


Abstract
Cyclic dinucleotides are second messenger molecules produced by both prokaryotes and eukaryotes in response to external stimuli. In bacteria, these molecules bind to RNA riboswitches and several protein receptors ultimately leading to phenotypic changes such as biofilm formation, ion transport and secretion of virulence factors. Some cyclic dinucleotide analogs bind differentially to biological receptors and can therefore be used to better understand cyclic dinucleotide mechanisms in vitro and in vivo. However, production of some of these analogs involves lengthy, multistep syntheses. Here, we describe a new, simple method for enzymatic synthesis of several 3′, 5′ linked cyclic dinucleotide analogs of c-di-GMP, c-di-AMP and c-AMP-GMP using the cyclic-AMP-GMP synthetase, DncV. The enzymatic reaction efficiently produced most cyclic dinucleotide analogs, such as 2′-amino sugar substitutions and phosphorothioate backbone modifications, for all three types of cyclic dinucleotides without the use of protecting groups or organic solvents. We used these novel analogs to explore differences in phosphate backbone and 2′-hydroxyl recognition between GEMM-I and GEMM-Ib riboswitches.
INTRODUCTION
Cyclic dinucleotides are second messengers found ubiquitously in prokaryotes and as part of the innate immune system in eukaryotes (1). Three cyclic dinucleotides have been identified in bacteria (Figure 1A)–cyclic-di-GMP (c-di-GMP), cyclic-di-AMP (c-di-AMP), and 3′,5′-cyclic-AMP-GMP (c-AMP-GMP)—and one cyclic dinucleotide in higher order eukaryotes—2′,5′-cyclic-AMP-GMP (2′,5′-cGAMP) (2,3). In bacteria, the levels of each of these cyclic dinucleotides fluctuate in response to external stimuli and result in lifestyle changes essential for bacterial survival and host infection. In general, differing levels of c-di-GMP are associated with biofilm formation and virulence factor production (4–8). c-di-AMP regulates potassium homeostasis, osmolarity, DNA integrity and biofilm formation (9–13). c-AMP-GMP levels are associated with changes in chemotaxis in several species of bacteria and exoelectrogenesis in Geobacter (14,15).
Figure 1.

Mechanism of DncV catalysis. (A) Chemical structure of three known natural cyclic dinucleotides. The adenine base is indicated by green and the guanine base is shaded purple. (B) Cartoon representation of base recognition and chemical mechanism of c-AMP-GMP 3′-5′-phosphodiester bond based on previous crystal structures (23,24,26). Residue, D193, of the catalytic triad is highlighted in red. Hydrogen bonds are indicated by dashed lines. Interactions between the triphosphate backbone of the ligand and divalent ion are represented by dashed, orange lines.
Homeostasis of each of these molecules is maintained by distinct proteins in prokaryotic cells. c-di-GMP, the most well-studied cyclic dinucleotide, is synthesized by diguanylate cyclases containing GGDEF domains (16) and it is hydrolyzed to either pGpG or GMP by phosphodiesterases containing HD-GYP or EAL domains (6,17). c-di-AMP is produced by diadenylate cyclases that specifically cyclize adenosine triphosphate (18). These molecules are broken down by phosphodiesterases containing DHH/DHHA or HD domains (19). Finally, c-AMP-GMP is synthesized by Hypr GGDEF proteins found in Geobacter (20) or DncV proteins (14) and broken down by phosphodiesterases containing HD-GYP-domains (21).
While diguanylate cyclases typically dimerize to form the necessary active site for nucleotide triphosphate cyclization (22), DncV contains a single active site (23–26). In vivo, this unique active site is thought to preferentially recognize the adenine of adenosine triphosphate (ATP) in an anti conformation in the ‘acceptor’ pocket, while the guanine of guanosine triphosphate (GTP) is recognized in a syn conformation in a ‘donor’ binding pocket (Figure 1B) (23). The resulting dinucleotide triphosphate is then released and rebound in the opposite orientation with guanosine bound at the acceptor pocket and adenosine bound at the donor pocket. As a consequence of this mechanism, both GTP and ATP are recognized by either pocket. For this reason, the enzyme can also cyclize two ATPs or two GTPs to form c-di-AMP or c-di-GMP, respectively (14,26).
Several groups have identified riboswitch and protein receptors that recognize cyclic dinucleotides in bacteria. For example, there are two different riboswitch classes that recognize c-di-GMP by different mechanisms, the GEMM-I and GEMM-II riboswitches (27–31). The GEMM-Ib riboswitches, which are structurally similar to the GEMM-I riboswitch, exclusively recognize c-AMP-GMP (15,32,33). Finally, a structurally distinct riboswitch, the YdaO riboswitch, recognizes c-di-AMP in two different binding sites in a single aptamer (34,35).
In order to understand the molecular and phenotypic mechanisms of the cyclic dinucleotide pathway, several groups have synthesized analogs of these second messengers (36–42). In addition to providing information about specific molecular interactions, some studies have found that certain analogs of cyclic dinucleotides bind differentially to RNA and protein receptors (36,38). It may be possible, therefore, to selectively target specific cyclic dinucleotide receptors by making such modifications. Some analogs, such as those containing a phosphorothioate backbone, bind to phosphodiesterases but resist cleavage (37). Therefore, cyclic dinucleotide analogs also show promise as tools to study cyclic dinucleotide pathways in vivo. Development of a one-flask synthesis method for producing phosphorothioate modifications has been used to streamline synthesis of many cyclic dinucleotide analogs (39,40). However, this process cannot produce all phosphorothioate stereoisomers and 2′-hydroxyl modifications (40).
Previous biochemical studies provided valuable insight into molecular interactions between the phosphate backbone and 2′-hydroxyl of c-di-GMP and its cognate riboswitches, GEMM-I and GEMM-II (36,43). However, c-di-GMP is a symmetrical molecule and can flip its orientation in the binding pocket. Therefore, some of the loss in affinity due to modification in these areas could not be definitively attributed to specific hydrogen bond disruptions. Here, we took advantage of the promiscuity of DncV, and further optimized its catalysis using different divalent ions, to create an expanded library of cyclic dinucleotide analogs modified at the sugar, base and phosphate backbone. We scaled up production of several c-AMP-GMP and c-di-GMP analogs and used these compounds to biochemically probe phosphate backbone and sugar recognition of these ligands with c-AMP-GMP and c-di-GMP riboswitches.
MATERIALS AND METHODS
Purification of DncV
Vibrio cholerae DncV was expressed in BL21 cells and purified as previously described (14,24). In brief, cells expressing pET-DUET1-DncV were grown to an OD of 0.6 at 37°C and then induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 18°C for 4 h. The cells were harvested and resuspended in 50 mM KH2PO4, pH 7.0, 300 mM NaCl and 5 mM β-mercaptoethanol (BME). Resuspended cells were lysed using a sonicating probe and clarified by centrifugation. The supernatant was incubated with Nickel-agarose and the protein was eluted with 20, 50 and 250 mM imidazole. The protein was dialyzed into 50 mM Tris–HCl, 100 mM NaCl, 10% glycerol and 5 mM β-mercaptoethanol and further purified by size exclusion chromatography (SEC) on a Superdex 75 column.
