

Abstract
An improved synthesis using microwave heating affords (C6H4PO(OCH3)2)2 in excellent isolated yield (95 %). The ligand properties of this bisphosphonateester were explored towards hard metal centers M 2+ (M = Ca, UO2) and M 3+ (M = La, Ce, Sm, Eu) resulting in coordination polymers, for which the reduction of ionic size of the central metal atom resulted in lower‐dimensional structural motifs as opposed to higher dimensional networks obtained for the larger ions. All coordination polymers were characterized by single‐crystal X‐ray diffraction, IR spectroscopy, and combustion analysis. The ligand was furthermore characterized with multinuclear NMR spectroscopy.
Keywords: Phosphorus, Uranium, Lanthanides, Calcium, Coordination modes
Introduction
Owing to their unique physical properties, lanthanide nitrate based coordination polymers have attracted substantial interest over several decades in addition to their interesting structural features.1, 2, 3, 4 Therefore such coordination polymers are relevant in a wide range of technological fields, such as medicine, LEDs, lanthanide separation, etc.5 The most frequently used binding sites in metal‐organic frameworks or coordination polymers are carboxylates or nitrogen bases.1, 6 In recent times phosphonates are being explored due to the possibility of accessing new metal phosphate morphologies.7 In contrast, neutral phosphonate esters have not been so widely explored as ligands.3, 8 Very recently we reported an optimized synthesis of such ditopic phosphonate esters giving improved yields at reduced reaction times using microwave heating.9 This stimulated us to explore the solvent free microwave assisted synthesis of (C6H4PO(OMe)2)2 which has been mentioned in literature before however with very limited characterization.10 So far the ligand properties have been explored with cadmium11 and manganese12 as central metal atoms, affording a two‐dimensional coordination polymer. Herein we further report the use of this ligand for complexation with lanthanides and earth alkaline metals, as well as uranium.
Results and Discussion
Starting from 4,4′‐dichloromethylbiphenyl and trimethyl phosphite we have been able to prepare compound L on a multigram scale in excellent yield using microwave heating (Scheme 1). The 31P NMR spectrum in solution shows a signal at δ = 28.9 ppm split to a multiplet owing to phosphorus proton coupling (2 J PH = 22 Hz) and is in a similar region as closely related compounds.9 Solid L shows two IR bands characteristic for phosphonates derivatives at 1246 and 1178 cm–1.8, 9
Scheme 1.
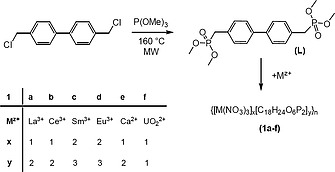
Synthesis of L and coordination polymers 1a–1f.
Compound L crystallizes in space group P 1. Owing to an inversion center located at the central C–C bond, only half of the ligand is present in the asymmetric unit resulting in co‐planar arrangement of the phenyl rings (Figure 1.). The P=O bond length is 1.4619(16) Å and within standard deviation for similar bonds.3, 9, 13 The P–O single bonds are 1.5812(16) and 1.5778(15) Å respectively. The angles around the phosphorus atom cover a range of 101.31(9) to 115.73(9)°. They show slight deviation from tetrahedral angles due to interactions between neighboring molecules.
Figure 1.
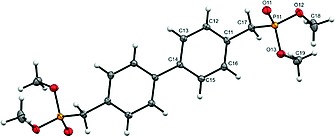
Solid state structure of ligand L. Ellipsoids are drawn at 30 % probability level.
The formation of coordination polymers can be achieved by slow evaporation of a methanol solution of L and a corresponding Ln(NO3)3 salt. Only in case of lanthanum, cerium, samarium, and europium it was possible to obtain single crystals of good quality for X‐ray analysis. Two new types of structures were found. In addition, all compounds were also characterized by elemental analysis and IR spectroscopy. The coordination polymers 1a and 1b show two almost identical vibrational bands at 1206 and 1172 cm–1 and are shifted towards lower wavenumbers compared to the uncoordinated ligand L. The bands for 1c and 1d are also almost identical at 1201 and 1164 cm–1 yet slightly shifted compared to 1a, 1b. The large shift of the P–O vibrational band in both free the ligand (1178 cm–1) and its complexes compared with the previously published ligand (C6H4PO(OiPr)2)2 (1105 cm–1) is due to the different steric situation as a consequence of the reduced size of the substituents at the oxygen atoms in 1a–1d.
