

Abstract
Dinoflagellates are a group of unicellular protists with immense ecological and evolutionary significance and cell biological diversity. Of the photosynthetic dinoflagellates, the majority possess a plastid containing the pigment peridinin, whereas some lineages have replaced this plastid by serial endosymbiosis with plastids of distinct evolutionary affiliations, including a fucoxanthin pigment-containing plastid of haptophyte origin. Previous studies have described the presence of widespread substitutional RNA editing in peridinin and fucoxanthin plastid genes. Because reports of this process have been limited to manual assessment of individual lineages, global trends concerning this RNA editing and its effect on the biological function of the plastid are largely unknown. Using novel bioinformatic methods, we examine the dynamics and evolution of RNA editing over a large multispecies data set of dinoflagellates, including novel sequence data from the peridinin dinoflagellate Pyrocystis lunula and the fucoxanthin dinoflagellate Karenia mikimotoi. We demonstrate that while most individual RNA editing events in dinoflagellate plastids are restricted to single species, global patterns, and functional consequences of editing are broadly conserved. We find that editing is biased toward specific codon positions and regions of genes, and generally corrects otherwise deleterious changes in the genome prior to translation, though this effect is more prevalent in peridinin than fucoxanthin lineages. Our results support a model for promiscuous editing application subsequently shaped by purifying selection, and suggest the presence of an underlying editing mechanism transferred from the peridinin-containing ancestor into fucoxanthin plastids postendosymbiosis, with remarkably conserved functional consequences in the new lineage.
Keywords: plastid, transcript editing, dinoflagellate, serial endosymbiosis, constructive neutral evolution
Introduction
Dinoflagellates, a group of unicellular protists, have unusual cellular processes and life cycles, making them of interest to ecologists, cell biologists, and evolutionary scientists alike. Dinoflagellates account for a substantial portion of global marine diversity (Le Bescot et al. 2016). These include photosynthetic members that are important primary producers (Taylor et al. 2008), of which some are capable of producing “blooms” large enough to be visible from space, with dramatic effects on the local environment (do Rosário Gomes et al. 2014), while others, such as Symbiodinium spp., are essential symbionts of marine organisms (Kopp et al. 2015). Their ecological role can also directly affect human health; neurotoxins produced by dinoflagellates can be absorbed by shellfish and cause food poisoning in consumers (Hackett, Anderson, et al. 2004).
Dinoflagellates belong to the highly diverse alveolate clade, within the SAR (Stramenopiles, Alveolates, and Rhizaria) supergroup (Adl et al. 2012; Janouškovec et al. 2017). Their unusual cell biology, including their unorthodox nuclear and organellar genomes, has been a prominent focus of study. Dinoflagellates have extremely large nuclear genomes with highly condensed chromatin, compacted by nonhistone nuclear proteins (Gornik et al. 2012). The mitochondrial genome is highly repetitive and can be fragmented (Jackson et al. 2007; Nash et al. 2007), and dinoflagellate mitochondria are supported by an unusual protein complement lacking many of the key protein import subunits and electron transport complexes conserved across other lineages (Butterfield et al. 2016; Dorrell et al. 2017). Finally, dinoflagellates have extraordinarily diverse photosynthetic life strategies and plastid types (Dorrell and Howe 2015; Gavelis et al. 2015). The majority of chloroplast-bearing dinoflagellate species harbour plastids containing the soluble accessory light-harvesting pigment peridinin, which are of ultimately red algal origin, though the exact nature of endosymbiotic events giving rise to the SAR clade plastids is still under debate (Dorrell and Howe 2015; Ševčíková et al. 2015; Waller et al. 2016). The peridinin plastid genome is the most reduced in terms of coding content within any photosynthetic eukaryote, with evidence for large-scale transfer of plastid genes to the nucleus (Bachvaroff et al. 2004; Hackett, Yoon, et al. 2004). The structure and expression of the peridinin plastid genome is also unusual; it has been fragmented entirely into small plasmid-like “minicircles,” generally containing only a single gene (Zhang et al. 1999; Mungpakdee et al. 2014), although in a few species minicircles containing multiple protein-coding genes, or combinations of protein-coding and transfer RNA genes, are known (Barbrook et al. 2001; Hiller 2001; Dorrell et al. 2017). Transcribed minicircles with no genes, or only containing pseudogene fragments, have also been identified (Barbrook et al. 2001, 2006; Hiller 2001; Green 2004). These minicircles have been inferred to be replicated and transcribed via rolling circle mechanisms (Leung and Wong 2009; Dang and Green 2010; Barbrook et al. 2012). The broader functional consequences of this fragmentation event for the organization and expression of the peridinin plastid genome remain poorly understood.
