

Abstract
Focal segmental glomerulosclerosis (FSGS) is a histologic lesion resulting from a variety of pathogenic processes that cause injury to the podocytes. Recently, mutations in more than 50 genes expressed in podocyte or glomerular basement membrane were identified as causing genetic forms of FSGS, the majority of which are characterized by onset in childhood. The prevalence of adult-onset genetic FSGS is likely to be underestimated and its clinical and histological features have not been clearly described. A small number of studies of adult-onset genetic FSGS showed that there is heterogeneity in clinical and histological findings, with a presentation ranging from sub-nephrotic proteinuria to full nephrotic syndrome. A careful evaluation of adult-onset FSGS that do not have typical features of primary or secondary FSGS (familial cases, resistance to immunosuppression and absence of evident cause of secondary FSGS) should include a genetic evaluation. Indeed, recognizing genetic forms of adult-onset FSGS is of the utmost importance, given that this diagnosis will have major implications on treatment strategies, selecting of living-related kidney donor and renal transplantation success.
Keywords: genetic FSGS, nephrotic syndrome, podocin, podocytopathies, steroid-resistant nephrotic syndrome
Introduction
Focal segmental glomerulosclerosis (FSGS) is a histologic lesion defined by the presence of sclerosis in part of some glomeruli seen on light microscopy (LM), immunofluorescence (IF) or electron microscopy (EM) examination on a kidney biopsy sample [1]. This histologic pattern is the result of a variety of pathogenic processes, which share as a common mechanism injury to podocytes that in turn leads to loss of selectivity of glomerular filtration barrier and proteinuria [2]. Primary FSGS is a podocytopathy due to a not yet identified circulating factor(s) that exerts a toxic effect on the podocytes. Widespread foot process effacement on EM is the morphologic expression of the podocyte injury in primary FSGS, clinically manifested by the development of severe proteinuria and nephrotic syndrome (NS) [3]. Primary FSGS typically responds to immunosuppressive treatment. On the other hand, secondary FSGS occurs as a result of glomerular hyperfiltration (e.g. reduction in renal mass) or due to drug toxicity, or as a result of a viral infection. Morphologically, podocyte damage secondary to glomerular hyperfiltration is characterized by segmental foot process effacement on EM and clinical manifestation of proteinuria in the absence of NS [4]. Treatment of secondary FSGS is focused on eliminating the offending agent, lowering hemodynamic stress on glomeruli (e.g. weight loss) and maximizing antiproteinuric strategies.
The term ‘genetic FSGS’ has been coined based on the discovery of genes that if mutated can cause monogenic forms of FSGS in humans [5]. All the encoded proteins are either expressed on the podocytes or the glomerular basement membrane (GBM) and as a result, if mutated, result in damage to the glomerular filtration barrier followed by proteinuria. Clinical presentation of genetic FSGS is extremely variable, with differences in age of onset, gene penetrance, presence or absence of NS and time of progression to end-stage renal disease (ESRD). Although genetic FSGS most commonly involves the pediatric population it can affect adults (age of onset after 18 years of age) as well. In fact, a causal mutation has been described in 43% of familial and 10% of sporadic adult cases who present with steroid-resistant nephrotic syndrome (SRNS) [6]. This highlights the importance of a better understanding of monogenic forms of FSGS in adults, in particular, given that this diagnosis will have major implications on treatment strategies and future renal transplantation success (lower risk of recurrence compared with primary FSGS). However, little is known regarding the clinical presentation and histological characteristics of adults with genetic forms of FSGS. In this review, we summarize genetic, clinical and histological features of genetic FSGS in adults and propose a screening strategy in this population.
Podocyte gene mutations and mode of inheritance in genetic FSGS
More than 50 genes have been described as potential sites of mutation responsible for monogenic forms of FSGS or SRNS [7] (Table 1). The encoded proteins belong to distinct structural protein complexes and signaling pathways, which are essential for glomerular filtration barrier integrity and function.
Table 1.
Genes mutated in SRNS/FSGS
| Gene | Chromosome | Protein | Inheritance | Age of onset | Eventual syndromic forms |
|---|---|---|---|---|---|
| Slight diaphragm proteins | |||||
| NPHS1 | 19q13.1 | Nephrin | AR | Congenital nephrotic syndrome; childhood FSGS | |
| NPHS2 | 1q25.2 | Podocin | AR | Early childhood, adolescence or adulthood | |
| CD2AP | 6p12 | CD2-associated protein | AD; rarely AR | Childhood or adulthood | |
| CRB2 | 9q33.4 | Crumbs homolog 2 | AR | Childhood FSGS | Association with cerebral ventriculomegaly |
| PLCE1 | 10q23.33 | Phospholipase C ε 1 | AR | Early-onset childhood | |
| TRPC6 | 11q22.1 | Transient receptor potential cation channel 6 | AD | Adulthood, rarely childhood | |
| Cytoskeleton structural and regulatory proteins | |||||
| ACTN4 | 19q13 | α-Actinin-4 | AD | Adulthood | |
| MYO1E | 15q22.2 | Non-muscle myosin 1E | AR | Childhood | |
| MYH9 | 22q12.3 | Myosin heavy chain 9 | AD | Childhood | Epstein–Fechtner syndrome (FSGS, deafness, cataracts, macrothrombocytopenia, leukocyte inclusions) |
| INF2 | 14q32.33 | Inverted formin 2 | AD | Adulthood | Association with Charcot–Marie–Tooth disease |
| ANLN | 7p15-p14 | Anillin | AD | Adulthood | |
| ARHGDIA | 17q25.3 | Rho GDP dissociation inhibitor α | AR | Congenital nephrotic syndrome/early childhood | |
| ARHGAP24 | 4q21.23 | RhoGTPase activating protein 24 | AD | Early childhood | |
| KANK 1 | 9p24.3 | Kidney ankyrin repeat- containing protein 1 | AR | Early adulthood | Association with intellectual disability |
| KANK 2 | 19p13.2 | Kidney ankyrin repeat- containing protein 2 | AR | Early childhood | |
| KANK 4 | 1p31.3 | Kidney ankyrin repeat- containing protein 4 | AR | Early childhood | Association with intellectual disability, facial dysmorphism and atrial septal defect |
| Adhesion proteins | |||||
| ITGA3 | 17q21.33 | Integrin α3 | AR | Early childhood | Association with epidermolysis bullosa and interstitial lung disease |
| ITGB4 | 17q11 | Integrin β4 | AR | Early childhood | Association with epidermolysis bullosa and pyloric atresia |
| LAMB2 | 3p21 | Laminin β2 | AR | Early childhood onset DMS or FSGS | Pierson syndrome (microcoria, neuromuscular junction defects) |
| Glomerular basement membrane proteins | |||||
| COL4A3 | 2q36-q37 | α3 type IV collagen | AR | Childhood, adulthood | Alport syndrome or familial/sporadic FSGS |
| COL4A4 | 2q35-q37 | α4 type IV collagen | AR | Childhood, adulthood | Alport syndrome or familial/sporadic FSGS |
| COL4A5 | Xq22 | α5 type IV collagen | X linked | Childhood, adulthood | Alport syndrome or familial/sporadic FSGS |
| LAMA 5 | 20q13.2-q13.3 | Laminin alpha 5 | AD | Adulthood | |
| Nuclear transcription factors | |||||
