality). High levels of ALA can cause vascular injury, resulting in focal oedema in susceptible regions of the brain, such as the hypothalamus [56]. The blood–brain barrier protects the brain from toxic agents, but certain areas, such as the hypothalamus (the site where ADH is released) do not have such protection. Pathological specimens of the hypothalamus in an experimental model of AIP highlighted the vacuolization of neurons in the supra-optic nucleus, leading to leakage of ADH into the circulation [57]. Although not a renal disease, the syndrome of inappropriate ADH secretion is considered a frequent cause of hyponatraemia in AIP and must be identified as a symptom of AIP crisis.
Conclusion and perspectives
Porphyrias and kidney diseases are two strongly interrelated processes with two phenotypic landscapes: PCT in ESRD patients and CKD in AIP patients. Due to progress in the care of ESRD patients under dialysis that has been made in the 1990s (including the use of high-flux membranes and recombinant erythropoietin), which allow a higher clearance of circulating porphyrins and a normal activity of uroporphyrin decarboxylase, PCT is no longer a frequent complication of ESRD. In turn, PAKD has been identified as a specific clinical entity with a well-defined clinical, biological and histological expression. In addition, the spectrum of the biological impact of porphyrin precursors, particularly ALA, on arterioles and tubules has recently been delineated. Nephrologists must be aware of this association because HMBS mutations are frequent and AIP remains underdiagnosed, particularly for patients with few or atypical acute symptoms.
Funding
The study was funded by Agence Nationale pour la Recherche and Agence de la Biomédecine.
Conflict of interest statement. None declared.
References
- 1. Tchernitchko D, Tavernier Q, Lamoril J. et al. A variant of peptide transporter 2 predicts the severity of porphyria-associated kidney disease. J Am Soc Nephrol 2017; 28: 1924–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poulos TL. Heme enzyme structure and function. Chem Rev 2014; 114: 3919–3962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Manceau H, Gouya L, Puy H.. Acute hepatic and erythropoietic porphyrias: from ALA synthases 1 and 2 to new molecular bases and treatments. Curr Opin Hematol 2017; 24: 198–207 [DOI] [PubMed] [Google Scholar]
- 4. Szlendak U, Bykowska K, Lipniacka A.. Clinical, biochemical and molecular characteristics of the main types of porphyria. Adv Clin Exp Med 2016; 25: 361–368 [DOI] [PubMed] [Google Scholar]
- 5. Puy H, Gouya L, Deybach JC.. Porphyrias. Lancet 2010; 375: 924–937 [DOI] [PubMed] [Google Scholar]
- 6. Bissell DM, Anderson KE, Bonkovsky HL.. Porphyria. N Engl J Med 2017; 377: 862–872 [DOI] [PubMed] [Google Scholar]
- 7. Karim Z, Lyoumi S, Nicolas G. et al. Porphyrias: a 2015 update. Clin Res Hepatol Gastroenterol 2015; 39: 412–425 [DOI] [PubMed] [Google Scholar]
- 8. Döring F, Walter J, Will J. et al. Delta-aminolevulinic acid transport by intestinal and renal peptide transporters and its physiological and clinical implications. J Clin Invest 1998; 101: 2761–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rud E, Gederaas O, Hogset A. et al. 5-aminolevulinic acid, but not 5-aminolevulinic acid esters, is transported into adenocarcinoma cells by system BETA transporters. Photochem Photobiol 2000; 71: 640–647 [DOI] [PubMed] [Google Scholar]
- 10. Palacin M, Estevez R, Bertran J. et al. Molecular biology of mammalian plasma membrane amino acid transporters. Physiol Rev 1998; 78: 969–1054 [DOI] [PubMed] [Google Scholar]
- 11. Laafi J, Homedan C, Jacques C. et al. Pro-oxidant effect of ALA is implicated in mitochondrial dysfunction of HepG2 cells. Biochimie 2014; 106: 157–166 [DOI] [PubMed] [Google Scholar]
- 12. Handler NS, Handler MZ, Stephany MP. et al. Porphyria cutanea tarda: an intriguing genetic disease and marker. Int J Dermatol 2017; 56: e106–e117 [DOI] [PubMed] [Google Scholar]
- 13. Woods JS. Regulation of porphyrin and heme metabolism in the kidney. Semin Hematol 1988; 25: 336–348 [PubMed] [Google Scholar]
- 14. Tracz MJ, Alam J, Nath KA.. Physiology and pathophysiology of heme: implications for kidney disease. J Am Soc Nephrol 2007; 18: 414–420 [DOI] [PubMed] [Google Scholar]