Synthesis of cyclic dinucleotides
Nucleotide triphosphate analogs were purchased from Trilink Biotechnologies, Biolog, Sigma-Aldrich or New England Biolabs. A total of 10 μM DncV was incubated with a total of 2 mM nucleotide triphosphate in 25 mM Tris–HCl, pH 7.5, 100 mM NaCl and 10 mM divalent ion. For divalent metal screening, 10 mM MgCl2 was replaced by 10 mM CaCl2, CdCl2, CoCl2, CuCl2, MnCl2 or ZnCl2. Reactions were incubated as specified at 37°C and heated to 70°C for 10 min to quench the reaction. Aliquots were separated by High Performance Liquid Chromatography (HPLC) using an Atlantis T3 3 μm, 4.6 × 150 mm column equilibrated in a mobile phase containing 10 mM ammonium acetate, pH 5.0 and acetonitrile as indicated in Supplementary Table S1. The analogs were separated using a gradient of up to 50% acetonitrile with a flow rate of 1.0 ml/min. Peak area percent was calculated using Agilent Chemstation based on absorbance signal at 254 nm. A limit of detection was set based on a signal-to-noise ratio of 10 for c-AMP-GMP at 254 nm and limit of quantitation was set based on a signal-to-noise ratio of 3. Major peaks were identified using electrospray ionization mass spectrometry in a mobile phase of formic acid and acetonitrile.
Scale-up of cyclic dinucleotide analog synthesis
Analog synthesis used for binding studies were scaled up in 200–500 μl reactions. Cyclic dinucleotides were purified on an Atlantis T3 3 μm, 9.6 × 300 mm column. Purified peaks were identified by electrospray mass spectrometry and their purity confirmed to be >95% using the analytical HPLC method described above. c-AMP-GMP, c-di-GMP and c-di-AMP analog concentrations were calculated based on extinction coefficients of 25 050 M−1cm−1 at 256 nm, 23 700 M−1cm−1 at 254 nm and 27 000 M−1cm−1 at 259 nm, respectively.
Synthesis of RNA
The sequence for GEMM-Ib riboswitch aptamer, Gs 1761, from Geobacter sulfurreducins flanked by a T7 promoter at the 5′ end and the restriction enzyme recognition sequence for BsaI on the 3′ end was cloned into pUC19. The RNA was synthesized in vitro by T7 polymerase from an EcoRV linearized plasmid and purified by denaturing gel electrophoresis followed by crush-and-soak. The GEMM-I riboswitch, VC2 GUAA, and the GEMM-II riboswitch aptamers were synthesized as previously described (30,44).
Gel shift assays
Radiolabeled c-AMP-GMP was prepared by incubating 17.5 μM GTP and α-32P-ATP with 10 μM DncV in 25 mM Tris–HCl, pH 7.5, 100 mM NaCl and 10 mM MnCl2 for 1 h. Radiolabeled c-di-GMP was prepared by incubating α-32P-GTP with the uninhibited mutant of a diguanylate cyclase from Thermatoga maratime, tDGC-R158A (45), for 1 h. Radiolabeled c-AMP-GMP was separated from c-di-AMP and c-di-GMP on a 20% native polyacrylamide gelelectrophoresis (PAGE) gel (100 mM Tris/HEPES, pH 7.5, 0.2 mM EDTA) at 4°C and extracted by soaking the gel slice in MilliQ water.
To measure direct binding of c-AMP-GMP to the Gs 1761 aptamer, native gel shifts were used as previously described (27). Briefly, increasing concentrations of RNA were incubated with trace radiolabeled c-AMP-GMP. Bound and unbound ligand were separated on a 12% native PAGE gel (100 mM Tris/HEPES, pH 7.5, 10 mM MgCl2, 0.2 mM ethylenediaminetetraacetic acid (EDTA)) at 4°C. Data were analyzed in ImageQuant and fit to a single site-specific binding model in GraphPad Prism.
A competition gel shift assay previously used for c-di-GMP and c-di-AMP riboswitches (36,46) was modified to measure the binding affinities of c-AMP-GMP and c-di-GMP analogs. A total of 20 nM Gs 1761 was folded in the presence of trace radiolabeled c-AMP-GMP and increasing concentrations of cold c-AMP-GMP analog. The binding reactions were incubated until they had reached equilibrium (at least 19 h) and the bound and unbound cyclic dinucleotide was separated on a native PAGE gel at 4°C. Data were analyzed as described above and the Kd of each analog was determined using the following equation:
 |
where 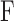 is the fraction bound,
is the fraction bound, 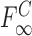 is the fraction bound at saturating concentrations of cold competitor,
is the fraction bound at saturating concentrations of cold competitor, 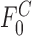 is the fraction bound in the absence of cold competitor,
is the fraction bound in the absence of cold competitor, 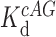 is the equilibrium dissociation constant of c-AMP-GMP bound to Gs 1761,
is the equilibrium dissociation constant of c-AMP-GMP bound to Gs 1761, 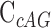 is the concentration of radiolabeled c-AMP-GMP (estimated to be 0.025 nM as previously calculated (36)),
is the concentration of radiolabeled c-AMP-GMP (estimated to be 0.025 nM as previously calculated (36)), 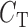 is the concentration of cold competitor,
is the concentration of cold competitor, 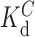 is the equilibrium dissociation constant of the competitor and
is the equilibrium dissociation constant of the competitor and 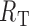 is the total concentration of Gs1761 RNA (20 nM). Competition assays for c-di-GMP analogs with GEMM-I and GEMM-II aptamers were performed with radiolabeled c-di-GMP as previously described (36).
is the total concentration of Gs1761 RNA (20 nM). Competition assays for c-di-GMP analogs with GEMM-I and GEMM-II aptamers were performed with radiolabeled c-di-GMP as previously described (36).
RESULTS AND DISCUSSION
DncV synthesizes several cyclic dinucleotides in the presence of magnesium
DncV is a promiscuous dinucleotide cyclase from Vibrio cholerae that preferentially synthesizes c-AMP-GMP in vivo (14). However, the enzyme can also make c-di-AMP and c-di-GMP in vitro when provided only ATP or GTP, respectively (14,26). In its preferred conformation, DncV is thought to recognize adenosine at the acceptor site by hydrogen bonding between S259 and the N6 of ATP as depicted in Figure 1 (23). The enzyme recognizes guanosine by hydrogen bonding to N1 and N2 of GTP via D348. DncV then catalyzes a 3′,5′-phosphodiester bond between the two nucleotide triphosphates via the catalytic triad D131, D133 and D193 (23,24). The resulting dinucleotide triphosphate is released and rebound in the opposite orientation with guanosine recognized by S259 and Y179 in the acceptor pocket and adenosine binding to the donor pocket (24). The second 3′,5′-linkage is then formed in order to complete the cyclic dinucleotide (23,24).
Because the acceptor and donor pockets are promiscuous for both ATP and GTP, we sought to determine if DncV could accept nucleotide triphosphates with base analogs. We first tested substrates with conservative base modifications, such as inosine triphosphate and 6-thio-guanosine triphosphate (Figure 2A). DncV synthesized cyclic dinucleotides with these modifications using magnesium as a divalent metal (Figure 2C). Because 6-thio-guanosine is capable of chemical crosslinking with proteins, we tried this reaction in the presence and absence of dithiothreitol (DTT) to reverse this reactivity, but found that the cyclic dinucleotide was formed in both cases (data not shown).
Figure 2.

Base substitutions to cyclic dinucleotides and divalent ion optimization. (A) Base substitutions included on nucleotide triphosphates. (B) Divalent ion screen of DncV using GTP as substrate. (C) Cyclization of base analog nucleotide triphosphates using different divalent ions.
Under these conditions, DncV could not efficiently synthesize cyclic dinucleotides containing more dramatic modifications. For example, we incubated DncV with purine triphosphate, which does not contain any exocyclic functional groups, or Z-nucleotide triphosphate (5-amino 4-imidazole carboxamide riboside 5′-triphosphate), the proposed bacterial alarmone and purine metabolite (47–49) which doesn’t contain a full pyrimidine ring (Figure 2A). In neither case did we observe a significant HPLC peak corresponding to a cyclized product. Finally, to see if DncV could recognize other bases besides purines, we also tried incubating the pyrimidine nucleotide triphosphates, cytidine triphosphate (CTP) and uridine triphosphate (UTP), with DncV to form cyclic-di-CMP and cyclic-di-UMP, but found that they were also not cyclized efficiently (Figure 2C).
