

Abstract
Chloroplast genomes have undergone tremendous alterations through the evolutionary history of the green algae (Chloroplastida). This study focuses on the evolution of chloroplast genomes in the siphonous green algae (order Bryopsidales). We present five new chloroplast genomes, which along with existing sequences, yield a data set representing all but one families of the order. Using comparative phylogenetic methods, we investigated the evolutionary dynamics of genomic features in the order. Our results show extensive variation in chloroplast genome architecture and intron content. Variation in genome size is accounted for by the amount of intergenic space and freestanding open reading frames that do not show significant homology to standard plastid genes. We show the diversity of these nonstandard genes based on their conserved protein domains, which are often associated with mobile functions (reverse transcriptase/intron maturase, integrases, phage- or plasmid-DNA primases, transposases, integrases, ligases). Investigation of the introns showed proliferation of group II introns in the early evolution of the order and their subsequent loss in the core Halimedineae, possibly through RT-mediated intron loss.
Keywords: mobile elements, freestanding ORFs, genome evolution, Bryopsidales, Chlrorophyta
Introduction
Chloroplasts are light-harvesting organelles of photosynthetic eukaryotes. Their origin can be traced back to a primary endosymbiosis event over a billion years ago, in which a heterotrophic eukaryotic cell captured a cyanobacterium that became stably integrated and evolved into a membrane-bound organelle (Gould et al. 2008; Keeling 2010; Ponce-Toledo et al. 2017). Over evolutionary time, the genome of the chloroplast was reduced by gene loss and gene transfer to the host nucleus, leading to closer integration with the host as an organelle (Timmis et al. 2004). Although chloroplasts typically retain a core set of genes involved in photosynthesis, ATP generation, transcription, and translation, they depend on nuclear-encoded, plastid-targeted proteins for the maintenance of several biochemical pathways and functions such as genome replication and gene expression (Green 2011; Lang and Nedelcu 2012). The Archaeplastida lineage resulting from this primary endosymbiosis event diversified into the green plants (Chloroplastida), the red algae (Rhodophyta), and the glaucophytes (Glaucocystophyta) (Rodríguez-Ezpeleta et al. 2005). This was followed by a complex history of chloroplast acquisition via eukaryote–eukaryote endosymbioses, resulting in the spread of plastids to other eukaryotic lineages (Keeling 2010).
Green algae have retained fewer genes in their chloroplast genome compared with the glaucophytes and red algae (Green 2011). The genomes are present in multiple copies per cell, are relatively small in size, and are uniparentally inherited. This makes them relatively easy to sequence with high-throughput methods and, as a consequence, they have established themselves as a useful tool for phylogenetic inference and a convenient model for evolutionary genomics (Fučíková et al. 2014; Sun et al. 2016).
The green algae comprise two clades, the Chlorophyta, including a wide diversity of marine, freshwater, and terrestrial algae, and the Streptophyta, including mostly freshwater algae (charophytic green algae) from which the land plants evolved (Leliaert et al. 2012). The plastid genomes in these two clades can differ in essential components (de Vries et al. 2017). Chloroplast genomes in the Chlorophyta vary extensively in architecture, including size, gene content, number of introns and repeats, nucleotide composition, and conformations that vary not just between the major green algal lineages but also within them (Brouard et al. 2010; de Vries et al. 2013; Lemieux et al. 2014; Turmel et al. 2015; Leliaert et al. 2016; Del Cortona et al. 2017). Given that the chloroplast genomes have undergone tremendous alterations across the main lineages of Chlorophyta, it would be desirable to get a more detailed view of the underlying genome dynamics within groups of relatively closely related species. This study focuses on the order Bryopsidales, a morphologically diverse group of marine macroalgae in the class Ulvophyceae for which a relatively large number of chloroplast genome sequences are available. These algae are characterized by a siphonous structure, meaning they consist of a single massive tubular cell (siphon) that branches to form more complex morphologies (Vroom and Smith 2003). The siphonous cell contains thousands of nuclei and chloroplasts and features cytoplasmic streaming (Mine et al. 2008).
To date, ten complete chloroplast genomes of Bryopsidales have been sequenced (supplementary table S1, Supplementary Material online) and they do not feature a quadripartite architecture (Lü et al. 2011; Leliaert and Lopez-Bautista 2015; Lam and Lopez-Bautista 2016; Marcelino et al. 2016; Verbruggen et al. 2017). Chloroplast genome sizes and gene arrangement differ considerably among taxa. In addition, freestanding open reading frames (ORFs) not associated with introns and not showing significant homology to conserved (standard) plastid genes as defined by Lang and Nedelcu (2012: table 3.1) have been reported (Lü et al. 2011; Leliaert and Lopez-Bautista 2015; Lam and Lopez-Bautista 2016). These features make the siphonous green algae a good candidate for a more in-depth analysis of chloroplast genome evolution.
The goal of this study is to evaluate the evolutionary dynamics of the chloroplast genome in siphonous green algae. We present five new chloroplast genomes, yielding a data set representing all but one family in the Bryopsidales. Besides characterizing the chloroplast genomes, we investigate how features such as genome size, gene content, introns, and diversity of nonstandard genes have changed during the evolution of the order using comparative phylogenetic methods.
Materials and Methods
DNA Isolation and Sequencing
Fragments of field-collected Bryopsis sp. (HV04063), Codium arenicola (HV04071), Caulerpa manorensis (HV04986), Rhipilia penicilloides (HV04325), and Chlorodesmis fastigiata (HV03865) were cleaned and desiccated in silica gel. Total genomic DNA was extracted using the modified cetyltrimethylammonium bromide (CTAB) protocol described in Cremen et al. (2016).
For Bryopsis sp., Codium arenicola, and Chlorodesmis fastigiata, a library was prepared from ca. 350-bp fragments using TruSeq Nanno LT Kit and sequenced on Illumina HiSeq 2000 (paired end, 100 bp) at Cold Spring Harbor Laboratory (Cold Spring Harbor, NY). For Caulerpa manorensis, the library was prepared from ca. 500 bp size fractions with a Kapa Biosystems Kit and sequenced on Illumina NextSeq 500 (paired end 150 bp) at the Georgia Genome Facility (Athens, GA). Finally, for Rhipilia penicilloides, the library was prepared from ca. 500-bp fragments using NEB Next Ultra DNA Library Kit and sequenced on the Illumina HiSeq (paired end 150 bp) at Novogene (Beijing, China).
Genome Assembly and Annotation
Assembly and annotation followed procedures described in Verbruggen and Costa (2015) and Marcelino et al. (2016), with some minor alterations. In brief, de novo assembly was performed from the paired-end Illumina reads using three different assembly programs: 1) CLC Genomics Workbench 7.5.1, 2) MEGAHIT 1.0.6 (Li et al. 2015), and 3) SPAdes 3.8.1 (Bankevich et al. 2012). Contigs were imported into Geneious 8.0.5, where completeness and circularity were evaluated by manually comparing the contigs generated from different assemblers. This process was guided by visual assessment of the SPAdes assembly graphs in Bandage v.0.8.0 (Wick et al. 2015). Average read coverage was assessed in Geneious by mapping the forward and reverse raw reads to each circular-mapping contig.
Preliminary annotations were obtained from DOGMA (Wyman et al. 2004), MFannot (Beck and Lang 2010), and ARAGORN (Laslett and Canback 2004) and imported into Geneious. The “annotate from” feature in Geneious was also used to transfer annotations from related genomes based on sequence similarity. Open reading frames (ORFs) were identified using Glimmer (Delcher et al. 2007) and “Find ORF” function in Geneious with the minimum size set at 300 bp using the bacterial genetic code. Identified ORFs were extracted and checked for similar protein sequences using BLASTx against nonredundant NCBI database. A separate BLASTx search was conducted but constrained to Viridiplantae (taxon ID: 33090) to check if any of the ORFs are homologous to other green plants. All annotations were vetted and a master annotation track was manually created from them. In the final annotation, conserved domains of both intronic and freestanding ORFs were determined using NCBI Conserved Domain database using default settings (Marchler-Bauer 2015).
Repeats and tandem repeats were detected using the Phobos v.3.3.11 (Mayer 2007) plugin in Geneious using the following settings: lengths between 15 and 1,000 bp; “perfect” search mode. Emboss (Rice et al. 2000) was used to detect palindromic sequences using default settings. Circular genome maps were drawn using OGDraw (Lohse et al. 2013).