| LMX1B | 9q34 | LIM homeobox transcription factor 1β | AD | Familial FSGS | Nail–Patella syndrome (hypoplastic or absent patella, dysplasia of elbows, nail abnormalities) |
| WT1 | 11p13 | Wilms tumor 1 | AD | Childhood, adolescence | Frasier syndrome (FSGS, male pseudohermaphroditism, gonadoblastoma), Deny–Drash syndrome (DMS, male pseudohermaphroditism, Wilms tumor) |
| SMARCAL1 | 2q34-36 | SMARCA-like protein | AR | Childhood | Schimke immuno-osseous dysplasia (immunodeficiency, skeletal dysplasia) |
| NXF5 | Xq22 | Nuclear RNA export factor 5 | X linked | Adulthood | Association with cardiac conduction disorders |
| Nuclear pore complex proteins | |||||
| NUP93 | 16q13 | Nucleoporin 93 kDa | AR | Childhood | |
| NUP205 | 7q33 | Nucleoporin 205 kDa | AR | Childhood | |
| XPO5 | 6p21.1 | Exportin 5 | AR | Childhood | |
| NUP107 | 12q15 | Nucleoporin 107 kDa | AR | Childhood | Association with microcephaly |
| Coenzyme Q10 biosynthesis | |||||
| ADCK4 | 19q13.2 | aarF domain containing kinase 4 | AR | Childhood, early adulthood | |
| COQ2 | 4q21.23 | Coenzyme Q2 hydroxybenzoate-polyprenyl transferase | AR | Childhood | Association with encephalopathy |
| COQ6 | 14q24.3 | Coenzyme Q6 monooxygenase | AR | Early childhood | Association with deafness |
| PDSS2 | 6q21 | Prenyl (decaprenyl) diphosphate synthase | AR | Congenital SRNS | Association with encephalomyopathy |
| Other | |||||
| MTTL1 | mtDNA | Mitochondrially encoded tRNA leucine 1 | Maternal | Adulthood | MELAS syndrome (mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes) |
| SCARB2 | 4q13-21 | Scavenger receptor class B member 2 | AR | Early adulthood | Action myoclonus-renal failure syndrome (ataxia, myoclonus, collapsing FSGS) |
| CUBN | 10p12.31 | Cubilin | AR | Childhood | Association megaloblastic anemia |
| DGKE | 17q22 | Diacylglycerol kinase | AR | Childhood | |
| PTRO | 12p13-p12 | Protein tyrosine phosphatase, receptor type O | AR | Childhood | |
| PMM2 | 16p13.3 | Phosphomannomutase 2 | AR | Childhood | |
| WDR73 | 15q22 | WD repeat domain 73 | AR | Childhood | Galloway–Mowat syndrome (microcephaly and developmental delay) |
| ALG1 | 16p13.3 | Asparagine-linked glycosylation 1 | AR | Congenital nephrotic syndrome | |
AR, autosomic recessive; AD, autosomic dominant; DMS, diffuse mesangial sclerosis; mtDNA, mitochondrial DNA.
Podocytes, GBM and endothelium compose the so-called glomerular filtration barrier, the structural element responsible for the selective permeability of renal filtration process. Podocytes are highly differentiated cells, with an arborized shape characterized by multiple projections, subdivided into larger major processes and finer pedicels called foot processes. Glomerular capillaries are wrapped by podocyte bodies and foot processes, which form an interdigitating pattern characterized by a unique type of inter-cellular junctions, called slit diaphragms. Nephrin (NHPS1) is an essential component of the glomerular slit diaphragm; mutations in this gene account for 40–60% of the congenital nephrotic syndrome (CNS) cases, developing in the first 3 months of life [8, 9]. Podocin (NPHS2) is a transmembrane protein involved in recruitment of nephrin at the slit diaphragm; homozygous or compound heterozygous mutations in NPHS2 are associated with childhood-onset FSGS [10]. Other mutations involving proteins of the slit diaphragm that have been described include: transient receptor potential cation channel 6 (TRPC6), which is a calcium channel localized at podocyte foot process [11], CD2-associated protein (CD2AP), which is a molecule that acts as a bridge between slit diaphragm and actin cytoskeleton, and phospholipase C epsilon 1 (PLCε1), which is another protein that interacts with nephrin at the slit diaphragm [12].
The physiological function of podocytes is closely related to their specialized shape, which is strictly dependent on a highly organized cytoskeleton, comprising of a core of parallel actin bundles surrounded by a branched subcortical actin network [13]. Loss of this characteristic structure has been shown in animal experimental models of FSGS. The mutations causing FSGS due compromise of the cytoskeleton can involve either structural (ACTN4, Myo1E, MYH9) or regulatory [IFN2, ARHGAP24, ARHGDIA, anilin (ANLN)] cytoskeleton proteins (Table 1) [14].
Podocytes must adhere tightly to the GBM to maintain the glomerular filtration barrier and mutations in adhesion proteins have been found in different syndromic forms of SRNS (laminin β2, integrin α3 and integrin β4), often in association with skin lesions such as epidermolysis bullosa [15–17]. Thus, mutations in structural components of GBM like type IV collagenic genes (COL4A3, COL4A4, COL4A5) and laminin alpha 5 (LAMA5) can also present with a FSGS [18, 19].
Genes encoding the proteins involved in nuclear function are also involved in genetic forms of syndromic or isolated FSGS/SRNS. Mutations in nuclear transcription factors, such as Wilms Tumor 1 (WT1) [20], SMARCA-like protein (SMARCAL1) [21] and LIM homeobox transcription factor 1β (LMX1β) [22], are related to different syndromic forms of SRNS (Nail-Patella syndrome, Denys-Drash syndrome, Frasier syndrome, Schimke immuno-osseous dysplasia). Furthermore, mutations in genes coding the nuclear pore complex proteins, such as nucleoporin 93 kDa (NUP93), nucleoporin 107 kDa (NUP 107), nucleoporin 205 kDa (NUP 205) and exportin 5 (XPO5), have been identified recently in some cases of isolated and syndromic familial childhood-onset FSGS [23, 24].
Finally, mutation in genes encoding proteins that belong to several cellular metabolic pathways can involve not only podocytes but also other cell types resulting in familial syndromic FSGS. Coenzyme Q10 (ubiquinone) is a component of cell membrane essential for normal function of the mitochondrial respiratory chain. Mutations in genes related to coenzyme Q10 are associated with childhood-onset FSGS and encephalomyopathy. There is data to suggest that early coenzyme Q10 supplementation can have beneficial effects in these patients [25].
Several susceptibility genes for FSGS have been described; likely an environmental factor (the so-called ‘second hit’) is necessary to develop the disease [26]. Apolipoprotein L1 (APOL1) gene is the best-known susceptibility gene: expression of G1 and G2 polymorphisms (which show the highest prevalence in the African American population) is associated with an increased risk of adult-onset FSGS, hypertensive nephropathy and HIV-associated nephropathy [27, 28].
Mutations described in genetic FSGS/SRNS involve both recessive and dominant genes. This distinction is important as it can have phenotypic correlation mainly related to the age of onset of the disease. Autosomal recessive mutations typically manifest during early childhood, with wide variability: NHPS1, LAMB2 and PLCE1 mutations are related to CNS, while mutations in NPHS2 are associated with a later onset in childhood. On the other hand, mutations in autosomal dominant genes (such as ACTN4, TRPC6 and INF2) commonly manifest as adult-onset FSGS. However, this distinction is not absolute: mutations in WT1 (autosomal dominant) can present as childhood-onset SRNS [20, 29], while compound heterozygous mutation in NPHS2 with polymorphism R229Q mutation often presents as adult-onset FSGS [30].