Dinoflagellate plastids are also associated with two unusual transcript processing pathways: 3′-polyuridylylation and substitutional RNA editing, (Zauner et al. 2004; Wang and Morse 2006). The plastid RNA editing observed can involve both transition and transversion substitutions, and may occur on significant numbers (>5%) of residues in certain dinoflagellate plastid transcripts, resulting in dramatic changes between genomic and transcript sequence content (Dorrell and Howe 2015). This RNA editing occurs prior to the translation of transcripts, but can have significant effects on the expression of transcript sequences, for example, via the removal of in-frame premature termination codons and other residues that, if translated, would compromise protein function (Jackson et al. 2013).
Both pathways are highly characteristic of peridinin dinoflagellate plastid lineages (fig. 1): 3′ plastid polyuridylylation is known in the basally divergent genus Amphidinium, and in the closely related, chromerid algae Chromera velia and Vitrella brassicaformis, and is inferred to be an ancestral feature of the peridinin plastid (Janouškovec et al. 2010, 2017; Barbrook et al. 2012; Dorrell et al. 2014). However, polyuridylylation has never been detected in any other plastid lineage, and is not known to occur either in dinoflagellate nuclei or mitochondria (Dorrell and Howe 2012; Cahoon et al. 2017). Plastid RNA editing has been detected in multiple distantly related dinoflagellate genera (Ceratium, Heterocapsa, Lingulodinium, Alexandrium, Symbiodinium) (Zauner et al. 2004; Wang and Morse 2006; Dang and Green 2009; Iida et al. 2009; Mungpakdee et al. 2014), but is not known in Amphidinium (Barbrook et al. 2012), and has only been detected to occur at very low frequencies in other plastid lineages, including those of chromerids and apicomplexans (fig. 1, Janouškovec et al. 2013; Dorrell et al. 2014; Nisbet et al. 2016). RNA editing is known in plant plastids, and in some species can occur at elevated frequencies (>1%; Oldenkott et al. 2014), but the editing events in these lineages are restricted to C-to-U interconversions, in contrast to the much more diverse editing events found in dinoflagellates (Dorrell and Howe 2015). Extensive and functionally promiscuous RNA editing has been detected in both the mitochondria (Lin et al. 2002; Nash et al. 2007; Jackson et al. 2012) and nuclei of dinoflagellates (Liew et al. 2017), although it is unknown how this is related to the RNA editing machineries found in other nuclear lineages across eukaryotes (Gray 2012; Smith and Keeling 2015).
Fig. 1.

—Summary of plastid transcript editing in dinoflagellates. This diagram denotes the relationship between taxa under study, plastid affiliation, and presence of plastid biological features including minicircles, transcript polyuridylation, and transcript editing. Symbols are denoted in figure inset. Dashed line surrounding editing symbol between Amphidinium spp. and fucoxanthin lineages represents uncertainty regarding editing in basal peridinin dinoflagellates and related taxa. Placement of dashed boxes for both C-to-G and G-to-U base conversions in two places represents two alternate evolutionary hypotheses, as discussed in the main text. The single polytomy represents uncertainty in peridinin dinoflagellate branching order.
Further complexity arises from the fact that dinoflagellates are the only algal lineage to contain confirmed cases of serial endosymbiosis, a process where a plastid within a photosynthetic eukaryote is replaced with a plastid of a different evolutionary lineage (Dorrell and Howe 2015). Serial endosymbiosis is observed in Lepidodinium, which possesses a green algal chloroplast (Kamikawa et al. 2015), the “dinotoms,” dinoflagellate species within the Peridiniaceae, with diatom endosymbionts (Imanian et al. 2010; Yamada et al. 2017), and dinoflagellates within the genera Karenia, Karlodinium, and Takayama, which possess a haptophyte endosymbiont containing the pigment fucoxanthin (Tengs et al. 2000; Dorrell and Howe 2015).
The evolution of the fucoxanthin plastid genome has been studied in particular detail. Nuclear phylogenies place fucoxanthin dinoflagellates as an early-diverging lineage within the peridinin dinoflagellates, indicating that they descended from an ancestor that possessed the plastid polyruidlyylation pathway, and may have performed plastid RNA editing (fig. 1, Saldarriaga et al. 2003; Hoppenrath and Leander 2010; Janouškovec et al. 2017). Fucoxanthin-containing plastids have a largely single-chromosome genome and, despite exhibiting gene loss relative to free-living haptophytes, a much larger gene complement than that in peridinin plastids. However, similar to peridinin plastids (Zhang et al. 1999), fucoxanthin plastid genomes contain highly divergent gene sequences, have been highly rearranged, and, in some cases, appear to be approaching fragmentation, with at least one minicircle containing the Hsp70 gene dnaK and a glutamyl-tRNA gene known in the fucoxanthin dinoflagellate Karlodinium veneficum (Gabrielsen et al. 2011; Espelund et al. 2012; Richardson et al. 2014).
