

Abstract
Muscle fibers lacking dystrophin undergo a long-term alteration of Ca2+ homeostasis, partially caused by a leaky Ca2+ release ryanodine (RyR) channel. S48168/ARM210, an RyR calcium release channel stabilizer (a Rycal compound), is expected to enhance the rebinding of calstabin to the RyR channel complex and possibly alleviate the pathologic Ca2+ leakage in dystrophin-deficient skeletal and cardiac muscle. This study systematically investigated the effect of S48168/ARM210 on the phenotype of mdx mice by means of a first proof-of-concept, short (4 wk), phase 1 treatment, followed by a 12-wk treatment (phase 2) performed in parallel by 2 independent laboratories. The mdx mice were treated with S48168/ARM210 at two different concentrations (50 or 10 mg/kg/d) in their drinking water for 4 and 12 wk, respectively. The mice were subjected to treadmill sessions twice per week (12 m/min for 30 min) to unmask the mild disease. This testing was followed by in vivo forelimb and hindlimb grip strength and fatigability measurement, ex vivo extensor digitorum longus (EDL) and diaphragm (DIA) force contraction measurement and histologic and biochemical analysis. The treatments resulted in functional (grip strength, ex vivo force production in DIA and EDL muscles) as well as histologic improvement after 4 and 12 wk, with no adverse effects. Furthermore, levels of cellular biomarkers of calcium homeostasis increased. Therefore, these data suggest that S48168/ARM210 may be a safe therapeutic option, at the dose levels tested, for the treatment of Duchenne muscular dystrophy (DMD).—Capogrosso, R. F., Mantuano, P., Uaesoontrachoon, K., Cozzoli, A., Giustino, A., Dow, T., Srinivassane, S., Filipovic, M., Bell, C., Vandermeulen, J., Massari, A. M., De Bellis, M., Conte, E., Pierno, S., Camerino, G. M., Liantonio, A., Nagaraju, K., De Luca, A. Ryanodine channel complex stabilizer compound S48168/ARM210 as a disease modifier in dystrophin-deficient mdx mice: proof-of-concept study and independent validation of efficacy.
Keywords: skeletal muscle, Duchenne muscular dystrophy, preclinical drug testing, murine model, rycals
Duchenne muscular dystrophy (DMD) is the most common X-linked inherited disease (1), present in 1 in 5000 live male births in all populations, with 30% of the cases resulting from de novo mutation (2). The disease is caused by a mutation in the dystrophin gene that leads to the absence of dystrophin protein in skeletal muscle (3). Dystrophin is a subsarcolemmal protein that forms part of the link between the contractile machinery and the extracellular matrix. In the absence of dystrophin, the transmembrane dystrophin–glycoprotein complex (DGC) components are lost, and the stability of the sarcolemma is reduced (4, 5). The loss of this force transmission system results in an increased fragility and susceptibility to damage of the dystrophic muscle fibers (6,–9). It is thought that mechanical injury in dystrophic muscle occurring during contraction can result in an abnormal influx of extracellular calcium via an anomalous function of calcium-permeable channels in the sarcolemma of dystrophic muscles (10). This calcium influx, combined with oxidative stress and altered mitochondrial function, leads to a vicious cycle, worsening the handling of cytosolic calcium, and contributing to muscle dysfunction and progressive necrosis (11,–14).
The disturbance of Ca2+ homeostasis in DMD was initially thought to be a result of the fact that the total Ca2+ (free and bound Ca2+) content is doubled in the muscles of mdx mice (a mouse model of DMD) and was subsequently blamed on the observation that the influx of Ca2+ through the cell membrane is increased (15). This alteration in Ca2+ influx is part of an altered mechanotransduction mechanism (16) that has been demonstrated by means of microspectrofluorimetry and patch-clamp experiments in myofibers from chronically exercised mdx mice (17, 18). Moreover, sarcoplasmic reticulum (SR) Ca2+ reuptake is slowed in mdx myofibers, and an SR Ca2+ leak of uncertain etiology has been reported (16, 19). The rate of Ca2+ sparks in dystrophic fibers is increased, further suggesting that a defect in SR Ca2+ release occurs in muscular dystrophy (20). An elevated cytoplasmic Ca2+ concentration has been implicated in the pathophysiology of protein degradation and cell death in mdx muscle (21, 22), leading to the hypothesis that dystrophin-lacking fibers have chronic Ca2+ overload that results in the activation of Ca2+-dependent proteases, such as calpains (23, 24).
Bellinger et al. (19) have shown that ryanodine receptor (RyR)-1, the skeletal muscle isoform of the SR Ca2+ release channel, contributes to altered Ca2+ homeostasis in dystrophic muscles in the mdx mouse model of DMD. In addition, RyR1 isolated from mdx skeletal muscle shows an age-dependent increase in S-nitrosylation, coincident with dystrophic changes, enhanced oxidative stress, and altered NO signaling in muscle. RyR1 S-nitrosylation depletes the channel complex of the stabilizing protein calstabin-1, resulting in “leaky” channels (19). Such a mechanism could contribute to deregulated cytosolic calcium and may also deplete the SR, leading to a lowered availability of calcium for contraction and, finally, weakness. The association of calstabin-1 with RyR1 can be preserved by stabilizing compounds called Rycals. S107, a Rycal-stabilizing calstabin-1, which binds to the nitrosylated channel and inhibits SR Ca2+ leaks. A reduction in the biochemical and histologic evidence of muscle damage, improved muscle function, and increased exercise performance have been observed in young mdx mice treated for the short term (2 wk) with S107 (19).
Restoring RyR-calstabin binding and inhibiting Ca2+ leakage could form a strategy for the treatment of DMD. The compound S48168/ARM210 has been selected from a library of Rycal candidates for preclinical advancement as a potential treatment for DMD, based on its ability to stabilize the RyR channel complex, its oral bioavailability, and its distribution in skeletal muscles. The present study is the first preclinical assessment of the effects of S48168/ARM210 in the mdx mouse model of DMD. In particular, we investigated both the short-term (4 wk) and long-term (12 wk) effects of daily oral administration of S48168/ARM210 at two different doses in 5-wk-old exercised mdx mice, with primary pathology-related endpoints: forelimb and hindlimb strength by in vivo grip test, plasma levels of creatine kinase (CK), muscle contraction parameters [i.e., isometric contraction of diaphragm (DIA) strips and isometric and eccentric contraction of the extensor digitorum longus (EDL) muscle ex vivo], and muscle histopathology evaluation. Plasma and tissue drug levels were also evaluated to estimate the exposure of the animals. In the short-term proof-of-concept study, secondary endpoints were also assessed to validate the drug’s mechanism of action, and in the 12 wk study, we aimed at verifying the long-term efficacy of the test compound. These two trials were independently performed by two laboratories, both adhering to internationally recognized standard operating procedures (SOPs) for preclinical tests in mdx mice, to verify the comparability of the results and to further validate them.
MATERIALS AND METHODS
Animals and in vivo studies
Male mdx mice (4–5-wk-old; C57Bl/10ScSn-Dmd mdx) and male C57Bl/10ScSn control mice [4–5-wk-old; corresponding to wild-type (WT) mice] were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Animals were acclimatized for 1 wk in the animal facility before the beginning of the study.
All animals were examined and weighed before the initiation of the study and sorted into cages to be assigned to different experimental groups. A <5% difference in mean body weight was present among the experimental groups. The cages of mice were then randomly assigned to each treatment group.
All the researchers were blinded to treatment and genotype. Regular light–dark cycles (based on the dawn and dusk phasing) were maintained. The animals were housed in a group of 2 per cage at the Department of Pharmacy–Drug Sciences, University of Bari (hereafter referred to as UNIBA) or 5 per cage at Agada Biosciences Inc. (hereafter referred to as AGA). The laboratory where each test was performed is indicated in parentheses throughout the text. The functional tests were performed during the light-cycle phase. The room temperature was maintained at 20–25°C, with a relative humidity of 40–65%. Chow was provided ad libitum for the duration of the study. Normally, a mean of 4–5 g/d per mouse was added. Food consumption was checked daily. The composition of the food used in the two laboratories involved in the study is provided in Supplemental Table 1S.
Animals were provided with untreated drinking water (spring water, the vehicle) or spring water that contained the test compound S48168/ARM210 at the doses given below. Compound-treated drinking water and untreated drinking water were made available ad libitum. S48168/ARM210 was supplied by Servier (Suresnes, France) in powder form. The mean water consumption per animal per week was calculated based on the volume of water measured at the end of each week divided by the number of animals in each cage, normalized to the mean body weight of the animals in the cage. The selection of the dose levels of S48168/ARM210 (10 and 50 mg/kg per day) was based on the results of previous pharmacological studies in nonexercised mdx mice or other animal models, the dose of 10 mg/kg per day having affected the parameters of muscle function and force (Servier, personal communication, 2013).
The treatment duration was 4 wk (phase 1) or 12 wk (phase 2). The ages of the mice at the terminal endpoints were 8–10 wk for phase 1 and 16–18 wk for phase 2. Phase 1 was a proof-of-concept study and was performed exclusively in Prof. De Luca’s laboratory at UNIBA. For this study, 3 groups of 10 mdx mice were randomly allocated to the vehicle or drug treatment group, with age-matched WT mice (C57Bl/10ScSn; n = 6) used for strain control. The phase 2 experiments to verify and validate the drug’s long-term efficacy were independently performed at both Prof. De Luca’s laboratory at UNIBA (4 groups of mice) and at AGA. The AGA experiments were supervised by Prof. Nagaraju at Agada Biosciences, Inc. Both trials were performed at about the same time, with both laboratories having the knowledge that the identical trial was being performed elsewhere. Three groups of 8 (UNIBA) and 15 (AGA) mdx mice were randomly allocated to the vehicle or drug treatment group, with age-matched WT mdx mice (C57Bl/10ScSn; n = 8 at UNIBA and n = 15 at AGA) as a second control. With respect to the possibility of laboratory-related differences, strict adherence to SOPs was ensured as far as experimental protocols and primary clinically relevant readouts were concerned (i.e., in vivo and ex vivo force measurements and histology). This approach was pivotal in fulfilling the requirement for independent validation of the preclinical results and for verifying that SOP adherence allowed for a better comparison of data obtained in different laboratories (25, 26). As part of this approach, attention was given to the comparison between the data for mdx vehicle-treated mice and WT mice for the two laboratories in terms of the aforementioned parameters (i.e., a phenotypic effect comparison between the two laboratories). The comparison was then made between the mdx vehicle-treated mice and the mdx mice treated with one of the two doses of S48168/ARM210.
Treadmill exercise was used to unmask the mdx mouse phenotype and was performed as has been published (27, 28).
In vivo grip strength and fatigability measurements
In the phase 1 study (UNIBA), only forelimb grip strength was assessed; in the phase 2 study, forelimb grip (UNIBA and AGA) and hindlimb grip (AGA) strengths were both measured using a grip strength meter (Columbus Instruments, Columbus, OH, USA) (27, 28). Force was detected weekly, at least 48 h after the last treadmill session. Five measurements were taken for each mouse. In the AGA study, the grip strength measurements were collected once over a 5 d period after 12 wk of treatment, whereas in the UNIBA studies, the same measurements were performed weekly on the same day of the week. The maximum values for each measurement were used for subsequent analysis.