Prevalence of genetic forms of FSGS
Congenital NS and childhood-onset FSGS are the most studied forms of genetic FSGS/SRNS. Mutations have been found in CNS (onset within first 3 months of life) with a frequency of 69.4–100% [6, 31, 32]. Nephrin (NPHS1) is the most frequent gene mutation in CNS, with a prevalence ranging from 40% to 80% of cases, followed by podocin (NPHS2), WT1 and Laminin β2 (LAMB2). Prevalence of monogenic forms of SRNS/FSGS decreases with increasing age. Sadowski et al. [31] studied the largest cohort of familial and sporadic SRNS cases (n = 2016) finding mutations in 27 genes known to cause of monogenic disease. Mutations were found in 49.7% of infantile onset cases (4–12 months), 25.3% of early childhood onset cases (13 months to 6 years), 17.8% of late childhood onset cases (6–12 years) and 10.8% of adolescent onset cases (13–18 years). Interestingly, distribution of causative genes differed according to the age of onset: NPHS2 was the most common gene mutated in non-congenital NS (onset from 3 months to 18 years), while NPHS1 mutation was almost absent in cases with age of onset after 1 year of age. Overall, more than 90% of childhood and adolescence onset cases have mutations involving a limited number of genes: NPHS1, NPHS2, WT1, LAMB2, PLCE1 and scavenger receptor class B member 2 (SCARB2). Renal biopsies, when performed, revealed FSGS as the exclusive histological lesion in patients with age of onset after 7 years of age, while ‘minimal change disease’ and diffuse mesangial sclerosis were the main renal biopsy findings in infantile and early childhood SRNS.
The real prevalence of the monogenic form of adult-onset FSGS is difficult to infer. There are very few studies available in the literature, with conflicting results. This is primarily due to the different panel of genes that were studied and to the variable inclusion criteria adopted in different studies (e.g. SRNS versus biopsy-proven FSGS, familial FSGS versus sporadic FSGS) (Table 2). Laurin et al. [35] studied 28 children and 37 adults with sporadic FSGS seeking monogenic causes of FSGS, by analyzing five genes (NPHS2, TRPC6, ACTN4, INF2 and PLCE1). Among adult-onset cases of sporadic FSGS no pathogenetic mutation was identified, while homozygous or compound heterozygous mutations in the NPHS2 gene were detected in 7.1% of children. These results led the authors to propose genetic screening only in childhood-onset sporadic FSGS and the gene evaluation only limited to the NPHS2 gene. These recommendations need to be considered with caution taking into account that this study evaluated only a handful of genes and that the study population may have been enriched with patients with primary FSGS (23 of the patients achieved remission after calcineurin inhibitor treatment) and patients were not necessarily resistant to immunosuppressive therapy. Moreover, the majority of patients were African-American (54% of adults) and 79.5% of them showed APOL1 gene risk alleles G1 and/or G2. Similar results were shown by Aucella et al. [36], who analyzed NPHS2 and ACTN4 in a cohort of 33 cases with sporadic adult-onset FSGS. No mutation in ACTN4 was found, while NPHS2 heterozygous for the R229Q allele mutation was found in three. Inclusion of cases of primary FSGS in these studies could have resulted in underestimation of the real prevalence of genetic form of adult-onset FSGS. Indeed, studies that utilized a more comprehensive genetic panel and larger cohorts that excluded primary FSGS showed different results (Table 2).
Table 2.
Prevalence of genetic forms of adult-onset FSGS
| Author | Year | Population studied | Number of cases (or families) | Gene screened | Prevalence of genetic forms |
|---|---|---|---|---|---|
| Gast et al. [18] | 2016 | Familial and sporadic FSGS | 69 patients | 39 genes (comprised COL4A3, COL4A4 and COL4A5) | 13% of definitely pathogenic mutations, 20% of definitely or probably pathogenic mutation, COL4A3-5 mutations were identified in 38% of familial FSGS and 3% of sporadic FSGS |
| Sadowski et al. [31] | 2015 | Sporadic and familial SRNS (onset between 0 and 25 years of age) | 1783 families (2016 individuals) | 27 different genes | 21.4% families with adult-onset SRNS |
| Sen et al. [33] | 2017 | Sporadic and familial SRNS | 46 adults | 11 genes (comprised COL4A3, COL4A4 and COL4A5) | 10% of all adult-onset SRNS cases, 20% of cases with familial history of SRNS, COL4A3-5 mutations were identified in 6% of cases |
| Santin et al. [6] | 2011 | Sporadic and familial SRNS | 48 families | NPHS1, NPHS2, TRPC6, CD2AP, PLCE1, INF2, WT1,ACTN4 | 43% in familial cases and 10% in sporadic cases |
| Büscher et al. [34] | 2012 | FSGS and NS cases with ESRD (data were obtained from a waiting list for renal transplantation); exclusion of patients with known causes of secondary FSGS | 26 cases | NPHS1, NPHS2, TRPC6, CD2AP, INF2, WT1, ACTN4 | 8% of cases |
| Laurin et al. [35] | 2014 | Sporadic FSGS (exclusion of patients with known causes of secondary FSGS) | 37 cases | NPHS2, TRPC6, ACTN4, PLCE1, INF2 | 0% of cases |
| Aucella et al. [36] | 2005 | Sporadic FSGS | 33 cases | NPHS2, ACTN4 | 0% of cases |
| Zhang et al. [37] | 2013 | Familial FSGS (exclusion of patients with known causes of secondary FSGS) | 80 patients | ACTN4, TRCP6 | 2.5% of cases (no ACTN4 mutations were detected) |
| Xie et al. [38] | 2015 | Familial and sporadic FSGS | 40 families + 50 sporadic cases | COL4A3/COL4A4 | 12.5% in familial FSGS and 2% in sporadic FSGS |
| Malone et al. [39] | 2014 | Familial FSGS | 70 families | COL4A3/COL4A4 | 10% of families |
| Barua et al. [40] | 2013 | Sporadic and familial FSGS | 912 familial cases (215 families) and 281 sporadic cases | INF2 | 0.7% in sporadic cases and 12% of familial cases (9% of studied families) |
| Gbadegesin et al. [41] | 2012 | Sporadic and familial FSGS with disease segregation pattern consistent with autosomal dominant transmission (exclusion of secondary causes of FSGS and known mutations) | 49 families and 31 sporadic cases | INF2 | 0% in sporadic cases and 16% in familial cases |
| Boyer et al. [42] | 2011 | Familial FSGS and sporadic FSGS (exclusion of patients with known causes of secondary FSGS) | 78 familial cases (54 families) and 84 sporadic cases | INF2 | 17% of studied families and 1% of sporadic cases |
| Santin et al. [43] | 2009 | Sporadic and familial SRNS | 52 cases | NPHS1 | 2% of cases |
| Machuca et al. [44] | 2009 | Sporadic and familial SRNS | 119 cases | NPHS2 (R229Q allele + other pathogenetic mutation) | 10% of cases |
| He et al. [45] | 2007 | Sporadic and familial FSGS (exclusion of patients with known causes of secondary FSGS) | 87 cases | NPHS2 | 1.15% of causes |
| Tsukaguchi et al. [30] | 2002 | Familial FSGS with disease segregation pattern consistent with autosomal recessive transmission and sporadic FSGS (exclusion of patients with known causes of secondary FSGS) | 30 families + 91 sporadic cases | NPHS2 | 30% of families studied and 12% of sporadic cases |
| Santin et al. [46] | 2009 | Familial and sporadic FSGS | 55 cases | TRPC6 | 3.5% of cases |
| Zhu et al. [47] | 2009 | Familial FSGS | 31 families | TRPC6 | 3% of families |
Sadowski et al. [31] found a monogenic cause in 21.4% of the cases with SRNS and onset between 19 and 25 years of age. All the biopsies from patients with a proven mutation showed a histologic pattern of FSGS [27]. According to this study, monogenic causes are not uncommon in young adults with steroid-resistant FSGS and performing genetic screening appears to be worthwhile. Similar data come from a Spanish study in which a pathogenic mutation was found in 14% of 48 adult SRNS cases [6]. When cases were classified into familial (n = 7) versus sporadic (n = 41), genetic mutations were found more frequently in familial cases (43%) versus sporadic cases (10%). Despite the small sample size, these epidemiological studies suggest a potential role for genetic screening in adults with SRNS and identifying the appropriate patient population even in the absence of a family history. These criteria include absence of an obvious cause of secondary FSGS and more importantly resistance to immunosuppressive therapy.
