

Abstract
To analyze the spectrum and founder effect of TMC1 mutations in patients with non‐syndromic deafness in the Xiamen area. Sporadic pedigrees were detected by targeted next‐generation sequencing, and 110 unrelated patients from Xiamen Special Education School were analyzed through Sanger sequencing for the TMC1 gene. In total, 53 SNPs were designed to analyze the haplotypes of the TMC1 c.2050G>C mutation. The probands of three families were found to be homozygous for TMC1 c.2050G>C, and their parents were all heterozygous for the TMC1 c.2050G>C mutation. In 110 unrelated patients from Xiamen Special Education School, four were found to carry compound heterozygotes of TMC1 c.2050G>C, which were compound heterozygotes of c.804G>A, c.1127T>C, c.1165C>T, and c.1396_1398delAAC, respectively. Three types of TMC1 polymorphisms (c.45C>T, c.1713C>T, c.2208+49C>T) and two heterozygotes of novel variants (c.1764‐4C>A, c.2073G>A[p.K691K]) were found in the remaining 100 patients. In total, four novel variants were detected in this study. These mutations and variants were not detected in 100 normal samples. The haplotypes of the probands of families with TMC1 c.2050G>C were identical. There were unique hotspots and spectra of TMC1 mutations in the Xiamen deaf population. Haplotype analysis is useful to understand the founder effect of the hot spot mutation.
Keywords: founder effect, haplotype, hearing impairment, single nucleotide polymorphisms
1. INTRODUCTION
TMC1 mutations have recently been found to be one of the major causes of profound recessive deafness worldwide (Ayesha et al., 2016). In humans, most TMC1 mutations are recessive mutations associated with congenital non‐syndromic severe to profound deafness DFNB7/11, whereas dominant alleles of TMC1 were reportedly associated with progressive loss of hearing DFNA36 (Kitajiri et al., 2007). Until now, there were 84 definite pathogenic sites of TMC1 reported in HGMD (http://www.hgmd.cf.ac.uk/ac/index.php); the pathogenicity of most of them was determined by a consanguineous marriage pedigree study, which showed a sporadic phenomenon. In this study, we discovered that a single site of TMC1 (c.2050G>C) from sporadic families in one region had a high incidence; single nucleotide polymorphisms (SNPs) were used to identify its founder effect.
The TMC1 gene has 24 exons and spans 314,551 base pairs on chromosome 9q21.13. The protein, which functions as an ion channel or transporter to mediate K+ homeostasis for the normal function of cochlear hair cells, consists of 760 amino acids containing six transmembrane (TM) domains (Xue et al., 2013). TMC1 c.2050G>C (p.D684H) is a missense variant in exon21. We used TMHMM2.0 to predict the structure of the TMC1 protein, including its six TMs with cytoplasmic N‐ and C‐termini. The results indicated that TMC1 c.2050G>C (p.D684H) occurs in the extracellular region between TM5 and TM6, and that this mutation may affect ion channel function in hair cells.
2. MATERIALS AND METHODS
2.1. Materials
2.1.1. Family recruitment and clinical evaluations
Three families were enrolled in this study. Deafness gene detection was performed over different periods in each family. The proband of the first family (XJA) was a student at the Xiamen Special Education School in Fujian province, whose common deafness gene detection (GJB2, SLC26A4, and mtDNA 12SrRNA) was negative. The other two probands (M126, M372) were outpatients at the Chinese PLA General Hospital. The three families were all from the same region of China: Southern Fujian. All probands had congenital severe to profound sensorineural hearing loss. Their standard audiometric evaluations and pedigrees are shown. (Figure 1).
Figure 1.