The exhaustion test was performed every 4 wk in each group for both the phase 1 and 2 studies (UNIBA). Groups of mice were made to run on a horizontal treadmill for 5 min at 5 m/min, followed by an increase in the speed of 1 m/min each minute. The total distance run by each mouse until exhaustion was measured. Exhaustion was defined as the inability of the mouse to continue running on the treadmill for 1 min, despite repeated gentle stimulation.
Ex vivo force measurement: contractile properties of EDL muscle and DIA
At the end of the in vivo phase, mice were deeply anesthetized with urethane (1.2 g/kg i.p.; UNIBA) or with ketamine and xylazine (80 and 10 mg/kg; AGA).
The EDL muscle of the right hindlimb was removed and securely tied with braided surgical silk at both tendon insertions. The entire DIA muscle and surrounding ribcage were rapidly excised and placed in a dish containing oxygenated Ringer’s solution at 25°C [pH 7.4; AGA: NaCl (137 mM), NaHCO3 (24 mM), Glucose (1 mM), KCl (5 mM), CaCl2 (2 mM), MgSO4 (1 mM), Tubocurarine (0.025 mM); UNIBA: NaCl (148 mM), KCl (4.5 mM), CaCl2 (2 mM), MgCl2 (1 mM), NaHCO3 (12 mM), NaHPO4 (0.44 mM), glucose (5.55 mM)] to microscopically isolate portions for examination. DIA strips composed of longitudinally arranged full-length muscle fibers were tied firmly with braided surgical silk at the central tendon and sutured through a portion of the rib bone affixed to the distal end of the strip. Then, each preparation was rapidly placed in an oxygenated muscle chamber (95% O2, 5% CO2) containing Ringer’s solution (pH 7.4) at 25°C (AGA) or 27°C (UNIBA) to perform the isometric and eccentric contraction recordings (29,–34).
In brief, after equilibration (30 min), each preparation was stretched to its optimal length (Lo, measured with an external caliper), the length producing the maximum, twitch in response to a 0.2 ms pulse of the maximum effective voltage (40–60 V). At Lo, the force developed was measured during trains of stimulation (EDL, 250 ms at AGA, 350 ms at UNIBA; DIA, 450 ms) with increasing frequencies, until the highest plateau was achieved. The force generated to obtain the highest plateau was established as the maximum force generated by the muscle.
At AGA, EDL muscle fatigue was measured with 60 isometric contractions for 300 ms each, once every 5 s, at 100 Hz. The force was recorded every minute during the repetitive contractions and at 5 and 10 min afterward to measure recovery. At UNIBA, EDL muscle fatigue was recorded with 2 min trains of 100 Hz tetani under isometric conditions and measured as the percentage of force reduction at the 5th and the 10th pulse, when compared with the first pulse. Full recovery from fatigue was assessed before starting the eccentric contraction protocol.
The eccentric contraction force drop in EDL muscle (UNIBA) was measured as described previously, using a mild protocol able to selectively impair muscle force in mdx muscle, while exerting no effect or only mild potentiation in WT muscle (30). An index of stiffness during each eccentric contraction was obtained by dividing the difference in stress produced between the isometric response and the peak stress resulting from the imposed stretch (i.e., change in stress) by the change in length of the muscle during the stretch [0.1 Lo](35).
Data were collected and analyzed using AcqKnowledge software v. 3.8 (Biopac System, Goleta, CA, USA) (UNIBA) or using the 605A: Dynamic Muscle Date Acquisition and Analysis System (Aurora Scientific, Richmond, BC, Canada) (UNIBA and AGA). At the end of the experiments, the samples were removed, trimmed of their tendons, blotted, and weighed on an analytical balance. Absolute values of tension were normalized by the cross-sectional area according to the equation sP = P/(Mass/Lf × D), where P is the absolute tension; Mass is the muscle mass from tendon to tendon; D is the density of the skeletal muscle, assumed to be 1.06 g/cm3; and Lf is determined by multiplying Lo by previously determined muscle length-to-fiber length ratios: 0.44 for the EDL and 1 for the DIA (30, 32).
Fura-2 microfluorescence analyses
Enzymatically isolated myofibers of the flexor digitorum brevis (FDB) muscle were used (UNIBA). After dissection, the FDB muscles were incubated in a Petri dish with Ringer’s solution (2 ml) and 0.8–1 mg/ml collagenase type XI-S (Millipore-Sigma, St. Louis, MO, USA) for 45 min at 37°C under constant agitation and continuous O2 flux. After incubation, the muscle was transferred to a tube containing 1 ml of Ringer’s and washed 3 times. Muscles were transferred on a Petri dish coated with Sylgard, and fibers were separated with gentle pipetting. The best fibers were chosen after microscopic observation, put on a microscope slide previously treated with Cultrex (R&D Systems, Minneapolis, MN, USA) and finally incubated at 37°C for 3 h. After that, 400 µl Fura-2 AM (5 μM) was added to the slide and incubated for 1 h at room temperature, with no light. Recording and analysis were performed according to Fraysse et al. (17).
Mechanical threshold
The EDL muscle was dissected intact and fixed by tendons in the recording chamber containing Ringer solution (T = 28°C; pH = 7.2–7.4) (UNIBA). The mechanical threshold (MT) of the individual myofibers was determined in the presence of tetrodotoxin (3 μM), by using a 2-microelectrode “point” voltage-clamp method, according to published reports (27). The threshold membrane potential (V, in millivolts) was read on a digital sample-and-hold millivoltmeter for each fiber at the various pulse durations (t, in milliseconds); the mean values at each t allowed the construction of a “strength–duration” curve. The rheobase voltage (R, in millivolts) was obtained by a nonlinear least-squares algorithm, according to the following equation: V = H − R exp(t/τ)/1 − exp(t/τ) (27, 31, 36, 37).
CK serum levels
CK was determined from serum taken from blood samples withdrawn by cardiac puncture (UNIBA and AGA). Blood was centrifuged at 10,000 rpm for 10 min at 4°C (AGA) or 3000 g for 10 min at 4°C (UNIBA) to obtain the serum. CK determination was performed by standard spectrophotometric analysis by using a CK diagnostic kit (30, 37).
Calpain activity
DIA muscle lysates were used to measure the activity of Ca2+-dependent calpains in DIA muscle lysates (UNIBA), with a commercially available fluorogenic kit (Calbiochem Calpain Activity Kit; Millipore-Sigma, Billerica, MA). Tissues were kept at −80°C until homogenized for calpain measurement. Total proteins were measured by the Bradford assay (Bio-Rad) (24).
Real-time PCR
Total RNA was isolated from GC muscles (UNIBA) with an RNeasy Fibrous Tissue Mini Kit (C.N.74704; Qiagen, Valencia, CA, USA) and quantified by spectrophotometry (ND-1000 NanoDrop; Thermo Fisher Scientific, Waltham, MA, USA). To perform reverse transcription, for each sample 400 ng total RNA was added to 1 μl of 10 mM dNTP mix [Roche, Basel, Switzerland; catalog number (C.N.) 11277049001] and 1 μl of 50 μM random hexamers (C.N. n808-0127; Thermo Fisher Scientific) and incubated at 65°C for 5 min. Then, 4 μl of 5× First Standard Buffer (C.N. Y02321; Thermo Fisher Scientific), 2 μl of 0.1 M DTT (C.N. Y00147; Thermo Fisher Scientific), and 1 μl of 40 U/μl recombinant RNasin ribonuclease inhibitor (C.N. N2511; Promega, Madison, WI, USA) was added and incubated at 42°C for 2 min. Then, 1 μl of 200 U/μl Super Script II reverse transcriptase (C.N. 18064-014; Thermo Fisher Scientific) was added, and the samples were incubated at 25°C for 10 min, at 42°C for 50 min, and at 70°C for 15 min. Real-time PCR was performed with the Applied Biosystems Real-Time PCR 7500 Fast system (Thermo Fisher Scientific). Each reaction was carried out in triplicate on a single-plex reaction. Reactions consisted of 8 ng of cDNA, 0.5 μl of TaqMan Gene Expression Assays, 5 μl of TaqMan Universal PCR Master Mix No AmpErase UNG (2×) (C.N. 4324018), and Nuclease-Free Water Not DEPC220-treated (C.N. AM9930; all from Thermo Fisher Scientific) to give a final volume of 10 μl. The RT-TaqMan-PCR conditions were step 1: 95°C for 20 s; step 2: 95°C for 3 s; and step 3: 60°C for 30 s. Steps 2 and 3 were repeated 40 times. Results were compared with a relative standard curve obtained from 5 points of 1:4 serial dilutions. The mRNA expression of the genes was normalized to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). TaqMan hydrolysis primer and probe gene expression assays were ordered with the following Assay IDs: glyceraldehyde-3-phosphate dehydrogenase: Mm99999915_g1; RyR1: Mm01175211_m1; calstabin (Fkb1a): Mm01243847_g1; utrophin (UTRN): Mm01168866_m1; myogenin (MIOG): Mm00446194_m1; follistatin (FSTN): Mm00514982_m1; Bcl-2 19 kDa interacting protein 3 (BNIP3): Mm01275600_g1; TGF-β: Mm01178820_m1; and NADPH oxidase (NOX)-2 (CYBB): Mm01287743_m1 (38, 39).
Histologic evaluation
DIA muscles and left gastrocnemius muscles were frozen (cooled isopentane in N2; AGA) or fixed in paraffin (UNIBA), sectioned at 6–10 μm, and stained with hematoxylin and eosin (H&E). Endpoints included the assessment of the following parameters: degeneration, centronucleated fibers (CNFs), normal fibers, and regeneration. Five random digital images were taken using a Axioplan II and Axiocam HRC Color Camera on an Image Capture microscope (Zeiss, Jena, Germany) and blinded analysis was performed on data from at least 6 animals per group with Image J (U.S. National Institutes of Health, Bethesda, MD, USA) software (28, 31, 32, 40, 41).
S48168 plasma and muscle levels
S48168 in plasma samples and GC muscles was measured by Servier using liquid chromatography (an Acquity HSS PFP analytical column, 50 × 2.1 mm, 1.8 μm; Waters, Guyancourt, France) and tandem mass spectrometry detection (API 4000) in ESI+, after protein precipitation on Ostro phospholipid removal plates (Waters) with Y2190 (SIL) as the internal standard. The working range was 5–500 ng/ml in plasma and 10–1000 ng/g in muscle tissue, using 20 μl of plasma or the equivalent 20 μg of tissue (see Table 5).
TABLE 5.