Gene expression pathways associated with the peridinin plastid have also been documented in fucoxanthin dinoflagellate plastids, despite their absence from studied haptophytes (Dorrell and Howe 2015). In 2012, Dorrell and Howe (2012) reported the presence of extensive RNA editing and polyuridylylation in the fucoxanthin plastid of Karenia mikimotoi, which was later also identified in the plastid of Karl. veneficum (Jackson et al. 2013; Richardson et al. 2014). These RNA processing pathways were proposed to have originated in the peridinin-containing ancestor of dinoflagellates, having been applied to the fucoxanthin plastid shortly following their endosymbiotic uptake. The plastid RNA editing observed in fucoxanthin and peridinin-containing dinoflagellates may have evolved independently and convergently (fig. 1), dependent on whether this pathway was established in the plastids of their common ancestor, but in either case it is not known to occur in haptophyte plastids (Dorrell and Howe 2012). The recruitment of these two unusual RNA processing pathways to the replacement plastid from a lineage with no known history of either, and the genomic changes associated with plastids in a dinoflagellate cell, represent a fascinating system for studying the organellar interactions involved in endosymbiosis. As molecular genetic techniques have not yet been successfully applied in dinoflagellates these studies have been restricted to sequence-based analyses, based on relatively small selections of plastid genes, with only two full genomes that include extensive plastid editing analyzed from one fucoxanthin and one peridinin dinoflagellate (Karl. veneficum and S. minutum, respectively) (Mungpakdee et al. 2014; Richardson et al. 2014).
In this study, we have focused on the evolution and function of RNA editing across peridinin and fucoxanthin dinoflagellate lineages in an effort to answer two questions: what are the observable effect(s) of editing, and why is the pathway maintained in extant taxa, including in serially acquired fucoxanthin plastids? To this end, we use bioinformatic tools to analyze properties of RNA editing at genome-wide scales, and apply these to a comprehensive data set spanning the diversity of dinoflagellates and including novel plastid transcriptomic and genomic data from the peridinin dinoflagellate Pyrocystis lunula and plastid genomic data from the fucoxanthin dinoflagellate Kare. mikimotoi. We identified biases associated with editing, including clustering of editing events along the length of genes, prevalence of editing in the first two codon positions, and an overall trend toward restoration of amino acid residues found in related organisms in the translation products of edited sequences. Furthermore, computer simulations suggest that the observed trends are unlikely when editing is modelled as a random process. We provide evidence that editing events in extant lineages are unlikely to represent differential retention of ancestral events, suggesting that editing is likely the result of lineage-specific applications of an otherwise conserved machinery, and that this application may be under different selective regimes between fucoxanthin and peridinin plastids. Overall, our results imply some level of conservation of pathway function across serial endosymbiosis, which has not previously been established.
Materials and Methods
Culturing, Extraction, Sequencing, and Assembly
Pyrocystis lunula (Schütt) UTEX 2271 was cultured autotrophically in 1 l of L1 medium (Guillard and Hargraves 1993) at room temperature under a 14/10 h light/dark cycle at an irradiance of ∼50 µE m−2 s−1. Cells were harvested for RNA and DNA extraction 6–8 weeks after inoculation during late exponential phase. One liter of P. lunula culture was pelleted in a clinical centrifuge and nucleic acids extracted using Qiagen’s (Valencia, CA) RNeasy and DNeasy kits, following the manufacturer’s instructions. RNA and DNA were quantified using a Nanodrop Lite (Thermo Scientific, Waltham, MA).
The P. lunula genome and transcriptome were sequenced by the University of Illinois sequencing center (Springfield, IL). The gDNA and total RNA (rRNA depleted) libraries had average insert sizes of 520 nt and 280 nt, respectively. They were sequenced on one lane for 100 cycles from each end on an Illumina HiSeq2000 platform to produce 100 nt reads. The gDNA library yield was 69,846,936 paired end reads and the RNA library was 141,434,468 paired end reads.
Transcriptomes were assembled from read data using SOAPdenovo v1.0.3 (Luo et al. 2012), Trinity v2.2.0 (Grabherr et al. 2011), and Velvet v1.2.10 (Zerbino and Birney 2008). For each assembler, the default k-mer size was used (63, 25, and 31, respectively). Putative chloroplast transcripts were identified from the assemblies using standalone tBLASTx (Altschul et al. 1997) to compare predicted proteins to all Genbank-archived plastome records (accessed April 2013). Contig translations that matched an existing coding region with an E-value of 0.001 or less were considered significant and binned.
Bacterial contaminants resulting from the xenic P. lunula cultures were removed by analyzing BLAST searches against the nr database (NCBI; downloaded June 25, 2013). Here, a more stringent E-value cutoff of 1×10−10 was employed. If the taxon label for the most statistically significant hit contained the string “bacter,” the contig was removed. Finally, the resulting contigs were sorted and separated into files based on the gene name of the top BLAST hit.
Putative mRNA sequences were selected from Trinity contigs using a pipeline of three Python scripts (available from https://github.com/DacksLab/RNAediting; last accessed March 21, 2018). The first extracts any potential ORF with a minimum length of 40 amino acids from the contig data. The second script executes a BLASTp search on each ORF found, comparing it to a custom database of 42 dinoflagellate plastid protein sequences, including all 12 proteins encoded in peridinin dinoflagellate plastids (Dorrell et al. 2017). The final script parses the output from the BLASTp searches and selects the most likely protein coding contig for a given gene based on e-value and contig length.