- 15. Agarwal A, Balla J, Alam J. et al. Induction of heme oxygenase in toxic renal injury: a protective role in cisplatin nephrotoxicity in the rat. Kidney Int 1995; 48: 1298–1307 [DOI] [PubMed] [Google Scholar]
- 16. Fowler BA, Kimmel CA, Woods JS. et al. Chronic low-level lead toxicity in the rat. III. An integrated assessment of long-term toxicity with special reference to the kidney. Toxicol Appl Pharmacol 1980; 56: 59–77 [DOI] [PubMed] [Google Scholar]
- 17. Woods JS, Fowler BA.. Altered regulation of mammalian hepatic heme biosynthesis and urinary porphyrin excretion during prolonged exposure to sodium arsenate. Toxicol Appl Pharmacol 1978; 43: 361–371 [DOI] [PubMed] [Google Scholar]
- 18. Woods JS, Fowler BA.. Renal porphyrinuria during chronic methyl mercury exposure. J Lab Clin Med 1977; 90: 266–272 [PubMed] [Google Scholar]
- 19. Hunter GA, Ferreira GC.. Molecular enzymology of 5-aminolevulinate synthase, the gatekeeper of heme biosynthesis. Biochim Biophys Acta 2011; 1814: 1467–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Laet C, Dionisi-Vici C, Leonard JV. et al. Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis 2013; 8: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shieh S, Cohen JL, Lim HW.. Management of porphyria cutanea tarda in the setting of chronic renal failure: a case report and review. J Am Acad Dermatol 2000; 42: 645–652 [PubMed] [Google Scholar]
- 22. Vasconcelos P, Luz-Rodrigues H, Santos C. et al. Desferrioxamine treatment of porphyria cutanea tarda in a patient with HIV and chronic renal failure. Dermatol Ther 2014; 27: 16–18 [DOI] [PubMed] [Google Scholar]
- 23. Seubert S, Seubert A, Rumpf KW. et al. A porphyria cutanea tarda-like distribution pattern of porphyrins in plasma, hemodialysate, hemofiltrate, and urine of patients on chronic hemodialysis. J Invest Dermatol 1985; 85: 107–109 [DOI] [PubMed] [Google Scholar]
- 24. Day RS, Eales L.. Porphyrins in chronic renal failure. Nephron 1980; 26: 90–95 [DOI] [PubMed] [Google Scholar]
- 25. Mamet R, Gafter U, Korzets A. et al. Decreased uroporphyrinogen decarboxylase activity in patients with end-stage renal disease undergoing hemodialysis. Nephron 1995; 70: 202–206 [DOI] [PubMed] [Google Scholar]
- 26. Yasuda G, Ikeda Y, Satta H. et al. Porphyrin metabolism abnormalities and its treatment in a uremic patient with porphyria cutanea tarda. Nephron 1993; 63: 235–236 [DOI] [PubMed] [Google Scholar]
- 27. Gebril M, Weinkove C, Ead R. et al. Plasma porphyrins in chronic renal failure. Nephron 1990; 55: 159–163 [DOI] [PubMed] [Google Scholar]
- 28. Carson RW, Dunnigan EJ, DuBose TD Jr. et al. Removal of plasma porphyrins with high-flux hemodialysis in porphyria cutanea tarda associated with end-stage renal disease. J Am Soc Nephrol 1992; 2: 1445–1450 [DOI] [PubMed] [Google Scholar]
- 29. Fontanellas A, Herrero JA, Moran MJ. et al. Efficiency of three different hemodialysis membranes for plasma porphyrin removal. Am J Kidney Dis 1995; 25: 30–33 [DOI] [PubMed] [Google Scholar]
- 30. Ryan Caballes F, Sendi H, Bonkovsky HL.. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int 2012; 32: 880–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van Meter JR, Tierney KR, Pittelkow MR.. Iron, genes, and viruses: the porphyria cutanea tarda triple threat. Cutis 2011; 88: 73–76 [PubMed] [Google Scholar]
- 32. Gafter U, Mamet R, Korzets A. et al. Bullous dermatosis of end-stage renal disease: a possible association between abnormal porphyrin metabolism and aluminium. Nephrol Dial Transplant 1996; 11: 1787–1791 [PubMed] [Google Scholar]
- 33. Anderson KE, Goeger DE, Carson RW. et al. Erythropoietin for the treatment of porphyria cutanea tarda in a patient on long-term hemodialysis. N Engl J Med 1990; 322: 315–317 [DOI] [PubMed] [Google Scholar]
- 34. Sarkell B, Patterson JW.. Treatment of porphyria cutanea tarda of end-stage renal disease with erythropoietin. J Am Acad Dermatol 1993; 29: 499–500 [DOI] [PubMed] [Google Scholar]
- 35. Ewing S, Crosby DL.. Renal transplantation for porphyria cutanea tarda. N Engl J Med 1997; 336: 811. [DOI] [PubMed] [Google Scholar]