S48168 concentrations in plasma and GC muscle in the 4 wk treatment groups and in the 12 wk treatment groups
| Matrix | Parameter | Unit | 10 mg/kg |
50 mg/kg |
||
|---|---|---|---|---|---|---|
| Mean | CV% | Mean | CV% | |||
| 4 Wk treatment by UNIBA | ||||||
| Plasma | Concentration | µg/ml | 0.881 | 31 | 3.023 | 34 |
| Skeletal muscle (GC) | Concentration | µg/g | 0.151 | 31 | 0.512 | 48.7 |
| KP | 0.18 | 28.7 | 0.174 | 27.9 | ||
| 12 Wk treatment by AGA | ||||||
| Plasma | Concentration | µg/ml | 6.125 | 44.4 | 18.775 | 25.7 |
| Skeletal muscle (GC) | Concentration | µg/g | 0.552 | 40.6 | 1.716 | 32 |
| KP | 0.093 | 15.3 | 0.094 | 28.6 | ||
| 12 Wk treatment by UNIBA | ||||||
| Plasma | Concentration | µg/ml | 0.893 | 54.6 | 5.889 | 75.2 |
| Skeletal muscle (GC) | Concentration | µg/g | 0.123 | 45.5 | 1.156 | 42 |
| KP | 0.139 | 21.7 | 0.259 | 70.3 | ||
KP, plasma partition coefficient.
Study approval
AGA study
This non–Good Laboratory Practice (GLP) study was performed according to a Dalhousie University Committee on Laboratory Animals–approved protocol (14-017) and in compliance with the Guide for the Care and Use of Laboratory Animals.
UNIBA study
All experiments were conducted in conformity with the Italian law for Guidelines for Care and Use of Laboratory Animals (D.L. 116/92), and European Directive 2010/63/UE. The study was approved by the National Ethics Committee for Research Animal Welfare of the Italian Ministry of Health (protocol number: DGSAF0024012).
Statistics
All values are expressed as means ± sem. Statistical analyses were conducted on all available data and were performed with Stata ver. 13 (College Station, TX, USA). Significance levels were set to α = 0.05 for main comparisons, α = 0.10 for interactions where applicable.
All outcomes were analyzed at each time point at which data were collected. Mean comparisons between the WT group and the mdx vehicle-treated group were performed with unpaired Student’s t tests and between mdx vehicle and each dose group using 1-way ANOVA with Dunnett’s multiple comparison tests on raw or transformed values. The recovery scores were calculated for quantitatively measurable parameters, because the effect of the treatment was not only measured as the difference between treated and untreated mice, but also relative to the extent of the difference between normal and affected animals. The recovery score indicates directly how much of the deficit (%) was recovered by the treatment.
The recovery score for the drug treatment was evaluated according to the SOP described on the Treat-NMD website (http://www.treat-nmd.eu/research/preclinical/DMD_SOPs) using Eq. 1:
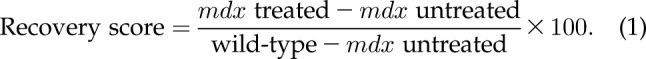 |
RESULTS
Proof-of-concept 4 wk phase 1 study
Animal dose regimen and body weight
The animals were weighed at the end of each week throughout the period of the phase 1 study. The mdx vehicle mice weighed significantly more than their WT counterparts at all time points except week 2 (W2). A comparison between the mdx vehicle-treated group and the treatment group showed no changes in body weight as a consequence of the treatment (Fig. 1A).
Figure 1.

A, B) Body weight measurement (A) and mean water consumption normalized to body weight (B) over the 4 wk of treatment for WT mice, mdx mice treated with vehicle, and mdx mice treated with S48168/ARM210 (hereafter referred to as S48168) at 10 or 50 mg/kg per day. *P < 0.05; **P < 0.01, mdx vehicle-treated vs. WT mice for all time points except 2 wk after treatment. C) Calculated dose of S48168 received by mice at both concentrations (mg/kg) over the 4 wk of treatment is shown. D–F) Baseline (t = 0) and posttreatment (t = 4) maximum forelimb grip strength (KGF; D), normalized forelimb strength to body weight (KGF/kg; E), and total distance run during an exhaustion test (F) for all experimental groups. All values are expressed as means ± sem (n = 6–10 animals/group). *P < 0.05; **P < 0.01; ***P < 0.001, WT vs. mdx vehicle-treated mice, by unpaired Student’s t test, and mdx vehicle-treated vs. S48168-treated groups, by 1-way ANOVA with Dunnett’s multiple comparison test, as indicated. Although not shown, there was a statistically significant difference between the mdx vehicle-treated and WT mice in (F).
The body weight determination was also important for calculation of the weekly dose of S48168/ARM210 that was provided with the drinking water. In turn, the estimation of the amount of water consumed by the mice at the end of each week allowed us to verify the dose of drug actually consumed by each mouse. No significant differences between mdx groups were observed mean water consumption normalized by body weight over time (Fig. 1B).
The dose of S48168/ARM210 consumed by the mice was in the expected range of 10–50 mg/kg per day, except for the first week of treatment (Fig. 1C). In fact, because of an underestimation of the amount of water drunk by these mice during the first week of treatment, the amount of drug consumed was initially higher than the expected dose of 50 mg/kg per day.
In vivo parameters
Forelimb grip strength measurement
The mdx mice had significantly lower values for absolute forelimb strength than did the WT mice, at both baseline (W0) and after 4 wk of treatment (W4). A protection against muscle weakness was observed in the mice treated with S48168/ARM210 at 50 mg/kg per day; this effect was statistically significant at W4 with respect to the mdx vehicle-treated group (Fig. 1D), with a recovery score of 70%. The onset of the effect of the drug on maximum absolute force was very rapid at both doses of S48168/ARM210 (Supplemental Table 2S). In the 10 mg/kg per day-treated mdx mice, a significant increase was observed at W1 by comparison to untreated mdx mice, whereas a significant increase was detected at W1 and W4 for the 50 mg/kg per day–treated mdx mice (Supplemental Table 2S). Because of differences in body weight between the WT and mdx mice, all forelimb strength values were normalized to body weight for both W0 and W4. A significant difference between WT and vehicle-treated mdx mice was maintained (Fig. 1E). Similar to what was observed with regard to absolute force, a dose-dependent improvement in the normalized forelimb strength was observed, with 50 mg/kg per day–treated mice showing a significant increase in normalized force when compared to the untreated mdx mice. The recovery scores also confirmed the dose-dependent effects (14 and 41% for 10 and 50 mg/kg per day, respectively).
Exhaustion test for in vivo fatigability
Another measure of neuromuscular function is the resistance to treadmill running (exhaustion test), evaluated as the total distance that each mouse is able to run until exhaustion. The test was performed in all groups at baseline (W0) and after W4. The total distance covered by the WT mice before exhaustion was similar at W0 and W4 (Fig. 1F). The mdx mice group showed a basal level of total distance run that was significantly lower than that of the WT mice. Moreover, the vehicle-treated mdx mice showed a decline in running performance from W0 to W4, with a significant decrease in the total distance run (−90 m; P < 0.05). Conversely, all the drug-treated mdx mice, although not showing statistically significant improvement, maintained the ability to cover the same total distance before exhaustion (W0 vs. W4). No dose-dependent effect was observed for this parameter, with the recovery score being the same for both treated groups (25%).
Ex vivo force measurement after 4 wk of treatment
DIA
In agreement with previous studies (30, 31, 40), the twitch and tetanic force values of the DIA of the WT mice were markedly higher than those of the mdx mice (Fig. 2A, B). The drug treatment was associated with a dose-dependent trend toward an increase in DIA strength in the mdx mice. This improvement was mostly evident in the maximum specific tetanic force, for which a clear dose-dependent relationship was observed. In particular, the specific tetanic force of the DIA of mice treated with 50 mg/kg per day was significantly higher than that of the vehicle-treated mdx mice (Fig. 2B). In parallel, faster contraction kinetics and a tendency toward a lower ratio of peak twitch force normalized to maximum tetanic force (sPtw/sPo) were also observed, in line with the action of the drug on Ca2+ handling mechanisms (data not shown).
Figure 2.

Specific twitch force (sPtw; A), specific tetanic force (sPo; B), and force–frequency curves (C) for DIA muscles in WT mice, mdx mice treated with vehicle, and mdx mice treated with S48168 at 10 or 50 mg/kg per day, after 4 wk of treatment. All values are expressed as means ± sem (n = 6–10/group). *P < 0.05, WT vs. mdx vehicle-treated mice, by unpaired Student’s t test, and mdx vehicle-treated vs. S48168-treated mice at 50 mg/kg per day, by 1-way ANOVA with Dunnett’s multiple comparison test.
The DIA force–frequency relationship data showed that at the highest frequencies (120 and 140 Hz), the mdx mice treated at 50 mg/kg per day had significantly higher values than did the mdx treated with vehicle (Fig. 2C). No genotype-related differences and no effect of S48168/ARM210 at any dose level were found upon DIA fatigue, nor upon recovery from fatigue (data not shown).
EDL
There was no effect of S48168/ARM210 on EDL muscle force after 4 wk of treatment. Both twitch and specific tetanic force in the vehicle-treated mdx mice were lower than in the WT mice (Supplemental Fig. 1SA, B). For both doses of S48168/ARM210, the values of the two parameters almost overlapped with those of the vehicle-treated mdx mice (Supplemental Fig. 1SA, B). No significant differences in the force–frequency relationship (Supplemental Fig. 1SC) or the force decrease (Supplemental Fig. 1SD) or recovery (data not shown) during fatigue or eccentric contraction were observed (data not shown) in the S48168/ARM210- vs. vehicle-treated mdx mice.
Effect of S48168 on excitation–contraction coupling and Ca2+ homeostasis
Considering that a leaky RyR can play a key role in the dysfunction of the excitation–contraction coupling mechanism in dystrophic muscle, we assessed the outcome of the S48168/ARM210 treatment on this function after 4 wk of treatment. For this purpose, the membrane voltage threshold for single myofiber contraction (MT) was measured in the EDL muscle by means of the microelectrode point voltage–clamp method. MT is an electrophysiological index of excitation–contraction coupling and an integrated measure of calcium-handling mechanisms, including RyR-mediated Ca2+ release (27). In line with previous evidence, mdx myofibers of the vehicle-treated group contracted at a more negative potential with respect to the WT group at all depolarizing pulses (P < 0.05). Treatment with S48168/ARM210 at 50 mg/kg per day led to a significant shift in the potential for fiber contraction toward WT values at all durations of the depolarizing pulses. Accordingly, the membrane potential–duration curves, as well as the calculated value of the fitted rheobase voltage, were shifted toward more positive membrane potentials (Fig. 3A, B). A slight but nonsignificant effect was observed with S48168/ARM210 at 50 mg/kg per day, suggesting a dose-dependent effect of the drug on the rheobase voltage. Unfortunately, MT recordings could not be performed in the DIA because of methodological problems and DIA morphology.
Figure 3.