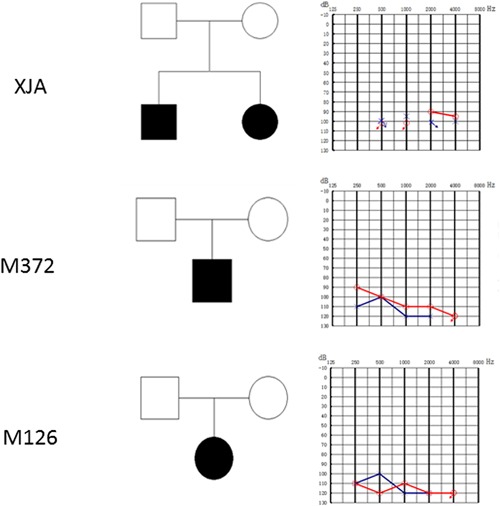
Pedigrees of the three families and pure tone audiometric evaluations. [Color figure can be viewed at http://wileyonlinelibrary.com]
2.1.2. Xiamen special education school
In total, 110 of 155 unrelated patients with hearing impairment (152 students, 3 teachers) from Xiamen Special Education School in Fujian province were not found to have a definite etiology in three common deafness genes by Sanger sequencing of all exons of GJB2, SLC26A4, and mtDNA 12SrRNA. The patients were then screened for the TMC1 gene (Yi et al., 2015). In addition, 100 individuals with normal hearing from the Xiamen area were also screened for the TMC1 gene.
2.2. Ethics statement and DNA samples
The protocol for this investigation was performed with the approval of the Ethics Committee of Chinese PLA General Hospital. All subjects or their guardians signed informed consent forms prior to blood sampling. Questionnaires included basic information such as name, age, address, family history, health records of the mother during pregnancy, and a clinical history of the patient such as infections and the use of aminoglycoside antibiotics. Physical examination, standard audiometric evaluations, and a computed tomography (CT) scan of the temporal bone of the probands were performed (Yi et al., 2015). DNA was extracted from peripheral blood leukocytes using a commercially available DNA extraction kit (Watson Biotechnologies Inc., Shanghai, China).
2.3. Methods
2.3.1. Targeted next‐generation sequencing
The next‐generation sequencing panel of MyGenostics was used for the three families (Ying et al., 2015). This panel was designed with biotinylated oligo‐probes to capture all exons, splice sites, and immediate flanking intron sequences of 109 deafness genes (Table S1, Supplementary Information), including all known genes for non‐syndromic hearing loss and some relatively common genes for syndromic hearing loss (at the time when the panel was designed). Captured DNA fragments were sequenced on Illumina HiSeq2000 Analyzers. Data analysis and bioinformatics processing were performed following standard Illumina procedures. Reads were aligned to NCBI37/hg19 assembly using the BWA Multi‐Vision software package. SNVs were detected and genotyped with the GATK UnifiedGenotyper in single‐sample mode. Indels were identified using the GATK Indel Genotyper. Potentially pathogenic variants were defined as nonsense, missense, splice‐site, and indel variants that have allele frequencies under 0.01 (determined by databases including NCBI dbSNP, NHLBI ESP, and 1000 Genomes).
2.3.2. TMC1 sequencing
All coding exons flanking 16–407 base pairs of the TMC1 gene were sequenced in patients from Xiamen Special Education School and the control group. The primers used for amplification and sequencing are listed in Supplementary Table S2. PCRs were set up in a reaction volume of 25 μl, containing 50 ng of genomic DNA, 200 nM of each primer, 200 μm of each dNTP, and 1.25 units of DNA polymerase (TAKARA) and 1X buffer. After initial denaturation at 96°C for 5 min, 30 thermal cycles of 96°C for 20 s, 60°C for 30 s, and 72°C for 1 min were carried out, followed by incubation at 72°C with a 5‐min hold (Veriti 96‐Well Thermal Cycler, Applied Biosystems, Foster City, CA). Sequencing was performed with an ABI 3730 DNA sequencer (ABI, Foster City, CA). Results were analyzed with Sequence Mutation Detection software (CapitalBio, Beijing, China) using Human Genome GRCh38.p7 Primary Assembly as a reference.