The information regarding the prevalence of genetic forms in those cases of FSGS non-associated with NS is extremely conflicting. Gast et al. [18] found definite or probable pathogenic mutations in 33% of cases; on the other hand, different authors report a prevalence ranging from 0 to 8% [34–36].
Adult-onset FSGS has been associated with mutations in a limited number of genes. NPHS2 is the gene most frequently involved, with an autosomal recessive pattern of inheritance and a reported prevalence that ranges from 4% to 30% in familial cases [30, 31, 44, 48] and from 0 to 11% in sporadic cases [30, 31, 44, 45, 48]. In the Western European population, the most common genotype associated with adult-onset FSGS is a compound heterozygosity in the NPHS2 gene, with combination of R229Q allele and pathogenic NPHS2 mutations accounting for almost 95% of cases of podocin gene mutation [49]. R229Q allele has a frequency of about 3% in the Western European population and may act as a disease modifier that would predispose individuals to developing NS after a renal insult [44]. In fact, early-onset FSGS has been described when the R229Q allele is associated with specific gene mutations such as L327F or A297V [50]. On the other hand, INF2 mutations represent the most common cause of autosomal dominant FSGS, accounting for up to 17% of familial cases an 1% of sporadic cases [40–42]. TRPC6 mutations have been detected with prevalence up to 15% in familial cases and 2.5% in sporadic cases [6, 31, 34, 37, 47]. Mutations in other genes, like ACTN4, CD2AP and ANLN, are rare causes of adult-onset FSGS and have been described almost exclusively in familial cases [51–55].
Genetic, clinical and renal histology correlations
Classification of FSGS into primary, secondary and genetic forms is the cornerstone of a correct clinical and therapeutic approach, allowing the physician to appropriately choose between immunosuppressive or conservative treatment. Distinction between primary and secondary forms of FSGS is based on clinical presentation, EM examination and presence or absence of causative factors. Clinically, primary FSGS is typically characterized by full NS (urine protein >3.5 g/24 h and serum albumin <3.5 g/dL). A similar clinical presentation may be observed in a small subset of secondary FSGS due to certain drugs or infections with toxic effects on podocytes. On the other hand, patients with secondary FSGS unrelated to drugs or infections, typically present with progressively increasing proteinuria that may be in the nephrotic range, but without developing NS [1]. Ultrastructural examination of foot process effacement on EM is also important in distinguishing primary from secondary FSGS. Deegens et al. [3] analyzed the differences in foot processes width between patients with primary versus secondary FSGS and found the effacement to be most severe in cases of primary FSGS, with foot processes relatively preserved in secondary cases and little overlap between the two. Thus, the degree of foot process effacement by EM is a crucial clue as to whether or not the patient has the primary versus secondary form of FSGS, with some exceptions of secondary FSGS such as cases of ‘collapsing’ FSGS due to HIV, interferon or pamidronate therapy and genetic forms (see below) where diffuse foot process effacement can be present on EM. The combination of clinical presentation and EM examination has been proposed as a diagnostic tool in order to discriminate between primary and secondary FSGS [1, 4, 56].
Clinical features of genetic FSGS have been extensively described in the childhood-onset form, which typically presents with full NS and is usually resistant to immunosuppression. In some cases, SRNS may be associated with extra-renal manifestation as can be seen with different syndromic forms of genetic FSGS (Table 1). On the other hand, the clinical and renal histology features of adult-onset genetic FSGS have not been clearly described. In the small number of studies that have evaluated the clinical features of adult-onset genetic FSGS there appears to be an extreme heterogeneity in clinical presentation. This is likely explained by the fact that the genes that are mutated have different functions in the podocyte and affect the glomerular filtration barrier differently. Table 3 and Figure 1 show the clinical and renal histology features of two cases of genetic FSGS, one with compound heterozygous NPHS2 mutation and one with heterozygous PLCE1 mutation. Clinically both cases presented very differently in that the first case presented with chronic kidney disease (CKD) Stage 4, NS and diffuse foot process effacement, whereas the second case presented with normal renal function in the absence of NS and only partial foot process effacement.
Table 3.
Characteristics of six cases of adult-onset genetic FSGS referred to Division of Nephrology and Hypertension, Mayo Clinic, Rochester
| Patient | Genotype | Age of onset (years) | Nephrotic syndrome | Proteinuria at presentation (g/day) | Foot process effacement on EM | Potential causes for secondary FSGS | Progression toward ESRD (last creatinine clearance) | Familial history of FSGS |
|---|---|---|---|---|---|---|---|---|
| Patient 1 | NPHS2 gene (compound heterozygous R229Q+ R285fx302X) | 22 | Yes | 5.8 | Diffuse (90%) | No | CKD Stage 4 at the age of 51 years (29 mL/min) | No |
| Patient 2 | Heterozygous mutation in PLCE1 (c.1495C>T) | 25 | No | 2.0 | Segmental | No | Normal renal function at the age of 39 years (84 mL/min) | Yes |
| Patient 3 | NPHS2 gene (compound heterozygous R229Q+ p.R286fs) | 58 | Yes | 4.8 | Diffuse (85%) | No | Normal renal function at the age of 66 years (70 mL/min) | Yes |
| Patient 4 | NPHS2 gene (compound heterozygous R229Q+ p.R286fs) | 47 | No | 2.7 | Segmental | No | Normal renal function at the age of 55 years (85 mL/min) | Yes |
| Patient 5 | INF2 gene (heterozygous mutation R213H on exon 4) | 32 | No | 7.0 | Segmental | No | Preemptive kidney transplant at the age of 48 years | Yes |
| Patient 6 | COL4A3 gene 4981C>T (Arg1661Cys) (heterozygous) + NPHS1 gene c.133G>C (Glu45Gln) (heterozygous) | 33 | No | 3.6 | Diffuse | No | CKD Stage 3 at the age of 43 years (48 mL/min) | Yes |
Fig. 1.
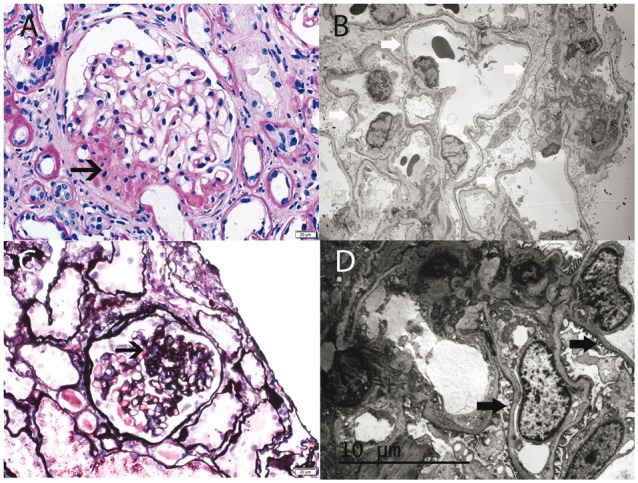
(A, B) Focal segmental glomerulosclerosis; 22 year old Caucasian woman with a clinical picture of NS (24 h urine protein 5.8 g, serum albumin 3.3 g/dL) and impaired renal function (serum creatinine 1.5 mg/dL). Genetic analysis showed compound heterozygous mutation in the NPHS2 gene (see Table 3, patient 1). (A) Light microscopy showing segmental sclerosis (see black arrow). (B) EM showing diffuse foot process effacement; white thick arrows point to areas of foot process effacement. (A, periodic acid–Schiff ×40, B ×3500). (C, D) Focal segmental glomerulosclerosis; 25 year old Caucasian man with normal renal function (serum creatinine 1.0 mg/dL), sub-nephrotic proteinuria (2 g/24 h) and normal serum albumin (4 g/dL). Genetic analysis showed heterozygous mutation in the PLCE1 gene (see Table 3, patient 2). (C) Light microscopy showing segmental sclerosis (see black arrow). (D) EM showing only minimal foot process effacement; thick black arrows point to preserved foot processes (C, periodic acid–Schiff ×40, D, ×6000).