The coordination polymers of lanthanum 1a and cerium 1b are isostructural. Therefore only the cerium structure will be described, which is representative for both. The polymer crystallizes in space group P21 and the asymmetric unit consists of one Ce(NO3)3 unit and two symmetry independent ligands L. The repeating unit of the polymer backbone is [Ce(NO3)3L2] which is further confirmed by elemental analysis. The coordination sphere around the central metal atom consists of three bidentate nitrate anions and four monodentate phosphonate units as part of ligand L, adding up to a coordination number of 10 (Figure 2.).
Figure 2.
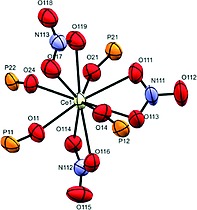
Coordination sphere of 1b. Ellipsoids are drawn at 30 % probability level.
The bond lengths from cerium to the nitrate oxygen atoms are in the range of 2.565(10) to 2.675(8) Å and do not deviate from similar coordination polymers. On the other hand the bond lengths of cerium to oxygen atoms belonging to phosphonate ester groups are shorter and in the range of 2.413(11) to 2.489(9) Å. The polymer forms a rhombic pattern within one sheet. The angle between vertices of the rhomb is 100.61° with distances between neighboring cerium atoms being 18.157 Å. The dihedral angles of biphenyl units are 32.1(4) and 24.2(4)°, respectively. The two neighboring sheets are interpenetrated (Figure 3.).
Figure 3.
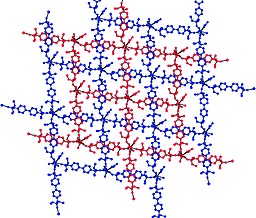
Solid state packing of 1b showing interpenetrated sheets.
Going down the line to samarium 1c and europium 1d, again isostructural coordination polymers have been found albeit with different binding motif compared to coordination polymers 1a, 1b. Only the structure of the samarium compound 1c will be described, which is also representative for 1d. The polymer crystallizes in space group P 1. Formally the asymmetric unit is built of one Sm(NO3)3 center and one and a half ligand. Consequently the repeating unit of the polymer is [[Sm(NO3)3]2L3], which is further corroborated by elemental analysis. The metal is coordinated to three bidentate nitrate units and three monodentate ligands rising up to a coordination number of 9 (Figure 4).
Figure 4.
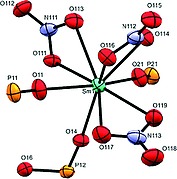
Coordination sphere of 1c. Ellipsoids are drawn at 30 % probability level.
The bond lengths between the samarium and nitrate oxygen atoms are in the range of 2.487(3) to 2.531(4) Å and slightly smaller than those found for the cerium derivative, which is in accordance with the smaller ionic radius of samarium compared to cerium. The bonds between samarium and the phosphonate ester oxygen atom are as well slightly shorter covering the range from 2.325(3) to 2.380(3) Å. Two ligands bridge two central metal atoms, thus forming a dimer. Those dimers are bridged by a third ligand forming a 1D chain (Figure 5). The biphenyl units within the dimers show a disorder in half of the phenyl units leading to dihedral angles of 33.6(3) and 44.4(3)° between the phenyl units. In contrast, in the bridging ligand the biphenyl unit is planar. The distance between the central metal atoms of the dimeric part is 15.488 Å, while that between two dimers amounts to 18.791 Å. The general topology of 1c and 1d is related to other 1D polymeric structures based on d‐block metals with macrocyclic arrangements containing lateral π‐systems.14, 15
Figure 5.