Once a best candidate was identified, a second draft mRNA sequence for each gene was produced using the best candidate transcript for a template-based assembly using Geneious v8.1 (Kearse et al. 2012). Second draft mRNAs were then used as template sequences to assemble gDNA contigs. After assembly of DNA reads to each transcript, the transcript template was removed and the consensus sequence saved as a first draft gDNA assembly. Each gDNA first draft was used as a template to repeat the assembly process and create gDNA second draft assemblies. Final gDNA and mRNA assemblies were translated using Virtual Ribosome (Wernersson 2006) or Geneious. tRNA screens were performed using tRNAScan and ARAGORN (Lowe and Eddy 1997; Laslett and Canback 2004). Details for each P. lunula gene, including mapped genomic and transcript reads, as well as the determined location of genes on minicircles, are provided (supplementary table S1, Supplementary Material online).
Karenia mikimotoi RCC1513 was grown in modified f/2 medium under a 12/12 h cycle of 30 µE m−2 s−1 as described in Dorrell and Howe (2012). Cells were harvested for DNA extraction 4 weeks after inoculation from ∼200 ml late exponential phase culture, and DNA was isolated using phenol chloroform extraction as described in Barbrook et al. (2012). About 400 ng crude DNA, as quantified using a Qubit fluorometer (Invitrogen) was used to generate a sequencing library with a NexteraXT tagmentation kit (Illumina), and sequenced over 500 cycles using a MiSeq sequencer. Reads were trimmed using the MiSeq reporter version 2.0.26, and assembled into 574,711 contigs using ELAND (Illumina). Contigs of probable plastid origin were identified using reciprocal BLASTn and tBLASTx searches, using as queries transcript sequences encoded within the Kare. mikimotoi plastid, previously confirmed experimentally (Dorrell et al. 2016), and a BLAST cutoff value of E-05. Genomic sequences were confirmed by alignment against the corresponding transcript sequences using the built-in alignment programme within Geneious v4.76 (Kearse et al. 2012) using default settings.
The sequences of 16 plastid genes, for which contigs spanning > 700 bp were obtained through the next-generation sequencing approaches, were verified by PCR using primer sequences designed against the contig in question, and Pfu High-Fidelity Polymerase (Thermo), following previously defined methodology (Dorrell et al. 2016). In select cases, specific products were amplified using nested rounds of PCR with multiple forward and/or reverse primers (supplementary table S1, Supplementary Material online). Each PCR was repeated twice, and each product obtained was independently purified with a Nucleospin DNA Cleanup Column (Macherey-Nagel), and submitted for Sanger sequencing (GATC Biotech, Germany) using both forward and reverse PCR primers. The products from these reactions were pooled with previous products obtained for a further 11 Kare. mikimotoi plastid genes confirmed by Sanger sequencing in previous studies (Dorrell and Howe 2012; Dorrell et al. 2016; supplementary table S1, Supplementary Material online). The assembled sequences were manually inspected using Geneious, and only positions with no visible ambiguities in the chromatogram trace files (hence no evidence for polymorphism) were analyzed for editing.
PsbA Alignment and Phylogeny
gDNA and mRNA psbA sequences for 15 peridinin dinoflagellate species were extracted from a previously constructed sequence library (Dorrell et al. 2017), alongside plastid genomic and transcript sequences for Kare. mikimotoi and Karl. veneficum. PsbA was selected as the number of species for which sequences are available is vastly greater than other dinoflagellate plastid-encoded genes, allowing a phylogenetically sensitive appraisal of the distribution of editing sites (Dorrell et al. 2017). gDNA and mRNA sequences from the same species were first aligned to one another, trimmed, and then globally aligned using the translation sequence, with Geneious v 4.76 (Kearse et al. 2012). A tabular form alignment, including annotations of all editing events observed, is provided (supplementary table S5, Supplementary Material online). RAxML v.8.2.10 and MrBayes v.3.2.6 trees were inferred for a 30 taxa×1,121 nucleotide alignment, consisting of all of the previously identified dinoflagellate plastid mRNA sequences, and an outgroup of 13 nondinoflagellate orthologues, using the corresponding programmes in-built into the CIPRES gateway (Miller et al. 2010; Ronquist et al. 2012; Stamatakis 2014), and the default conditions.
SNP Analysis of Nucleotide Variability
Available raw reads for P. lunula were filtered using MOCAT2 (Kultima et al. 2016) with length and quality cut-offs of 45 and 30. Filtered reads were subsequently mapped to consensus gDNA sequences using BWA (Li and Durbin 2009), and filtered for unique mappers with at least 95% similarity and 40 nt. Nucleotide variability analysis and SNP detection was then performed using metaSNV (Costea et al. 2017) with filtering parameters m = 1, d = 0.1, b = 1 and c = 1.