In childhood, mutations of podocin (NPHS2) lead to a widespread damage of the slit diaphragm, due to the crucial function of this protein in nephrin recruitment. Therefore, SRNS with onset in childhood is the typical presentation of homozygote missense or nonsense mutation in the NPHS2 gene [49]. On the other hand, compound heterozygote mutation with the R229Q variant is usually associated with onset in adulthood. The clinical pattern of adult-onset disease induced by this compound heterozygosity is characterized by SRNS and diffuses foot process effacement by EM [44, 57]. This genotype–phenotype correlation can be, however, extremely variable: Table 3 and Figure 2 provide a description of two siblings referred to our Clinic (patients 3 and 4). Despite the same genotype, they had different clinical features (NS with diffuse foot process effacement in one versus non-nephrotic proteinuria and partial foot process effacement in the other). This example highlights how the complex relation between genotype, environment and likely epigenetic phenomena is responsible for a wide variability in the phenotype.
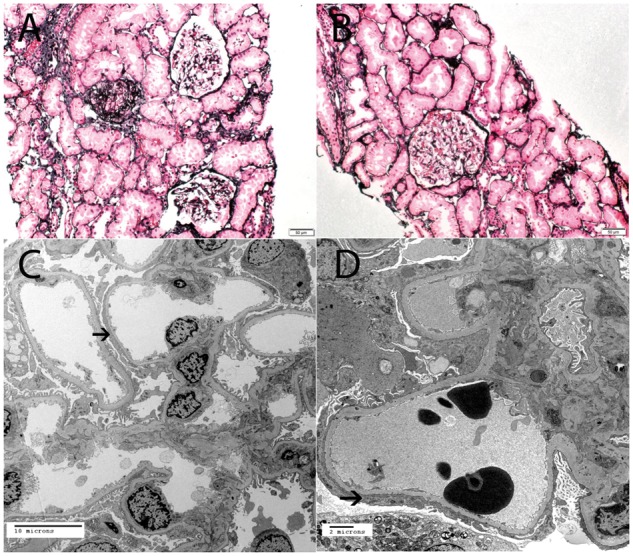
Fig. 2.

The figure shows EM from two siblings with genetic FSGS who share the same genotype (compound heterozygous R229Q + p.R286fs in the NPHS2 gene). (A, B) Focal segmental glomerulosclerosis; 58 year old Caucasian man with normal renal function (serum creatinine 1.1 mg/dL) and NS (urine protein 4.8 g/24 h, serum albumin 3.4 g/dL). EM shows diffuse foot process effacement, involving more than 80% of capillary loops; thin black arrows point to foot process effacement. See Table 3 for clinical information (patient 3) (A, ×6000 and B, ×5000). (C, D) Focal segmental glomerulosclerosis; 47 year old Caucasian woman with normal renal function (serum creatinine 0.8 mg/dL), sub-nephrotic proteinuria (2.7 g/24 h) and normal albumin (4.0 g/dL). EM shows segmental podocyte foot processes; thin black arrows point to foot process effacement and thick black arrow points to preserved foot processes. See Table 3 for clinical information (patient 4) (C, ×2900 and D, ×4800).
Mutations that involve other genes expressed at the slit diaphragm are associated with a less well-defined phenotype. Studies on TRPC6 mutations show heterogenous presentations: some cases series describe a significant prevalence of full NS [46, 58, 59], while nephrotic-range proteinuria without NS in the only clinical pattern described in cases from Hofstra et al. [60] and Zhu et al. [47]. Interestingly, those few case reports that provide a description of EM in cases of TRPC6 mutations revealed diffuse foot process effacement [61, 62]. Mutations in PLCE1 are almost exclusively described in childhood-onset SRNS, with an autosomal recessive pattern of inheritance. However, our patient with a PLCE1 mutation, described in Table 3 (patient 2), showed FSGS without NS and completely preserved renal function even 14 years after the onset and with only segmental foot process effacement on EM. Interestingly, his family history was positive for FSGS leading to ESRD (two siblings), with an autosomal dominant pattern of inheritance. In our case, PLCE1 mutation was heterozygous, which was similar to that described in a case of adult-onset FSGS by Laurin et al. [35].
INF2 and ACTN4 products are regulatory and structural cytoskeleton proteins, respectively. Their mutation leads to podocyte cytoskeleton damage and subsequent slit diaphragm alteration. The indirect mechanism that induces slit diaphragm damage is likely responsible for the partial foot process effacement that has been described in patients with INF2 [40–42, 61] and ACTN4 [62–64] mutations. Henderson et al. [63] evaluated the degree of foot process effacement in cases of ACTN4-related FSGS versus primary FSGS versus secondary FSGS. While primary FSGS cases showed the highest degree of foot process effacement, both ACTN4-related FSGS and adaptive FSGS cases presented with segmental foot process effacement [63]. Moreover, in patients who had cytoskeleton protein mutation, some peculiar features such as irregularly aggregated electron-dense material in the podocyte cytoplasm were noted on EM [63]. Clinically, the majority of these forms of genetic FSGS present with nephrotic-range proteinuria and show a low prevalence of NS [40, 63]. Figure 3 shows the renal pathology from a case of INF2-related FSGS (patient 5 in Table 3), with segmental foot process effacement on EM. The patient had a strong family history of FSGS (father and brother) suggestive of autosomal dominant pattern of inheritance. At the age of 32 years, this patient developed nephrotic-range proteinuria (7 g/day) with normal serum albumin (4 g/dL) and normal renal function (creatinine clearance 90 mL/min). Genetic tests revealed heterozygous mutation R213H on exon 4 of the INF2 gene. Despite a maximized anti-proteinuric approach, he had progressive loss of renal function and ultimately developed ESRD, at which point he underwent preemptive kidney transplantation at 48 years of age.
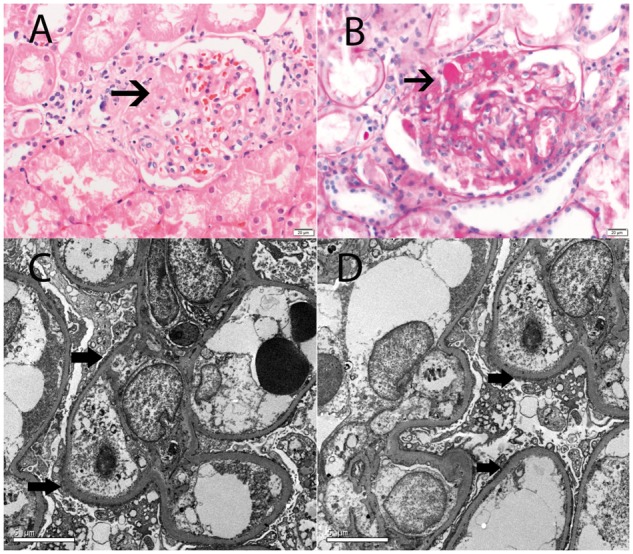
Fig. 3.
Focal global glomerulosclerosis; 32 year old Caucasian man with nephrotic-range proteinuria (7 g/day), normal serum albumin (4 g/dL) and normal renal function (creatinine clearance 90 mL/min). Genetic tests revealed heterozygous mutation R213H on exon 4 of the INF2 gene (see Table 3, patient 5). (A, B) Light microscopy showing mild focal global glomerulosclerosis (silver methenamine A, ×20, B, ×40). (C, D) EM showing segmental foot process effacement; black arrow points to segmental foot process effacement (C, ×3500 and D, ×6000).