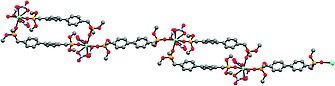
Ball and stick model of 1c showing the polymeric motif. Hydrogen atoms are omitted for clarity.
For the sake of comparison the calcium analog 1e was investigated as well. The calcium coordination polymer crystallizes in space group I 42d. In the asymmetric unit the calcium atom is found on a fourfold axis with half of a nitrate and ligand. Due to the high symmetry the repeating unit of the coordination polymer is [Ca(NO3)2L2]. The coordination environment of one calcium ion consists of two bidentate nitrate and four phosphonate units as ligands, which adds up to a total coordination number of 8. The bond length between the calcium and oxygen atoms of nitrate is 2.572(8) Å and thus in a similar range as in 1b. As expected the contact between the calcium ion and the oxygen atom of the phosphonate ester unit is shorter than in the previously discussed coordination polymers, 2.331(11) Å. The structural motif consists of a square pattern within one sheet, with distances between neighboring calcium atoms of 18.237 Å. The next sheet is intertwined with the first one forming a mesh network (Figure 6).
Figure 6.
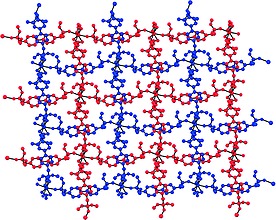
Packing of 1e showing interpenetrated mesh network.
Using uranium as coordination center, starting from uranyl nitrate, the respective coordination polymer 1f gives a one‐dimensional zigzag type polymer backbone (Figure 7). In the solid state 1f crystallizes in space group P 1, where in the asymmetric unit the uranium atom is located on an inversion center. This corresponds formally to an occupation of half an uranyl unit with one bidentate nitrate ligand and in addition one half of ligand L. Therefore, the repeating unit of coordination polymer 1f can be described as [UO2(NO3)2L], which is further corroborated by the composition based on elemental analysis. The coordination number of uranium atom is 8, for which a hexagonal bipyramidal arrangement is observed (Figure 8.).
Figure 7.
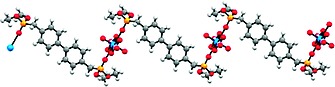
Ball and stick model of the zig‐zag chain in 1f.
Figure 8.
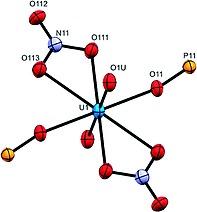
Coordination sphere of 1f showing hexagonal bipyramidal coordination of the central uranium atom. Ellipsoids are draw at 30 % probability level.
The bond lengths of uranium to oxygen atoms of nitrate unit are 2.525(4) and 2.509(4) Å. The bond length between the uranium and the phosphonate‐based oxygen atom is 2.365(3) Å which, in agreement with other examples, is shorter than the corresponding contact to the nitrate. The distance between two uranium atoms in the polymer backbone is 14.152 Å, which is considerably shorter than the intermetallic distances of polymers 1a–1e. Given the unique luminescence behavior of some of the elements involved, we explored the photophysical properties of ligand L and the europium and uranyl coordination polymers 1d, f in the solid state. The ligand itself shows an emission quantum yield of 29 % at the excitation wavelength of 366 nm with an emission maximum at 417 nm (Figure 9).
Figure 9.
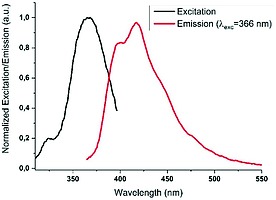
Normalized excitation and emission of solid ligand L.