Automated Analysis of Editing Events
Automated editing event detection was carried out using custom Python scripts (available from https://github.com/DacksLab/RNAediting; last accessed March 21, 2018). Sequence translation used the standard genetic code, as there is no evidence for systematic differences in the genetic code applied to internal residues in plastid transcript sequences in previous studies of peridinin and fucoxanthin dinoflagellates (Dorrell et al. 2016, 2017), from plastid genome sequences of free-living haptophytes (Puerta et al. 2005; Hovde et al. 2014), or in our data set (supplementary table S2, Supplementary Material online). Alignments were built from our initial sequences using MUSCLE v.3.8.31 (Edgar 2004) with default settings. Nucleotide alignments were scanned from the first aligned base to the last, whereas amino acid alignments were taken to start and end when at least five of ten overlapping bases were identical. The aligned region was then scanned to identify differences between genomic and transcript sequences. For this and all subsequent analyses, we note that we are unable to differentiate between single versus multiple editing events at a position, as only the initial genomic and final transcript sequences were compared.
Sliding Window Correlation Analysis
Automated sliding window analysis involved segmenting the aligned region of genomic, transcript, and references sequences into a number of overlapping substrings of length W, the “window” size (W = 60 to remain consistent with Richardson et al. 2014). Editing events and genomic/reference sequence similarity (based on nucleotide identity and amino acid similarity) were calculated across each substring. We scored amino acid similarity as previously described: identical amino acids scored 1.0, those with positive scores in the Blosum62 substitution matrix (Henikoff and Henikoff 1992) scored 0.5, and all other amino acids scored 0.0. Finally, the Pearson correlation across all windows was calculated using a standard formula:
Where and are the -th element of each set, and and are the mean of each set. In order to score amino acid similarity in sequences with different lengths following translation the sequences were aligned using an internal pairwise alignment algorithm with a three-matrix dynamic programming approach and affine gap penalties (gap open = 11, gap extend = 1) and the Blosum62 substitution matrix (Henikoff and Henikoff 1992):
Where and are amino acids to be aligned from each sequence, is the gap penalty, and , , and are matrices corresponding to match and gap states.
Automated trimming of indels in aligned sequences was carried out to remove as much unconserved sequence (present either in the reference, or in the genomic/transcript sequence) as possible while maintaining the reading frame of all sequences. The resulting output sequences do not possess indels >5 bp, that is, two codons.
Analysis of Editing Effect on Amino Acid Sequences
The editing score was quantified as the difference in Blosum62 substitution matrix (Henikoff and Henikoff 1992) score between the genomic and transcript residues, and the corresponding reference residue. Comparison of editing score in alignments with more than one reference sequence involved calculating the average of editing scores for each genomic/transcript and reference sequence pairing, as described earlier.
Positional Entropy Calculations
Positional entropy, a measure of amino acid variability (Sander and Schneider 1991; Valdar 2002), was calculated as follows:
Where PE is the positional entropy, g is the proportion of gaps at a given position, and ai is the proportion of each amino acid at a given position. In this scheme, gaps are penalized and the score is normalized to fall between 0 (maximum entropy) and 1 (complete conservation). Positions within alignments with >50% gap (“-”) characters were not used for calculations.
Simulations to Assess Cluster Significance
For each N-length gene, a sequence of equal length comprising a random sequence of nucleotide bases was generated using the Python random module. For each generation in the simulation, the same number of editing events as observed in the relevant gene was applied randomly across this sequence. Editing events were not allowed to occur in the same position multiple times. Clustering of editing events would lead to larger deviations from the mean rate of editing across the length of the sequence, and hence a larger variance in observed values between simulated and real data. About 1,000 simulations were run for each gene and variance in observed editing rates across all windows (W = 60) was calculated using the levene function of the SciPy library v0.15.1 (Jones et al. 2001) to compare median values between experimental and simulated data; equivalent to the Brown–Forsythe test. A frequency of significant (P value < 0.05) tests >50% was taken to indicate editing more clustered than expected by chance.
Statistical analysis of editing among adjacent codons involved randomly distributed the 3,518 editing events across the 23,415 codons with a uniform probability between codons but taking into account the positional bias within codons. This model was repeated 100 times and the mean and SD for the number of codons edited once, twice, and three times as well as the proportion of edits on each position close to another edit (either the same or adjacent codon) was extracted. The probability of the observed value was computed assuming a normal distribution.
Simulation to Assess Corrective Editing Significance
For each sequence the codon position, base conversion, and frequency associated with all possible editing events was determined. Then, for each generation in the simulation, the same number of editing events as observed in the relevant gene sequence was applied to the original genomic sequence. For each editing event, the combination of codon position, starting base, and edited base were chosen using a weighted random choice scheme based on observed data. A position meeting the selected criteria was selected randomly along the length of the gene, not allowing changes to the same position twice. Finally, the sequences were translated and compared with the relevant orthologue from E. huxleyi using the editing score metric. After 1,000 simulations, significance was assessed by comparison of scores for all editing events between experimental and simulated data using the ranksums function of the SciPy library v0.15.1 (Jones et al. 2001). A frequency of significant (P value < 0.05) tests >50% was taken to indicate editing more corrective than expected by chance.