A, B) Effect of 4 wk of treatment with S48168 on the mechanical threshold (MT) of mdx EDL muscle fibers; the curves for membrane voltages for myofiber contraction in relation to the duration of the depolarizing pulse (strength–duration curves) are shown in A. The MT rheobase voltage values obtained from the fit of the curves are shown in B–D, which show the resting intracellular Ca2+ concentration of FDB muscle fibers and calpain activity in DIA, respectively. Each graph shows the values from WT mice, mdx mice treated with vehicle, and mdx mice treated with S48168 at 10 or 50 mg/kg per day. A–C) Data are expressed as means ± sem from 40 to 11 values (n = 4–5 mice. D) All values are expressed as means ± sem from 5 animals per group. B–D) **P < 0.01; ***P < 0.001, WT vs. mdx vehicle-treated group, by unpaired Student’s t test.
Other indices of Ca2+ homeostasis were also assessed in this proof-of-concept 4 wk treatment study. Figure 3C shows the level of free cytosolic Ca2+ recorded by Fura-2 microfluorescence in single isolated myofibers of the FDB muscle. In line with the results obtained for intact myofiber bundles (17, 18), the isolated mdx myofibers had a significantly higher level of Ca2+ than did the WT myofibers. A 25% reduction in cytosolic Ca2+ was observed in the mdx myofibers treated with 50 mg/kg per day S48168/ARM210 when compared to the mdx vehicle-treated myofibers. There was no difference in the level of cytosolic Ca2+ as the result of the 10 mg/kg per day S48168/ARM210 treatment, when compared to the mdx vehicle group.
Calpain activity was also measured in this study by using the DIA lysates to assess a possible reduction in Ca2+-dependent proteolytic events. Activity was significantly higher in the mdx DIA muscles than those of the WT mice (Fig. 3D), in line with other results (24). A trend toward a reduction in calpain activity was found in the S48168/ARM210-treated groups, but no significant difference was observed, possibly because of the high variability between samples.
Histologic evaluation and biochemical biomarkers
The histologic assessment of H&E–stained sections for percentage of inflammation and other parameters (percentage of CNFs, regeneration, and degeneration) was performed on both the DIA and GC muscles (Fig. 4).
Figure 4.

Representative images of H&E-stained DIA and GC muscles, exhibiting mild-to-severe phenotypic variances of mdx mice treated with vehicle or with S48168 at 10 or 50 mg/kg per day after 4 wk of treatment. Scale bars, 200 µm.
In both vehicle- and S48168/ARM210-treated mice, the DIA and GC muscles clearly showed typical dystrophic features, such as an alteration in muscle architecture, with the presence of areas of necrosis, infiltrates, and nonmuscle areas, most likely resulting from a deposition of fibrotic and adipose tissue. A large variability in fiber size and the presence of CNFs were also clearly detectable (Fig. 4). The morphometric analysis showed a dose-dependent decrease in the percentage of total area of damage (the sum of the necrotic and nonmuscle areas) in both muscle types, except for the DIA in mice treated with 10 mg/kg per day S48168/ARM210 (Fig. 5A, B). Because of the high interindividual variability, the drug effect was mostly not statistically significant for either the DIA or GC muscles. However, significant increases and decreases were observed in the percentage of regenerating area and total damage, respectively, in the GC from mdx mice treated with 50 mg/kg per day S48168/ARM210. In the DIA, the 50 mg/kg per day S48168/ARM210-treated mice showed a reduction in the percentage of total damage and an increase in the percentage of regeneration, although neither was significant because of the high variability (Fig. 5A, B). No marked differences were observed in the number of CNF vs. normal fibers in the DIA or GC muscles at either dose of S48168/ARM210, when compared with the vehicle group (data not shown), probably reflecting a balance between a reduction in damage and a parallel increase in regeneration.
Figure 5.

Area of total damage and regeneration in GC muscle (A) and DIA (B) from mdx mice treated with vehicle or S48168 at 10 or 50 mg/kg per day after 4 wk of treatment. All values are expressed as means ± sem (n = 6 animals per group). *P < 0.05; ***P < 0.001, vehicle- vs. S48168-treated mice, by 1-way ANOVA with Dunnett’s multiple comparison test.
No significant change in the serum level of CK, a biomarker of sarcolemmal fragility, was observed after 4 wk of treatment at either dosage when compared to vehicle-treated mdx mice (Supplemental Fig. 3SA).
Effect of S48168 on target gene expression
To gain insight into the possible mechanism of action of S48168/ARM210 on dystrophic muscle, real-time PCR analysis of relevant genes was performed in GC muscle. RyR1 and calstabin genes were selected as potential targets of Rycal action, TGF-β as a marker of fibrosis, follistatin as a gene involved in control of muscle mass, myogenin as a marker of muscle regeneration, cytochrome B-245 β-chain [CYBB, known as NADPH oxidase (NOX)-2] as a marker of reactive oxygen species (ROS) production, B-cell lymphoma-2–interactive protein (BNIP)-3 as a marker of muscle autophagy, and utrophin as a marker of myofiber regeneration.
Figure 6 shows the effect of treatment with 10 or 50 mg/kg per day S48168/ARM210 on the ratio between the target genes and the housekeeping gene (GAPDH). Treatment with S48168/ARM210 at 50 mg/kg per day produced a significant increase in the gene expression of RyR1 when compared with WT mice. No significant changes were observed in the expression of RyR1, FKB1α, UTRN, MIOG, FSTN, TGF-β, or CYBB in the treated mice, when compared to the vehicle-treated mdx mice. A trend toward an increase in the BNIP3 gene expression level when compared to the vehicle-treated mdx mice was found in the mice treated at 50 mg/kg per day. This result suggests that S48168/ARM210 improves autophagy mechanisms.
Figure 6.

Gene expression profiling of RyR1, FKB1α, UTRN, MIOG, FSTN, BNIP3, TGF-β, and CYBB normalized to GAPDH for WT mice, mdx mice treated with vehicle, or mdx mice treated with S48168 at 10 or 50 mg/kg per day after 4 wk of treatment. All values are expressed as means ± sem (n = 6 animals per group). *P < 0.05; **P < 0.01 WT vs. mdx vehicle-treated mice, by unpaired Student’s t test, and WT mice vs. mdx treated mice, by 1-way ANOVA with Dunnett’s multiple comparison test between as indicated.
Phase 2 study: independent long-term validation of efficacy
Considering the positive outcome of the 4 wk proof-of-concept study, we thought it important to assess whether the drug could maintain or even enhance efficacy if the duration of the treatment was prolonged. This validation was focused on drug effects on various endpoints, both in vivo and ex vivo, including histomorphology, and was performed in parallel by the two independent sites adhering to the same SOPs.
Animal dose regimen and body weight
The animals were weighed at the end of each week throughout the entire trial period during the phase 2 study. The mdx vehicle-treated mice weighed significantly more than their WT counterparts at all the time points in both laboratories (Fig. 7A, D). A comparison between the mdx vehicle-treated group and the S48168/ARM210-treated group showed no changes in body weight at any time point (Fig. 7A, D).
Figure 7.

Body weight measurements from UNIBA (A) and AGA (D) and mean water consumption normalized to body weight from UNIBA (B) and AGA (E) over the 12 wk of treatment for WT mice, mdx mice treated with vehicle, and mdx mice treated with S48168 at 10 or 50 mg/kg per day. The calculated doses of S48168 received by the mice at both concentrations (mg/kg per day) over the 12 wk of treatment are shown in UNIBA (C) and AGA (F) All values are expressed as means ± sem (n = 8 to 15 animals per group).
As described for the 4 wk study, the body weight determination was important for the weekly calculation of the doses of S48168/ARM210 to be provided in the drinking water of the animals. In turn, the estimation of the amount of water consumed by the mice at the end of each week allowed us to verify the dose of drug actually consumed by each mouse. No differences between groups were observed in mean water consumption, as normalized to body weight (Fig. 7B, E). The level of S48168/ARM210 taken by the mice was within the specified range of 50 and 10 mg/kg per day, except for the first week, with levels higher than expected (Fig. 7C, F). As for the phase 1 study, the higher levels were the result of underestimation of the amount of water drunk by these mice. The three parameters measured by the two independent laboratories appeared to replicate each other, indicating the reproducible and reliable character of the data.
In vivo parameters
Forelimb GSM
The effect of S48168/ARM210 on forelimb force after 12 wk of treatment was less evident than after 4 wk of treatment (phase 1), although slight differences were observed, most likely related to the fact that different experimenters performed the force determinations in the two laboratories.
At both UNIBA and AGA, the forelimb GSM during the 12 wk protocol was significantly lower in the mdx vehicle-treated mice than in the WT mice, consistent with other reports (28, 30, 32, 42). Slight differences were observed in the absolute and normalized values of grip strength between the two laboratories. This difference was probably because of differences in protocols (see Materials and Methods) and well-known experimenter-related influences. More important, the degree of impairment was similar in the two laboratories, as shown for the percentage changes of the normalized values in comparative Table 1. In the UNIBA study, the effect of S48168/ARM210 on normalized forelimb force at week 12 was significant for the 10 mg/kg group [5 ± 0.23 kilogram force (KGF)/kg vs. 4.06 ± 0.20 KGF/kg for the vehicle-treated mice; P < 0.01]. This effect was greater than that for the highest dose (recovery scores of 35% vs. 12%, respectively). However, it is worth mentioning that the two doses showed a similar efficacy up to 11 wk of treatment (Supplemental Fig. 2SA). In addition, significant improvement in the strength (P < 0.001) was observed at weeks 10–12, when the mdx mice treated with 10 mg/kg per day were compared to the vehicle-treated mice, and at week 5 and weeks 10 and 11 (P < 0.005), when the mice treated with 50 mg/kg per day were compared to the vehicle-treated mice.
TABLE 1.
Comparison of in vivo parameters in two independent 12 wk studies
| Treatment | Comparative group | Summary data |
|||
|---|---|---|---|---|---|
| UNIBA |
AGA |
||||
| N | Percentage change | N | Percentage change | ||
| Vehicle | WT | 8 | −40*** | 15 | −35*** |
| 10 mg/kg/d | MDX-VEHICLE | 8 | +23* | 15 | +3 |
| 50 mg/kg/d | MDX-VEHICLE | 8 | +8 | 14 | +10** |
The outcome measure was the percentage change in normalized forelimb grip strength resulting from disease and from treatment with S48168 at 10 or 50 mg/kg per day, measured at the two laboratories over the 12 wk of the study. *P < 0.05; **P < 0.01, mdx vehicle-treated vs. S48168-treated mice, by 1-way ANOVA with Dunnett’s multiple comparison test; and ***P < 0.001 vs. WT and mdx vehicle-treated mice, by unpaired Student’s t test.
In the AGA study, a dose effect was observed for the normalized forelimb grip strength during the overall treatment period. The highest dose level of 50 mg/kg per day had a statistically significant effect when compared to the mdx vehicle treatment (3.99 ± 0.15 KGF/kg vs. 3.62 ± 0.14 KGF/kg; P < 0.05), whereas the dose level of 10 mg/kg per day had no statistically significant effect on the normalized forelimb grip strength of the mdx mice. A comparison of the changes in normalized forelimb force in the two laboratories is shown in Table 1.