Optimization of divalent metals in DncV increases cyclic dinucleotide production
Replacement of magnesium with other divalent ions is known to change the promiscuity and reaction rate for many nucleic acid enzymes (50–53). This is especially well documented in restriction enzymes that are increasingly promiscuous in the presence of manganese rather than magnesium (54). We therefore screened a number divalent metals to determine if any could alter the activity of DncV. We used GTP as a substrate and tested Ca2+, Cd2+, Co2+, Cu2+, Mn2+, Zn2+ for compatibility with DncV. As expected based upon other enzymatic systems, Ca2+ inhibited cyclic-di-GMP formation most likely due to its increased atomic radius (Figure 2B). Zn2+, Cd2+ and Cu2+ also did not readily facilitate cyclic dinucleotide formation. However, the enzyme could cyclize GTP to c-di-GMP in the presence of Mn2+ and Co2+ (Figure 2B).
To determine if additional cyclic dinucleotides could be prepared in the presence of these alternative metals, we repeated the enzymatic synthesis of cyclic-di-PuMP (from purine triphosphate), cyclic-di-UMP (from uridine triphosphate), cyclic-di-CMP (from cytosine triphosphate) and cyclic-di-ZMP (from z-nucleotide triphosphate) in the presence of either cobalt or manganese. All of these cyclic dinucleotide analogs, except cyclic-di-ZMP (Supplementary Figure S1), could be synthesized within a few hours in the presence of cobalt and the percent peak areas for all analogs were substantially increased when the enzymatic reaction was performed in the presence of manganese (Table 1, Figure 2C and Supplementary Figure S1). Because DncV successfully synthesized both purine and pyrimidine cyclic dinucleotide analogs while failing to prepare one with a Z-nucleotide triphosphate, it appears that there simply needs to be a complete pyrimidine ring present for nucleotide recognition in this enzyme.
Table 1. Synthesis of cyclic dinucleotide base analogs by DncV.
| Compound | % Peak areaa | Incubation time (h) | Predicted major fragment m/z | Observed major fragment m/z |
|---|---|---|---|---|
| c-di-GMP | 91 | 1 | 689.1 | 688.8 |
| c-di-AMP | 98 | 1 | 657.1 | 656.9 |
| c-AMP-GMP | 84 | 1 | 673.1 | 672.9 |
| c-di-CMP | 63 | 1 | 609.1 | 609.0 |
| c-di-UMP | 75 | 1 | 611.3 | 610.7 |
| c-di-IMP | 100 | 1 | 659.4 | 658.9 |
| c-di-PuMP | 68 | 2.5 | 627.1 | 626.8 |
| c-di-ZMP | <LOD | N/A | 640.0 | N/A |
| c-di-6TGMP | 100 | 1 | 722.1 | 720.8 |
aAverage of two reactions based on A254 signal in the presence of MnCl2.
LOD: limit of detection (0.1 μM/2 mM substrate).
N/A: not applicable.
We also retested the synthesis of cyclic-di-AMP, cyclic-di-GMP, cyclic-di-IMP and cyclic-di-6TGMP to see if the reaction efficiency could be increased by using a different metal. All cyclic dinucleotide syntheses showed a slight increase in the product peak area when the divalent metal was changed from magnesium to cobalt or manganese, with the exception of cyclic-di-6TGMP (Figure 2C). Interestingly, cobalt inhibited synthesis of cyclic-di-6TGMP while manganese promoted its synthesis.
DncV synthesizes 2′-modified analogs of cyclic dinucleotides
No residues appear to directly interact with the 2′-hydroxyl of nucleotide triphosphates as observed within the crystal structures of DncV with its substrates (23–26) (Figure 1). We tested if DncV could synthesize cyclic dinucleotides modified at this position in the presence of MnCl2 using nucleotide triphosphate analogs. DncV reactivity was tested with ATP or GTP analogs that had either a fluorine (2′F), a hydrogen (deoxy, abbreviated with the suffix ‘d’), a methoxy (2′OMe) or an amino group (2′NH2) (Table 2). c-AMP-GMP analogs were prepared by adding 1:1 ratios of the ATP and GTP analogs. c-di-GMP and c-di-AMP analogs were prepared using only the ATP or GTP substrates, respectively.
Table 2. Synthesis of 2′-hydroxyl analogs of cyclic dinucleotides by DncV.

|
Although the DncV does not appear to make direct 2′-OH contacts, the synthesis of 2′-substituted analogs was only possible in the presence of manganese. In the presence of this divalent metal, the enzyme synthesized 2′-fluoro and 2′-amino analogs of all three cyclic dinucleotides (Table 2 and Supplementary Figure S2). However, while nearly all of the 2′-fluoro nucleotide triphosphates were converted to cyclic dinucleotide analogs (c-di-2′F-GMP, c-di-2′F-AMP and c-2′F-AMP-2′F-GMP), much less of the 2′-amino analogs (c-di-2′NH2-GMP, c-di-2′NH2-AMP and c-2′NH2-AMP-2′NH2-GMP) were produced from the respective nucleotide triphosphate analogs (Table 2 and Supplementary Figure S2). DncV also prepared 2′-amino monosubstitutions of cyclic-di-GMP and cyclic-AMP-GMP within an hour when incubated with 1:1 ratios of modified and unmodified nucleotide triphosphates. Although the peak area for these monosubstituted products appears to be lower than many disubstitutions, the enzyme simultaneously synthesized other possible cyclic dinucleotides. For example, DncV converted approximately 90% of substrates into products when incubated with equal amounts of unmodified GTP and modified 2′-NH2-GTP. This included three products: the unsubstituted c-di-GMP cyclic dinucleotide, the disubstituted c-di-NH2-GMP analog and the monosubstituted c-2′NH2-G-GMP (Table 2 and Supplementary Figure S2). The largest peak in the chromatogram from this reaction was c-di-GMP and the smallest peak was c-di-NH2-GMP. This suggests that c-di-GMP is the most favorable product while the disubstituted product is the least favorable.
There are limitations to the diversity of substitutions at the 2′-position that are accepted by DncV. The 2′-O-methyl was the bulkiest modification tested and no cyclic 2′-O-methylated dinucleotide products were observed even after incubation with the enzyme for >20 h. In addition, DncV could not cyclize significant quantities of dGTP or dATP to form c-di-dGMP and c-di-dAMP, respectively (Supplementary Figure S4 and Table 2). In both cases, large peaks formed earlier than would be expected for the 2′-deoxy cyclic dinucleotide analogs and none of them corresponded to m/z expected for these analogs (Supplementary Figure S5). Both early-eluting peaks had major fragments with an m/z >800 suggesting that they are likely to be dinucleotide triphosphate intermediates. This implies the enzyme was able to complete the first, but not the second step of the dimerization reaction. Interestingly, DncV was able to synthesize c-dAMP-dGMP when provided with equal amounts of dATP and dGTP, though the efficiency of this reaction was low. The reaction had to be incubated overnight (>20 h) and even then, only about a quarter of the substrate was converted to product (Table 2 and Supplementary Figure S3).
DncV synthesizes phosphorothioate analogs of all three cyclic dinucleotides
Phosphorothioate backbone-modified cyclic-di-AMP and cyclic-di-GMP are typically prepared using a one-flask synthesis method (40). However, this method only allows synthesis of RpRp and RpSp phosphorothioate cyclic dinucleotides. A more complicated synthetic procedure, requiring additional purification steps, is necessary to prepare the SpSp phosphorothioate (55).