- 36. Stevens BR, Fleischer AB Jr, Piering F. et al. Porphyria cutanea tarda in the setting of renal failure. Response to renal transplantation. Arch Dermatol 1993; 129: 337–339 [PubMed] [Google Scholar]
- 37. Green JJ, Manders SM.. Pseudoporphyria. J Am Acad Dermatol 2001; 44: 100–108 [DOI] [PubMed] [Google Scholar]
- 38. Pallet N, Mami I, Schmitt C. et al. High prevalence of and potential mechanisms for chronic kidney disease in patients with acute intermittent porphyria. Kidney Int 2015; 88: 386–395 [DOI] [PubMed] [Google Scholar]
- 39. Church SE, McColl KE, Moore MR. et al. Hypertension and renal impairment as complications of acute porphyria. Nephrol Dial Transplant 1992; 7: 986–990 [PubMed] [Google Scholar]
- 40. Andersson C, Lithner F.. Hypertension and renal disease in patients with acute intermittent porphyria. J Intern Med 1994; 236: 169–175 [DOI] [PubMed] [Google Scholar]
- 41. Chang CJ, Lee YH, Yang JY. et al. Pilot in vitro toxicity study of 5-ALA and Photofrin in microvascular endothelial cell cultures. J Clin Laser Med Surg 1997; 15: 83–87 [DOI] [PubMed] [Google Scholar]
- 42. Middelburg TA, de Bruijn HS, Tettero L. et al. Topical hexylaminolevulinate and aminolevulinic acid photodynamic therapy: complete arteriole vasoconstriction occurs frequently and depends on protoporphyrin IX concentration in vessel wall. J Photochem Photobiol B 2013; 126: 26–32 [DOI] [PubMed] [Google Scholar]
- 43. Lithner F. Could attacks of abdominal pain in cases of acute intermittent porphyria be due to intestinal angina? J Intern Med 2000; 247: 407–409 [DOI] [PubMed] [Google Scholar]
- 44. Kupferschmidt H, Bont A, Schnorf H.. Transient cortical blindness and bioccipital brain lesions in two patients with acute intermittent porphyria. Ann Intern Med 1995; 123: 598–600 [DOI] [PubMed] [Google Scholar]
- 45. Lai CW, Hung TP, Lin WS.. Blindness of cerebral origin in acute intermittent porphyria. Report of a case and postmortem examination. Arch Neurol 1977; 34: 310–312 [DOI] [PubMed] [Google Scholar]
- 46. Çelik M, Forta H, Türker Dalkılıç BG.. MRI reveals reversible lesions resembling posterior reversible encephalopathy in porphyria. Neuroradiology 2002; 44: 839–841 [DOI] [PubMed] [Google Scholar]
- 47. Soysal A, Dogan P, Dayan C. et al. Reversible MRI findings of porphyric encephalopathy. A report of two cases. Neuroradiol J 2008; 21: 655–659 [DOI] [PubMed] [Google Scholar]
- 48. Nochy D, Daugas E, Droz D.. The intrarenal vascular lesions associated with primary antiphospholipid syndrome. J Am Soc Nephrol 1999; 10: 507–518 [DOI] [PubMed] [Google Scholar]
- 49. Marsden JT, Chowdhury P, Wang J. et al. Acute intermittent porphyria and chronic renal failure. Clin Nephrol 2008; 69: 339–346 [DOI] [PubMed] [Google Scholar]
- 50. Sardh E, Andersson DE, Henrichson A. et al. Porphyrin precursors and porphyrins in three patients with acute intermittent porphyria and end-stage renal disease under different therapy regimes. Cell Mol Biol 2009; 55: 66–71 [PubMed] [Google Scholar]
- 51. Nunez DJ, Williams PF, Herrick AL. et al. Renal transplantation for chronic renal failure in acute porphyria. Nephrol Dial Transplant 1987; 2: 271–274 [PubMed] [Google Scholar]
- 52. Barone GW, Gurley BJ, Anderson KE. et al. The tolerability of newer immunosuppressive medications in a patient with acute intermittent porphyria. J Clin Pharmacol 2001; 41: 113–115 [DOI] [PubMed] [Google Scholar]
- 53. El-Haggan W, Lobbedez T, Ryckelynck JP. et al. Sirolimus tolerability in a kidney transplant recipient with acute intermittent porphyria. Nephrol Dial Transplant 2002; 17: 1147. [DOI] [PubMed] [Google Scholar]
- 54. Tebar MT, Aguilera L.. [Acute intermittent porphyria and inappropriate ADH syndrome]. Rev Esp Anestesiol Reanim 2010; 57: 311–313 [DOI] [PubMed] [Google Scholar]
- 55. Lopez Montes A, Lorenzo I, Perez Martinez J.. [Porphyria and inappropriate antidiuretic hormone syndrome]. Nefrologia 2004; 24: 85–88 [PubMed] [Google Scholar]
- 56. Usalan C, Erdem Y, Altun B. et al. Severe hyponatremia due to SIADH provoked by acute intermittent porphyria. Clin Nephrol 1996; 45: 418. [PubMed] [Google Scholar]
- 57. Tschudy DP, Valsamis M, Magnussen CR.. Acute intermittent porphyria: clinical and selected research aspects. Ann Intern Med 1975; 83: 851–864 [DOI] [PubMed] [Google Scholar]