2.3.3. SNPs and MassARRAY SNP genotyping
The SNPs used in the study were identified based on the NCBI database. SNPs for haplotypes were chosen as follows: (i) CHB (Han Chinese in Beijing, China) heterozygosity in the 1000 Genomes database; (ii) MAF (minimum allele frequency) of CHB of 0.3–0.7; and (iii) upstream, downstream, and internal SNPs of TMC1 (Figure 2). Analysis of the identified SNPs was performed using the MassARRAY SNP genotyping system (Sequenom, San Diego, CA). Genotyping assays (primers for PCR amplification and extension) were automatically designed by the Sequenom MassARRAY Assay Design 4.0 software (Supplementary Table S3). The steps of the PCR cycling program, SAP (shrimp alkaline phosphatase) cleanup procedure, primer extension, extended reaction resin cleanup, and genotyping were performed as per the protocol (Stacey, Liuda & Diana, 2009).
Figure 2.
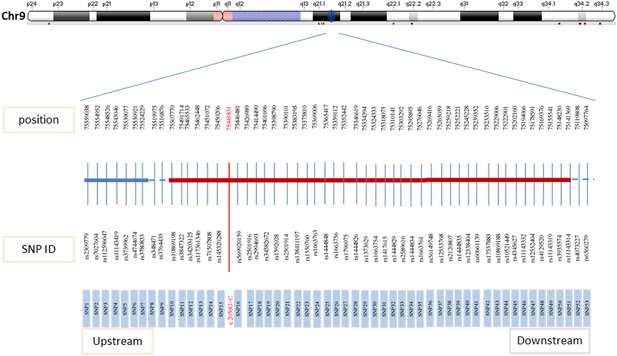
Single nucleotide polymorphisms of TMC1 for haplotypes. [Color figure can be viewed at http://wileyonlinelibrary.com]
3. RESULTS
3.1. NGS of families
Three families were tested by targeted next‐generation sequencing (NGS) of the deafness gene panel. The coverage of targeted regions exceeded 99.0%, and the average depth was 387.4‐fold. Details of the bioinformatics analysis followed American College of Medical Genetics and Genomics standards and guidelines.
Candidate variants are summarized in Supplementary Table S4. Consistent with the recessive inheritance of the families, bi‐allelic mutations were identified in these probands. Ruling out the mutations that result in homozygous or compound heterozygotes in the normal hearing database (in‐house NGS database of our own lab), a homozygous TMC1 c.2050G>C was found through NGS in two deaf children in a XJA family with two heterozygous normal hearing parents.
3.2. Xiamen special education school
One‐hundred fifty‐five unrelated patients with hearing impairment (152 students and 3 teachers) from Xiamen Special Education School in Fujian province were tested for three common deafness genes (GJB2, SLC26A4, and mtDNA 12SrRNA) of China. There were 20 patients (12.9%) confirmed to have GJB2 deafness‐causing mutations, 18 (11.61%) confirmed to be carrying two SLC26A4 pathogenic mutations, and 6 (3.87%) who carried a mtDNA 12SrRNA mutation. Except for the proband of the XJA family, 110 unrelated patients without positive genetic testing results from Xiamen Special Education School in Fujian province and an additional 100 negative samples from Xiamen with normal hearing were screened for the TMC1 gene. Four patients were compound heterozygotes for TMC1 mutations (Table 1), including c.2050G>C/c.804G>A, c.2050G>C/c.1127T>C, c.2050G>C/c.1165C>T, and c.2050G>C/c.1396_1398delAAC combinations. Only the parents of XT087 sent their blood samples for this study. The subject's father was heterozygous for the TMC1 c.2050G>C mutation, and the subject's mother was heterozygous for the TMC1 c.1396_1398delAAC mutation. The other three compound heterozygotes of TMC1 mutations were not verified due to the lack of parental DNA. These mutations were not detected in the 100 normal samples.
Table 1.