Hindlimb GSM
Hindlimb force was measured only in the 12 wk study performed at AGA. Absolute hindlimb grip strength was significantly reduced in the mdx vehicle-treated mice when compared to the WT mice, consistent with previous reports (32, 42). There were no differences between the treatment groups, when compared to the mdx vehicle-treated group with respect to absolute hindlimb grip strength. However, when the normalized values were considered, a significant dose effect was observed during the overall treatment period, with the 50 mg/kg per day S48168/ARM210 treatment having a statistically significant ameliorative effect (Supplemental Fig. 2SB).
Ex vivo force measurement after 12 wk of treatment
DIA
At this time point, the DIA muscles from the mdx vehicle-treated mice showed a significantly lower specific tetanic force and specific twitch force than did the DIA muscles from WT mice, a difference of ∼50% in both laboratories (Table 2 and Supplemental Table 3S). The drug treatment showed a considerable improvement in the DIA twitch force output in the mdx mice, but only in the results from AGA, with the 10 mg/kg per day dose showing a higher force output than the 50 mg/kg per day dose. A similar, although smaller, trend toward improvement was observed for tetanic force in the AGA study for 10 mg/kg per day dose (Table 2 and Supplemental Table 4S). The dose-dependent relationship observed in the phase 1 study was not seen in the phase 2 study in either of the two laboratories, with the drug showing a slightly lower efficacy than after 4 wk of treatment.
TABLE 2.
Comparison of the effects of pathology and treatments on ex vivo contractile parameters of the DIA in 2 independent 12 wk studies
| Treatment | Comparative group | Summary data |
|||
|---|---|---|---|---|---|
| UNIBA |
AGA |
||||
| n | % change | n | % change | ||
| Twitch force study | |||||
| Vehicle | WT | 8 | −47* | 11 | −58* |
| 10 mg/kg per day | MDX-VEHICLE | 8 | +2 | 13 | +22 |
| 50 mg/kg per day | MDX-VEHICLE | 8 | −9 | 12 | +16 |
| Tetanus force study | |||||
| Vehicle | WT | 8 | −46† | 12 | −45† |
| 10 mg/kg per day | MDX-VEHICLE | 8 | +2 | 14 | +14 |
| 50 mg/kg per day | MDX-VEHICLE | 8 | −7 | 12 | −11 |
Percentage changes in the DIA twitch and tetanic force values resulting from disease and from 12 wk of treatment with S48168 at 10 or 50 mg/kg per day, measured at the two laboratories. *P < 0.001, WT mice vs. mdx vehicle-treated mice, by unpaired Student’s t test; and †P < 0.001, mdx vehicle-treated vs. S48168-treated mice, by 1-way ANOVA, with Dunnett’s multiple comparison test.
EDL
Ex vivo force contraction measurements showed that the EDL muscles from the mdx vehicle-treated mice had a significantly lower specific tetanic force and specific twitch force than did the EDL muscles from the WT mice. This decrease in twitch and tetanic force related to genotype was similar between the two laboratories (Table 3 and Supplemental Table 3S). We then asked whether a longer duration of the treatment would unveil the effect of S48168/ARM210 on EDL muscle force. None of the treated mdx groups in either laboratory showed a statistically significant difference from the mdx vehicle-treated group. However, a clear trend toward a dose-related force increase in treated muscle was observed, with a similar level of force production obtained by both laboratories (Table 3 and Supplemental Table 4S). Slight, if any, genotype-related differences were observed in the time to peak and half relaxation time, and no effect of S48168/ARM210 was found on the muscle-contraction kinetics (data not shown).
TABLE 3.
Comparison of the effects of pathology and treatments on ex vivo contractile parameters of the EDL muscle, measured in 2 independent 12 wk studies
| Treatment | Comparative group | Summary data |
|||
|---|---|---|---|---|---|
| UNIBA |
AGA |
||||
| n | % change | n | % change | ||
| Twitch force | |||||
| Vehicle | WT | 8 | −36* | 13 | −25† |
| 10 mg/kg per day | MDX-VEHICLE | 8 | −1 | 14 | −2 |
| 50 mg/kg per day | MDX-VEHICLE | 8 | +14 | 13 | +6 |
| Tetanus Force | |||||
| Vehicle | WT | 8 | −42‡ | 13 | −36§ |
| 10 mg/kg per day | MDX-VEHICLE | 8 | +8 | 14 | +11 |
| 50 mg/kg per day | MDX-VEHICLE | 8 | +18 | 13 | +13 |
The table shows the percentage changes in EDL muscle twitch and tetanic force values resulting from pathology and from the 12 wk treatment with S48168 at 10 or 50 mg/kg per day, measured at AGA and UNIBA. *P < 0.01; †P < 0.001, WT mice vs. mdx vehicle-treated mice, by unpaired Student’s t test; and ‡P < 0.01; §P < 0.001, mdx vehicle-treated vs. S48168-treated mice, by 1-way ANOVA with Dunnett’s multiple comparison test.
Similarly, there were no differences in the fatigue-related decline in force or in recovery from fatigue between the treated mdx groups and the mdx vehicle-treated group (data not shown). No protective effect of S48168/ARM210 at either dose was observed on the force decrease in mdx EDL muscle as a consequence of eccentric contraction stimulation (Supplemental Fig. 2SC). Stiffness is another parameter that can discriminate between WT and mdx muscles. The mdx EDL muscles were less compliant to stretch than were the WT muscles, as shown by the lower value obtained by calculating the difference in force before and after the stretch. A slight but not significant amelioration of stiffness was observed in the 50 mg/kg per day–treated group (Supplemental Fig. 2SD).
Histologic evaluation, biochemical biomarkers, and gene expression after 12 wk of treatment
Evaluation of the DIA and GC muscle histopathology of mdx mice showed results consistent with those in published reports for this age range and was similar for the two investigation sites. There was a significant increase in the percentage of inflammation per muscle, percentage degenerating muscle fibers per field, percentage regenerating muscle fibers per field, and percentage CNFs/field in the mdx vehicle-treated mice, when compared to the WT mice. Typical dystrophic features were also observed in both muscles types from mdx mice treated with S48168/ARM210 at either 10 or 50 mg/kg per day (Fig. 8). The morphometric analysis showed a reduction in the total muscle damage in DIA, which was comparable between the two laboratories (Table 4), although statistical significance was found only for AGA’s data. However, in the UNIBA study, the morphometric analysis revealed a significant dose-dependent reduction in CNFs, from 72.3 ± 1.6 to 62.4 ± 2.9% at 10 mg/kg (P < 0.01) and 53.3 ± 2.5% at 50 mg/kg (P < 0.01), with a parallel significant increase in normal fibers. These results are indicative of a reduction in the degeneration–regeneration cycle. In the AGA study, a dose-dependent effect was observed when single components of damage were evaluated (inflammation, necrosis, and centronucleation), suggesting an overall improvement in the morphology of the DIA muscle. All the histologic parameters measured at AGA showed substantial but nonsignificant dose-dependent reductions in inflammation, regeneration, and degeneration and necrosis in both the GC and DIA muscles (>20% reduction when compared to the mdx vehicle-treated group in both muscles). In both laboratories, the drug did not have a protective effect on GC in terms of total damage (Table 4), despite a trend toward improvement in several of the histology parameters mentioned earlier (data not shown).
Figure 8.

Representative images of H&E-stained DIA and GC muscles exhibiting mild-to-severe phenotypic variations in mdx mice treated with vehicle or with S48168 at10 or 50 mg/kg per day for 12 wk. Scale bar, 200 µm.
TABLE 4.
Effects of drug treatment on DIA histology, measured in 2 independent 12 wk studies
| Treatments | Comparative group | Summary data |
|||
|---|---|---|---|---|---|
| UNIBA |
AGA |
||||
| n | Change in % damaged | n | Change in % damaged | ||
| Total damaged DIA (%) | |||||
| 10 mg/kg per day | MDX-VEHICLE | 8 | −9 | 15 | −14** |
| 50 mg/kg per day | MDX-VEHICLE | 8 | −19 | 14 | −10* |
| Total damaged gastrocnemius (%) | |||||
| 10 mg/kg per day | MDX-VEHICLE | 8 | 14 | 15 | 1 |
| 50 mg/kg per day | MDX-VEHICLE | 8 | 20 | 14 | 5 |
Percentage changes in the total damage in response to the drug treatment with S48168 at 10 or 50 mg/kg per day over 12 wk of treatment, measured at the laboratories. *P < 0.05; **P < 0.01, mdx vehicle-treated vs. S48168-treated mice, by 1-way ANOVA with Dunnett’s multiple comparison test.
As observed in the phase 1 study, there were no significant changes in CK level in either the UNIBA or AGA study at 12 wk of treatment (Supplemental Fig. 3SB, C). Treatment did not lower the CK levels in these mice.
In addition, qPCR experiments were performed on GC muscle from the 12 wk study, with the genes that were modified in the 4 wk study. No significant changes were observed in the expression level of RyR1, calstabin, or BNIP3 at any dose, suggesting a reduced sensitivity of the muscle to the compound in the long term, compared with the short-term treatment (Supplemental Fig. 3SD).
Evaluation of animal drug exposure and the search for potential toxic effects: animal exposure in the phase 1 and 2 studies
The overall results suggest that S48168/ARM210 exerts short-term effects on function, with evidence of ameliorated histomorphology after 4 wk of treatment; however, prolongation of the treatment did not evidently result in either an increase in efficacy or a broader action. It was then important to evaluate the degree of animal exposure to the drug to verify the maintenance of an effective level in both the plasma and muscle sites.
In the phase 1 study, the S48168/ARM210 level in plasma was assessed from blood collected about 2 h after the drinking water was withdrawn from the mice. The mean plasma concentrations were dose dependent, as they were in GC muscle (Table 5).
In the phase 2 study, the S48168/ARM210 level in plasma were assessed from blood collected 20–30 min (AGA) or 2 h (UNIBA) after drinking water was withdrawn. In both studies, a clear dose-dependent exposure was observed in both the plasma and GC muscle. As expected, the drug levels were higher in the AGA study because of the shorter period after water withdrawal. In the UNIBA study, the dose dependency observed in phase 1 was confirmed. In addition, the mean plasma concentrations were similar to those found after 4 wk of treatment, whereas muscle levels were higher.
Assessment of potential toxic effect: organs and muscle mass
Supplemental Figure 4S shows the mass of various organs, expressed as values normalized to the body weight to evaluate potential gross toxicity after 4 wk of treatment (phase 1). No significant drug effects were observed, except for a lower normalized mean value for the heart in mdx animals treated with S48168/ARM210 at 10 mg/kg per day, when compared to vehicle-treated mice. As far as other organs (spleen and liver) were concerned, only differences between the mdx and WT groups were observed. Similarly, no drug effect on the mass of various skeletal muscles was observed.