The excitation spectrum of polymer 1d shows a hypsochromic shift of the ligand based excitation to 332 nm. The europium based excitation remains unchanged compared to Eu(NO3)3 that was used as a starting material. The emission spectra obtained from ligand centered excitation at 332 nm can be assigned to all seven literature known europium emissions suggesting an antenna like effect energy transfer from ligand to metal, with quantum yield of 32 % (Figure 10). The photophysical properties of the europium coordination polymer revealed a similar antenna effect as in related coordination polymers quantum yields have been observed.9 The uranium coordination polymer 1f shows characteristic uranium‐centered emissions (spectra provided in Supporting Information). Ligand‐centered excitation at 283 nm results in both uranyl‐ and ligand‐centered emission suggesting a very poor antenna effect in this case. On the other hand metal‐centered excitation at 415 nm results exclusively in uranyl‐centered emission and no radiative energy transfer to the ligand is observed. Independent of the excitation wavelength (415 nm or others), the uranium‐centered emissions show very low quantum yields (<1 %) suggesting higher vibrational relaxation in case of phosphonate ester coordination.16, 17
Figure 10.
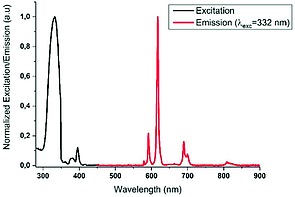
Normalized excitation and emission of solid 1d.
Conclusions
To conclude, we have explored the ligand properties of (C6H4PO(CH3)2)2 towards selected lanthanides, calcium, and uranium for the first time. In case of the lanthanide coordination polymers the reduction of ionic size of the central metal atom resulted in lower‐dimensional structural motifs of the polymer. In case of lanthanum and cerium a two‐dimensional interpenetrated sheet polymer was obtained. On the other hand samarium and europium form a one‐dimensional polymer with macrocyclic units containing laterally assembled π‐systems within the polymeric chain. Calcium and uranium showed similar affinity for phosphonate ester coordination as the lanthanides mentioned before forming 1D coordination polymers as well. In the future we would like to take advantage of the structural variety of phosphonates to further explore the ligand behavior towards f‐block metals in analogy to the rich coordination chemistry of the isolobal silanetriols.18, 19, 20, 21
Experimental Section
All chemical were purchased from Sigma‐Aldrich or TCI and used without further purification. NMR measurements were done on Varian 400 MHz and 500 MHz spectrometers. Infrared (IR) spectra were recorded with a Bruker ATR IR spectrometer with diamond probe. The microwave assisted synthesis was performed in a CEM DISCOVER‐SP reactor in pyrex vessels closed with Activent‐lids. Excitation und emission spectra as well as luminescent quantum yields (absolute method) measurements were carried out with a Hamamatsu C11347 system. For the refinement of the data ORIGIN 2017 was used. Combustion analyses were performed at the Institut für Chemie, Universität Kassel on a HEKAtech Euro EA elemental analyzer. Samples were prepared in a Sn cup and analyzed with added V2O5 to ensure complete combustion. X‐ray diffraction was performed with a STOE IPDS 2 with image plate (diameter 34 cm) using Mo‐GENIX source (λ = 0.71073 nm) and a STOE StadiVari with DECTRIS PILATUS 200K using Mo‐GENIX source (λ = 0.71073 nm). Structures were solved using the dual space method (SHELXT22) and are refined against F 2 with SHELXL‐2014.23 All non‐hydrogen atoms were refined anisotropically. Hydrogen atoms were placed on adjacent atoms using a riding model. Further programs used in structure analysis are the WinGX,24 Mercury,25 and Platon.26 Details of the structure determinations and refinement for L and 1a–1f are summarized in Table 1.
Table 1.