All Bryopsidales chloroplast genomes used in this study, including those downloaded from GenBank, are listed in supplementary table S1, Supplementary Material online. Chloroplast genome sequences generated in this study are available in GenBank under accession numbers KY0819063–KY0819066, and KY0819068. The sequence of Bryopsis hypnoides (NC_013359) was reannotated following Leliaert and Lopez-Bautista (2015).
Phylogenomic Analyses
Alignments of named chloroplast protein-coding genes were inferred using TranslatorX (Abascal et al. 2010), which translates sequences to amino acids, uses MAFFT (Katoh and Standley 2013) to align the amino acid sequences and generates the corresponding nucleotide alignments. Individual gene alignments were manually checked in Geneious. For those that could not be reliably aligned, GBlocks (a program which eliminates poorly aligned positions and divergent regions of DNA alignments) was used. If GBlocks removed >60% of the alignment position for each individual gene, the entire gene was excluded from the phylogenetic reconstruction. This was the case for ftsH, rpoA, rpoB, rpoC1, rpoC2, rps18, tilS (=ycf62), and ycf1. The rpoB and rpoC genes excluded on this bases are known to be subject to coding-region expansion, which can mislead phylogenetic reconstruction because of violation of the assumptions of substitution models (Novis et al. 2013). The concatenated alignment comprising of 70 genes was generated at the nucleotide level. Poorly aligned positions were removed using the GBlocks server (Castresana 2000), forcing it to keep codons intact and with the least stringent settings, which allowed smaller final blocks, gap positions within the final block, less strict flanking positions, and many contiguous nonconserved positions. Using these settings, GBlocks reduced the 70-gene alignment from 45,645 to 39,183 positions.
Maximum Likelihood (ML) analyses were carried out using RAxML (Stamatakis 2014) with a GTRGAMMAI model as suggested by jModelTest2 (Guindon and Gascuel 2003; Darriba et al. 2012) using 1, 000 replicates for bootstrap support.
We included only Bryopsidales in our study because chloroplast genome size and structure varies extensively between orders of the Ulvophyceae, from excessively large (> 1 Mb) and repeat-rich genomes in the Dasycladales (Leible et al. 1989; De Vries et al. 2013) to highly fragmented genomes consisting of single-stranded hairpin chromosomes in Cladophorales (Del Cortona et al. 2017), and we did not want to risk our analyses being biased by this enormous variation seen in related orders. In the absence of outgroups from other orders, we determined the root position of our tree otherwise. The relationships among the main lineages of Bryopsidales have been studied in great detail using chloroplast genomes (Verbruggen et al. 2017), and irrespective of which other orders of Ulvophyceae were chosen as outgroups in that study, the Ostreobineae were consistently sister to the remaining Bryopsidales. Therefore, we performed unrooted ML analyses and manually rooted the tree between the Ostreobineae and the remaining Bryopsidales.
Genome Size and Intron Content
Chloroplast genome size and intron content (group I and group II introns) were separately mapped onto the ML tree. The following R packages were used: contMap function of phytools (Revell 2012) for genome size analysis and ape (Paradis et al. 2004), geiger (Harmon et al. 2008), and phytools (Revell 2012) for intron content. Visualization was done using TreeGradients (Verbruggen 2012) or phytools.
Evolution of Freestanding ORFs
To assess putative origins and evolutionary histories of freestanding ORFs (> 300 bp) we applied a combination of BLAST similarity searches and phylogenetic analyses. To test if certain groups of freestanding ORFs have a common evolutionary history within Bryopsidales, we identified freestanding ORFs that showed high similarity among different chloroplast genomes of Bryopsidales using BLASTp searches (E-value threshold < 10E-6) against a custom BLAST database including all freestanding ORFs of published Bryopsidales genomes (supplementary table S1, Supplementary Material online). Groups of similar freestanding ORFs from two or more Bryopsidales species were supplemented with sequences from BLASTp searches (E-value threshold < 10E-6) against NCBI’s nonredundant protein database (nr) and a custom BLAST database including all CDSs of green algal chloroplast genomes available in GenBank (June 1, 2017) (supplementary table S2, Supplementary Material online). In each group, amino acid sequences were aligned with ClustalW using the Blosum matrix with gap open penalty 10 and gap extension penalty 0.05. Maximum likelihood trees were generated using RAxML (Stamatakis 2014) with 100 replicates for bootstrap support. Best-fit amino acid substitution models (supplementary table S3, Supplementary Material online) were used under BIC criterion as suggested in ModelFinder (Kalyaanamoorthy et al. 2017).
Chloroplast Genome Alignment and Rearrangements
Chloroplast genome alignment was done using the Mauve plug-in in Geneious (Darling 2004). This alignment shows locally collinear blocks (LCBs)—homologous regions in the sequences that are free from major rearrangements. The beginning of the 16S rRNA gene was selected as starting position for the Mauve alignment. The progressive Mauve algorithm was used with default settings: automatically calculate seed weight and minimum LCB score, compute LCBs, full alignment.
To calculate the number of genome rearrangements along the branches of the bryopsidalean phylogeny, the MGRA v.2 webserver was used (Avdeyev et al. 2016), using the phylogenomic topology and the collinear blocks generated with Mauve as inputs. Finally, the Double-cut-and-join (DCJ) model in UniMog (Hilker et al. 2012) was used to calculate the number of rearrangements among the pairwise aligned sequences.
Results and Discussion
Five New Bryopsidales Chloroplast Genomes
The assembly of the Illumina reads for the five newly sequenced species yielded complete circular-mapping chloroplast genomes that corresponded to a single contig (supplementary figs. S1–S5, Supplementary Material online) without ambiguous regions. The read coverage was homogeneous within species and ranged from 1,693× to 7,514× between species (supplementary table S1, and fig. S6, Supplementary Material online). The gene and intron content and various other genome features are listed in supplementary table S4, Supplementary Material online.
Consistent with previously published chloroplast genomes in Bryopsidales, all newly sequenced genomes lack a large inverted repeat (IR), suggesting it was lost in the ancestor of the order. Other members of Ulvophyceae do have an IR, for example, Ignatiales (Turmel et al. 2017), Oltmannsiellopsidales (Pombert et al. 2006), and some Ulvales/Ulotrichales (e.g., Pseudeneochloris marina, Pseudendoclonium akinetum, Chamaetrichon capsulatum, Trichosarcina mucosa; Pombert 2005; Turmel et al. 2017). Certain Ulvales, Ulotrichales, Chlorophyceae, Trebouxiophyceae, and prasinophytes also lack a large IR, suggesting that loss of the IR has been common across many lineages of the Chlorophyta (Turmel, Otis, et al. 2009; Brouard et al. 2010; Melton et al. 2015; Turmel et al. 2015, 2016).
The concatenated chloroplast gene data resolved the relationships among the bryopsidalean species with full support (100% bootstrap support for all branches) with the exception of the relationship between Halimedaceae, Rhipiliaceae, and Udoteaceae (84% bootstrap support for the branch joining Halimeda and Rhipilia) (fig. 1). Overall, the phylogeny recovered here is in line with previous studies (Verbruggen et al. 2009, 2017; Marcelino et al. 2016) and provides a useful framework to study the evolutionary dynamics of genome features.
Fig. 1.

—Maximum likelihood phylogeny of Bryopsidales based on the concatenated alignment of 70 protein-coding genes of the chloroplast genomes. Numbers on the node are bootstrap support values. Numbers above the branch lengths represent the number of rearrangement inferred from MGRA2.
Genome Size
The median chloroplast genome size across the order Bryopsidales is 105 kb, but there is considerable variation across lineages (fig. 2 and supplementary table S4, Supplementary Material online). Except within the Ostreobineae, which all have small chloroplast genomes, there appears to be little phylogenetic conservatism of genome sizes. The Bryopsidineae and Halimedineae show extensive variation in genome size, and both show instances of reduction (Codium arenicola and Chlorodesmis fastigiata) and expansion (Bryopsis hypnoides, Caulerpa lineage, Halimeda discoidea).
Fig. 2.

—Phylogenetic mapping shows variation of chloroplast genome size across lineages. The amount of conserved plastid protein-coding regions and ribosomal + transfer RNAs is fairly constant among species, and differences in genome size are mostly accounted for by intergenic space, introns, and freestanding ORFs.