GRAVY Score Calculation
Consistent with previous studies (Mungpakdee et al. 2014) the relative hydrophobicity of protein sequences was calculated using GRAVY score (Kyte and Doolittle 1982). Positions not corresponding to standard amino acids in one or both sequences, such as STOP codons and indels, were removed prior to calculations. Calculation of GRAVY score and molecular weight were both performed using the Bio.SeqUtils module in Biopython v1.64 (Cock et al. 2009).
Motif Analysis
To identify possible motifs associated with editing sites, we repeated a methodology adapted from previous studies (Liew et al. 2017). For this, flanking regions surrounding each editing event were extracted (up to 200 nt) and grouped by conversion. Each sequence set was randomly divided into a test and training set. De novo identification of motifs in the training set used MEME (Bailey and Elkan 1994) with a maximum of 10 motifs, each of maximum width 60 nucleotides. These motifs were then searched for in the test set using MAST (Bailey and Gribskov 1998). As a control, each motif was also used to search a data set of 10,000 sequences comprising the same distribution of lengths and base distributions as the training set. Analyses were carried out using the MEME suite v4.11.3 (Bailey et al. 2015). All input and output files are available from https://github.com/DacksLab/RNAediting, last accessed March 21, 2018.
Calculation of dN/dS
The number of nonsynonymous and synonymous edits was extracted from the data, considering edits independently within each codon, and the number of nonsynonymous and synonymous sites computed by multiplying the number of each codon by its number of nonsynonymous and synonymous sites. The dN/dS ratio was corrected by taking into account both the observed positional bias, that is, considering the biased distribution of edits among codon position, and the “mutational” bias. An approximate “mutational rate” was computed considering only edits occurring in the two first positions, in order to reduce the impact of increased synonymous changes in the third position due to a potential bias of nonsynonymous mutations compared with synonymous ones at this position.
Statistical Analysis
Unless stated otherwise, all statistical analyses were performed using R v3.3.2, all scripts are available from https://github.com/DacksLab/RNAediting, last accessed March 21, 2018 and further details are provided along with the statistical results. Normal distribution of variables was tested using the Shapiro–Wilk test and a visual inspection of the distribution was performed before applying any further tests. In case of normality two-tailed Student’s t-test or ANOVA were used, if not, Wilcoxon tests were performed. When the data were not independent, post hoc Tukey’s test was used on mixed linear models. Statistical correlations were tested with Spearman’s rank tests (for molecular weight vs. hydrophobicity analysis, as to remain consistent with Mungpakdee et al. 2014) or Pearson correlation tests (all other analyses) as they were performed on normally distributed quantitative data.
Results
Acquisition of New Sequence Data for the Dinoflagellates Pyrocystis lunula and Karenia mikimotoi
As previous studies based on single taxon sampling points yielded inconclusive results regarding editing function, we first obtained genomic and transcriptomic data for two additional dinoflagellates, the peridinin dinoflagellate P. lunula and the fucoxanthin dinoflagellate Kare. mikimotoi (Materials and Methods).
We identified 11 full-length P. lunula plastome-derived coding regions (atpA, atpB, petB, petD, psaA, psaB, psbA, psbB, psbC, psbD, and psbE) by comparison to a dinoflagellate-specific database, following removal of bacterial and nuclear contaminants (Materials and Methods; supplementary fig. S1 and table S1, Supplementary Material online). In the transcriptome assemblies, all 11 genes appeared monocistronic. Comparison of genomic and transcriptomic sequences revealed that four genes had conventional AUG start codons while three had AUU, two UUG, one UCG, and one CUU (supplementary table S1, Supplementary Material online). Bioinformatic searches for tRNA genes (Materials and Methods) yielded no putative hits.
The plastomes of peridinin dinoflagellates typically occur as minicircles (fig. 1), and three P. lunula minicircle sequences are archived in GenBank for psbA (AF490365), psbC (AF490366), and rpl28-rpl33 (AF490367). Our assemblies matched the psbA and psbC entries with the exception of SNPs that may be strain-specific and discrepancies in length. Our psbC assembly is 1,701 nucleotides compared with 4,811 nucleotides for AF490366, and differs from AF490366 in upstream noncoding content, suggesting these two sequences may represent different copies of the same gene, potentially with different subcellular localizations (Laatsch et al. 2004; Owari et al. 2014). Compared with AF490365, which is 657 nucleotides in length and encodes a partial psbA gene, our psbA coding region is 1,325 nucleotides and encodes the full-length gene. We were unable to find rpl28-rpl33 either separately or together, corroborating previous findings that plastid-encoded rpl28-rpl33 sequences are likely to be artifacts or highly strain-specific (Dorrell et al. 2017). To test for the presence of minicircles in the P. lunula plastid genome, primers were designed to perform outward facing PCR that would produce an amplicon only if the sequence formed a minicircle. Six (those encoding atpB, petB, psaA, psbA, psbB, and psbC) produced amplicons, suggesting the plastid genome may occur as multiple minicircles (supplementary table S1, Supplementary Material online).