Crystallographic data and structure refinement for L and 1a–1f
| L | 1a | 1b | 1c | 1d | 1e | 1f | |
|---|---|---|---|---|---|---|---|
| Empirical formula | C18H24O6P2 | C36H48LaN3O21P4 | C36H48CeN3O21P4 | C27H36N3O18P3Sm | C27H36EuN3O18P3 | C36H48CaN2O18P4 | C18H24N2O14P2U |
| Formula weight | 398.31 | 1121.56 | 1122.77 | 933.85 | 935.46 | 960.72 | 792.36 |
| Crystal description | colorless plate | colorless plate | colorless block | colorless plate | colorless plate | colorless block | colorless block |
| Crystal size /mm | 0.220 × 0.160 × 0.040 | 0.160 × 0.060 × 0.020 | 0.230 × 0.127 × 0.070 | 0.140 × 0.060 × 0.020 | 0.110 × 0.060 × 0.010 | 0.250 × 0.200 × 0.200 | 0.160 × 0.120 × 0.060 |
| Crystal system | triclinic | monoclinic | monoclinic | triclinic | triclinic | tetragonal | triclinic |
| Space group | P 1 | P21 | P21 | P 1 | P 1 | I 42d | P 1 |
| Radiation and λ /Å | Mo‐K α 0.71073 | Mo‐K α 0.71073 | Mo‐K α 0.71073 | Mo‐K α 0.71073 | Mo‐K α 0.71073 | Mo‐K α 0.71073 | Mo‐K α 0.71073 |
| Monochromator | Graded multilayer mirror | plane graphite | plane graphite | plane graphite | plane graphite | plane graphite | plane graphite |
| Temperature /K | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) |
| Unit cell dimensions | |||||||
| a /Å | 6.6655(6) | 9.5823(9) | 9.6779(7) | 8.7345(5) | 8.6833(7) | 12.8952(5) | 6.6921(6) |
| b /Å | 8.1466(6) | 23.451(2) | 23.5561(9) | 10.0870(6) | 21.923(2) | 12.8952(5) | 8.3224(8) |
| c /Å | 9.4308(8) | 9.9136(9) | 10.0275(6) | 21.8040(14) | 10.0974(9) | 25.7346(11) | 11.9449(10) |
| α /° | 74.575(6) | 90 | 90 | 89.380(5) | 89.783(8) | 90 | 81.331(7) |
| β /° | 74.846(7) | 91.913(7) | 92.229(6) | 89.143(5) | 114.055(6) | 90 | 85.401(7) |
| γ /° | 74.344(6) | 90 | 90 | 66.245(4) | 90.593(7) | 90 | 85.992(8) |
| Volume /ų | 465.32(7) | 2226.5(4) | 2284.3(2) | 1758.06(19) | 1755.2(3) | 4279.3(4) | 654.42(10) |
| Z | 1 | 2 | 2 | 2 | 2 | 4 | 1 |
| Calculated density | 1.421 | 1.673 | 1.632 | 1.764 | 1.770 | 1.491 | 2.011 |
| F(000) | 210 | 1140 | 1142 | 940 | 942 | 2008 | 380 |
| Linear absorption coefficient μ /mm–1 | 0.266 | 1.188 | 1.219 | 1.889 | 2.006 | 0.374 | 6.393 |
| Absorption correction | integration | integration | integration | integration | integration | integration | integration |
| Unit cell determination | STOE X‐area | STOE X‐area | STOE X‐area | STOE X‐area | STOE X‐area | STOE X‐area | STOE X‐area |
| Diffractometer | StadiVari | STOE IPDS 2 | STOE IPDS 2 | STOE IPDS 2 | STOE IPDS 2 | STOE IPDS 2 | STOE IPDS 2 |
| Radiation source | Mo Genix | Genix Mo HF′ | Genix Mo HF | Genix Mo HF | Genix Mo HF | Genix Mo HF | Genix Mo HF |
| Scan type | Omega scan | Omega scan | Omega scan | Omega scan | Omega scan | Omega scan | Omega scan |
| Θ range for data collection | 2.29–31.67 | 1.74–26.44 | 1.73–26.28 | 1.87–26.25 | 1.86–25.14 | 1.59–26.19 | 1.73–26.28 |