The amount of space taken up by standard plastid protein-coding genes is fairly constant (61.1 ± 2.2 kb), as is the amount of tRNA and rRNA (6.7 ± 0.5 kb), and genome size variation is mainly accounted for by a combination of the amount of intergenic space, introns, and freestanding ORFs (fig. 2). This trend transcends the major phylogenetic groups, with the relatively large chloroplast genomes of Bryopsis hypnoides and Caulerpa cliftonii both containing large intergenic spaces and many freestanding ORFs. In addition, Bryopsis hypnoides also has several repeats in the intergenic space. On the opposite end of the spectrum, intergenic spaces are very short in the compact chloroplast genomes of Ostreobium quekettii, Ostreobium sp., Chlorodesmis fastigiata, and Codium arenicola.
Similar to our findings, previous works have also attributed expansion of algal chloroplast genome size to increased intergenic space (Turmel et al. 2005; Brouard et al. 2010; Muñoz-Gómez et al. 2017), introns (Muñoz-Gómez et al. 2017), repeats (Maul et al. 2002; Smith and Lee 2009; Brouard et al. 2010), or a combination of factors (Pombert 2005). The underlying causes of genome size variation are still a matter of debate (Lynch 2006; Lynch et al. 2006; Schubert and Vu 2016). It has been argued that rates of DNA deletion normally exceed rates of insertions, resulting in a pervasive deletion bias and consequent genome shrinkage (Mira et al. 2001; Kuo and Ochman 2010; Wolf and Koonin 2013). Although genome sizes can be largely explained by neutral processes (Lynch 2006), natural selection can favour compact genomes where resources and/or time for replication are limited (Giovannoni et al. 2005; Hessen et al. 2010). This appears to be the case for the small genomes observed in the Ostreobineae, a lineage that is considered to have experienced streamlining as an adaptation to the very low light habitat in which these organisms live (Marcelino et al. 2016). On the other hand, Codium arenicola, which also has a small chloroplast genome, would not be expected to experience the same limitations, suggesting that the causes for genome reduction in Bryopsidales are diverse. Genome expansion has been attributed to the proliferation of selfish and junk DNA as transposable elements, which can be deleterious, neutral, or beneficial to their host (Doolittle and Sapienza 1980; Orgel and Crick 1980; Kidwell and Lisch 2001). This could be the case for the large genomes observed in some bryopsidalean genomes where nonstandard genes involved in mobile functions abound. Transposable elements are an important source of evolutionary innovation for their host (Kidwell and Lisch 2001). Although genome reduction is a gradual and slow process, genome expansion is thought to occur in bursts alongside evolutionary transitions (Wolf and Koonin 2013).
Conserved Gene Content
The gene repertoire of chloroplast genomes is quite homogeneous within Bryopsidales and similar to that of other Ulvophyceae. A total of 96 chloroplast protein coding genes including three ribosomal RNAs and 25 transfer RNAs are shared by all members of Bryopsidales and other ulvophycean taxa (supplementary table S5, Supplementary Material online).
In comparison with other core Chlorophyta (clade comprising the Ulvophyceae, Trebouxiophyceae, Chlorophyceae, Pedinophyceae, and Chlorodendrophyceae), Bryopsidales have two genes encoding for components of the sulphate ABC transport system (cysA and cysT) found in other green algae (trebouxiophytes, and Pedinomonas) but lost in other ulvophycean chloroplast genomes. Two tRNAs (trnF(aaa) and trnN(auu)) are found in all Ulvophyceae except the Bryopsidales (supplementary table S5, Supplementary Material online). The organelle division inhibitor factor gene (minD) are only found in Oltmannsiellopsis viridis and Pseudendoclonium akinetum (supplementary table S5, Supplementary Material online) and absent in Bryopsidales, Ulva spp. and Cladophorales (Del Cortona et al. 2017). Loss of minD has been associated with the evolution of polyplastidy (de Vries and Gould 2017), a feature present in the Bryopsidales, Cladophorales, and Dasycladales.
The chloroplast envelope membrane protein (cemA) gene was lost twice in the Bryopsidales—once in the lineage leading to Ostreobium and a second time in the lineage leading to Avrainvillea mazei (see also Marcelino et al. 2016; Verbruggen et al. 2017). The ycf47 gene was lost on three occasions within the Halimedineae (Avrainvillea mazei, Caulerpa lineage, Halimeda discoidea). The ribosomal protein L12 (rpl12) gene was lost at the base of the core Halimedineae. Several other genes were lost in individual species, that is, ycf20 in Bryopsis hypnoides, psb30 (ycf12) in Chlorodesmis fastigiata, and rpl32 in Halimeda discoidea. Loss of several tRNAs within the bryopsidalean lineage was also observed (supplementary fig. S7, Supplementary Material online).
The genes that were lost in different bryopsidalean lineages have diverse functions including inorganic carbon dioxide uptake into chloroplasts (cemA), photosynthesis (psb30), translation (rpl12, rpl32), and proteins of unknown function (ycf20, ycf47). Knockout experiments on Chlamydomonas reinhardtii have shown that cemA is not essential for photosynthesis or the viability of the cell but its absence increases light sensitivity of the cell (Rolland 1997), and that psb30 is required for the optimal functionality of the PSII complex in high light (Inoue-Kashino et al. 2011).
Comparative studies of chloroplast genome sequences indicate frequent losses of nonessential gene have been observed in chloroplast genomes of various algal lineages (Martin et al. 1998). In addition, loss of rpl12, rpl32, ycf20, and ycf47 are not unique to Bryopsidales as these genes have been lost in some members of the streptophytes (Lemieux et al. 2016) and the chlorophycean Stigeoclonium helveticum (Bélanger et al. 2006). The possibility that these genes have been transferred to the nucleus cannot be ruled out.
Fragmentation of tilS and rpoB
In the bryopsidalean chloroplast genomes, the tRNA Ile-lysidine synthetase (tilS = ycf62) and RNA polymerase b-subunit (rpoB) genes are fragmented. Fragmentation of these two genes can be subdivided in three types: 1) gene with an intron; 2) gene fragmented with an insertion that is not associated with sequences typical of group I or group II introns; 3) gene with an in-frame stop codon; and 4) gene with a frame shift (fig. 3).
Fig. 3.
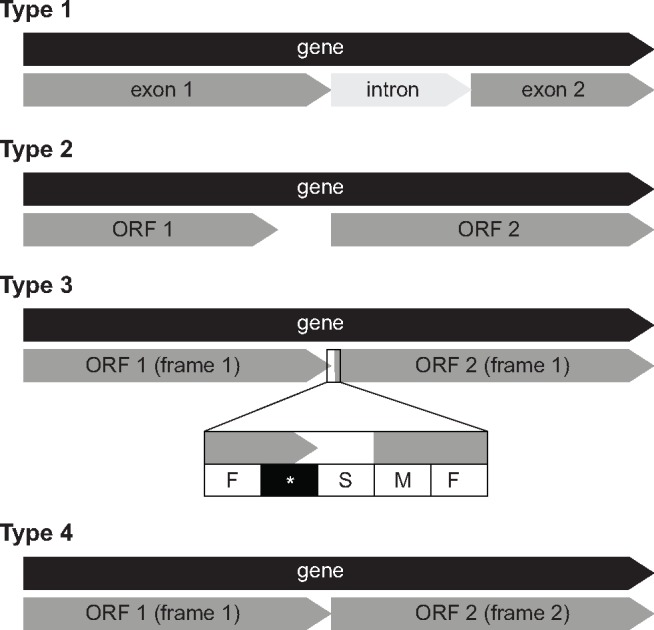
—Fragmentation pattern of tilS and rpoB genes in Bryopsidales. Type 1: gene separated by an intron; Type 2: gene fragmented with an insertion that is not associated with sequences typical of group I or group II intron; Type 3: gene with an in-frame stop codon (inset highlights the position of the stop codon in black); Type 4: gene with a frame shift.
Previous studies of bryopsidalean genomes have annotated tilS as a pseudogene as it contains either a stop codon or indels in the middle of the gene (Zuccarello et al. 2009). In our newly sequenced taxa, tilS also consists of two subsequent short ORFs that both have sequence similarity to canonical tilS. Although tilS was reported to be absent in Caulerpa cliftonii (Marcelino et al. 2016), reinvestigation revealed that the tilS gene is present as two putative exons (orf180 and orf144) separated by an intron (type 1), which contained an ORF (orf116) with a group II reverse transcriptase/intron maturase motif. In Derbesia sp., tilS has an in-frame stop codon (type 3). In Bryopsis plumosa and B. hypnoides, the tilS gene was previously reported to have an insertion (Leliaert and Lopez-Bautista 2015), but reinvestigation of the data revealed fragmentation of tilS with a frame shift (type 4). The position of the intron in C. cliftonii is at the same position as the frame shifts observed in other bryopsidalean taxa (supplementary fig. S8a, Supplementary Material online). Fragmentation of tilS has also been reported in some representatives of core Trebouxiophyceae (Turmel et al. 2015). In these species however, tilS does not exhibit a frame shift, but the two ORFs are either found in different regions of the genomes (Watanabea reniformis and Xychloris irregularis), or are separated by a 224-bp insertion not associated with group I or group II introns (Paradoxia multiseta).