Compound heterozygotes of TMC1 in Xiamen Special Education School
| Number | Mutation (cDNA) | Amino acid changes | Type of sequence variation | Exon | Category | Reference |
|---|---|---|---|---|---|---|
| XT078 | c.2050G>C | p.D684H | Missense | 21 | Pathogenic | Tahir et al. (2015) |
| c.804G>A | p.W268X | Stop gain | 13 | Novel | This study | |
| XT090 | c.2050G>C | p.D684H | Missense | 21 | Pathogenic | Tahir et al. (2015) |
| c.1127T>C | p.L376P | SNV | 15 | Novel | This study | |
| XT161 | c.2050G>C | p.D684H | Missense | 21 | Pathogenic | Tahir et al. (2015) |
| c. 1165C>T | p.R389X | Stop gain | 15 | Pathogenic | Meyer et al. (2005) | |
| XT087 | c.2050G>C | p.D684H | Missense | 21 | Pathogenic | Tahir et al. (2015) |
| c.1396_1398delAAC | p.466del | Deletion | 16 | Likely pathogenic | Fangzhu et al. (2014) |
SNV, single nucleotide variation.
We found three types of TMC1 polymorphisms (c.45C>T, c.1713C>T, c.2208+49C>T) and two heterozygotes of novel variants (c.1764‐4C>A, c.2073G>A[p.K691K]) in this group. In total, the percentage of TMC1 mutations that account for the genetic etiology in Xiamen Special Education School was 3.2% (5/155), and the allele frequency of TMC1 was 3.2% (10/310). The allele frequency of TMC1 c.2050G>C was 1.9% (6/310).
3.3. Mutation analysis
There were four novel variants not reported before. We predicted their pathogenicity with software.
3.3.1. c.804G>A(p.W268X)
The pathogenicity of this mutation was analyzed by MutationTaster software and SPIDEX (http://tools.genes.toronto.edu/), with the results suggesting “disease causing automatic” with a MutationTaster score of 1, and a SPIDEX score of −61.61 with the meaning of pathogenic possibility. An alignment of the amino acid sequence of nine species from UCSC Genome Browse suggested the evolutionary conservation of c.804G>A. The variant of c.804G>A (p.W268X) was predicted to be a disease‐causing mutation.
3.3.2. c.1127T>C(p.L376P)
The pathogenicity of this mutation was analyzed by SIFT, Polyphen‐2, and MutationTaster software. The results suggested c.1127T>C was “damaging” with a SIFT score of 1, “possibly damaging” with a Polyphen‐2 score of 0.999, and “disease causing” with a MutationTaster score of 1. An alignment of the amino acid sequence of nine species from UCSC Genome Browse suggested the evolutionary conservation of c.1127T>C. The variant c.1127T>C(p.L376P) was predicted to be a disease‐causing mutation.
3.3.3. c.1764‐4C>A
The score of the splicing variants from SPIDEX was −10.47, which means low pathogenicity. This variant was predicted to be a polymorphism.
3.3.4. c.2073G>A(p.K691K)
This variant causes no change in amino acid, and its SPIDEX score was −0.21, which means low pathogenicity. This variant was also predicted to be a polymorphism.
3.3.5. Haplotype of TMC1 c.2050G
Four families (XJA, M126, M372, and XJ87) with TMC1 c.2050G>C mutations were haplotype analyzed with SNPs. A total of 53 SNPs was used to construct haplotypes for TMC1 c.2050G>C, and two generations of these families were tested. The proband of each family was divided into two haplotypes from parents. According to the results of SNP testing, the haplotype of TMC1 c.2050G>C was identical for the four families (Figure 3).
Figure 3.