The heart and muscle mass values after 12 wk of treatment (phase 2) are shown in Supplemental Fig. 5S. No significant difference between the genotypes was observed in the heart mass, either in the absolute mass or the mass normalized to body weight. The mdx mice had a greater muscle mass than did the WT mice for the tibialis anterior and quadriceps, in line with results in phase 1. A significant increase in both the absolute and normalized values of the quadriceps mass was found in the 10 mg/kg per day–treated mice (UNIBA data). In the AGA study, the changes in the organ and muscle mass were in line with findings in the UNIBA study. No differences were observed between the treatment groups, when compared to the vehicle-treated group. However, significant genotype differences were still evident in the skeletal muscle mass.
As for phase 1, the liver, kidney, and spleen maintained genotype-related differences, with minor if any drug effects (data not shown), corroborating the lack of specific organ toxicity with prolongation of the treatment. However, in the UNIBA study, a further increase in the liver mass was observed in the 50 mg/kg per day–treated mice with respect to the vehicle-treated mdx mice [46.51 ± 0.81 (g/g × 1000) vs. 56.39 ± 2.61 (g/g × 1000); P < 0.005, n = 8], which was not observed in the AGA study.
DISCUSSION
In DMD, damaged skeletal muscle fibers undergo a characteristic degenerative process that is followed by regeneration (43). The progressive muscle wasting is related to a complex network of pathologic events, with dysregulation of Ca2+ homeostasis playing a key role (43,–46). Earlier studies with S107, a Rycal targeting the skeletal muscle isoform of the SR Ca2+ release channel (RyR1), support a role of leaky hypernitrosylated RyRs in the alteration of Ca2+ homeostasis and in turn in sustaining functional and morphologic alteration of dystrophic muscles (19). S48168/ARM210, a novel Rycal derivative, has shown good oral bioavailability, together with the capacity to effectively re-establish calstabin binding to PKA-phosphorylated RyR channels in SR vesicles from either cardiac or skeletal muscles (data not shown) and a broader action on RyR1/RyR2 channels. The purpose of the present study was to perform a proof-of-concept assessment and to evaluate the efficacy of S48168/ARM210 in the most severe model of the exercised mdx mouse.
The 4 wk proof-of-concept study disclosed the ability of S48168/ARM210 to improve muscle force, increase DIA force, and ameliorate the DIA histologic profile. It is of interest that we observed that the mechanical threshold of the EDL myofibers improved in a dose-dependent manner, corroborating a role for leaky RyRs in the alteration of excitation–contraction coupling in the dystrophic myofibers and confirming the effect of the drug on myofiber function. In addition, indices of Ca2+ homeostasis, such as intracellular Ca2+ and calpain activity, were ameliorated in FDB and DIA muscle, respectively, corroborating the mechanism of action of S48168/ARM210. The reason that the amelioration of excitation–contraction coupling did not directly correlate with an improvement in the whole EDL muscle contractile efficiency may be a function of the more complex physiology of intact muscle. In contrast, the greater efficacy observed in the DIA could be a result of muscle-specific drug action and differences in muscle structure and metabolism, as well as the degree of tenderness and vascularization. Unfortunately, it was not feasible to obtain MT recordings in the DIA because of the less stable microelectrode impalement and voltage control related to DIA morphology; performing this recording would have yielded better insight into the correlation between function and excitation–contraction coupling. We then hypothesized that amelioration of hindlimb muscle function, directly solicited by the exercise protocol, would require longer drug exposure. Consistent with this view, 4 wk treatment with S48168/ARM210 improved the histopathology of the GC, while increasing gene expression of RyR1 and BNIP3, a marker of autophagy, which is known to be impaired in dystrophic muscle (39, 47).
Based on the 4 wk results, we then performed a longer assessment in parallel in the two independent sites. The reproducibility of results from independently performed scientific experiments is emphasized in various guidelines as good practice (25, 26) and has been put forth by regulatory agencies (e.g., the U. S. Food and Drug Administration) for moving any compound from preclinical studies to clinical trials, especially for rare diseases, where clinical trials are hampered by the low number of patients. Our study was performed maintaining standard experimental conditions, according to internationally recognized SOPs for preclinical testing of therapeutics in DMD, as implemented by TREAT-NMD. SOPs for rare neuromuscular diseases are dynamically revised to include innovative methods in the identification of promising therapeutics (42, 48,–50).
The phase 2 study showed that similar results could be obtained and then validated by two independent laboratories, as far as the activity of compound S48168/ARM210 is concerned. The reproducibility of this study was mainly attributed to the common procedures performed by the two laboratories according to SOPs, whereas the differences likely reflect the variability introduced by slightly different protocols or environmental conditions (e.g., the chow composition; Supplemental Table 1S).
First, the comparison was made between mdx vehicle-treated mice and WT mice for the two laboratories. This comparison provides the phenotype effect comparison needed to determine the level of pathologic alteration of the mdx vehicle-treated mice vs. the WT mice in the two laboratories. Both laboratories showed comparable results, with similar levels of percentage decrease in functional performance in the mdx vehicle-treated mice, when compared to WT mice in all the parameters measured (normalized forelimb strength, DIA and EDL twitch and tetanic force). Also, a similar histopathologic profile was found, despite the high interindividual variability in pathology and analysis.
The comparison was then made between mdx mice treated with vehicle and mdx mice treated with 1 of 2 doses of S48168/ARM210. At both doses (10 and 50 mg/kg per day), the two laboratories had the same trends in terms of change in normalized forelimb strength, as well as for DIA and EDL twitch and tetanic force, except for the DIA ex vivo force parameters at 50 mg/kg per day, which showed an opposite trend between the two laboratories. Again, this difference may be related to several factors. Considering that the changes observed were not significantly different, they may just be related to the high interindividual variability of the genotype as well as to the environmental setting.
Given the progressive nature of DMD, it is essential, not only to determine whether a drug is rapidly efficacious in improving the muscle phenotype, but also whether the treatment maintains or even increases the beneficial effects over time. The results of the studies presented herein suggest that S48168/ARM210 is more effective in the short-term than in the long-term treatment. However, in the long-term treatment, S48168/ARM210 maintained a certain level of compound efficacy. In fact, the pharmacokinetic assessment clearly showed that dose-dependent drug exposure was maintained over time in both the plasma and muscle in both laboratories, allowing us to rule out any possible adaptation to long-term treatment. Similarly, no gross macroscopic alteration was seen in the vital organs that could lead to the occurrence of a toxic effect masking the therapeutic one.
The reason for this apparent reduction in efficacy with prolonged treatment must be better understood. A likely hypothesis is that the Rycal actually corrects the failing binding of RyR1 to calstabin but does not correct the main cause of RyR1 leakiness (i.e., nitrosylation and altered channel modulation). In fact, neither effect of S48168/ARM210 has been observed on NOX2 expression, or on ROS-mediating pathways such as TGF-β or TNF-α. Given the high level of TGF-β expression (∼3-fold) in both treated and untreated mdx muscles, blocking TGF-β in conjunction with Rycal treatment may further enhance the efficacy of S48168/ARM210. TGF-β overexpression may indeed play a pathogenic role with respect to RyR1 oxidation and nitrosylation, as well as subsequent dissociation from calstabin. Moreover, TGF-β is a recognized player in fibrotic and trans-differentiation processes (51).
Identification of a druggable, muscle-specific component of the TGF-β pathway would offer a promising approach to DMD therapy, especially in combination with S48168/ARM210. In addition, this mechanism follows the pathologic progression of DMD, which can be worsened by exercise. In particular, recent studies have shown that long-term exercise in mdx mice may affect the expression of the molecules involved in metabolic protection against mechanical stressors, leading to an unbalanced effect of damaging signals and the consequent reinforcement of inflammation and oxidative stress (39). In this case, it would also be of interest to assess the effect of S48168/ARM210 in combination with drugs that control oxidative stress, such as the gold-standard steroids or other compounds that are effective in oxidative stress-related muscle damage, such as apocynin, N-acetylcysteine (NAC), and taurine (52,–54). Incidentally, in vitro application of NAC has been found to have an effect similar to that of S107 on the leaky RyR2 channels of dystrophic heart (53). Also, the importance of crosstalk has been described in the modulation of cardiac RyR2 by oxidation and phosphorylation in mdx mice (55), supporting the contention that the concomitant targeting of posttranslational events may reinforce the effects of Rycal.
In the present research, an improvement in functional test outcomes for grip strength supports the general improvement in the phenotype of mdx mice. The overall profile of the DIA muscle was improved by S48168/ARM210 in the phase 1 study, and this effect was confirmed in the phase 2 AGA study, as far as function and histology profile are concerned, suggesting that the mice are experiencing better respiratory function. Considering that the impairment in respiratory function is a major characteristic of patients with DMD, these effects may have important therapeutic implications. GC histologic features were also improved by S48168/ARM210, which may have contributed to the amelioration observed in the grip strength measurements in the treated mice. In addition, the experiments at UNIBA showed that the effect of S48168/ARM210 on EDL muscle were slightly stronger after the long-term study than after the short-term investigation, although this difference did not reach statistical significance. Studies that employed exon skipping as a potential treatment for DMD showed an ∼10–20% improvement in specific force values (56, 57). This therapy targets the primary defect of the disease: dystrophin deficiency. Having compounds that help to alleviate the secondary symptoms in all patients, irrespective of mutation, and that achieve results comparable to those obtained by gene correction is of interest. The lack of efficacy of the drug in terms of the drop in eccentric contraction force is not surprising, considering that the mechanism of action does not involve a sarcolemmal reinforcement, either directly, as a result of DGC rescue, or as a result of reduction in oxidative stress, as with NAC (58, 59). Accordingly, no effect of the drug was observed on the CK plasma level, lending credence to the conclusion that the study and the endpoints evaluated allow a clear demonstration of specific beneficial effects.
It is widely acknowledged that the mdx mouse model shows significant interanimal variability, which contributes to the difficulty in observing statistically significant treatment effects in some tests, particularly histology (42). Greater effects are preferable in preclinical assays, also considering that the drug may be less effective when tested in human settings as the result of an even higher variability in disease course and modifiers. Although the lack of statistical significance should not be ignored, the interpretation of our results needs to take this variation into account. In this case, an estimation of the percentage differences in a large sample population may help determine the effect of the drug, while the recovery score is another important index for defining drug action in translational research (SOP) (25, 26). The identification of other biomarkers of efficacy has to be considered, to make informed decisions and in light of recent disappointing results obtained in clinical trials for drugs that were very effective in preclinical tests.
As anticipated, the levels of CK measured in mdx mice were significantly higher than those in WT mice. In both the phase 1 and 2 studies, no effect of the drug was observed on CK levels. The question remains as to whether the serum CK level is a true predictor of skeletal muscle damage. Several studies have indicated that, although CK levels are used as a clear diagnostic marker, especially in disease states such as DMD, the levels do not always correlate with the degree of structural damage (60,–63). As mentioned earlier, a decrease in CK levels can more easily be observed after treatment with drugs that are able to reinforce the sarcolemma or to decrease inflammation and oxidative stress (37, 40, 41), and the lack of effect on this endpoint in the present setting is therefore not surprising.