A similar situation was found for the rpoB gene, which was fragmented in all species except Bryopsis hypnoides. In Ostreobineae, the rpoB gene is interrupted by a group II intron (type 1). In Bryopsidineae and Avrainvillea mazei, the gene exhibits type 2 fragmentation with the insertion ranging between 302 and 414 bp and are AT-rich (75–86%). In the core Halimedineae, the rpoB gene of Rhipilia penicilloides and Tydemania expeditionis has an in-frame stop codon (type 3), whereas in Caulerpa cliftonii, C. manorensis, Chlorodesmis fastigiata, and Halimeda discoidea the gene exhibits type 4 fragmentation. Unlike in the tilS gene, the fragmentation of the rpoB gene is found at different positions in different species (supplementary fig. S9a, Supplementary Material online). Ostreobineae, Bryopsidineae, and Avrainvillea mazei share the same fragmentation site. In the core Halimedineae, the positions of frame shifts are in the same region for all species except in Caulerpa cliftonii. Fragmentation of rpoB has also been reported in Chlamydomonas reinhardtii (Maul et al. 2002), Scenedesmus obliquus (de Cambiaire et al. 2006), several other chlorophycean taxa (Novis et al. 2013) and the trebouxiophyte Leptosira terrestris (de Cambiaire et al. 2007). However, the size of the insertion in S. obliquus (1,017 bp), C. reinhardtii (617 bp), and L. terrestris (1,196 bp) are much larger than in the core Halimedineae (between 6 and 43 bp).
Amino acid alignments of tilS and rpoB genes showed that the sequences are conserved across all lineages except for the highly divergent sequence of rpoB in B. hypnoides (supplementary figs. S8b and S9b, Supplementary Material online). The fact that sequence conservation persists beyond the in-frame stop codon suggests that there is functional coding sequence on both sides of the stop codon. One possible explanation is that the stop codon does not lead to termination of protein extension or is altered by RNA editing, leading to translation of the entire gene. However, the frame shifts observed in tilS genes of most species would suggest that this is unlikely. Another possible scenario is that the original gene has been fragmented into two subunits, but further work is needed to evaluate this possibility. The latter seems to be the case for the frame shifts observed in rpoB gene of Caulerpa cliftonii, C. manorensis, Chlorodesmis fastigiata, and Halimeda discoidea.
Diversity and Evolution of Nonstandard Genes
Aside from standard plastid genes, 153 freestanding ORFs of >300 bp long were found across the 14 bryopsidalean chloroplast genomes. Most of these freestanding ORFs occur in clusters of two to nine genes in regions 3–13.5 kb long, whereas other freestanding ORFs were found solitary. In 65 freestanding ORFs, structural and functional domains were found (table 1 and supplementary table S6, Supplementary Material online), whereas the remaining 88 freestanding ORFs showed no significant sequence similarity to known proteins. The most common motifs are DNA methyltransferase (MTase), group II intron reverse transcriptase/maturase, family A DNA polymerase, phage- or plasmid-associated DNA primase, and integrase.
Table 1.
Conserved Protein Domains Detected in the 153 Freestanding ORFs of 14 Bryopsidales Chloroplast Genomes
| Conserved Domain | No. of ORFs |
|---|---|
| Methyltransferase | 19 |
| Group II intron reverse transcriptase/maturase | 18 |
| DNA polymerase A | 6 |
| Phage- or plasmid-associated DNA primase | 6 |
| Integrase | 4 |
| NAD+ dependent DNA ligase | 3 |
| Rhs family protein | 2 |
| AGE domain | 1 |
| HNH endonuclease | 1 |
| Histidine carboxylase PI chain | 1 |
| Nonproteinogenic amino acid hydroxylase | 1 |
| Trimeric dUTPase | 1 |
| psbE | 1 |
| DNA primase | 1 |
| No conserved domain | 88 |
DNA MTases in prokaryotes are components of the restriction-modification systems, which protect the host cell against infection of foreign DNA (Jeltsch 2002; Ponger and Li 2005), and they participate in DNA replication and gene regulation (Buryanov and Shevchuk 2005). MTases have also been described as selfish mobile elements, inducing genome rearrangements such as amplifications, insertions, and transpositions (Furuta et al. 2010). DNA MTases have only rarely been reported in chloroplast genomes (Turmel et al. 2013, 2015; Leliaert and Lopez-Bautista 2015). We identified different types of MTases in the chloroplast genomes of Bryopsidales, including cytosine-C5-specific DNA MTase, adenine-specific MTase, and Type I restriction-modification system DNA methylase.
Group II intron reverse transcriptases/maturases are multifunctional proteins mostly encoded in bacterial and organellar group II introns, and are involved in splicing of these mobile genetic elements (Matsuura et al. 2001). They are also abundantly found in green algal chloroplast genomes (Brouard et al. 2016). In bryopsidalean chloroplast genomes, we identified group II intron reverse transcriptase/maturase domains in both group II intron-encoded proteins (IEPs) and freestanding ORFs. Likewise, ORFs with group II intron reverse transcriptase/maturase domain are present in introns and in intergenic regions in some trebouxiophycean and chlorophycean green algae (Turmel et al. 2015; McManus et al. 2017).
Family A DNA polymerases are found primarily in prokaryotes, and are involved in filling DNA gaps that arise during DNA repair, recombination, and replication (Garcia-Diaz and Bebenek 2007). These polymerases have so far only been found in chloroplast genomes of the Bryopsidales.
Phage- or plasmid-associated DNA primase (Ziegelin and Lanka 1995) have been reported in various green algal lineages, including prasinophytes (Turmel et al. 1999; Turmel, Gagnon, et al. 2009), Chlorophyceae (Brouard et al. 2016), desmids (Lemieux et al. 2016), and Bryopsidales (Leliaert and Lopez-Bautista 2015). Integrases, along with transposases catalyze the movement and integration of DNA copies to new locations within and between genomes (Rice and Baker 2001). A putative transposase has up till now only been identified in the bryopsidalean Tydemania (Leliaert and Lopez-Bautista 2015).
Although rare, nonstandard genes are being discovered in an increasing number of organellar genomes (Huang and Yue 2013; Mackiewicz et al. 2013), including green algal plastid genomes (Turmel et al. 1999, 2013, 2015; Brouard et al. 2008; McManus et al. 2017). The evolutionary origins of these genes, however, remain elusive. They may be remnants of the cyanobacterial ancestor of plastids, which were differentially lost in the chloroplast genomes of all other algal lineages. However, with the exception of group 4 freestanding ORFs, the bryopsidalean freestanding ORFs did not show close affinities with cyanobacterial genes (supplementary table S6 and fig. S10, Supplementary Material online). Alternative scenarios for the presence of nonstandard plastid genes have been hypothesized, including that they are vestiges of viral infections (Turmel et al. 2013), were acquired from bacterial donors (Leliaert and Lopez-Bautista 2015), or are remnants of introns originally present in standard plastid genes (Turmel et al. 2015).
Chloroplast genomic data from densely sampled lineages, such as the Trebouxiophyceae, have shown that nonstandard plastid genes are not conserved over long evolutionary timescales, suggesting that they are selfish genetic elements that provide no selective advantage (Turmel et al. 2015). Conversely, our study indicates that several freestanding ORFs with conserved protein domain show some level of conservation within bryopsidalean chloroplast genomes. BLASTp searches (E-value threshold < 10E-6) resulted in the delimitation of nine groups of freestanding ORFs showing similarity between two or more bryopsidalean chloroplast genomes (table 2), along with other sequences, mainly from plastid intronic and bacterial origin. Despite applying a relatively conserved E-value threshold, amino acid similarities within these groups are low (table 2), and therefore the results of the phylogenetic analyses (supplementary fig. S10, Supplementary Material online) should be interpreted with caution.
Table 2.