Type IV collagen gene (COL4A3, COL4A4, COL4A5) mutations have been recently detected in up to 38% of familial forms of FSGS and 3% of sporadic forms [18]. Despite the fact that these genes are well known to be associated with Alport’s syndrome, in the study by Gast et al. [18] on the basis of clinical and histological features, only one case (out of eight) was suspected to have Alport’s syndrome. Clinical presentation was characterized by nephrotic-range proteinuria, and only one patient presented with NS. Even though microhematuria was present in the majority of the patients (62.5%), hearing loss at presentation was uncommon (one case) and EM showed the characteristic thickening, fraying and laminations of the GBM in only one case. Recently, Sen et al. [33] published results from 302 patients referred to their Institution for diagnostic gene analysis. Of the 302 patients, 267 were referred for NS and 35 for proteinuria and hematuria with suspicion for Alport syndrome; 11 genes were studied, including COL4A3, COL4A4 and COL4A5. Among the 46 patients with adult-onset SRNS, genetic testing was positive in 21.7%. Moreover, 6% of the patients with adult-onset SRNS and family history of renal disease who had a renal biopsy suggestive of FSGS had genetic testing that was positive for mutations in COL4A3, COL4A4 or COL4A5. In these cases, Alport syndrome was not suspected clinically or based on the renal biopsy findings. Unfortunately, histological data (including degree of foot process effacement on EM) and complete clinical data such as degree of proteinuria and serum albumin levels were not provided [33]. These data support the need to extend genetic testing in FSGS patients to include type IV collagen genes, considering that more than 5% of patients had genetic mutations in collagen IV genes that were not suspected based on clinical or histological findings. Patient 6 (Table 3) represents an interesting case of familial adult-onset FSGS, where genetic analysis showed heterozygote mutation in the COL4A3 gene for the c.4981C>T variant. This variant has been reported to be pathogenic for autosomal recessive Alport syndrome when present together with another pathogenic variant. Malone et al. [39] have reported the same heterozygote variant in a family with FSGS. In patient 6, the interpretation of the genotype–phenotype correlation was even more challenging since there was also a heterozygous mutation in the NPHS1 gene (c.133G>C). This variant has never been reported to cause disease and based on three different prediction programs, functional evidence of its pathogenicity has been suspected but not proven. This case highlights the possibility that heterozygote mutations in two different genes could represent two different ‘hits’ that when combined are sufficient to induce diffuse podocyte injury (see Figure 4).
Fig. 4.
Focal segmental glomerulosclerosis; 33 year old Caucasian man with impaired renal function (serum creatinine 1.8 mg/dL), nephrotic-range proteinuria (3.6 g/24 h) and normal albumin (4.3 g/dL). Genetic analysis showed heterozygosis in the COL4A3 gene for the variant c.4981C>T and heterozygous in the NPHS1 for a sequence variant designated c.133G>C. (A, B) Light microscopy showing segmental glomerulosclerosis sclerosis (thin black arrow: A, hematoxylin and eosin ×40 and B, periodic acid–Schiff ×40). (C, D) EM showing widespread foot process effacement; thick black arrow points to foot process effacement (C, D ×4500) (see Table 3, patient 6).
Genetic screening in FSGS: when, why and how?
As highlighted above, the prevalence of the monogenic form of adult-onset FSGS is likely underestimated. The clinical and histological phenotype of adult-onset genetic FSGS is widely variable and rarely specific. Therefore, identifying patients with a likelihood of genetic cause of FSGS remains a challenge for the clinician, highlighting the need for an inclusive evaluation of clinical and pathological features. A positive familial history of renal disease is the most obvious clue to suspect a genetic cause of FSGS. The autosomal dominant pattern of inheritance (with the exception of NPHS2-related forms) is typically associated with a positive family history in patients with adult-onset genetic FSGS. However, a significant number of patients with adult-onset genetic FSGS do not have a known family history of renal disease, because many of these mutations have an incomplete penetrance [41, 46], highlighting the fact that absence of family history should not dissuade the clinician to pursue genetic testing if there are other clinical features that suggest a potential genetic cause of FSGS.
Genetic FSGS should be suspected if the patient’s clinical picture is not typical for either primary or secondary FSGS. For example, in a patient suspected to have primary FSGS based on clinical presentation of NS, the absence of response to immunosuppression should raise the suspicion of a genetic form of FSGS. Resistance to steroids is widely described in the genetic forms, while response to calcineurin inhibitor in adult-onset genetic FSGS has been reported only anecdotally [43, 65]. Moreover, these studies reported only partial clinical, histological and genetic features of those rare patients who responded to calcineurin inhibitor. Santin et al. [43] described a 27 year-old patient, with NPHS1 mutation, who had a partial response to immunosuppression (proteinuria decreased to 3 g/24 h). EM and proteinuria at the time of diagnosis were not reported in this patient. Similarly, Büscher et al. [34] reported complete or partial remission in six cases of genetic SRNS treated with cyclosporin but the exact phenotype of the responders and the EM findings were not described. Therefore, assessment of the true response of immunosuppression in these patients is difficult. The role of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in the treatment of proteinuria in genetic form of FSGS has not been widely studied. The paucity of information comes from small case series and supports a role for these medications in the management of proteinuria [6]. On the other hand, in patients who are suspected to have secondary FSGS (lack of NS and partial foot process effacement on EM), absence of an evident cause should be a clue to the clinician to suspect a genetic form of FSGS. We therefore recommend screening patients for genetic causes in the following scenarios:
Adult-onset FSGS with a family history of FSGS.
Primary FSGS resistant to immunosuppressive treatment.
Secondary FSGS without an obvious cause being identified.
Adult-onset FSGS, with clinical and pathological features that do not support a primary or a secondary form.
Genetic testing can be expensive and one may deliberate whether to pursue genetic testing in patients who are suspected to have a genetic form of FSGS. We believe that identifying whether a patient has a genetic form of FSGS is of utmost importance. First, it would provide solid grounds to avoid immunosuppressive therapy, which is unlikely to be effective and may be associated with significant toxicity. Secondly, another major clinical benefit of diagnosing a monogenic form of FSGS is its implication in kidney transplantation. The rate of recurrence of FSGS post-renal transplantation is reported to be approximately 30%. These data would be even higher with rigorous selection of patients classified as primary FSGS, with recurrence rate reaching up to 75% especially in those who progress to ESRD within 3 years of diagnosis [66, 67]. On the other hand, recurrence of disease after transplantation in genetic forms of FSGS is extremely rare. This has been shown in several studies, with recurrence rate ranging from 0 to 2.5% [44, 60, 68–70]. In addition, recognizing that the patient has a genetic form of FSGS will have an impact on identifying the appropriate donor as family members may need to be screened prior to donation. In fact, studies have reported cases of donors developing FSGS in the remaining kidney after donation [70], emphasizing the importance of genetic screening in the patients and their potential donors who are suspected to have a genetic form of FSGS [71].
In patients with adult-onset FSGS and a positive family history, the prevalence of a genetic cause can reach up to 40%. Therefore, several authors propose a first-level genetic screening that should include NPHS2, ACTN4, TRPC6 and INF2 in this patient population [6, 71]
In our opinion, for the patients with positive family history (group 1) and for those without a family history (groups 2–4) a more inclusive genetic screening, which includes all genes associated with FSGS, should be considered. Progress in genetic testing has resulted in the development of cheaper commercial tests for genes associated with FSGS. A negative genetic screening in a case with strong clinical suspicion of genetic FSGS should not deter the clinician from further evaluating the patient. These patients should be candidates for whole-exome sequencing examination. In a recent study, Warejko et al. [72] used whole-exome sequencing to detect monogenic cause of SRNS in a cohort of 300 families previously studied using panel sequencing. Interestingly, a causative mutation in a known SRNS gene was detected in 28.5% of cases (similar results were obtained using panel sequencing) and one or more potential novel candidates genes were identified in 28% of patients. Considering the increasing number of new candidate genes identified in the recent past, use of whole-exome sequencing could be a cost effective alternative to continuous updating of genetic panels. Improving whole exome sequencing technique and identifying novel genes will allow this rapidly expanding field to move forward [73] (Table 4).