For Kare. mikimotoi, we obtained genomic sequences for 27 plastid genes using a combination of next-generation and Sanger sequencing (Materials and Methods). We identified two alternative translation initiation codons (one UUG and one AUU), and additionally note that the stop codon for psbX is absent initially from the genomic sequence and is generated through an editing event (supplementary table S1, Supplementary Material online).
Editing Events in Karenia mikimotoi and Pyrocystis lunula
Previous analyses of plastid transcript editing in dinoflagellates relied on manual annotation, which is prohibitive for larger systematic studies. We developed an automated framework for comparing genomic and transcript sequences to detect editing events (Materials and Methods). Comparison of manual and automated editing event detection for psbB, a gene present in the majority of our study taxa, demonstrated perfect agreement across the aligned region in terms of codon position and base conversion, suggesting that our automated methods accurately recapitulate manual assessment (supplementary table S2, Supplementary Material online).
We applied this automated analysis framework to characterize the editing landscape of Kare. mikimotoi and P. lunula, and compared our results to similar analyses of other available sequence data (table 1 and supplementary table S3, Supplementary Material online). We analyzed 27 Kare. mikimotoi genes totaling 15,758 nucleotides, and detected 858 editing events for an overall editing rate of 5.18% (table 1). Although this rate was slightly higher than that of the related Karl. veneficum (4.48%), comparison of 22 genes for which editing data are also available in both lineages suggest the editing rate in Kare. mikimotoi is not significantly higher (P value = 0.37, t-test). Similarly, we analyzed 11 P. lunula genes totaling 13,827 nucleotides and identified 747 edited residues, for an overall editing rate of 4.86% (table 1). The editing rate in P. lunula is significantly higher than in S. minutum (4.86% vs. 2.89%, P value = 0.043, t-test) (Mungpakdee et al. 2014), and is similar to Karl. veneficum (4.48%) (Richardson et al. 2014).
Table 1.
Summary of Plastid Transcript Editing across Taxa
| Organism | # Genes | Length (bp) | Length (aa) | # Edits | Avg % Edits | Avg % Nonsyn | Avg % aa Change |
|---|---|---|---|---|---|---|---|
| C. horridum | 3 | 2,988 | 994 | 196 | 6.41 ± 1.25 | 93.72 ± 1.44 | 16.34 ± 3.47 |
| Heterocapsa triquetra | 10 | 11,681 | 3,887 | 24 | 0.31 ± 0.35 | 89.29 ±18.21 | 0.85 ± 1.09 |
| Karenia mikimotoi | 27 | 15,758 | 5,248 | 858 | 5.18 ± 2.79 | 81.80 ± 25.45 | 12.34 ± 7.73 |
| Karlodinium veneficum | 62 | 26,938 | 8,747 | 1,087 | 4.48 ± 2.66 | 92.46 ± 14.04 | 11.52 ± 6.18 |
| Lingulodinium polyedrum | 1 | 1,037 | 345 | 11 | 1.06 | 9.09 | 0.29 |
| Pyrocystis lunula | 11 | 13,827 | 4,608 | 747 | 4.86 ± 2.16 | 87.83 ± 9.56 | 11.62 ± 5.13 |
| Symbiodinium minutum | 12 | 13,395 | 4,465 | 389 | 2.89 ± 1.86 | 95.32 ± 4.64 | 7.68 ± 4.67 |
Assessment of editing events in consensus sequences could be complicated by three factors: variability of genomic sequences for the same gene within a single genome, incompletely processed or unprocessed transcripts, and different base conversions at the same position. To understand the extent to which consensus sequences might mask this underlying variability, we carried out SNP analysis of filtered genomic and transcript reads (Materials and Methods). We identified 490 editing events in P. lunula with high quality mapped reads for both genomic and transcript sequences. Of these, 35 positions had genomic reads containing the edited base, yet only at nine positions did this account for >10% of the reads. Furthermore, transcript reads harboured only two alleles corresponding to the original genomic and edited bases; for the majority (416/490) of positions the edited base accounted for >50% of the reads, suggesting the presence of some incompletely processed/unprocessed transcripts (supplementary table S4, Supplementary Material online). The small fraction of positions in which the editing event accounts for <50% of the reads is likely due to the stringent filtering of reads prior to SNP analysis (Materials and Methods).