Nine Groups of Freestanding ORFs Showing Significant Homology between Two or More Bryopsidalean Chloroplast Genomes
| Group | Protein Conserved Domain | Bryopsidales Free-Standing ORFsa | Amino Acid Percent Identityb |
|---|---|---|---|
| group1c | group II intron reverse transcriptase/maturase | Bhyp (orf552), Ccli (orf519) | 38–49 |
| group1a | group II intron reverse transcriptase/maturase | Amaz (orf442), Bhyp (orf7), Bplu (orf7), Bsp (orf429), Ccli (orf347), Cman (orf341), Cfas (orf373), Care (orf294), Dsp (orf401), Oque (orf470), Osp (orf451), Rpen (orf387), Texp (orf3) | 24–33 |
| group2 | Integrase | Bsp (orf180), Care (orf484), Dsp (orf279) | 32–41 |
| group3 | Rhs family protein | Bhyp (orf2015), Bplu (orf3) | 31–31d |
| group4 | no conserved domain | Bhyp (orf5), Bplu (orf5) | 58–60 |
| group5 | Various: DNA polymerase family A domain, phage- or plasmid-associated DNA primase, and bacterial Rhs-family proteins | Bhyp (orf376), Bplu (orf3), Ccli (orf148, orf196, orf275, orf656, orf781), Cman (orf267, orf331, orf764, orf810), Hdis (orf164, orf1108), Rpen (orf556, orf787), Texp (orf15, orf16) | 25–51 |
| group6 | Methyltransferases: Type I restriction-modification system DNA methylase subunit, and adenine-specific methyltransferase | Ccli (orf829), Cman (orf606, orf823, orf839), Rpen (orf191) | 46–67 |
| group7 | Methyltransferase: cytosine-C5-specific DNA MTase | Ccli (orf156; orf242), Cman (orf598), Hdis (orf604), Texp (orf9) | 31–43 |
| group8 | Integrase/transposase | Ccli (orf141), Hdis (orf104), Texp (orf13) | 29–46 |
| group9 | NAD+ dependent DNA ligase | Cfas (orf120, orf139, orf217), Hdis (orf725) | 28–36 |
Only freestanding ORFs from the 14 examined bryopsidalean cp genomes are listed: Avrainvillea mazei (Amaz); Bryopsis hypnoides (Bhyp); Bryopsis plumosa (Bplu); Bryopsis sp. (Bsp); Caulerpa cliftonii (Ccli); Caulerpa manorensis (Cman); Chlorodesmis fastigiata (Cfas); Codium arenicola (Care); Derbesia sp. (Dsp); Halimeda discoidea (Hdis); Ostreobium quekettii (Oque); Ostreobium sp. (Osp); Rhipilia penicilloides (Rpen); Tydemania expeditionis (Texp). The phylogenetic trees showing all sequences are available in supplementary figure S9, Supplementary Material online.
Percent identity between bryopsidalean sequences and most closely related nonbryopsidalean sequences. This was based on BLASTp searches of bryopsidalean free-standing ORFs against nr.
group1 excluding group1a.
Similarity was mostly due to repeats.
Freestanding ORFs in group 1 include a group II intron reverse transcriptase/maturase specific domain, and are related to group II intronic ORFs from various algal chloroplast and mitochondrial genomes. Our data indicate mobility of these ORFs among and within organellar genomes, and multiple transfers from group II introns to intergenic regions. ORFs with a reverse transcriptase/maturase specific domain have been identified within and outside group II introns in a number of other green algal chloroplast genomes (Turmel et al. 2015; McManus et al. 2017), and have been suggested to be remnants of group II introns that have been transferred to intergenic regions by intragenomic proliferation of mobile introns, degeneration of a duplicated intron-containing genes, genomic rearrangement, or horizontal transfer of mobile introns (Turmel et al. 2015). The presence of a reverse transcriptase domain in these ORFs indicates that their transfer may be mediated by retrotransposition (Zimmerly and Semper 2015). Similar mechanisms may have resulted in the proliferation of group II introns in the green alga Gloeotilopsis, some of which occur in the untranslated regions of genes (Turmel et al. 2016). In subgroup 1a, the freestanding ORFs are conserved in all 14 chloroplast genomes of Bryopsidales and are likely vertically transmitted, as evidenced by the high congruence between the freestanding ORF phylogeny and chloroplast phylogeny (supplementary fig. S10a, Supplementary Material online).
The freestanding ORFs in groups 2–9 are less conserved within Bryopsidales compared with group 1a. Groups 2, 3, 4, and 5a are shared among species of Bryopsidineae, whereas groups 5 b, 6, 7, 8, and 9 are restricted to species of the core Halimedineae (fig. 4). In groups 5 b, 6, and 9, the freestanding ORFs occur in multiple copies, suggesting intragenomic proliferation. Apart from these groups, several other freestanding ORFs have apparently proliferated within certain genomes: similar freestanding ORFs are found in Avrainvillea mazei (orf254, orf275, and orf244), Caulerpa cliftonii (orf131 and orf781), Caulerpa manorensis (orf182, orf661, orf639), and Halimeda discoidea (orf184, orf304). The mode of proliferation of these ORFs remains elusive, but the presence of protein domains that are associated with mobile functions (phage- or plasmid-associated DNA primase, Rhs-family proteins, and NAD+ dependent DNA ligase) may explain their mobility and propagation within the chloroplast genome.
Fig. 4.

—Phylogenetic distribution of freestanding ORFs in Bryopsidales.
Understanding the affinities and origins of the high diversity of nonstandard genes in bryopsidalean chloroplast genomes will need further investigation, especially since there are no known mechanisms for DNA acquisition in plastids. Wider sampling of both chloroplast and nuclear genomes in green algae may provide further clues for the evolution of these genes.
Intron Content
A total of 29 genes were found to contain introns, and 11 of them contain intronic ORFs (supplementary table S7, Supplementary Material online). Ancestral reconstruction of intron content (fig. 5) revealed that group II introns may have been abundant early in the evolution of Bryopsidales. This situation is still observed in the Ostreobineae, Bryopsidineae, and Dichotomosiphonaceae, but group II introns were largely lost in the core Halimedineae. Instead, this lineage showed a proliferation of group I introns, which were likely rare or absent in the early evolution of the Bryopsidales.
Fig. 5.

—Ancestral reconstruction based on presence/absence of introns. (A) Group II introns and (B) Group I introns. Unshaded circles indicate absence of intronic ORFs, whereas shaded circles indicate presence of intronic ORFs and the corresponding conserved domains—LHE, LAGLIDADG homing endonuclease; HNH, nuclease; RT, reverse transcriptase; IM, intron maturase.
In bryopsidalean chloroplast genomes, the majority of group II introns are found in protein-coding genes and their IEPs (when present) contain a reverse transcriptase (RT) and/or intron maturase (IM) domain, and sometimes a H-N-H nuclease domain (fig. 5 and supplementary table S7, Supplementary Material online). The psbC IEP of Caulerpa manorensis is the only protein where all three domains were inferred to be present. In contrast, the majority of group I introns are found in the large subunit rRNA gene and their IEPs (when present) all encode LAGLIDADG homing endonuclease (LHE). One of the IEPs in the large subunit rRNA of Caulerpa manorensis and two in Caulerpa cliftonii and Halimeda discoidea contain two LHE motifs.
Group II intron mobility is by retrohoming (Lambowitz and Zimmerly 2011) while group I introns accomplish this by homing (Haugen et al. 2005). For group II introns, splicing and mobility are promoted by IEPs with multiple domains present—RTs, maturases, and HNH endonucleases (Lambowitz and Zimmerly 2011). In cases where IEPs are absent, host-encoded proteins are recruited for splicing (Bonen and Vogel 2001; Lambowitz and Zimmerly 2011). Although all group II introns in the two Ostreobium spp. included in this study lack IEPs, both have a freestanding ORF (orf470 and orf451) encoding a group II intron RT/maturase that may promote splicing of the introns. A similar case is observed for IEP-lacking group II introns in Rhipilia penicilloides where a freestanding ORF (orf387) encodes for IM. All of the motifs mentioned have group II intron origins and could promote splicing of the introns present in their respective taxa. Similarly, mobility of group I introns are promoted by IEPs that encodes DNA endonucleases. In some cases, the IEPs are also adapted to function in splicing (Lambowitz et al. 1999). It has been reported that IEPs with two motifs of LAGLIDADG have maturase activity which can also function for splicing (Lambowitz and Belfort 1993).