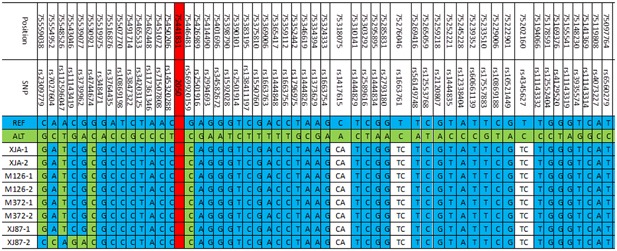
Haplotype of the four families. REF, reference sequence; ALT, alteration sequence; −1, haplotypes from the father; −2, haplotypes from the mother. [Color figure can be viewed at http://wileyonlinelibrary.com]
4. DISCUSSION
Most reports about mutations of TMC1 were from the special pedigree study of families with consanguineous marriages. Kalay reported TMC1 mutations in 6% (4/65) of the GJB2‐negative ARNSHL families and 4% (4/93) of all ARNSHL families from the northeast and east of Turkey (Kalay et al., 2005). TMC1 mutations account for 3.4% (19/557) of recessive deafness in Pakistani ARNSHL families, 3.5% (3/85) in Tunisian families (Tlili et al., 2008), and 1.6% in India (Ganapathy et al., 2014). The novel mutations are all from families with consanguineous marriages. The hot spot in Pakistan was p.R34X (1.8%,10/557). Kitajiri et al. (2007) designed three short‐tandem repeats (STRs) and one SNP to test 10 families with p.R34X; the results strongly suggested a single ancestral origin of this mutation. In past years, there were few reports of hot spots; most reports were about novel mutations of TMC1, suggesting that TMC1 is a highly conserved gene and its variants are rarely tolerated (Bademci et al., 2016).
In China, recent studies on TMC1 mutations such as c.589G>A, c.1171C>T (Xue et al., 2013), c.1209G>C (Tao et al., 2013Tao, Xiaoming, Yongchuan, Lei, & Hao, 2013), c.1253T>A (Yali et al., 2014), c.1714G>A (Xue et al., 2015), and c.1979C>T (Jiongjiong et al., 2016) have been reported. All mutations were analyzed by genetic testing of sporadic families without regional specificity. In this study, a high incidence of TMC1 mutations in Xiamen was observed in the genetic testing of sporadic families and epidemiologic investigation in a deaf population revealing a hot spot phenomenon in this area.
South Fujian, including Xiamen with the dialect of Hokkien (Southern Min), showed special mutation sites in common deafness‐causing genes. The second most common mutation of SLC26A4 was c.2168A>G (p.H723R) in most areas of China, while it was c.1079C>T (p.A360V) in the Xiamen patient cohort, which has rarely been reported in other areas of China (Yi et al., 2015). A similar phenomenon was found in TMC1 screening of the deaf population in the Xiamen area.
Ruling out the mutations that result in homozygous or compound heterozygotes in the normal hearing database (in‐house NGS database of our own lab), a homozygous TMC1 c.2050G>C was found through NGS in two deaf children in a XJA family with two heterozygous normal hearing parents. Because the proband of XJA was from Xiamen Special Education School in Fujian province, we analyzed the other 110 patients from this school who lacked positive pathogenic mutations of three common deafness genes (GJB2, SLC26A4, and mtDNA 12SrRNA). There were four compound heterozygotes of TMC1 c.2050G>C, which means that the mutation allele frequency of TMC1 was 1.9% (6/310) in this group. The detection rate of this mutation in 100 normal Xiamen people was zero. Two other families (M126, M372) with hearing‐impaired children and normal parents were found to have homozygous TMC1 c.2050G>C in the proband and heterozygous TMC1 c.2050G>C in the parents. These two families were also from South Fujian. The haplotype of TMC1 c.2050G>C shows the founder effect of the four families (XJA, M126, M372, and XJ87). Meanwhile, the haplotype of TMC1 c.1396_1398delAAC was different from the one of TMC1 c.2050G>C. In China, close‐relative marriages are forbidden, so the high incidence of the same mutation, such as TMC1 c.2050G>C, in one area may be explained by its founder effect in very early ancestors.
In the investigation of the Xiamen Special Education School, four novel mutations of TMC1 were found that were not identified by previous studies on TMC1 mutations in China. Pathogenicity analysis of c.804G>A (p.W268X) and c.1127T>C (p.L376P) suggested possible damaging changes in the protein structure, and according to the structural prediction for these proteins of human wild‐type sequenced evolutionary conservation, we predicted the pathogenicity of these two novel mutations to be high. The TMC1 mutation detection rate was 3.2% in Xiamen Special Education School, which means TMC1 could be the fourth most common deafness gene in this area.