Table 4.
|
In summary, genetic forms of FSGS presenting in adults are not as uncommon as once thought and clinicians should maintain a high level of suspicion for a genetic cause when evaluating and treating patients with FSGS. If a patient has NS and is resistant to therapy, or a patient is suspected to have secondary FSGS but no clear cause can be identified and certainly in cases with a positive family history, genetic testing must be pursued. Only by persistently pursuing genetic tests in these patients will we be able to assess the true role of genetic mutation in cases of adult-onset FSGS.
Funding
Supported in part by the Dieter H. and Eva Kruger Research Fund.
Authors’ contributions
All authors were involved and approved the final manuscript.
Conflict of interest statement
None declared.
References
- 1. Sethi S, Glassock RJ, Fervenza FC.. Focal segmental glomerulosclerosis: towards a better understanding for the practicing nephrologist. Nephrol Dial Transplant 2015; 30: 375–384 [DOI] [PubMed] [Google Scholar]
- 2. D’Agati VD, Kaskel FJ, Falk RJ.. Focal segmental glomerulosclerosis. N Engl J Med 2011; 365: 2398–2411 [DOI] [PubMed] [Google Scholar]
- 3. Deegens JKJ, Dijkman HBPM, Borm GF. et al. Podocyte foot process effacement as a diagnostic tool in focal segmental glomerulosclerosis. Kidney Int 2008; 74: 1568–1576 [DOI] [PubMed] [Google Scholar]
- 4. Hommos MS, De Vriese AS, Alexander MP. et al. The incidence of primary versus secondary focal segmental glomerulosclerosis: a clinicopathological study. Mayo Clin Proc 2017; 92: 1772–1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lovric S, Ashraf S, Tan W. et al. Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol Dial Transplant 2016; 31: 1802–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Santin S, Bullich G, Tazon-Vega B. et al. Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 2011; 6: 1139–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brown EJ, Pollak MR, Barua M.. Genetic testing for nephrotic syndrome and FSGS in the era of next-generation sequencing. Kidney Int 2014; 85: 1030–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kestilä M, Lenkkeri U, Männikkö M. et al. Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell 1998; 1: 575–582 [DOI] [PubMed] [Google Scholar]
- 9. Machuca E, Benoit G, Nevo F. et al. Genotype–phenotype correlations in non-Finnish congenital nephrotic syndrome. J Am Soc Nephrol 2010; 21: 1209–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hinkes B, Vlangos C, Heeringa S. et al. Specific podocin mutations correlate with age of onset in steroid-resistant nephrotic syndrome. J Am Soc Nephrol 2008; 19: 365–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Winn MP, Conlon PJ, Lynn KL. et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005; 308: 1801–1804 [DOI] [PubMed] [Google Scholar]
- 12. Gbadegesin R, Hinkes BG, Hoskins BE. et al. Mutations in PLCE1 are a major cause of isolated diffuse mesangial sclerosis (IDMS). Nephrol Dial Transplant 2008; 23: 1291–1297 [DOI] [PubMed] [Google Scholar]
- 13. Shirato I, Sakai T, Kimura K. et al. Cytoskeletal changes in podocytes associated with foot process effacement in Masugi nephritis. Am J Pathol 1996; 148: 1283–1296 [PMC free article] [PubMed] [Google Scholar]
- 14. Rheault MN, Gbadegesin RA.. The genetics of nephrotic syndrome. J Pediatr Genet 2016; 5: 15–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Has C, Spartà G, Kiritsi D. et al. Integrin alpha3 mutations with kidney, lung, and skin disease. N Engl J Med 2012; 366: 1508–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matejas V, Hinkes B, Alkandari F. et al. Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum Mutat 2010; 31: 992–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kambham N, Tanji N, Seigle RL. et al. Congenital focal segmental glomerulosclerosis associated with beta4 integrin mutation and epidermolysis bullosa. Am J Kidney Dis 2000; 36: 190–196 [DOI] [PubMed] [Google Scholar]
- 18. Gast C, Pengelly RJ, Lyon M. et al. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol Dial Transplant 2016; 31: 961–970 [DOI] [PubMed] [Google Scholar]
- 19. Chatterjee R, Hoffman M, Cliften P. et al. Targeted exome sequencing integrated with clinicopathological information reveals novel and rare mutations in atypical, suspected and unknown cases of Alport syndrome or proteinuria. PLoS One 2013; 8: e76360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barbaux S, Niaudet P, Gubler MC. et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet 1997; 17: 467–470 [DOI] [PubMed] [Google Scholar]
- 21. Boerkoel CF, Takashima H, John J. et al. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet 2002; 30: 215–220 [DOI] [PubMed] [Google Scholar]
- 22. Sweeney E, Fryer A, Mountford R. et al. Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet 2003; 40: 153–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Braun DA, Sadowski CE, Kohl S. et al. Mutations in nuclear pore genes NUP93, NUP205 and XPO5 cause steroid-resistant nephrotic syndrome. Nat Genet 2016; 48: 457–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rosti RO, Sotak BN, Bielas SL. et al. Homozygous mutation in NUP107 leads to microcephaly with steroid-resistant nephrotic condition similar to Galloway–Mowat syndrome. J Med Genet 2017; 54: 399–403 [DOI] [PubMed] [Google Scholar]
- 25. Montini G, Malaventura C, Salviati L.. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N Engl J Med 2008; 358: 2849–2850 [DOI] [PubMed] [Google Scholar]
- 26. Yu HY, Artomov M, Braehler S. et al. A role for genetic susceptibility in sporadic focal segmental glomerulosclerosis (Retraction of vol. 126, pg 1067, 2016). J Clin Invest 2016; 126: 1603–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sampson MG, Robertson CC, Martini S. et al. Integrative genomics identifies novel associations with APOL1 risk genotypes in black NEPTUNE subjects. J Am Soc Nephrol 2016; 27: 814–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kopp JB, Winkler CA, Zhao X. et al. Clinical features and histology of apolipoprotein L1-associated nephropathy in the FSGS clinical trial. J Am Soc Nephrol 2015; 26: 1443–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hall G, Gbadegesin RA, Lavin P. et al. A novel missense mutation of Wilms’ Tumor 1 causes autosomal dominant FSGS. J Am Soc Nephrol 2015; 26: 831–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tsukaguchi H, Sudhakar A, Le TC. et al. NPHS2 mutations in late-onset focal segmental glomerulosclerosis: R229Q is a common disease-associated allele. J Clin Invest 2002; 110: 1659–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sadowski CE, Lovric S, Ashraf S. et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 2015; 26: 1279–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hinkes BG, Mucha B, Vlangos CN. et al. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics 2007; 119: e907–19 [DOI] [PubMed] [Google Scholar]
- 33. Sen ES, Dean P, Yarram-Smith L. et al. Clinical genetic testing using a custom-designed steroid-resistant nephrotic syndrome gene panel: analysis and recommendations. J Med Genet 2017; 54: 795–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Büscher AK, Konrad M, Nagel M. et al. Mutations in podocyte genes are a rare cause of primary FSGS associated with ESRD in adult patients. Clin Nephrol 2012; 78: 47–53 [DOI] [PubMed] [Google Scholar]
- 35. Laurin LP, Lu M, Mottl AK. et al. Podocyte-associated gene mutation screening in a heterogeneous cohort of patients with sporadic focal segmental glomerulosclerosis. Nephrol Dial Transplant 2014; 29: 2062–2069 [DOI] [PubMed] [Google Scholar]