| Index ranges | –9 ≤ h ≤ 9 | –11 ≤ h ≤ 11 | –11 ≤ h ≤ 11 | –10 ≤ h ≤ 10 | –10 ≤ h ≤ 10 | –14 ≤ h ≤ 4 | –6 ≤ h ≤ 8 |
| –11 ≤ k ≤ 11 | –28 ≤ k ≤ 28 | –28 ≤ k ≤ 28 | –10 ≤ k ≤ 12 | –26 ≤ k ≤ 26 | –15 ≤ k ≤ 15 | –10 ≤ k ≤ 10 | |
| –13 ≤ l ≤ 13 | –12 ≤ l ≤ 12 | –12 ≤ l ≤ 12 | –26 ≤ l ≤ 26 | –12 ≤ l ≤ 12 | –31 ≤ l ≤ 27 | –14 ≤ l ≤ 14 | |
| Refl. collected / unique | 6402 / 2617 | 15655 / 8357 | 16684 / 8577 | 13146 / 6603 | 13153 / 6614 | 5414 / 1934 | 4705 / 2448 |
| Significant unique refl. | 2056 | 7103 | 7095 | 5530 | 3844 | 1782 | 2430 |
| R(int), R(sigma) | 0.0370, 0.0470 | 0.0941, 0.0703 | 0.0477, 0.0505 | 0.0339, 0.0409 | 0.1150, 0.1390 | 0.0495, 0.0339 | 0.0307, 0.0277 |
| Completeness to Θ = 26.0° | 0.839 | 0.968 | 0.995 | 0.990 | 0.984 | 0.996 | 0.992 |
| Refinement method | SHELXL‐2014/7 | SHELXL‐2014/7 | SHELXL‐2014/7 | SHELXL‐2014/7 | SHELXL‐2014/7 | SHELXL‐2014/7 | SHELXL‐2014/7 |
| Data/parameters/restraints | 2617 / 120 / 0 | 8357 / 516 / 1 | 8577 / 516 / 1 | 6603 / 533 / 0 | 6614 / 494 / 0 | 1934 / 111 / 0 | 2448 / 171 / 0 |
| Goodness‐of‐fit on F 2 | 1.092 | 1.097 | 1.060 | 1.064 | 1.024 | 1.078 | 1.145 |
| Final R indices [I > 2σ(I)] | 0.0453 | 0.0962 | 0.0610 | 0.0369 | 0.0878 | 0.1111 | 0.0259 |
| R indices (all data) | 0.0633 | 0.1138 | 0.0744 | 0.0480 | 0.1470 | 0.1160 | 0.0267 |
| Largest difference peak/hole /e·Å–3 | 0.763 / –0.330 | 2.581 / –2.899 | 1.590 / –1.188 | 1.512 / –1.495 | 1.369 / –2.378 | 1.282 / –0.827 | 0.922 / –1.253 |
Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre, CCDC, 12 Union Road, Cambridge CB21EZ, UK. Copies of the data can be obtained free of charge on quoting the depository numbers CCDC‐1587319, CCDC‐1587320, CCDC‐1587321, CCDC‐1587322, CCDC‐1587323, CCDC‐1587324, and CCDC‐1587325 (Fax: +44‐1223‐336‐033; E‐Mail: deposit@ccdc.cam.ac.uk, http://www.ccdc.cam.ac.uk).
Synthesis of 4,4′ Bis(dimethylphosphorylmethyl)biphenyl (L): In a microwave flask 4,4'‐ bis(chloromethyl)biphenyl (1.5 g, 6 mmol) was placed together with trimethyl phosphite (10 mL, 84 mmol). The reaction mixture was heated in a microwave reactor at 160 °C (200 W) under autogenous pressure for 0.5 h. After cooling to room temperature the flask was left open for 5 min and was placed back in the microwave reactor for 1 h under same conditions. The resulting solution was concentrated under reduced pressure, which resulted in precipitation of a white solid. The resulting solid was filtered and washed with cold n‐pentane and dried under reduced pressure. The product was recrystallized from acetone giving 2.3 g of crystalline product affording 95 % yield based on bis(chloromethyl)biphenyl. 1H NMR (400 MHz, CD3OD): δ = 7.54 (d, J = 8.0 Hz, 4 H), 7.35 (m, 4 H), 3.68 (d, J = 10.9 Hz, 12 H), 3.27 (d, J = 21.8 Hz, 4 H). 31P NMR [1H] (202 MHz, CD3OD): δ = 28.9 (m br) ppm. APCI‐MS: m/z = 399.07 [M + H]+. IR (ATR): ν̃ = 1178 cm–1 (m, P–O), 1246 cm–1 (st, P=O).