5. CONCLUSIONS
In this study, special hotspots and spectra of TMC1 mutations in the Xiamen deaf population were revealed. Haplotype analysis is useful to understand the founder effect of hot spot mutations, and this information will be helpful in designing the protocol for genetic testing for deafness and achieving an accurate molecular diagnosis in this area.
CONFLICTS OF INTEREST
The authors have no financial interests in companies or other entities, and the funders of all grants supporting this article played no role in the study design, data collection, analysis, decision to publish, or preparation of the manuscript. The authors have declared that no competing interests exist.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
Table S1. The 109 deafness genes.
Table S2. Primers used for amplification and sequencing of TMC1.
Table S3. MassARRAY SNPs primers for PCR amplification and extension.
Table S4. All variants identified by targeted NGS of XJA, M126, M372 families.
ACKNOWLEDGMENTS
The authors sincerely thank all of the subjects for their participation and cooperation in this study. This work was supported by State Key Program of National Natural Science Foundation of China (81230020), National Key Research and Development Program of China (2016YFC1000700, 2016YFC1000704, 2016YFC1000706), Fujian Medical Innovation Project (2016‐CX‐5).
Jiang Y, Gao S, Wu L, et al. Mutation spectra and founder effect of TMC1 in patients with non‐syndromic deafness in Xiamen area, China. Am J Med Genet Part B. 2018;177B: 301–307. https://doi.org/10.1002/ajmg.b.32603
Yi Jiang, Song Gao, and Lihua Wu contributed equally to this work.
The English in this document has been checked by two professional editors who are native speakers of English.
Contributor Information
Dongyi Han, Email: hdy301@263.net.
Huafang Gao, Email: ericgao168@gmail.com.
Pu Dai, Email: daipu301@vip.sina.com.
REFERENCES
- Ayesha, I. , Azra, M. , Atteeq, U. R. , Robert, J. M. , Jeffrey, R. H. , Thomas, B. F. , & Sadaf, N. (2016). Recessive mutations of TMC1 associated with moderate to severe hearing loss. Neurogenetics 17:115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bademci, G. , Foster, J. , Mahdieh, N. , Bonyadi, M. , Duman, D. , Cengiz, F. B. , … Tokgoz‐Yilmaz, S. (2016). Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort. Genetics in Medicine, 18, 364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fangzhu, L. , Dejun, L. , Ping, W. , Dongyan, F. , Ji, D. , … Wei, Z. (2014). Autosomal recessive non‐syndromic hearing loss is caused by novel compound heterozygous mutations in TMC1 from a Tibetan Chinese family. International Journal of Pediatric Otorhinolaryngology, 78, 2216–2221. [DOI] [PubMed] [Google Scholar]
- Ganapathy, A. , Pandey, N. , Srisailapathy, C. R. , Jalvi, R. , Malhotra, V. , Venkatappa, M. , … Agarwal, A. K. (2014). Non‐Syndromic hearing impairment in India: High allelic heterogeneity among mutations in TMPRSS3, TMC1, USHIC, CDH23, and TMIE. PLoS ONE, 9, e84773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiongjiong, H. , Fei, L. , Wenjun, X. , Lili, H. , Jun, L. , Zhenghua, Z. , … Zhaoxin, M. (2016). Exome sequencing identifies a mutation in TMC1 as a novel cause of autosomal recessive nonsyndromic hearing loss. Journal of Translational Medicine, 14, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalay, E. , Karaguzel, A. , Caylan, R. , Heister, A. , Cremers, F. P. , Cremers, C. W. , … Kremer, H. (2005). Four novel TMC1 (dfnb7/dfnb11) mutations in Turkish patients with congenital autosomal recessive nonsyndromic hearing loss. Human Mutation, 26, 591. [DOI] [PubMed] [Google Scholar]