- 36. Aucella F, De Bonis P, Gatta G. et al. Molecular analysis of NPHS2 and ACTN4 genes in a series of 33 Italian patients affected by adult-onset nonfamilial focal segmental glomerulosclerosis. Nephron Clin Pract 2005; 99: c31–c36 [DOI] [PubMed] [Google Scholar]
- 37. Zhang Q, Ma J, Xie J. et al. Screening of ACTN4 and TRPC6 mutations in a Chinese cohort of patients with adult-onset familial focal segmental glomerulosclerosis. Contrib Nephrol 2013; 181: 91–100 [DOI] [PubMed] [Google Scholar]
- 38. Xie J, Wu X, Ren H. et al. COL4A3 mutations cause focal segmental glomerulosclerosis. J Mol Cell Biol 2015; 7: 184. [DOI] [PubMed] [Google Scholar]
- 39. Malone AF, Phelan PJ, Hall G. et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int 2014; 86: 1253–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Barua M, Brown EJ, Charoonratana VT. et al. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int 2013; 83: 316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gbadegesin RA, Lavin PJ, Hall G. et al. Inverted formin 2 mutations with variable expression in patients with sporadic and hereditary focal and segmental glomerulosclerosis. Kidney Int 2012; 81: 94–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boyer O, Benoit G, Gribouval O. et al. Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. J Am Soc Nephrol 2011; 22: 239–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Santin S, Garcia-Maset R, Ruiz P. et al. Nephrin mutations cause childhood- and adult-onset focal segmental glomerulosclerosis. Kidney Int 2009; 76: 1268–1276 [DOI] [PubMed] [Google Scholar]
- 44. Machuca E, Hummel A, Nevo F. et al. Clinical and epidemiological assessment of steroid-resistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney Int 2009; 75: 727–735 [DOI] [PubMed] [Google Scholar]
- 45. He N, Zahirieh A, Mei Y. et al. Recessive NPHS2 (Podocin) mutations are rare in adult-onset idiopathic focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 2007; 2: 31–37 [DOI] [PubMed] [Google Scholar]
- 46. Santin S, Ars E, Rossetti S. et al. TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol Dial Transplant 2009; 24: 3089–3096 [DOI] [PubMed] [Google Scholar]
- 47. Zhu B, Chen N, Wang ZH. et al. Identification and functional analysis of a novel TRPC6 mutation associated with late onset familial focal segmental glomerulosclerosis in Chinese patients. Mutat Res 2009; 664: 84–90 [DOI] [PubMed] [Google Scholar]
- 48. Tonna SJ, Needham A, Polu K. et al. NPHS2 variation in focal and segmental glomerulosclerosis. BMC Nephrol 2008; 9: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bouchireb K, Boyer O, Gribouval O. et al. NPHS2 mutations in steroid-resistant nephrotic syndrome: a mutation update and the associated phenotypic spectrum. Hum Mutat 2014; 35: 178–186 [DOI] [PubMed] [Google Scholar]
- 50. Phelan PJ, Hall G, Wigfall D. et al. Variability in phenotype induced by the podocin variant R229Q plus a single pathogenic mutation. Clin Kidney J 2015; 8: 538–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weins A, Kenlan P, Herbert S. et al. Mutational and biological analysis of alpha-actinin-4 in focal segmental glomerulosclerosis. J Am Soc Nephrol 2005; 16: 3694–3701 [DOI] [PubMed] [Google Scholar]
- 52. Gbadegesin RA, Hall G, Adeyemo A. et al. Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J Am Soc Nephrol 2014; 25: 1991–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gigante M, Pontrelli P, Montemurno E. et al. CD2AP mutations are associated with sporadic nephrotic syndrome and focal segmental glomerulosclerosis (FSGS). Nephrol Dial Transplant 2009; 24: 1858–1864 [DOI] [PubMed] [Google Scholar]
- 54. Tsvetkov D, Hohmann M, Anistan YM. et al. A CD2AP mutation associated with focal segmental glomerulosclerosis in young adulthood. Clin Med Insights Case Rep 2016; 9: 15–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kaplan JM, Kim SH, North KN. et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 2000; 24: 251–256 [DOI] [PubMed] [Google Scholar]
- 56. Sethi S, Zand L, Nasr SH. et al. Focal and segmental glomerulosclerosis: clinical and kidney biopsy correlations. Clin Kidney J 2014; 7: 531–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ardiles LG, Carrasco AE, Carpio JD. et al. Late onset of familial nephrotic syndrome associated with a compound heterozygous mutation of the podocin-encoding gene. Nephrology (Carlton) 2005; 10: 553–556 [DOI] [PubMed] [Google Scholar]
- 58. Heeringa SF, Möller CC, Du J. et al. A novel TRPC6 mutation that causes childhood FSGS. PLoS One 2009; 4: e7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liakopoulos V, Huerta A, Cohen S. et al. Familial collapsing focal segmental glomerulosclerosis. Clin Nephrol 2011; 75: 362–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hofstra JM, Lainez S, van Kuijk WHM. et al. New TRPC6 gain-of-function mutation in a non-consanguineous Dutch family with late-onset focal segmental glomerulosclerosis. Nephrol Dial Transplant 2013; 28: 1830–1838 [DOI] [PubMed] [Google Scholar]
- 61. Xie J, Hao X, Azeloglu EU. et al. Novel mutations in the inverted formin 2 gene of Chinese families contribute to focal segmental glomerulosclerosis. Kidney Int 2015; 88: 593–604 [DOI] [PubMed] [Google Scholar]
- 62. Pollak MR, Alexander MP, Henderson JM.. A case of familial kidney disease. Clin J Am Soc Nephrol 2007; 2: 1367–1374 [DOI] [PubMed] [Google Scholar]
- 63. Henderson JM, Alexander MP, Pollak MR.. Patients with ACTN4 mutations demonstrate distinctive features of glomerular injury. J Am Soc Nephrol 2009; 20: 961–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mathis BJ, Kim SH, Calabrese K. et al. A locus for inherited focal segmental glomerulosclerosis maps to chromosome 19q13. Kidney Int 1998; 53: 282–286 [DOI] [PubMed] [Google Scholar]
- 65. Buscher AK, Beck BB, Melk A. et al. Rapid response to cyclosporin A and favorable renal outcome in nongenetic versus genetic steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 2016; 11: 245–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cosio FG, Cattran DC.. Recent advances in our understanding of recurrent primary glomerulonephritis after kidney transplantation. Kidney Int 2017; 91: 304–314 [DOI] [PubMed] [Google Scholar]
- 67. Hickson LJ, Gera M, Amer H. et al. Kidney transplantation for primary focal segmental glomerulosclerosis: outcomes and response to therapy for recurrence. Transplantation 2009; 87: 1232–1239 [DOI] [PubMed] [Google Scholar]
- 68. Jungraithmayr TC, Hofer K, Cochat P. et al. Screening for NPHS2 mutations may help predict FSGS recurrence after transplantation. J Am Soc Nephrol 2011; 22: 579–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Weber S, Gribouval O, Esquivel EL. et al. NPHS2 mutation analysis shows genetic heterogeneity of steroid-resistant nephrotic syndrome and low post-transplant recurrence. Kidney Int 2004; 66: 571–579 [DOI] [PubMed] [Google Scholar]
- 70. Conlon PJ, Lynn K, Winn MP. et al. Spectrum of disease in familial focal and segmental glomerulosclerosis. Kidney Int 1999; 56: 1863–1871 [DOI] [PubMed] [Google Scholar]
- 71. Rood IM, Deegens JK, Wetzels JF.. Genetic causes of focal segmental glomerulosclerosis: implications for clinical practice. Nephrol Dial Transplant 2012; 27: 882–890 [DOI] [PubMed] [Google Scholar]
- 72. Warejko JK, Tan W, Daga A. et al. Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 2017; doi: 10.2215/CJN.04120417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. De Vriese AS, Sethi S, Nath KA et al. Differentiating primary, genetic and secondary focal segmental glomerulosclerosis in adults: a clinico-pathological approach. J Am Soc Nephrol 2018 (in press) [Google Scholar]