We tested if DncV could convert α-phosphate-modified stereoisomers into the cyclic dinucleotide phosphorothioates. We incubated different α-thio-GTP analogs with DncV and assigned products based upon the chemically-synthesized standards of c-(RpRp)-di-Gps and c-(RpSp)-di-Gps that had been previously reported (36,40). When a 1:1 mixture of Sp-GTP-α-S and Rp-GTP-α-S was incubated with the DncV enzyme in the presence of MnCl2, a single peak matching the retention time and m/z value of c-(RpSp)-di-Gps appeared after 1 h (Table 3 and Supplementary Figure S4). Because we did not see peaks corresponding to other possible stereoisomers, c-(RpRp)-di-Gps and c-(SpSp)-di-Gps, we attempted to prepare these individually. α-Sp-GTP-α-S was incubated with DncV in the presence of either MnCl2 or CoCl2. In the presence of CoCl2, a significant peak matching the retention time of the chemically synthesized c-(RpRp)-di-Gps emerged (Supplementary Figure S4). This sample was evaluated by mass spectrometry and had an m/z of 721, consistent with a diphosphorothioate analog of c-di-GMP. This inversion of stereochemistry is consistent with previous enzymatic synthesis reactions (56,57). Finally, Rp-GTP-α-S was incubated with the enzyme and either CoCl2 or MnCl2 to prepare c-(SpSp)-di-Gps. After 20 h, a small peak emerged at ∼6.5 min in the presence of MnCl2. We identified this peak as c-(SpSp)-di-Gps because the molecule had an m/z value of 721 like the other two c-di-GMP diphosphorothioates and because the retention time was in between that of c-di-GMP and c-(RpSp)-di-Gps. This same order of elution was previously reported for the three c-di-GMP diphosphorothioate stereoisomers (55).
Table 3. Synthesis of phosphorothioate analogs of cyclic dinucleotides by DncV.

|
Taken together, it is not unexpected that the c-(RpSp)-Gps analog is the sole product in a mixture of stereoisomeric starting materials. c-(RpRp)-Gps is not readily formed in the presence of MnCl2 (Supplementary Figure S4). While it was possible to form the c-(SpSp)-di-Gps diphosphorothioate in the presence of this divalent ion, the reaction was not efficient and required overnight incubation. These observations further support the sole production of the c-(RpSp)-Gps analog, rather than either of the two stereoisomers, when a mixture of Sp-GTP-α-S and Rp-GTP-α-S are incubated with the enzyme in the presence of MnCl2.
We performed a similar experiment by incubating α-thio-ATP analogs incubated with DncV in the presence of either MnCl2 or CoCl2 and observed the same patterns for stereochemical inversion as seen for c-di-GMP. We used the previously characterized diphosphorothioate analogs (46) c-(RpRp)-di-Aps and c-(RpSp)-di-Aps as standards. When Sp-ATP-α-S was incubated with DncV in the presence of CoCl2, a peak matching the retention time and m/z of c-(RpRp)-di-Aps was produced. When a mixture of Sp-ATP-α-S and Rp-ATP-α-S was incubated in the presence of MnCl2, a single peak matching that of c-(RpSp)-di-Aps was visible. This is consistent with the efficient formation of the homologous c-(RpSp)-Gps analog described above. When Rp-ATP-α-S was incubated with DncV in the presence of MnCl2, a peak with an m/z value matching a diphosphorothioate analog of c-di-AMP emerged. This peak had a retention time between c-di-AMP and c-(RpSp)-di-Aps which is consistent with the elution order of a c-(SpSp)-di-Aps analog as seen for the c-di-GMP diphosphorothioates (55).
As with c-di-GMP phosphorothioates, the divalent metal identity seems to be especially important for production of RpRp and SpSp phosphorothioates of c-di-GMP and c-di-AMP. The crystal structures of DncV show divalent ion coordination with the α-pro-Rp oxygen of ATP and GTP (23,24) in the donor pocket (Supplementary Figure S5). Replacement of these oxygens with sulfurs most likely disrupts this coordination. Both Co2+ and Mn2+ are thiophilic and should accommodate the phosphorothioate. However, CoCl2 is the preferred divalent ion for production of RpRp diphosphorothioates while MnCl2 is preferred for SpSp diphosphorothioates (Figure 3 and Supplementary Figure S4). Further structural experiments would be necessary to fully understand the preference of MnCl2 or CoCl2 in these reactions.
Figure 3.

Enzymatic synthesis of phosphorothioate c-di-AMP analogs. (A) Chemical structure of c-di-AMP phosphorothioate analogs. Conformation of the thiol groups is shown in red. (B) Chromatogram of chemically synthesized c-di-AMP phosphorothioates. (C) Chromatogram of enzymatically synthesized c-(RpSp)-Aps in the presence of MnCl2 from α-Rp-ATP and α-Sp-ATP. (D) Chromatograms of enzymatically synthesized c-(RpRp)-Aps from α-Sp-ATP in the presence of MnCl2 (blue trace) and CoCl2 (red trace). (E) Chromatograms of enzymatically synthesized c-(SpSp)-Aps from α-Rp-ATP in the presence of MnCl2 (blue trace) and CoCl2 (red trace).
There are four possible stereoisomers of c-AMP-GMP phosphorothioates (Supplementary Figure S6). DncV synthesizes all of these analogs when given appropriate substrates (Table 3). Contrary to the symmetrical cyclic dinucleotides, synthesis of the c-(RpRp)-ApsGps analog did not require CoCl2 as the metal cofactor. We enzymatically synthesized c-(SpRp)-ApsGps and c-(RpSp)-ApsGps and determined that they were different compounds due to the difference in starting material and product peak retention times. We used Rp-ATP-α-S + Sp-GTP-α-S to produce c-(SpRp)-ApsGps and Sp-ATP-α-S + Rp-GTP-α-S to produce c-(RpSp)-ApsGps. These reactions resulted in product peaks eluting at retention times of 6.8 and 7.0 min for c-(SpRp)-ApsGps and c-(RpSp)-ApsGps, respectively. Both analogs had the same observed major fragment by mass spectrometry and had relatively good yield of greater than 70% peak area in 1 h as seen in Table 3. The synthesis of the c-(SpSp)-ApsGps analog was the least efficient of the four possible analogs, taking up to 2.5 h to convert about half of the starting material into product. When we scaled up these reactions starting from 400 μg substrate, we were able to obtain 13, 12, 19 and 5% yield following HPLC purification for c-(RpRp)-ApsGps, c-(RpSp)-ApsGps, c-(SpRp)-ApsGps, c-(SpSp)-ApsGps, respectively. These values are within range of chemical synthesis of c-di-GMP phosphorothioates (40,55).