- Kitajiri, S. I. , McNamara, R. , Makishima, T. , Husnain, T. , Zafar, A. U. , Kittles, R. A. , … Griffith, A. J. (2007). Identities, frequencies and origins of TMC1 mutations causing DFNB7/B11 deafness in Pakistan. Clinical Genetics, 72, 546–550. [DOI] [PubMed] [Google Scholar]
- Meyer, C. G. , Gasmelseed, N. M. , Mergani, A. , Magzoub, M. M. , Muntau, B. , Thye, T. , & Horstmann, R. D. (2005). TMC1 structural and splice variants associated with congenital nonsyndromic deafness in a Sudanese pedigree. Human Mutatation, 25, 100. [DOI] [PubMed] [Google Scholar]
- Stacey, G. , Liuda, Z. , & Diana, T . (2009). SNP Genotyping Using the Sequenom UNIT 2.12 MassARRAY iPLEX Platform. Current Protocols in Human Genetics, 60, 2.12.1–2.12.18. [DOI] [PubMed] [Google Scholar]
- Tahir, A. , Huseyin, O. , Ayca, A. , Guney, B. , Tayfun, K. , … Ferda, O . (2015). Comprehensive analysis of deafness genes in families with autosomal recessive nonsyndromic hearing loss. PLoS ONE, 10, e0142154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao, Y. , Xiaoming, W. , Yongchuan, C. , Lei, L. , & Hao, W . (2013). Genetic etiology study of the non‐syndromic deafness in Chinese Hans by targeted next‐generation sequencing. Orphanet Journal of Rare Diseases, 14, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlili, A. , Rebeh, I. B. , Aifa‐Hmani, M. , Dhouib, H. , Moalla, J. , Tlili‐Chouchène, J. , … Ghorbel, A. (2008). TMC1 but Not TMC2 is Responsible for autosomal recessive nonsyndromic hearing impairment in Tunisian families. Audiology and Neurotology, 13, 213–218. [DOI] [PubMed] [Google Scholar]
- Xue, G. , Sha‐Sha, H. , Yong‐Yi, Y. , Guo‐Jian, W. , Jin‐Cao, X. , Yu‐Bin, J. , … Pu, D . (2015). Targeted gene capture and massively parallel sequencing identify TMC1 as the causative gene in a six‐generation chinese family with autosomal dominant hearing loss. American Journal of Medical Genetics Part A, 167A, 2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, G. , Yu S., Li‐Ping G., Yong‐Yi, Y. , Sha‐Sha, H. , Yu, L ., … Pu, D . (2013). Novel compound heterozygous TMC1 mutations associated with autosomal recessive hearing loss in a chinese family. PLoS ONE, 8, e63026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yali, Z. , Dayong, W. , Liang, Z. , Feifan, Z. , Liping, G. , Peng, Z. , … Qiuju, W . (2014). A novel DFNA36 mutation in TMC1 orthologous to the Beethoven (Bth) mouse associated with autosomal dominant hearing loss in a Chinese family. PLoS ONE, 9, e97064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, J. , Shasha, H. , Tao, D. , Lihua, W. , Juan, C. , Dongyang, K. , … Pu, D . (2015). Mutation spectrum of common deafness‐causing genes in patients with non‐syndromic deafness in the Xiamen area, China. PLoS ONE, 108, e0135088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying, C. , Zhentao, W. , Zhaoyan, W. , Dongye, C. , Yongchuan, C. , Xiuhong, P. , … Hao, W . (2015). TArgeted next‐generation sequencing in uyghur families with non‐syndromic sensorineural hearing loss. PLoS ONE, 10, e0127879. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article.
Table S1. The 109 deafness genes.
Table S2. Primers used for amplification and sequencing of TMC1.
Table S3. MassARRAY SNPs primers for PCR amplification and extension.
Table S4. All variants identified by targeted NGS of XJA, M126, M372 families.