Synthesis of Lanthanide(III) Coordination Polymers 1a–1d: The ligand (100 mg, 0.25 mmol) was dissolved in 5 mL of MeOH. To this solution of the ligand a solution of Ln(NO3)3 ·H2O (0.25 mmol) in the same solvent was added dropwise under heavy stirring at 20 °C. Slow evaporation of the solvent yields colorless crystalline material. (Yield: 1a: 40 mg, 29 %; 1b: 80 mg, 58 %; 1c: 50 mg, 32 %; 1d: 50 mg, 32 %).
Synthesis of Calcium Coordination Polymer 1e: The ligand (100 mg, 0.25 mmol) was dissolved in 5 mL of MeOH. To this solution of the ligand a solution of CaCl2 (0.25 mmol) in the same solvent was added under heavy stirring at 20 °C. To the mixture 1 mL of HNO3 conc. was added. Slow evaporation of the solvent yields colorless crystalline material. (Yield: 2 mg, 2 %)
Synthesis of Uranyl Coordination Polymer 1f: The ligand (100 mg, 0.25 mmol) was dissolved in 5 mL of MeOH. To this solution of the ligand a solution of UO2(NO3)2 ·H2O (0.25 mmol) in the same solvent was added under heavy stirring at 20 °C. Slow evaporation of the solvent yields colorless crystalline material.
Analysis of 1a: IR (ATR): ν̃ = 1172 cm–1 (m, P–O), 1206 cm–1 (st, P=O). Elemental analysis calcd. C 38.55, H 4.31, N 3.75 %; found: C 38.477, H 4.030, N 3.915 %
Analysis of 1b: IR (ATR): ν̃ = 1172 cm–1 (m, P–O), 1206 cm–1 (st, P=O). Elemental analysis calcd. C 38.51, H 4.31, N 3.74 %; found: C 38.557, H 4.339, N 3.774 %
Analysis of 1c: IR (ATR): ν̃ = 1164 cm–1 (m, P–O), 1201 cm–1 (st, P=O). Elemental analysis calcd. C 34.73, H 3.89, N 4.50 %; found: C 34.693, H 3.945, N 4.864 %
Analysis of 1d: IR (ATR): ν̃ = 1164 cm–1 (m, P–O), 1201 cm–1 (st, P=O). Elemental analysis calcd. C 34.67, H 3.88, N 4.49 %; found: C 35.360, H 3.903, N 4.561 %
Supporting Information (see footnote on the first page of this article): NMR spectra of ligand L and absorption and emission spectra of 1f in the UV‐vis range.
Supporting information
Supporting information for this article is available on the WWW under https://doi.org/10.1002/zaac.201700424 or from the author.
Supporting Information
Acknowledgements
The authors would like to acknowledge Dr. Clemens Bruhn and Astrid Pilz for X‐ray measurements, Dr. Martin Maurer for NMR measurements and the Isotope Lab at University of Kassel for use of their facilities. We would also like to thank the EU ITN SHINE for financial support and the EU‐COST network CM1302 “Smart Inorganic Polymers” (SIPs).