2′-amino analogs of c-AMP-GMP provide insight into cyclic dinucleotide recognition by riboswitches
Previous studies have reported differential binding of 2′-modified cyclic-di-GMP analogs between the GEMM-I and GEMM-II riboswitches (36) and even among GEMM-I riboswitches from different bacterial species (43). The crystal structure of the GEMM-I riboswitch, VC2, shows a non-canonical Watson–Crick/Hoogsteen interaction between a guanine of c-di-GMP, known as the Gα, and G20 of the riboswitch (27). The other guanine of c-di-GMP, known as Gβ, binds to C92 of the riboswitch through a Watson–Crick base pair. This orientation allows for other points of contact between the riboswitch bases and the 2′-hydroxyl of the ligand (27). In particular, the 2′-hydroxyl of Gα hydrogen bonds to the phosphate backbone of A47 while the 2′-hydroxyl of Gβ is coordinated to a water molecule (Figure 4A). The GEMM-II riboswitch forms two non-canonical base pairs between both the Gα and Gβ of c-di-GMP, but makes fewer contacts with other functional groups of the ligand (30). For this reason, many c-di-GMP analogs show preferential binding to the GEMM-II rather than the GEMM-I riboswitch. However, the effect of an electropositive group, such as an amino, at the 2′ position of cyclic dinucleotides has not previously been tested. We synthesized 2′-amino analogs of c-di-GMP and c-AMP-GMP using DncV and tested the effects of this modification for binding to the two classes of c-di-GMP riboswitches (GEMM-I and GEMM-II) and a c-AMP-GMP (GEMM-Ib) riboswitch.
Figure 4.

2′-hydroxyl and phosphate backbone recognition of c-di-GMP and c-AMP-GMP in GEMM-I and GEMM-Ib riboswitches. (A) Hydrogen bonding to 2′-hydroxyl and phosphate backbone of c-di-GMP in the GEMM-I riboswitch, VC2, from pdb file 3muh (44). The riboswitch is pictured in gray and cyclic-di-GMP is shown in purple. (B) Hydrogen bonding to 2′-hydroxyl and phosphate backbone in GEMM-Ib riboswitch, Gs1761, from pdb file 4yaz (33). The riboswitch is pictured in gray and cyclic-AMP-GMP is shown in cyan.
We measured the binding of the c-di-2′NH2-GMP, which contains two 2′-amino modifications, to the GEMM-I and GEMM-II riboswitch using a competition gel shift assay. As expected, there was a negligible 1.6-fold decrease in affinity to the GEMM-II riboswitch. However, this same modification resulted in a significant 1700-fold loss in affinity to the GEMM-I riboswitch (Table 4). This large effect cannot be explained by disruption of hydrogen bonding or electrostatics. In its deprotonated state, the 2′-amino should maintain the hydrogen bond donor properties of the 2′-hydroxyl. If the 2′-amino is protonated, it should stabilize electrostatic interactions with the negatively charged phosphate backbone of A47 in the GEMM-I riboswitch rather than disrupt it. However, a large binding defect (1800-fold) was also reported for binding of the 2′-deoxy analog, c-di-dGMP, to the GEMM-I riboswitch (36). The reported result for the 2′-deoxy analog was attributed to a change in sugar pucker from the 3′-endo conformation seen in c-di-GMP to a 2′-endo sugar pucker upon replacement of the 2′-hydroxyl with a hydrogen in c-di-dGMP (36). A 2′-amino group is also thought to alter the ribose sugar pucker of nucleotides from the 3′-endo conformation to the 2′-endo (58). Therefore, this conformational change upon introduction of a 2′-amino group is likely responsible for perturbing the binding affinity of the ligand.
Table 4. Binding affinity of 2′ modified cyclic dinucleotides to riboswitches.
| Compound | Riboswitch | K d (nM) | Fold change | ΔΔG (kcal/mol) |
|---|---|---|---|---|
| c-di-GMP | GEMM-I (GUAA VC2) | 2.2 ± 0.8a | N/A | N/A |
| c-2′NH2-G-GMP | GEMM-I (GUAA VC2) | 68 ± 10 | 30 | 2.0 |
| c-di-2′NH2-GMP | GEMM-I (GUAA VC2) | 3800 ± 800 | 1700 | 4.4 |
| c-di-GMP | GEMM-II | 2.2 ± 0.2a,b | N/A | N/A |
| c-di-2′NH2-GMP | GEMM-II | 3.5 ± 0.4 | 1.6 | 0.3 |
| c-AMP-GMP | GEMM-Ib (Gs 1761) | 1.1 ± 0.04a,c | N/A | N/A |
| c-2′NH2-A-GMP | GEMM-Ib (Gs 1761) | 21 ± 2 | 19 | 1.3 |
| c-AMP-2′NH2-G | GEMM-Ib (Gs 1761) | 9.0 ± 1.8 | 8.2 | 0.8 |
| c-2′NH2-AMP-2′NH2-GMP | GEMM-Ib (Gs 1761) | 2500 ± 230 | 2300 | 4.1 |
In order to understand the contributions of a single 2′-amino substitution, we also tested binding of c-2′NH2-G-GMP to the GEMM-I riboswitch. This analog was prepared by scaling up the reaction of a 1:1 ratio of GTP and 2′NH2-GTP with DncV. Although only the 2′-hydroxyl group of Gα makes direct contact with the riboswitch, this monosubstituted analog still resulted in a 30-fold decrease in binding affinity, corresponding to a 2.0 kcal/mol loss and roughly half of the cost of the disubstituted analog, c-di-2′NH2-GMP. The significant binding defect suggests that a single 2′-amino substitution is enough to alter the sugar pucker and likely disrupts the constraints of the entire ligand in the binding pocket of the riboswitch.
Because c-di-GMP is a symmetric molecule and can flip its orientation in the binding pocket, the c-2′-NH2-G-GMP analog is expected to bind in the orientation that has the lowest energetic effect on the binding affinity. To eliminate the ambiguity of this possibility, we also tested analog binding to a GEMM-1b riboswitch, Gs 1761 (Supplementary Figure S7). This is a cyclic dinucleotide riboswitch variant that folds in a similar structure to the GEMM-I riboswitch, but specifically recognizes c-AMP-GMP. This riboswitch has also been crystallized (33) and the Aα of c-AMP-GMP binds to A14 and Gβ forms a Watson–Crick pair with C75 (Supplementary Figure S7). However, the orientation of c-AMP-GMP allows both 2′-hydroxyls to be in direct contact with the riboswitch (Figure 4B). The 2′-hydroxyl of the Aα hydrogen bonds to the phosphate backbone of A41 similar to interactions in the GEMM-I riboswitch. In addition, the Gβ 2′-hydroxyl of c-AMP-GMP forms a hydrogen bond with N7 of A11 in the riboswitch. Because c-AMP-GMP is an asymmetric molecule, it is possible to make specific modifications to the 2′-hydroxyl groups of the α and β sites to individually probe their interactions with the riboswitch.
We measured the binding affinity of native and modified ligands to the Gs 1761 GEMM-Ib riboswitch using direct binding gel shifts and competition gel shifts, respectively. In the direct binding assay, c-AMP-GMP bound to the riboswitch with an affinity of 1.1 nM. This is in agreement with the previously-reported, 530 pM, as measured by inline probing (32). When we measured binding affinity of c-2′NH2-AMP-2′NH2-GMP, which has 2′-amino substitutions at both hydroxyls, we found a 2300-fold weaker binding affinity, corresponding to 4.1 kcal/mol (Table 4). This value is similar to what was observed for c-di-2′NH2-GMP binding to the GEMM-I riboswitch (ΔΔG=4.4 kcal/mol) suggesting that the effect of disubstitution is the same in both riboswitches.
To determine the contribution of 2′-hydroxyl recognition at the two different binding sites, we next tested binding of monosubstituted 2′-amino-modified cyclic-AMP-GMP analogs. The c-2′NH2-A-GMP, which contains a modification of the 2′-hydroxyl at the Aα site, showed a 19-fold change in binding affinity (1.3 kcal/mol), while c-AMP-2′NH2-G, which is modified at the 2′-hydroxyl at the Gβ site, showed only an ∼8-fold loss in binding affinity (0.8 kcal/mol). Even though both 2′-hydroxyls make contact with the riboswitch (Figure 4B), these results indicate that the 2′-hydroxyl at the Aα site is slightly more important for recognition than the 2′-hydroxyl at the Gβ site.