Leaky RyR2 is considered to be responsible for cardiac failure and remodeling in mdx mice, allowing us to predict the potential usefulness of S48168/ARM210 in treating the cardiac symptoms of DMD. It would be interesting to assess the potential cardioprotective effects of the drug by echocardiography and histologic evaluation of fibrosis in mdx mice treated until at least 9–10 mo of age, when cardiomyopathy appears, or alternatively in a more severe disease model with an earlier cardiac involvement. In parallel, the drug levels in the hearts of mdx mice should be assessed. An early amelioration at the cellular level has been observed in mdx hearts treated with S107, suggesting that it may be worth using a preventive treatment. A longer treatment of older mdx mice or mice with the mutated gene on a more severe background would also help to address this important point.
Finally, our results disclosed for the first time the ability of a novel Rycal, S48168/ARM210, to exert beneficial effects on pathology progression in the more severe, exercised mdx mouse model. Pathology-related parameters and calcium-dependent parameters were markedly sensitive to the action of the drug, supporting our working hypothesis as well as the importance of an independent validation of the same drug in two independent research groups.
The observed decline in efficacy of the drug over time may be related to the need to control, in parallel, other mechanisms accounting for RyR1 dysfunction.
In conclusion, the rapid functional improvement induced by S48168/ARM210 in mdx mice, together with its oral bioavailability, low toxicity, and the potential pharmacologic effect on cardiac function, further corroborate the interest in this Rycal compound for future evaluation in the treatment of DMD.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
The authors thank Dr. Deborah McClellan for editing the manuscript. The compound used for this study was provided by Servier (Suresnes, France) and Armgo Pharma (Tarrytown, NY, USA), who are jointly developing S48168/ARM210 for indications including DMD. This study was supported in part by funds from Muscular Dystrophy Association Grant 228338 (Chicago, IL, USA); by U.S. National Institutes of Health Grant K26OD011171 (to K.N.); and by funds from the Dutch Duchenne Parent Project (NL-DPP) (to A.D.L.). K. N. and A. D. L. are senior coauthors. K. N. is a cofounder of Reveragen Biopharma, which is engaged in the development of therapeutic products for DMD. The remaining authors declare no conflicts of interest.
Glossary
- AGA
Agada Biosciences
- BNIP
B-cell lymphoma (BCL)2–interactive protein
- CK
creatine kinase
- CN
catalog number
- CNF
centronucleated fiber
- DGC
dystrophin glycoprotein complex
- DIA
diaphragm
- DMD
Duchenne muscular dystrophy
- EDL
extensor digitorum longus
- FDB
flexor digitorum brevis
- GC
gastrocnemius
- GSM
grip strength measurement
- H&E
hematoxylin and eosin
- KGF
kilogram force
- Lo
optimal length
- Lf
fiber length
- MT
mechanical threshold
- NAC
N-acetylcysteine
- ROS
reactive oxygen species
- RyR
ryanodine receptor
- SOP
standard operating procedure
- SR
sarcoplasmic reticulum
- UNIBA
University of Bari
- WT
wild type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
R. F. Capogrosso, P. Mantuano, K. Uaesoontrachoon, A. Cozzoli, K. Nagaraju, and A. De Luca, designed the study and wrote the paper; R. F. Capogrosso, P. Mantuano, A. Cozzoli, A. Giustino, T. Dow, S. Srinivassane, M. Filipovic, C. Bell, J. Vandermeulen, A. M. Massari, M. De Bellis., E. Conte, G. M. Camerino, and A. Liantonio performed experiments; K. Uaesoontrachoon, R. F. Capogrosso, P. Mantuano, K. Nagaraju, and A. De Luca reviewed the manuscript.
REFERENCES
- 1.Roses A. D., Herbstreith M. H., Appel S. H. (1975) Membrane protein kinase alteration in Duchenne muscular dystrophy. Nature 254, 350–351 [DOI] [PubMed] [Google Scholar]
- 2.Hoffman E. P., Schwartz L. (1991) Dystrophin and disease. Mol. Aspects Med. 12, 175–194 [DOI] [PubMed] [Google Scholar]
- 3.Hoffman E. P., Brown R. H. Jr., Kunkel L. M. (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928 [DOI] [PubMed] [Google Scholar]
- 4.Ervasti J. M., Ohlendieck K., Kahl S. D., Gaver M. G., Campbell K. P. (1990) Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 345, 315–319 [DOI] [PubMed] [Google Scholar]
- 5.Ohlendieck K., Campbell K. P. (1991) Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J. Cell Biol. 115, 1685–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hutter O. F., Burton F. L., Bovell D. L. (1991) Mechanical properties of normal and mdx mouse sarcolemma: bearing on function of dystrophin. J. Muscle Res. Cell Motil. 12, 585–589 [DOI] [PubMed] [Google Scholar]
- 7.Menke A., Jockusch H. (1991) Decreased osmotic stability of dystrophin-less muscle cells from the mdx mouse. Nature 349, 69–71 [DOI] [PubMed] [Google Scholar]
- 8.Menke A., Jockusch H. (1995) Extent of shock-induced membrane leakage in human and mouse myotubes depends on dystrophin. J. Cell Sci. 108, 727–733 [DOI] [PubMed] [Google Scholar]
- 9.Pasternak C., Wong S., Elson E. L. (1995) Mechanical function of dystrophin in muscle cells. J. Cell Biol. 128, 355–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petrof B. J., Shrager J. B., Stedman H. H., Kelly A. M., Sweeney H. L. (1993) Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 90, 3710–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emery A. E., Burt D. (1980) Intracellular calcium and pathogenesis and antenatal diagnosis of Duchenne muscular dystrophy. BMJ 280, 355–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fong P. Y., Turner P. R., Denetclaw W. F., Steinhardt R. A. (1990) Increased activity of calcium leak channels in myotubes of Duchenne human and mdx mouse origin. Science 250, 673–676 [DOI] [PubMed] [Google Scholar]
- 13.Turner P. R., Westwood T., Regen C. M., Steinhardt R. A. (1988) Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature 335, 735–738 [DOI] [PubMed] [Google Scholar]
- 14.Deconinck N., Dan B. (2007) Pathophysiology of duchenne muscular dystrophy: current hypotheses. Pediatr. Neurol. 36, 1–7 [DOI] [PubMed] [Google Scholar]
- 15.Gailly P. (2002) New aspects of calcium signaling in skeletal muscle cells: implications in Duchenne muscular dystrophy. Biochim. Biophys. Acta 1600, 38–44 [DOI] [PubMed] [Google Scholar]
- 16.Goonasekera S. A., Lam C. K., Millay D. P., Sargent M. A., Hajjar R. J., Kranias E. G., Molkentin J. D. (2011) Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J. Clin. Invest. 121, 1044–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraysse B., Liantonio A., Cetrone M., Burdi R., Pierno S., Frigeri A., Pisoni M., Camerino C., De Luca A. (2004) The alteration of calcium homeostasis in adult dystrophic mdx muscle fibers is worsened by a chronic exercise in vivo. Neurobiol. Dis. 17, 144–154 [DOI] [PubMed] [Google Scholar]
- 18.Rolland J. F., De Luca A., Burdi R., Andreetta F., Confalonieri P., Conte Camerino D. (2006) Overactivity of exercise-sensitive cation channels and their impaired modulation by IGF-1 in mdx native muscle fibers: beneficial effect of pentoxifylline. Neurobiol. Dis. 24, 466–474 [DOI] [PubMed] [Google Scholar]
- 19.Bellinger A. M., Reiken S., Carlson C., Mongillo M., Liu X., Rothman L., Matecki S., Lacampagne A., Marks A. R. (2009) Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 15, 325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X., Weisleder N., Collet C., Zhou J., Chu Y., Hirata Y., Zhao X., Pan Z., Brotto M., Cheng H., Ma J. (2005) Uncontrolled calcium sparks act as a dystrophic signal for mammalian skeletal muscle. Nat. Cell Biol. 7, 525–530 [DOI] [PubMed] [Google Scholar]
- 21.Robert V., Massimino M. L., Tosello V., Marsault R., Cantini M., Sorrentino V., Pozzan T. (2001) Alteration in calcium handling at the subcellular level in mdx myotubes. J. Biol. Chem. 276, 4647–4651 [DOI] [PubMed] [Google Scholar]
- 22.Turner P. R., Fong P. Y., Denetclaw W. F., Steinhardt R. A. (1991) Increased calcium influx in dystrophic muscle. J. Cell Biol. 115, 1701–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spencer M. J., Croall D. E., Tidball J. G. (1995) Calpains are activated in necrotic fibers from mdx dystrophic mice. J. Biol. Chem. 270, 10909–10914 [DOI] [PubMed] [Google Scholar]
- 24.Burdi R., Didonna M. P., Pignol B., Nico B., Mangieri D., Rolland J. F., Camerino C., Zallone A., Ferro P., Andreetta F., Confalonieri P., De Luca A. (2006) First evaluation of the potential effectiveness in muscular dystrophy of a novel chimeric compound, BN 82270, acting as calpain-inhibitor and anti-oxidant. Neuromuscul. Disord. 16, 237–248 [DOI] [PubMed] [Google Scholar]
- 25.Willmann R., De Luca A., Benatar M., Grounds M., Dubach J., Raymackers J. M., Nagaraju K.; TREAT-NMD Neuromuscular Network (2012) Enhancing translation: guidelines for standard pre-clinical experiments in mdx mice. Neuromuscul. Disord. 22, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willmann R., Luca A., Nagaraju K., Rüegg M. A. (2015) Best practices and standard protocols as a tool to enhance translation for neuromuscular disorders. J. Neuromuscul. Dis. 2, 113–117 [DOI] [PubMed] [Google Scholar]
- 27.De Luca A., Pierno S., Liantonio A., Cetrone M., Camerino C., Fraysse B., Mirabella M., Servidei S., Rüegg U. T., Conte Camerino D. (2003) Enhanced dystrophic progression in mdx mice by exercise and beneficial effects of taurine and insulin-like growth factor-1. J. Pharmacol. Exp. Ther. 304, 453–463 [DOI] [PubMed] [Google Scholar]
- 28.Spurney C. F., Gordish-Dressman H., Guerron A. D., Sali A., Pandey G. S., Rawat R., Van Der Meulen J. H., Cha H. J., Pistilli E. E., Partridge T. A., Hoffman E. P., Nagaraju K. (2009) Preclinical drug trials in the mdx mouse: assessment of reliable and sensitive outcome measures. Muscle Nerve 39, 591–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mele A., Camerino G. M., Calzolaro S., Cannone M., Conte D., Tricarico D. (2014) Dual response of the KATP channels to staurosporine: a novel role of SUR2B, SUR1 and Kir6.2 subunits in the regulation of the atrophy in different skeletal muscle phenotypes. Biochem. Pharmacol. 91, 266–275 [DOI] [PubMed] [Google Scholar]