Intron proliferation is not uncommon in green algal chloroplast genomes. For example, the chlorophycean Oedogonium cardiacum (Brouard et al. 2016) and several ulvophycean chloroplast genomes (Turmel et al. 2016, 2017) have been shown to contain large numbers of group II introns. Group II introns in the ulvophycean chloroplast genomes were found to have originated from different species and insertion sites (Turmel et al. 2017). In all these cases, intragenomic proliferation of these introns was attributed to retrohoming. On the other hand, the introns (27 in total) in the chloroplast genome of the ulvophycean Pseudendoclonium akinetum were all identified to be group I introns (Pombert et al. 2005). The similarity of the introns and the homing endonucleases they encode suggests that they resulted from intragenomic proliferation (Pombert et al. 2005).
Reverse transcriptase-mediated intron loss and genomic deletions are a few mechanisms attributed to intron loss (Roy and Gilbert 2005; Cohen et al. 2012; Odom and Herrin 2013). RT-mediated intron loss suggests reverse transcription of processed or semiprocessed mRNA by RT followed by the integration of the resulting cDNA by homologous recombination (Cohen et al. 2012). This mechanism has resulted in loss of group II intron in psbA gene of Chlamydomonas species (Odom and Herrin 2013) and may also explain the loss of several group II introns in the core Halmedineae.
Introns make up only a small portion of the bryopsidalean chloroplast genomes (1.7–13.8%). There was no clear trend observed between the number of introns and genome size. The relatively large genome of Rhipilia penicilloides only has 1.7% of its genome accounted for by introns. In contrast, introns account for 4% of the compact Ostreobium sp. genome.
Synteny and Rearrangement
Whole-genome alignment of 14 chloroplast genomes using Progressive Mauve resulted in small LCBs and suggests high levels of rearrangements across the siphonous green algae (supplementary fig. S11, Supplementary Material online). Analyses of the ancestral order of syntenic blocks showed a total of 127 rearrangements occurred along the Bryopsidales phylogeny (fig. 1). Rearrangements observed in the Bryopsidineae are minimal (total of 22) compared with the core Halimedineae (total of 93). A similar result was also observed on DCJ analyses (supplementary table S8, Supplementary Material online).
Despite the many rearrangements, there are a handful of gene clusters (three or more genes) that are conserved across all Bryopsidales: 1) psaM-psb30-psbK-psbN-trnM; 2) ccs1-cysA-psbB-psbT-psbH; 3) chlI-tufA-trnT; 4) rpl23-rpl2-rps19-rps3-rpl16-rpl14-rpl5-rps8-infA-rpl36-rps11-rpoA; 5) atpI-atpH-atpF-atpA; and 6) psbE-psbF-psbL-psbJ (supplementary fig. S12, Supplementary Material online). The latter three are also conserved in other members of the Ulvophyceae (based on Turmel, Otis, et al. 2009; supplementary fig. S13, Supplementary Material online). Conservation of these gene clusters could mean that they are transcriptional units essential for the group of organisms concerned.
Loss of IR and/or abundance of repeats have been correlated with increased genome rearrangements in green algal species like Stigeoclonium helveticum (Bélanger et al. 2006) and Leptosira (de Cambiaire et al. 2007). Loss of IR was also attributed to the genomic rearrangement in some land plants (Chumley et al. 2006; Wolf et al. 2010; Yap et al. 2015). In these cases, it has been hypothesized that intramolecular recombination between short dispersed repeats is enhanced by the loss of IR (Palmer 1991). However, since IR has been lost earlier in the evolution of the Bryopsidales and given the fact that extensive genome rearrangements are more prominent in the core Halimedineae, different factors might be the causing these observed rearrangements.
In the Zygnematales, Lemieux et al. (2016) suggested that early insertions of viral genes might have contributed to the instability of the IR. In addition, Civáň et al. (2014) suggested that ancient retroelement activities (as indicated by the presence of integrases-like and RT-like elements in the zygnematalean genus Roya) could have caused the extensive genomic rearrangement for the lineage. Considering that reverse transcriptases are found across all bryopsidalean taxa, this particular mobile genetic element is probably not cause for the rearrangement observed in the core Halimedineae. However, other mobile genetic elements (DNA polymerase, phage- or plasmid-associated DNA primase, methyltransferase, integrase/transposase, and ligases) restricted to the core Halimedineae might have played a role in the extensive rearrangement in this lineage as it did in the Zygnematales.
Conclusions
By using comparative phylogenetic analyses on chloroplast genome features of siphonous green algae, we have gained insights on the evolutionary dynamics of this ecologically and economically important group of green algae. Analyses of the freestanding ORFs highlight the diversity of these nonstandard, foreign genes based on their conserved protein domains and showed some level of conservation and intragenomic proliferation in the bryopsidalean chloroplast genomes.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by the Australian Biological Resources Study (RFL213-08), the Australian Research Council (DP150100705), the University of Melbourne (MIRS/MIFRS to M.C.M.C. and V.R.M.), and by use of the computational resources at Melbourne Bioinformatics (project UOM0007) and the Nectar Research Cloud, a collaborative Australian research platform supported by the National Collaborative Research Infrastructure Strategy (NCRIS). We thank Stephanie Muller (CSHL), Roger Nielsen (GGF), and Tingting Wang (Novogene) for the assistance they provided during the sequencing of the samples, Claude Payri for facilitating fieldwork in PNG, and the people from the Verbruggen lab especially Chris Jackson and Joana Costa for fruitful discussions. The authors declare no conflict of interest.
Literature Cited
- Abascal F, Zardoya R, Telford MJ.. 2010. TranslatorX: multiple alignment of nucleotide sequence guided by amino acid translations. Nucleic Acids Res. 38(suppl_2):W7–W13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdeyev P, et al. , 2016. Reconstruction of ancestral genomes in presence of gene gain and loss. J Comput Biol. 23(3):150–164. [DOI] [PubMed] [Google Scholar]
- Bankevich A, et al. , 2012. SPAdes: a new genome assembly algorithm and Its applications to single-cell sequencing. J Comput Biol. 19(5):455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck N, Lang B.. 2010. MFannot, organelle genome annotation webserver. Available from: http://megasun.bch.umontreal.ca/cgi-bin/mfannot/mfannotInterface.pl; last accessed November 2017.
- Bélanger A-S, et al. , 2006. Distinctive architecture of the chloroplast genome in the chlorophycean green alga Stigeoclonium helveticum. Mol Genet Genomics 276(5):464–477. [DOI] [PubMed] [Google Scholar]
- Bonen L, Vogel J.. 2001. The ins and outs of group II introns. Trends Genet. 17(6):322–331. [DOI] [PubMed] [Google Scholar]
- Brouard J-S, et al. , 2008. Chloroplast DNA sequence of the green alga Oedogonium cardiacum (Chlorophyceae): unique genome architecture, derived characters shared with the Chaetophorales and novel genes acquired through horizontal transfer. BMC Genomics 9:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouard J-S, et al. , 2010. The exceptionally large chloroplast genome of the green alga Floydiella terrestris illuminates the evolutionary history of the Chlorophyceae. Genome Biol Evol. 2(1):240–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouard J-S, et al. , 2016. Proliferation of group II introns in the chloroplast genome of the green alga Oedocladium carolinianum (Chlorophyceae). PeerJ 4:e2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buryanov Y, Shevchuk T.. 2005. The use of prokaryotic DNA methyltransferases as experimental and analytical tools in modern biology. Anal Biochem. 338(1):1–11. [DOI] [PubMed] [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17(4):540–552. [DOI] [PubMed] [Google Scholar]
- Chumley TW, et al. , 2006. The complete chloroplast genome sequence of Pelargonium × hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol Evol. 23(11):2175–2190. [DOI] [PubMed] [Google Scholar]
- Civáň P, et al. , 2014. Analyses of Charophyte chloroplast genomes help characterize the ancestral chloroplast genome of land plants. Genome Biol Evol. 6(4):897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen NE, Shen R, Carmel L.. 2012. The role of reverse transcriptase in intron gain and loss mechanisms. Mol Biol Evol. 29(1):179–186. [DOI] [PubMed] [Google Scholar]
- Cremen MCM, et al. , 2016. Taxonomic revision of Halimeda (Bryopsidales, Chlorophyta) in south-western Australia. Aust Syst Bot. 29(1):41–54. [Google Scholar]