Taken together, these data suggest a difference in ligand recognition of the 2′-hydroxyl not evident from the static crystal structures of the GEMM-I and GEMM-Ib riboswitch. Because the Gβ 2′-hydroxyl of c-di-GMP does not make direct contact with the riboswitch (Figure 4A), it is expected that the single 2′-amino group in the monosubstituted analog is oriented at this site. However, it still resulted in a 2 kcal/mol energetic loss in affinity, which was attributed to a change in the sugar pucker. In contrast, modification to the analogous location in c-AMP-GMP resulted in only a 0.8 kcal/mol change. This suggests that the GEMM-Ib riboswitch is more tolerant of conformational changes at this site.
Phosphorothioate-modified c-AMP-GMP analogs bind to a c-AMP-GMP riboswitch
We next sought to understand the interactions between the phosphate backbone of c-AMP-GMP and the GEMM-Ib riboswitch, Gs1761, using the expanded set of phosphorothioate substituted analogs. The crystal structure of the GEMM-I riboswitch shows hydrated divalent metals and other water molecules coordinating to at least one of the pro-Rp non-bridging oxygens. However, the crystal structure of the GEMM-Ib bound to c-AMP-GMP does not contain an equivalent set of interactions in the binding site (Figure 4).
Previous studies on the GEMM-I riboswitch reported decreased binding affinities for all phosphorothioate analogs compared to c-di-GMP and attributed this result to disruption of electrostatic interactions in the binding pocket (36). However, it is unclear how phosphorothioate modifications are oriented in the binding pocket of the GEMM-I riboswitch due to the symmetry of c-di-GMP. It is possible to map Sp and Rp phosphorothioate modifications on c-AMP-GMP within the GEMM-Ib binding pocket. Given that there unexpected differences between GEMM-I and GEMM-Ib riboswitches when the 2′-hydroxyl at the β site was modified to an amino group, we considered if modification of the phosphate backbone would also reveal differences in conformation or hydrogen bonding between the two riboswitches.
To elucidate the interactions between the riboswitch and the phosphate backbone, we tested binding of c-AMP-GMP analogs with monosubstitutions of oxygen to sulfur at each non-bridging oxygen of c-AMP-GMP to the GEMM-Ib riboswitch. Reactions of modified and unmodified nucleotide triphosphates were scaled up to prepare each of the four different possibilities: c-(Rp)-Aps-GMP, c-AMP-(Rp)-Gps, c-(Sp)-Aps-GMP and c-AMP-(Sp)-Gps. c-(Rp)-Aps-GMP and c-(Sp)-Aps-GMP are stereoisomers of sulfur substitution for the non-bridging oxygens at the Aα site. c-AMP-(Rp)-Gps and c-AMP-(Sp)-Gps are modified at the non-bridging oxygens at the Gβ site.
Neither of the analogs with modifications to the pro-Rp oxygens, c-(Rp)-Aps-GMP and c-AMP-(Rp)-Gps, demonstrated large changes in binding affinity (only 1.4- to 1.9-fold changes in Kd). These values are similar to the Kd reported for the analogous c-(Rp)-Gps-GMP bound to the GEMM-I construct, which shows only a 4-fold change in Kd when compared to c-di-GMP (36). These data are supported by the crystal structures of GEMM-I and GEMM-Ib that show pro-Rp oxygens of the c-di-GMP and c-AMP-GMP ligands pointed toward the solvent rather than making direct contact to their respective riboswitches (Figure 4). We also measured the binding affinity of monophosphorothioate modifications to the pro-Sp oxygens. Both of these analogs showed significant losses in binding affinity when compared to c-AMP-GMP. The c-AMP-(Sp)-Gps analog showed a 10-fold loss in Kd compared to c-AMP-GMP. c-(Sp)-Aps-GMP showed the largest defect in binding affinity (a 43-fold loss) confirming that this oxygen is especially important for recognition. This is in agreement with the crystal structure of the GEMM-Ib riboswitch (33) that shows the pro-Sp oxygen of Aα making hydrogen bonds with A41 and A12 (Figure 4B).
We next tested if phosphorothioate modifications were additive or if there might be other factors implicated in recognition of disubstituted analogs as seen for the 2′-amino modifications. All diphosphorothioate modifications had a significant effect on riboswitch binding. The c-(RpRp)-ApsGps analog bound the tightest out of the diphosphorothioates (Table 5), but still had a 29-fold effect on binding affinity. The c-(RpSp)-ApsGps and c-(SpRp)-ApsGps analogs showed even weaker binding when compared to c-AMP-GMP, 140- and 330-fold effects, respectively. Both of these analogs contain at least one pro-Sp oxygen modified to a sulfur and oriented toward the binding pocket. The pro-Sp oxygen at the Aα of c-AMP-GMP shows the most dramatic loss in binding affinity when changed to a sulfur as evidenced by the c-(Sp)-Aps-GMP analog. This modification is also present in the c-(SpRp)-ApsGps diphosphorothioate and further supports the importance of this oxygen for ligand recognition.
Table 5. Binding affinity of c-AMP-GMP and phosphorothioate analogs to Gs1761.
| Compound | K d (nM) | Fold change | ΔΔG (kcal/mol) |
|---|---|---|---|
| c-AMP-GMP | 1.1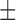 0.04a 0.04a
|
N/A | N/A |
| c-(Rp)-Aps-GMP | 2.1 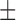 0.1 0.1 |
1.9 | 0.4 |
| c-AMP-(Rp)-Gps | 1.5 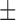 0.2 0.2 |
1.4 | 0.2 |
| c-(Sp)-Aps-GMP | 48 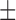 5 5 |
43 | 2.2 |
| c-AMP-(Sp)-Gps | 11  2 2 |
10 | 1.4 |
| c-(RpRp)-ApsGps | 32  3 3 |
29 | 2.0 |
| c-(RpSp)-ApsGps | 150  20 20 |
140 | 2.9 |
| c-(SpRp)-ApsGps | 360  60 60 |
330 | 3.4 |
| c-(SpSp)-ApsGps | 1200  130 130 |
1100 | 4.1 |
aDenotes measurements made by direct binding.
N/A: not applicable.
Because both Sp modifications showed weaker binding than the Rp phosphorothioate analogs, it is not surprising that the c-(SpSp)-ApsGps analog bound with the weakest affinity with a 1100-fold effect on binding (Table 5). Interestingly, c-(SpSp)-ApsGps shows a nearly additive effect when comparing affinity loss to individual monophosphorothioates. The combined energy lost with each individual monophosphorothioate, c-(Sp)-Aps-GMP, and c-AMP-(Sp)-Gps, is 3.6 kcal/mol and the ΔΔG of c-(SpSp)-ApsGps is 4.1 kcal/mol. However, all other diphosphorothioates present a discrepancy of at least 1 kcal/mol in comparison to the individual monophosphorothioates. The largest difference is seen in c-(RpRp)-ApsGps that has an extra 1.4 kcal/mol loss in binding affinity that cannot be explained by simply adding the ΔΔG values of the individual pro-Rp monosubstitutions. Therefore, it is likely that other interactions and/or alternative ligand conformations beyond the resolution of our experiments are involved in recognition of the pro-Rp oxygens in the disubstituted analog.