- 30.Capogrosso R. F., Mantuano P., Cozzoli A., Sanarica F., Massari A. M., Conte E., Fonzino A., Giustino A., Rolland J. F., Quaranta A., De Bellis M., Camerino G. M., Grange R. W., De Luca A. (2017) Contractile efficiency of dystrophic mdx mouse muscle: in vivo and ex vivo assessment of adaptation to exercise of functional end points. J. Appl. Physiol. (1985) 122, 828–843 [DOI] [PubMed] [Google Scholar]
- 31.Burdi R., Rolland J. F., Fraysse B., Litvinova K., Cozzoli A., Giannuzzi V., Liantonio A., Camerino G. M., Sblendorio V., Capogrosso R. F., Palmieri B., Andreetta F., Confalonieri P., De Benedictis L., Montagnani M., De Luca A. (2009) Multiple pathological events in exercised dystrophic mdx mice are targeted by pentoxifylline: outcome of a large array of in vivo and ex vivo tests. J. Appl. Physiol. (1985) 106, 1311–1324 [DOI] [PubMed] [Google Scholar]
- 32.Sali A., Guerron A. D., Gordish-Dressman H., Spurney C. F., Iantorno M., Hoffman E. P., Nagaraju K. (2012) Glucocorticoid-treated mice are an inappropriate positive control for long-term preclinical studies in the mdx mouse. PLoS One 7, e34204. 10.1371/journal.pone.0034204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uaesoontrachoon K., Quinn J. L., Tatem K. S., Van Der Meulen J. H., Yu Q., Phadke A., Miller B. K., Gordish-Dressman H., Ongini E., Miglietta D., Nagaraju K. (2014) Long-term treatment with naproxcinod significantly improves skeletal and cardiac disease phenotype in the mdx mouse model of dystrophy. Hum. Mol. Genet. 23, 3239–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tricarico D., Rolland J. F., Cannone G., Mele A., Cippone V., Laghezza A., Carbonara G., Fracchiolla G., Tortorella P., Loiodice F., Conte Camerino D. (2012) Structural nucleotide analogs are potent activators/inhibitors of pancreatic β cell KATP channels: an emerging mechanism supporting their use as antidiabetic drugs. J. Pharmacol. Exp. Ther. 340, 266–276 [DOI] [PubMed] [Google Scholar]
- 35.Grange R. W., Gainer T. G., Marschner K. M., Talmadge R. J., Stull J. T. (2002) Fast-twitch skeletal muscles of dystrophic mouse pups are resistant to injury from acute mechanical stress. Am. J. Physiol. Cell Physiol. 283, C1090–C1101 [DOI] [PubMed] [Google Scholar]
- 36.Cozzoli A., Liantonio A., Conte E., Cannone M., Massari A. M., Giustino A., Scaramuzzi A., Pierno S., Mantuano P., Capogrosso R. F., Camerino G. M., De Luca A. (2014) Angiotensin II modulates mouse skeletal muscle resting conductance to chloride and potassium ions and calcium homeostasis via the AT1 receptor and NADPH oxidase. Am. J. Physiol. Cell Physiol. 307, C634–C647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Luca A., Nico B., Liantonio A., Didonna M. P., Fraysse B., Pierno S., Burdi R., Mangieri D., Rolland J. F., Camerino C., Zallone A., Confalonieri P., Andreetta F., Arnoldi E., Courdier-Fruh I., Magyar J. P., Frigeri A., Pisoni M., Svelto M., Conte Camerino D. (2005) A multidisciplinary evaluation of the effectiveness of cyclosporine a in dystrophic mdx mice. Am. J. Pathol. 166, 477–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pierno S., Camerino G. M., Cannone M., Liantonio A., De Bellis M., Digennaro C., Gramegna G., De Luca A., Germinario E., Danieli-Betto D., Betto R., Dobrowolny G., Rizzuto E., Musarò A., Desaphy J. F., Camerino D. C. (2013) Paracrine effects of IGF-1 overexpression on the functional decline due to skeletal muscle disuse: molecular and functional evaluation in hindlimb unloaded MLC/mIgf-1 transgenic mice. PLoS One 8, e65167. 10.1371/journal.pone.0065167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Camerino G. M., Cannone M., Giustino A., Massari A. M., Capogrosso R. F., Cozzoli A., De Luca A. (2014) Gene expression in mdx mouse muscle in relation to age and exercise: aberrant mechanical-metabolic coupling and implications for pre-clinical studies in Duchenne muscular dystrophy. Hum. Mol. Genet. 23, 5720–5732 [DOI] [PubMed] [Google Scholar]
- 40.Capogrosso R. F., Cozzoli A., Mantuano P., Camerino G. M., Massari A. M., Sblendorio V. T., De Bellis M., Tamma R., Giustino A., Nico B., Montagnani M., De Luca A. (2016) Assessment of resveratrol, apocynin and taurine on mechanical-metabolic uncoupling and oxidative stress in a mouse model of duchenne muscular dystrophy: a comparison with the gold standard, α-methyl prednisolone. Pharmacol. Res. 106, 101–113 [DOI] [PubMed] [Google Scholar]
- 41.De Luca A., Nico B., Rolland J. F., Cozzoli A., Burdi R., Mangieri D., Giannuzzi V., Liantonio A., Cippone V., De Bellis M., Nicchia G. P., Camerino G. M., Frigeri A., Svelto M., Camerino D. C. (2008) Gentamicin treatment in exercised mdx mice: Identification of dystrophin-sensitive pathways and evaluation of efficacy in work-loaded dystrophic muscle. Neurobiol. Dis. 32, 243–253 [DOI] [PubMed] [Google Scholar]
- 42.Grounds M. D., Radley H. G., Lynch G. S., Nagaraju K., De Luca A. (2008) Towards developing standard operating procedures for pre-clinical testing in the mdx mouse model of Duchenne muscular dystrophy. Neurobiol. Dis. 31, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carlson B. M. (1973) The regeneration of skeletal muscle: a review. Am. J. Anat. 137, 119–149 [DOI] [PubMed] [Google Scholar]
- 44.Chargé S. B., Rudnicki M. A. (2004) Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84, 209–238 [DOI] [PubMed] [Google Scholar]
- 45.Armstrong R. B. (1990) Initial events in exercise-induced muscular injury. Med. Sci. Sports Exerc. 22, 429–435 [PubMed] [Google Scholar]
- 46.Grefte S., Kuijpers-Jagtman A. M., Torensma R., Von den Hoff J. W. (2007) Skeletal muscle development and regeneration. Stem Cells Dev. 16, 857–868 [DOI] [PubMed] [Google Scholar]
- 47.Pal R., Palmieri M., Loehr J. A., Li S., Abo-Zahrah R., Monroe T. O., Thakur P. B., Sardiello M., Rodney G. G. (2014) Src-dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nat. Commun. 5, 4425. 10.1038/ncomms5425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Willmann R., Possekel S., Dubach-Powell J., Meier T., Ruegg M. A. (2009) Mammalian animal models for Duchenne muscular dystrophy. Neuromuscul. Disord. 19, 241–249 [DOI] [PubMed] [Google Scholar]
- 49.Duan D., Rafael-Fortney J. A., Blain A., Kass D. A., McNally E. M., Metzger J. M., Spurney C. F., Kinnett K. (2016) Standard operating procedures (SOPs) for evaluating the heart in preclinical studies of Duchenne muscular dystrophy. J. Cardiovasc. Transl. Res. 9, 85–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wacker S. J., Jurkowski W., Simmons K. J., Fishwick C. W., Johnson A. P., Madge D., Lindahl E., Rolland J. F., de Groot B. L. (2012) Identification of selective inhibitors of the potassium channel Kv1.1-1.2((3)) by high-throughput virtual screening and automated patch clamp. ChemMedChem 7, 1775–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waning D. L., Mohammad K. S., Reiken S., Xie W., Andersson D. C., John S., Chiechi A., Wright L. E., Umanskaya A., Niewolna M., Trivedi T., Charkhzarrin S., Khatiwada P., Wronska A., Haynes A., Benassi M. S., Witzmann F. A., Zhen G., Wang X., Cao X., Roodman G. D., Marks A. R., Guise T. A. (2015) Excess TGF-β mediates muscle weakness associated with bone metastases in mice. Nat. Med. 21, 1262–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Terrill J. R., Radley-Crabb H. G., Grounds M. D., Arthur P. G. (2012) N-Acetylcysteine treatment of dystrophic mdx mice results in protein thiol modifications and inhibition of exercise induced myofibre necrosis. Neuromuscul. Disord. 22, 427–434 [DOI] [PubMed] [Google Scholar]
- 53.Fauconnier J., Thireau J., Reiken S., Cassan C., Richard S., Matecki S., Marks A. R., Lacampagne A. (2010) Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 107, 1559–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pierno S., Tricarico D., Liantonio A., Mele A., Digennaro C., Rolland J. F., Bianco G., Villanova L., Merendino A., Camerino G. M., De Luca A., Desaphy J. F., Camerino D. C. (2014) An olive oil-derived antioxidant mixture ameliorates the age-related decline of skeletal muscle function. Age (Dordr.) 36, 73–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Q., Wang W., Wang G., Rodney G. G., Wehrens X. H. (2015) Crosstalk between RyR2 oxidation and phosphorylation contributes to cardiac dysfunction in mice with Duchenne muscular dystrophy. J. Mol. Cell. Cardiol. 89(Pt B), 177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malerba A., Sharp P. S., Graham I. R., Arechavala-Gomeza V., Foster K., Muntoni F., Wells D. J., Dickson G. (2011) Chronic systemic therapy with low-dose morpholino oligomers ameliorates the pathology and normalizes locomotor behavior in mdx mice. Mol. Ther. 19, 345–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharp P. S., Bye-a-Jee H., Wells D. J. (2011) Physiological characterization of muscle strength with variable levels of dystrophin restoration in mdx mice following local antisense therapy. Mol. Ther. 19, 165–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whitehead N. P., Pham C., Gervasio O. L., Allen D. G. (2008) N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J. Physiol. 586, 2003–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Whitehead N. P., Yeung E. W., Froehner S. C., Allen D. G. (2010) Skeletal muscle NADPH oxidase is increased and triggers stretch-induced damage in the mdx mouse. PLoS One 5, e15354. 10.1371/journal.pone.0015354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Van der Meulen J. H., Kuipers H., Drukker J. (1991) Relationship between exercise-induced muscle damage and enzyme release in rats. J. Appl. Physiol. (1985) 71, 999–1004 [DOI] [PubMed] [Google Scholar]
- 61.Komulainen J., Vihko V. (1994) Exercise-induced necrotic muscle damage and enzyme release in the four days following prolonged submaximal running in rats. Pflugers Arch. 428, 346–351 [DOI] [PubMed] [Google Scholar]
- 62.Fridén J., Lieber R. L. (2001) Serum creatine kinase level is a poor predictor of muscle function after injury. Scand. J. Med. Sci. Sports 11, 126–127 [DOI] [PubMed] [Google Scholar]
- 63.Dahlqvist J. R., Voss L. G., Lauridsen T., Krag T. O., Vissing J. (2014) A pilot study of muscle plasma protein changes after exercise. Muscle Nerve 49, 261–266 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.