- Darling ACE. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14(7):1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D, et al. , 2012. jModelTest 2 : more models, new heuristics and parallel computing CircadiOmics : integrating circadian genomics, transcriptomics, proteomics. Nat Methods 9(8):772.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cambiaire JC, et al. , 2006. The complete chloroplast genome sequence of the chlorophycean green alga Scenedesmus obliquus reveals a compact gene organization and a biased distribution of genes on the two DNA strands. BMC Evol Biol. 6(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cambiaire JC, et al. , 2007. The chloroplast genome sequence of the green alga Leptosira terrestris: multiple losses of the inverted repeat and extensive genome rearrangements within the Trebouxiophyceae. BMC Genomics 8:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries J, Gould SB.. 2017. The monoplastidic bottleneck in algae and plant evolution. J Cell Sci. 0:1–13. doi:10.1242/jcs.203414. [DOI] [PubMed] [Google Scholar]
- de Vries J, et al. , 2013. Is ftsH the key to plastid longevity in sacoglossan slugs? Genome Biol Evol. 5(12):2540–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries J, Archibald JM, Gould SB.. 2017. The carboxy terminus of YCF1 contains a motif conserved throughout >500 myr of streptophyte evolution. Genome Biol Evol. 9(2):473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Cortona A, et al. , 2017. The plastid genome in cladophorales green algae is encoded by hairpin plasmids. Curr Biol. 27(24):3771–3782. [DOI] [PubMed] [Google Scholar]
- Delcher AL, et al. , 2007. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23(6):673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF, Sapienza C.. 1980. Selfish genes, the phenotype paradigm and genome evolution. Nature 284(5757):601–603. [DOI] [PubMed] [Google Scholar]
- Fučíková K, et al. , 2014. New phylogenetic hypotheses for the core Chlorophyta based on chloroplast sequence data. Front Ecol Evol. 2:63. [Google Scholar]
- Furuta Y, Abe K, Kobayashi I.. 2010. Genome comparison and context analysis reveals putative mobile forms of restriction-modification systems and related rearrangements. Nucleic Acids Res. 38(7):2428–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Diaz M, Bebenek K.. 2007. Multiple functions of DNA polymerases. Crit Rev Plant Sci. 26(2):105–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni SJ, et al. , 2005. Genome streamlining in a cosmopolitan oceanic bacterium. Science 309(5738):1242–1245. [DOI] [PubMed] [Google Scholar]
- Gould SB, Waller RF, McFadden GI.. 2008. Plastid evolution. Annu Rev Plant Biol. 59(1):491–517. [DOI] [PubMed] [Google Scholar]
- Green BR. 2011. Chloroplast genomes of photosynthetic eukaryotes. Plant J. 66(1):34–44. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O.. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 52(5):696–704. [DOI] [PubMed] [Google Scholar]
- Harmon LJ, et al. , 2008. GEIGER: investigating evolutionary radiations. Bioinformatics 24(1):129–131. [DOI] [PubMed] [Google Scholar]
- Haugen P, Simon DM, Bhattacharya D.. 2005. The natural history of group I introns. Trends Genet. 21(2):111–119. [DOI] [PubMed] [Google Scholar]
- Hessen DO, et al. , 2010. Genome streamlining and the elemental costs of growth. Trends Ecol Evol. 25(2):75–80. [DOI] [PubMed] [Google Scholar]
- Hilker R, et al. , 2012. UniMoG-a unifying framework for genomic distance calculation: and sorting based on DCJ. Bioinformatics 28(19):2509–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Yue J.. 2013. Horizontal gene transfer in the evolution of photosynthetic eukaryotes. J Syst Evol. 51(1):13–29. [Google Scholar]
- Inoue-Kashino N, Kashino Y, Takahashi Y.. 2011. psb30 is a photosystem II reaction center subunit and is required for optimal growth in high light in Chlamydomonas reinhardtii. J Photochem Photobiol B 104(1–2):220–228. [DOI] [PubMed] [Google Scholar]
- Jeltsch A. 2002. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem 3(4):274–293. [DOI] [PubMed] [Google Scholar]
- Kalyaanamoorthy S, et al. , 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14(6):587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ. 2010. The endosymbiotic origin, diversification and fate of plastids. Philos Trans R Soc Lond B Biol Sci. 365(1541):729–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell MG, Lisch DR.. 2001. Perspective: transposable elements, parasitic DNA, and genome evolution. Evolution 55(1):1–24. [DOI] [PubMed] [Google Scholar]
- Kuo CH, Ochman H.. 2010. Deletional bias across the three domains of life. Genome Biol Evol. 1(0):145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam DW, Lopez-Bautista JM.. 2016. Complete chloroplast genome for Caulerpa racemosa and compartive analyses of siphonous green seaweeds plastomes. Cymbella 2:23–32. [Google Scholar]
- Lambowitz A, et al. , 1999. Group I and group II ribozymes as RNPs: clues to the past and guides to the future In: Gesteland R, Cech TR, Atkins JF, editors. The RNA world. Cold Spring Harbor (NY: ): Cold Spring Harbor Laboratory Press; p. 451–486. [Google Scholar]
- Lambowitz AM, Belfort M.. 1993. Introns as mobile genetic elements. Annu Rev Biochem. 62:587–622. [DOI] [PubMed] [Google Scholar]
- Lambowitz AM, Zimmerly S.. 2011. Group II introns: mobile ribozymes that invade DNA. CSH Perspect Biol. 3(8):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang BF, Nedelcu AM.. 2012. Plastid genomes of algae In: Bock R, Knoop V, editors. Genomics of chloroplasts and mitochondria. Advances in photosynthesis and respiration. Dordrecht: Springer Netherlands; p. 59–87. [Google Scholar]
- Laslett D, Canback B.. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32(1):11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leible MB, Berger S, Schweiger HG.. 1989. The plastome of Acetabularia mediterranea and Batophora oerstedii: inter-and intraspecific variability and physical properties. Curr Genet. 15(5):355–361. [Google Scholar]
- Leliaert F, et al. , 2012. Phylogeny and molecular evolution of the green algae. Crit Rev Plant Sci. 31(1):1–46. [Google Scholar]
- Leliaert F, et al. , 2016. Chloroplast phylogenomic analyses reveal the deepest-branching lineage of the Chlorophyta, Palmophyllophyceae class. nov. Sci Rep. 6:25367.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leliaert F, Lopez-Bautista JM.. 2015. The chloroplast genomes of Bryopsis plumosa and Tydemania expeditiones (Bryopsidales, Chlorophyta): compact genomes and genes of bacterial origin. BMC Genomics 16:204.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux C, Otis C, Turmel M.. 2014. Chloroplast phylogenomic analysis resolves deep-level relationships within the green algal class Trebouxiophyceae. BMC Evol Biol. 14(1):211.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux C, Otis C, Turmel M.. 2016. Comparative chloroplast genome analyses of streptophyte green algae uncover major structural alterations in the Klebsormidiophyceae, Coleochaetophyceae and Zygnematophyceae. Front Plant Sci. 7:697.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, et al. , 2015. MEGAHIT: an ultra-fast single node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31(10):1674–1676. [DOI] [PubMed] [Google Scholar]
- Lohse M, et al. , 2013. OrganellarGenomeDRAW – a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41(Web Server Issue):W575–W581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lü F, et al. , 2011. The Bryopsis hypnoides plastid genome: multimeric forms and complete nucleotide sequence. PLoS One 6(2):e14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2006. Streamlining and simplification of microbial genome architecture. Annu Rev Microbiol. 60(1):327–349. [DOI] [PubMed] [Google Scholar]
- Lynch M, Koskella B, Schaack S.. 2006. Mutation pressure and evolution of organelle genomic architecture. Science 311(5768):1727–1730. [DOI] [PubMed] [Google Scholar]
- Mackiewicz P, Bodył A, Moszczyński K.. 2013. The case of horizontal gene transfer from bacteria to the peculiar dinoflagellate plastid genome. Mob Genet Elem. 3(4):e25845.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelino VR, et al. , 2016. Evolutionary dynamics of chloroplast genomes in low light: a case study of the endolithic green alga Ostreobium quekettii. Genome Biol Evol. 8(9):2939–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A. 2015. CDD: nCBI’s conserved domain database. Nucleic Acids Res. 43(Database Issue):D222–D226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin W, et al. , 1998. Gene transfer to the nucleus and the evolution of chloroplasts. Nature 393(6681):162–165. [DOI] [PubMed] [Google Scholar]
- Matsuura M, Noah JW, Lambowitz AM.. 2001. Mechanism of maturase-promoted group II intron splicing. EMBO J. 20(24):7259–7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul JE, et al. , 2002. The Chlamydomonas reinhardtii plastid chromosome: islands of genes in a sea of repeats. Plant Cell 14(11):2659–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C. 2007. Phobos: a tandem repeat search tool. Available from: http://www.geneious.com/plugins/phobos-plugin, last accessed November 2017.