CONCLUSION
The binding partner for the GEMM-I riboswitch was identified as c-di-GMP almost a decade ago (29). More recently, a subset of these RNA receptors were shown to selectively bound c-AMP-GMP (15,32). While these two riboswitches are globally similar, there are nuances to their molecular recognition that were previously ambiguous. The enzymatic synthesis method presented in this study allowed us to synthesize novel cyclic dinucleotide analogs to reveal differences in the sugar recognition in these two riboswitches, specifically at the Gβ site, that is not evident from the crystal structures. The data also suggest a conformational change in the diphosphorothioate cyclic dinucleotide analogs possibly through constraints on the cyclized dinucleotide backbone. While specific modifications to the phosphate and base of these second messenger molecules were used to probe targeted receptor recognition, still other modifications could be made using enzymatic synthesis in order to identify protein and RNA binding partners in bacterial cyclic dinucleotide pathways.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Roger Jones for generously providing chemically synthesized c-di-AMP and c-di-GMP phosphorothioate analogs. The plasmid containing DncV was a generous gift of John Mekalanos. The authors are most grateful to David Hiller and Caroline Reiss for providing helpful comments on this manuscript and other members of the Strobel Laboratory for invaluable discussion. This research made use of the West Campus Analytical Core at Yale University.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [P01 GM022778 to S.A.S., F32 GM115097 to K.D.L-F]. Funding for open access charge: National Institutes of Health [F32 GM115097].
Conflict of interest statement. None declared.
REFERENCES
- 1. Krasteva P.V., Sondermann H.. Versatile modes of cellular regulation via cyclic dinucleotides. Nat. Chem. Biol. 2017; 13:350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bowie A.G. Innate sensing of bacterial cyclic dinucleotides: more than just STING. Nat. Immunol. 2012; 13:1137–1139. [DOI] [PubMed] [Google Scholar]
- 3. Diner E.J., Burdette D.L., Wilson S.C., Monroe K.M., Kellenberger C.A., Hyodo M., Hayakawa Y., Hammond M.C., Vance R.E.. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 2013; 3:1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hengge R. Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 2009; 7:263–273. [DOI] [PubMed] [Google Scholar]
- 5. Tischler A.D., Camilli A.. Cyclic diguanylate (c-di-GMP) regulates Vibrio cholerae biofilm formation. Mol. Microbiol. 2004; 53:857–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Simm R., Morr M., Kader A., Nimtz M., Römling U.. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol. Microbiol. 2004; 53:1123–1134. [DOI] [PubMed] [Google Scholar]
- 7. Tamayo R., Pratt J.T., Camilli A.. Roles of cyclic diguanylate in the regulation of bacterial pathogenesis. Annu. Rev. Microbiol. 2007; 61:131–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jenal U., Reinders A., Lori C.. Cyclic di-GMP: second messenger extraordinaire. Nat. Rev. Microbiol. 2017; 15:271–284. [DOI] [PubMed] [Google Scholar]
- 9. Corrigan R.M., Gründling A.. Cyclic di-AMP: another second messenger enters the fray. Nat. Rev. Microbiol. 2013; 11:513–524. [DOI] [PubMed] [Google Scholar]
- 10. Gándara C., Alonso J.C.. DisA and c-di-AMP act at the intersection between DNA-damage response and stress homeostasis in exponentially growing Bacillus subtilis cells. DNA Repair. 2015; 27:1–8. [DOI] [PubMed] [Google Scholar]
- 11. Kim H., Youn S.-J., Kim S.O., Ko J., Lee J.-O., Choi B.-S.. Structural studies of potassium transport protein KtrA regulator of conductance of K+ (RCK) C domain in complex with cyclic diadenosine monophosphate (c-di-AMP). J. Biol. Chem. 2015; 290:161393–161402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oppenheimer-Shaanan Y., Wexselblatt E., Katzhendler J., Yavin E., Ben-Yehuda S.. c-di-AMP reports DNA integrity during sporulation in Bacillus subtilis. EMBO Rep. 2011; 12:594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gundlach J., Herzberg C., Kaever V., Gunka K., Hoffmann T., Weiß M., Gibhardt J., Thürmer A., Hertel D., Daniel R. et al. Control of potassium homeostasis is an essential function of the second messenger cyclic di-AMP in Bacillus subtilis. Sci. Signal. 2017; 10:eaal3011. [DOI] [PubMed] [Google Scholar]
- 14. Davies B.W., Bogard R.W., Young T.S., Mekalanos J.J.. Coordinated Regulation of Accessory Genetic Elements Produces Cyclic Di-Nucleotides for V. cholerae Virulence. Cell. 2012; 149:358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nelson J.W., Sudarsan N., Phillips G.E., Stav S., Lünse C.E., McCown P.J., Breaker R.R.. Control of bacterial exoelectrogenesis by c-AMP-GMP. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:5389–5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ryjenkov D.A., Tarutina M., Moskvin O.V., Gomelsky M.. Cyclic diguanylate is a ubiquitous signaling molecule in bacteria: insights into biochemistry of the GGDEF protein domain. J. Bacteriol. 2005; 187:1792–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Galperin M.Y., Natale D.A., Aravind L., Koonin E.V.. A specialized version of the HD hydrolase domain implicated in signal transduction. J. Mol. Microbiol. Biotechnol. 1999; 1:303–305. [PMC free article] [PubMed] [Google Scholar]
- 18. Romling U. Great times for small molecules: c-di-AMP, a second messenger candidate in bacteria and archaea. Sci. Signal. 2008; 1:pe39. [DOI] [PubMed] [Google Scholar]
- 19. Commichau F.M., Dickmanns A., Gundlach J., Ficner R., Stülke J.. A jack of all trades: the multiple roles of the unique essential second messenger cyclic di‐AMP. Mol. Microbiol. 2015; 97:189–204. [DOI] [PubMed] [Google Scholar]
- 20. Hallberg Z.F., Wang X.C., Wright T.A., Nan B., Ad O., Yeo J., Hammond M.C.. Hybrid promiscuous (Hypr) GGDEF enzymes produce cyclic AMP-GMP (3′, 3′-cGAMP). Proc. Natl. Acad. Sci. U.S.A. 2016; 113:1790–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao J., Tao J., Liang W., Zhao M., Du X., Cui S., Duan H., Kan B., Su X., Jiang Z.. Identification and characterization of phosphodiesterases that specifically degrade 3′3′-cyclic GMP-AMP. Cell Res. 2015; 25:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schirmer T. C-di-GMP synthesis: Structural aspects of evolution, catalysis and regulation. J. Mol. Biol. 2016; 428:3683–3701. [DOI] [PubMed] [Google Scholar]
- 23. Kato K., Ishii R., Hirano S., Ishitani R., Nureki O.. Structural basis for the catalytic mechanism of DncV, bacterial homolog of cyclic GMP-AMP synthase. Structure. 2015; 23:843–850. [DOI] [PubMed] [Google Scholar]
- 24. Kranzusch P.J., Lee A.S.Y., Wilson S.C., Solovykh M.S., Vance R.E., Berger J.M., Doudna J.A.. Structure-Guided reprogramming of human cGAS dinucleotide linkage specificity. Cell. 2014; 158:1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu D., Wang L., Shang G., Liu X., Zhu J., Lu D., Wang L., Kan B., Zhang J., Xiang Y.. Structural biochemistry of a vibrio cholerae dinucleotide cyclase reveals cyclase activity regulation by folates. Mol. Cell. 2014; 55:931–937. [DOI] [PubMed] [Google Scholar]