Dedicated to Professor Dietrich Gudat on the Occasion of his 60th Birthday
References
- 1. James S. L., Chem. Soc. Rev., 2003, 32, 276.14518181 [Google Scholar]
- 2. Kitagawa S., Kitaura R. and Noro S., Angew. Chem. Int. Ed., 2004, 43, 2334–2375. [DOI] [PubMed] [Google Scholar]
- 3. Zhang J.‐W., Zhao C.‐C., Zhao Y.‐P., Xu H.‐Q., Du Z.‐Y. and Jiang H.‐L., CrystEngComm, 2014, 16, 6635. [Google Scholar]
- 4. Guillou O., Daiguebonne C., Calvez G. and Bernot K., Acc. Chem. Res., 2016, 49, 844–856. [DOI] [PubMed] [Google Scholar]
- 5. Murugesu M. and Schelter E. J., Inorg. Chem., 2016, 55, 9951–9953. [DOI] [PubMed] [Google Scholar]
- 6. Daiguebonne C., Kerbellec N., Guillou O., Bünzli J.‐C., Gumy F., Catala L., Mallah T., Audebrand N., Gérault Y., Bernot K. and Calvez G., Inorg. Chem., 2008, 47, 3700–3708. [DOI] [PubMed] [Google Scholar]
- 7. Gupta S. K., Langley S. K., Sharma K., Murray K. S. and Murugavel R., Inorg. Chem., 2017, 56, 3946–3960. [DOI] [PubMed] [Google Scholar]
- 8. Zhang L., Dou W., Liu W., Xu C., Jiang H., Chen C., Guo L., Tang X. and Liu W., Inorg. Chem. Commun., 2015, 59, 53–56. [Google Scholar]
- 9. Krekic K., Klintuch D. and Pietschnig R., Chem. Commun., 2017, 53, 11076–11079. [DOI] [PubMed] [Google Scholar]
- 10. Guo P. C., Xuan X. T., Wu S. and Zhang J., Yingyong Huagong, 2010, 35, 2. [Google Scholar]
- 11. Zhang X.‐D., Ge C.‐H., Zhang X.‐Y., Shi C.‐Y., He C. and Yin J., Inorg. Chem. Commun., 2008, 11, 1224–1226. [Google Scholar]
- 12. Zhang X., Ge C., Yin J., Zhao Y. and He C., Chin. J. Chem., 2009, 27, 1195–1198. [Google Scholar]
- 13. Stock N., Guillou N., Senker J., Férey G. and Bein T., Z. Anorg. Allg. Chem., 2005, 631, 575–581. [Google Scholar]
- 14. Nohra B., Graule S., Lescop C. and Réau R., J. Am. Chem. Soc., 2006, 128, 3520–3521. [DOI] [PubMed] [Google Scholar]
- 15. Nohra B., Yao Y., Lescop C. and Réau R., Angew. Chem. Int. Ed., 2007, 46, 8242–8245. [DOI] [PubMed] [Google Scholar]
- 16. Hnatejko Z., Lis S. and Stryła Z., J. Therm. Anal. Calorim., 2010, 100, 253–260. [Google Scholar]
- 17. Liu C., Yang W., Qu N., Li L.‐J., Pan Q.‐J. and Sun Z.‐M., Inorg. Chem., 2017, 56, 1669–1678. [DOI] [PubMed] [Google Scholar]
- 18. Lorenz V., Fischer A., Bruser W., Edelmann F. T., Jacob K., Gelbrich T. and Jones P. G., Chem. Commun., 1998, 2217–2218. [Google Scholar]
- 19. Lorenz V., Fischer A., Gießmann S., Gilje J. W., Gun′ko Y., Jacob K. and Edelmann F. T., Coord. Chem. Rev., 2000, 206–207, 321–368. [Google Scholar]
- 20. Spirk S., Belaj F., Baumgartner J. and Pietschnig R., Z. Anorg. Allg. Chem., 2009, 635, 1048–1053. [Google Scholar]
- 21. Pietschnig R. and Spirk S., Coord. Chem. Rev., 2016, 323, 87–106. [Google Scholar]
- 22. Sheldrick G., Acta Crystallogr., Sect. A, 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sheldrick G., Acta Crystallogr., Sect. C, 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Farrugia L. J., J. Appl. Crystallogr., 2012, 45, 849–854. [Google Scholar]
- 25. Macrae C. F., Edgington P. R., McCabe P., Pidcock E., Shields G. P., Taylor R., Towler M. and van de Streek J., J. Appl. Crystallogr., 2006, 39, 453–457. [Google Scholar]
- 26. Spek A. L., Acta Crystallogr., Sect. D, 2009, 65, 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information for this article is available on the WWW under https://doi.org/10.1002/zaac.201700424 or from the author.
Supporting Information