- McManus HA, Sanchez DJ, Karol KG.. 2017. Plastomes of the green algae Hydrodictyon reticulatum and Pediastrum duplex (Sphaeropleales, Chlorophyceae). PeerJ 5:e3325.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melton JT, et al. , 2015. The complete chloroplast and mitochondrial genomes of the green macroalga Ulva sp. UNA00071828 (Ulvophyceae, Chlorophyta). PLoS One 10(4):e0121020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mine I, Menzel D, Okuda K.. 2008. Morphogenesis in giant-celled algae. Int Rev Cell Mol Biol. 266:37–83. [DOI] [PubMed] [Google Scholar]
- Mira A, Ochman H, Moran NA.. 2001. Deletional bias and the evolution of bacterial genomes. Trends Genet. 17(10):589–596. [DOI] [PubMed] [Google Scholar]
- Muñoz-Gómez SA, et al. , 2017. The new red algal subphylum Proteorhodophytina comprises the largest and most divergent plastid genomes known. Curr Biol. 27(11):1677–1678. [DOI] [PubMed] [Google Scholar]
- Novis PM, et al. , 2013. Inclusion of chloroplast genes that have undergone expansion misleads phylogenetic reconstruction in the Chlorophyta. Am J Bot. 100(11):2194–2209. [DOI] [PubMed] [Google Scholar]
- Odom OW, Herrin DL.. 2013. Reverse transcription of spliced psbA mRNA in Chlamydomonas spp. and its possible role in evolutionary intron loss. Mol Biol Evol. 30(12):2666–2675. [DOI] [PubMed] [Google Scholar]
- Orgel LE, Crick FHC.. 1980. Selfish DNA: the ultimate parasite. Nature 284(5757):604–607. [DOI] [PubMed] [Google Scholar]
- Palmer JD. 1991. Plastid chromosomes: structure and evolution In: Bogorad L, editor. Molecular biology of plastids. Orlando (FL: ): Academic Press; p. 5–53. [Google Scholar]
- Paradis E, Claude J, Strimmer K.. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20(2):289–290. [DOI] [PubMed] [Google Scholar]
- Pombert J-F, et al. , 2005. The chloroplast genome sequence of the green alga Pseudendoclonium akinetum (Ulvophyceae) reveals unusual structural features and new insights into the branching order of chlorophyte lineages. Mol Biol Evol. 22(9):1903–1918. [DOI] [PubMed] [Google Scholar]
- Pombert J-F, Lemieux C, Turmel M.. 2006. The complete chloroplast DNA sequence of the green alga Oltmannsiellopsis viridis reveals a distinctive quadripartite architecture in the chloroplast genome of early diverging ulvophytes. BMC Biol. 4(1):3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce-Toledo RI, et al. , 2017. An early-branching freshwater Cyanobacterium at the origin of plastids. Curr Biol. 27(3):386–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponger L, Li WH.. 2005. Evolutionary diversification of DNA methyltransferases in eukaryotic genomes. Mol Biol Evol. 22(4):1119–1128. [DOI] [PubMed] [Google Scholar]
- Revell LJ. 2012. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol Evol. 3(2):217–223. [Google Scholar]
- Rice P, Longden I, Bleasby A.. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16(6):276–277. [DOI] [PubMed] [Google Scholar]
- Rice PA, Baker TA.. 2001. Comparative architecture of transposase and integrase complexes. Nat Struct Biol. 8(4):302–307. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Ezpeleta N, et al. , 2005. Monophyly of primary photosynthetic eukaryotes: green plants, red algae, and glaucophytes. Curr Biol. 15(14):1325–1330. [DOI] [PubMed] [Google Scholar]
- Rolland N, et al. , 1997. Disruption of the plastid ycf10 open reading frame affects uptake of inorganic carbon in the chloroplast of Chlamydomonas. EMBO J. 16(22):6713–6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy SW, Gilbert W.. 2005. Rates of intron loss and gain: implications for early eukaryotic evolution. Proc Natl Acad Sci U S A. 102(16):5773–5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert I, Vu GTH.. 2016. Genome stability and evolution: attempting a holistic view. Trends Plant Sci. 21(9):749–757. [DOI] [PubMed] [Google Scholar]
- Smith DR, Lee RW.. 2009. The mitochondrial and plastid genomes of Volvox carteri: bloated molecules rich in repetitive DNA. BMC Genomics 10(1):132.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, et al. , 2016. Chloroplast phylogenomic inference of green algae relationships. Sci Rep. 6:20528.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmis JN, et al. , 2004. Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat Rev Genet. 5(2):123–135. [DOI] [PubMed] [Google Scholar]
- Turmel M, Gagnon MC et al. , 2009. The chloroplast genomes of the green algae Pyramimonas, Monomastix, and Pycnococcus shed new light on the evolutionary history of prasinophytes and the origin of the secondary chloroplasts of euglenids. Mol Biol Evol. 26(3):631–648. [DOI] [PubMed] [Google Scholar]
- Turmel M, Otis C, Lemieux C.. 1999. The complete chloroplast DNA sequence of the green alga Nephroselmis olivacea: insights into the architecture of ancestral chloroplast genomes. Proc Natl Acad Sci U S A. 96(18):10248–10253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmel M, Otis C, Lemieux C.. 2005. The complete chloroplast DNA sequences of the charophycean green algae Staurastrum and Zygnema reveal that the chloroplast genome underwent extensive changes during the evolution of the Zygnematales. BMC Biol. 3(1):22.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmel M, Otis C, Lemieux C.. 2009. The chloroplast genomes of the green algae Pedinomonas minor, Parachlorella kessleri, and Oocystis solitaria reveal a shared ancestry between the Pedinomonadales and Chlorellales. Mol Biol Evol. 26(10):2317–2331. [DOI] [PubMed] [Google Scholar]
- Turmel M, Otis C, Lemieux C.. 2013. Tracing the evolution of streptophyte algae and their mitochondrial genome. Genome Biol Evol. 5(10):1817–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmel M, Otis C, Lemieux C.. 2015. Dynamic evolution of the chloroplast genome in the green algal classes Pedinophyceae and Trebouxiophyceae. Genome Biol Evol. 7(7):2062–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmel M, Otis C, Lemieux C.. 2016. Mitochondrion-to-chloroplast DNA transfers and intragenomic proliferation of chloroplast group II introns in Gloeotilopsis green algae (Ulotrichales, Ulvophyceae). Genome Biol Evol. 8(9):2789–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmel M, Otis C, Lemieux C.. 2017. Divergent copies of the large inverted repeat in the chloroplast genomes of ulvophycean green algae. Sci Rep. 7(1):994.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbruggen H. 2012. TreeGradients version 1.03. Available from: http://www.phycoweb.net/software, last accessed November 2017.
- Verbruggen H, Costa JF.. 2015. The plastid genome of the red alga Laurencia. J Phycol. 51(3):586–589. [DOI] [PubMed] [Google Scholar]
- Verbruggen H, et al. , 2009. A multi-locus time-calibrated phylogeny of the siphonous green algae. Mol Phylogenet Evol. 50(3):642–653. [DOI] [PubMed] [Google Scholar]
- Verbruggen H, et al. , 2017. Phylogenetic position of the coral symbiont Ostreobium (Ulvophyceae) inferred from chloroplast genome data. J Phycol. 53(4):790–803. [DOI] [PubMed] [Google Scholar]
- Vroom PS, Smith CM.. 2003. Life without cells. Biologist 50:222–226. [Google Scholar]
- Wick RR, et al. , 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31(20):3350–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf PG, Roper JM, Duffy AM.. 2010. The evolution of chloroplast genome structure in ferns. Genome 53(9):731–738. [DOI] [PubMed] [Google Scholar]
- Wolf YI, Koonin EV.. 2013. Genome reduction as the dominant mode of evolution. BioEssays 35(9):829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman SK, Jansen RK, Boore JL.. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20(17):3252–3255. [DOI] [PubMed] [Google Scholar]
- Yap JYS, et al. , 2015. Complete chloroplast genome of the wollemi pine (Wollemia nobilis): structure and evolution. PLoS One 10(6):e0128126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegelin G, Lanka E.. 1995. Bacteriophage P4 DNA replication. FEMS Microbiol Rev. 17(1–2):99–107. [DOI] [PubMed] [Google Scholar]
- Zimmerly S, Semper C.. 2015. Evolution of group II introns. Mob DNA 6(1):7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccarello GC, et al. , 2009. Analysis of a plastid multigene data set and the phylogenetic position of the marine macroalga Caulerpa filiformis (chlorophyta). J Phycol. 45(5):1206–1212. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.