- 26. Ming Z., Wang W., Xie Y., Ding P., Chen Y., Jin D., Sun Y., Xia B., Yan L., Lou Z.. Crystal structure of the novel di-nucleotide cyclase from Vibrio cholerae (DncV) responsible for synthesizing a hybrid cyclic GMP-AMP. Cell Res. 2014; 24:1270–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smith K.D., Lipchock S.V., Ames T.D., Wang J., Breaker R.R., Strobel S.A.. Structural basis of ligand binding by a c-di-GMP riboswitch. Nat. Struct. Mol. Biol. 2009; 16:1218–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee E.R., Baker J.L., Weinberg Z., Sudarsan N., Breaker R.R.. An allosteric self-splicing ribozyme triggered by a bacterial second messenger. Science. 2010; 329:845–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sudarsan N., Lee E.R., Weinberg Z., Moy R.H., Kim J.N., Link K.H., Breaker R.R.. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science. 2008; 321:411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith K.D., Shanahan C.A., Moore E.L., Simon A.C., Strobel S.A.. Structural basis of differential ligand recognition by two classes of bis-(3′-5′)-cyclic dimeric guanosine monophosphate-binding riboswitches. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:7757–7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kulshina N., Baird N.J., Ferré-D’Amaré A.R.. Recognition of the bacterial second messenger cyclic diguanylate by its cognate riboswitch. Nat. Struct. Mol. Biol. 2009; 16:1212–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kellenberger C.A., Wilson S.C., Hickey S.F., Gonzalez T.L., Su Y., Hallberg Z.F., Brewer T.F., Iavarone A.T., Carlson H.K., Hsieh Y.-F. et al. GEMM-I riboswitches from Geobacter sense the bacterial second messenger cyclic AMP-GMP. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:5383–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ren A., Wang X.C., Kellenberger C.A., Rajashankar K.R., Jones R.A., Hammond M.C., Patel D.J.. Structural basis for molecular discrimination by a 3′, 3′-cGAMP sensing riboswitch. Cell Rep. 2015; 11:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gao A., Serganov A.. Structural insights into recognition of c-di-AMP by the ydaO riboswitch. Nat. Chem. Biol. 2014; 10:787–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ren A., Patel D.J.. c-di-AMP binds the ydaO riboswitch in two pseudo-symmetry–related pockets. Nat. Chem. Biol. 2014; 10:780–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shanahan C.A., Gaffney B.L., Jones R.A., Strobel S.A.. Differential analogue binding by two classes of c-di-GMP riboswitches. J. Am. Chem. Soc. 2011; 133:15578–15592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shanahan C.A., Gaffney B.L., Jones R.A., Strobel S.A.. Identification of c-di-GMP derivatives resistant to an EAL domain phosphodiesterase. Biochemistry (Mosc.). 2013; 52:365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luo Y., Zhou J., Watt S.K., Lee V.T., Dayie T.K., Sintim H.O.. Differential binding of 2′-biotinylated analogues of c-di-GMP with c-di-GMP riboswitches and binding proteins. Mol. Biosyst. 2012; 8:772–778. [DOI] [PubMed] [Google Scholar]
- 39. Wang C., Sinn M., Stifel J., Heiler A.C., Sommershof A., Hartig J.S.. Synthesis of all possible canonical (3′–5′-Linked) cyclic dinucleotides and evaluation of riboswitch interactions and immune-stimulatory effects. J. Am. Chem. Soc. 2017; 139:16154–16160. [DOI] [PubMed] [Google Scholar]
- 40. Gaffney B.L., Veliath E., Zhao J., Jones R.A.. One-flask syntheses of c-di-GMP and the [Rp, Rp] and [Rp, Sp] thiophosphate analogues. Org. Lett. 2010; 12:3269–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kline T., Jackson S.R., Deng W., Verlinde C.L.M.J., Miller S.I.. Design and synthesis of bis-carbamate analogs of cyclic bis-(3′-5′)-diguanylic acid (c-di-GMP) and the acyclic dimer PGPG. Nucleosides Nucleotides Nucleic Acids. 2008; 27:1282–1300. [DOI] [PubMed] [Google Scholar]
- 42. Furukawa K., Gu H., Sudarsan N., Hayakawa Y., Hyodo M., Breaker R.R.. Identification of ligand analogs that control c-di-GMP riboswitches. ACS Chem. Biol. 2012; 7:1436–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Luo Y., Zhou J., Wang J., Dayie T.K., Sintim H.O.. Selective binding of 2′-F-c-di-GMP to Ct-E88 and Cb-E43, new class I riboswitches from clostridium tetani and clostridium botulinum respectively. Mol. Biosyst. 2013; 9:1535–1539. [DOI] [PubMed] [Google Scholar]
- 44. Smith K.D., Lipchock S.V., Livingston A.L., Shanahan C.A., Strobel S.A.. Structural and biochemical determinants of ligand binding by the c-di-GMP riboswitch. Biochemistry (Mosc.). 2010; 49:7351–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rao F., Pasunooti S., Ng Y., Zhuo W., Lim L., Liu A.W., Liang Z.-X.. Enzymatic synthesis of c-di-GMP using a thermophilic diguanylate cyclase. Anal. Biochem. 2009; 389:138–142. [DOI] [PubMed] [Google Scholar]
- 46. Meehan R.E., Torgerson C.D., Gaffney B.L., Jones R.A., Strobel S.A.. Nuclease-Resistant c-di-AMP derivatives that differentially recognize RNA and protein receptors. Biochemistry (Mosc.). 2016; 55:837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bochner B.R., Ames B.N.. ZTP (5-amino 4-imidazole carboxamide riboside 5′-triphosphate): a proposed alarmone for 10-formyl-tetrahydrofolate deficiency. Cell. 1982; 29:929–937. [DOI] [PubMed] [Google Scholar]
- 48. Ducker G.S., Rabinowitz J.D.. ZMP: a master regulator of one-carbon metabolism. Mol. Cell. 2015; 57:203–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sabina R.L., Holmes E.W., Becker M.A.. The enzymatic synthesis of 5-amino-4-imidazolecarboxamide riboside triphosphate (ZTP). Science. 1984; 223:1193–1195. [DOI] [PubMed] [Google Scholar]
- 50. Boots J.L., Canny M.D., Azimi E., Pardi A.. Metal ion specificities for folding and cleavage activity in the Schistosoma hammerhead ribozyme. RNA. 2008; 14:2212–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Speckhard D.C., Wu F.Y.H., Wu C.-W.. Role of the intrinsic metal in RNA polymerase from Escherichia coli. In vivo substitution of tightly bound zinc with cobalt. Biochemistry (Mosc.). 1977; 16:5228–5234. [DOI] [PubMed] [Google Scholar]
- 52. El-Deiry W.S., Downey K.M., So A.G.. Molecular mechanisms of manganese mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 1984; 81:7378–7382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kumarevel T., Mizuno H., Kumar P.K.R.. Characterization of the metal ion binding site in the anti-terminator protein, HutP, of Bacillus subtilis. Nucleic Acids Res. 2005; 33:5494–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wei H., Therrien C., Blanchard A., Guan S., Zhu Z.. The Fidelity Index provides a systematic quantitation of star activity of DNA restriction endonucleases. Nucleic Acids Res. 2008; 36:e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhao J., Veliath E., Kim S., Gaffney B.L., Jones R.A.. Thiophosphate analogs of c-di-GMP: impact on polymorphism. Nucleosides Nucleotides Nucleic Acids. 2009; 28:352–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Eckstein F. Phosphorothioate analogs of Nucleotides—tools for the investigation of biochemical processes. Angew. Chem. Int. Ed. Engl. 1983; 22:423–439. [Google Scholar]
- 57. Griffiths A.D., Potter B.V., Eperon I.C.. Stereospecificity of nucleases towards phosphorothioate-substituted RNA: stereochemistry of transcription by T7 RNA polymerase. Nucleic Acids Res. 1987; 15:4145–4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Aurup H., Tuschl T., Benseler F., Ludwig J., Eckstein F.. Oligonucleotide duplexes containing 2′-amino-2′-deoxycytidines: thermal stability and chemical reactivity. Nucleic Acids Res. 1994; 22:20–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.