We additionally searched for evidence of promiscuous or incomplete editing in Sanger sequences of cloned individual transcripts, generated during previous investigations of the plastid transcriptomes of Kare. mikimotoi and Karl. veneficum (Dorrell and Howe 2012; Richardson et al. 2014; Dorrell et al. 2016). The sequences obtained through these reactions not only serve as a secondary control for the Kare. mikimotoi RNA-seq data but may also provide specific insights into transcripts that occur at low abundance in total RNA pools, but were specifically amplified through the RT-PCR experiments performed, for example, transcripts that extend past the 3′ poly(U) site of the corresponding gene, which typically are at much lower abundance, and have diminished editotypes compared with polyuridylylated transcripts (Dang and Green 2009; Dorrell and Howe 2012; Dorrell et al. 2016). We screened 26,699 nt aligned transcript sequences, corresponding to 4,361-bp genomic sequence from Kare. mikimotoi, and 2,217 nt aligned transcript sequences, corresponding to 684-bp genomic sequence from Karl. veneficum, against the corresponding genomic and consensus mRNA sequences for each organism, to identify abnormal editing events (supplementary table S4, Supplementary Material online).
We found only very limited evidence for incomplete editing of sites that are edited in the consensus mRNA sequence, with only 85/1,187 (6.0%) editing events predicted from consensus mRNA sequences missing from individual cloned transcripts in Kare. mikimotoi, and only 4/51 (6.6%) editing events missing from cloned transcripts in Karl. veneficum (supplementary fig. S2, Supplementary Material online). In addition, we could only find three positions across all the transcripts screened in which editing events not present in the consensus transcript sequence, that is, potential mis-editing events, were detected in >50% of the cloned transcript sequences of the gene (supplementary table S4, Supplementary Material online).
These analyses suggest that, although some limited variability is present at positions in both genomic and transcript sequences, this is unlikely to impact significantly the global analysis of editing function. Hence, we focussed on the dominant editotype for further analysis, that is, that of the most frequently present genomic base to the most frequently present transcript base.
Editing Is Not Distributed Based on Phylogenetic Affiliation
We considered whether specific editing events were conserved across orthologous plastid genes from multiple dinoflagellate species. For this, we compared global editing events across a 17 species (1,121 nt) alignment of psbA sequences, including both peridinin and fucoxanthin dinoflagellates, for which we could access both a gDNA and mRNA sequence from both GenBank and other published transcriptome resources (supplementary fig. S3 and table S5, Supplementary Material online) (Keeling et al. 2014; Dorrell et al. 2017). We identified 336 suspected editing events across the entire alignment. Of these, only 40 specific events (i.e., the same interconversion at the same position) were found to occur in more than one species (supplementary fig. S3A, Supplementary Material online), and only 35 editing events were found to have homologues (defined as any editing event occurring at the same position) within a majority of the members of an individual dinoflagellate clade, as inferred using a single-gene psbA phylogeny (supplementary figs. S3A and S4, Supplementary Material online). By comparing these two analyses, we only found two cases where the same specific editing interconversion was found in multiple related dinoflagellate species, and therefore might represent conserved events. These were a G-to-C editing event, identified in three members of the Symbiodiniaceae (S. minutum, spp. PSP1-05 and spp. C15) and an A-to-C/G editing event identified in three members of the Gonyaulacales (L. polyedrum, Alexandium catenella, and A. tamarense; supplementary fig. S3B, Supplementary Material online). Thus, the vast majority of editing events within the alignment were found to be species-specific, and do not possess clear homologues in related or unrelated species.
Quantitative Trends in Editing
Having established the likely species-specific nature of individual editing events within dinoflagellate plastids, we sought to determine whether general features of editing are conserved across dinoflagellates, which might suggest that editing follows similar principles and potentially involves conserved machinery.
All possible base conversions occur across our data set. A-to-G (∼42%) and T-to-C (∼25%) events were most frequent and some events appear taxonomically restricted, notably C-to-G events in fucoxanthin dinoflagellates and G-to-U events in P. lunula (table 2). Editing is concentrated in first and second codon positions in all taxa apart from L. polyedrum (ranging between 76.19% in H. triquetra and 93.40% in C. horridum), and this distribution in each lineage is significantly different from that expected by chance (P value < 0.05, χ2 test). This effect resulted in primarily nonsynonymous amino acid changes in gene translation products (ranging between 81.80% in Kare. mikimotoi and 95.32% in S. minutum, table 1), but without a large effect on codon usage (supplementary fig. S5, Supplementary Material online). Consistent with these observations, editing results in GC content increase in all taxa apart from L. polyedrum, and the increase is significantly greater in fucoxanthin than peridinin lineages (mean increase of 3.63% vs. 2.01%, P value = 7.74e-05, t-test). GC content increase is significant in codon positions one and two, but not three (fig. 2), and these trends are conserved across taxa (supplementary fig. S6 and table S3, Supplementary Material online).
Table 2.
Summary of Observed Base Conversion Frequencies across Taxa
| Organism | A/U | A/G | A/C | T/A | T/G | T/C | G/A | G/U | G/C | C/A | C/U | C/G |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C. horridum | 0 | 37.76 | 2.04 | 0 | 0 | 29.08 | 11.73 | 0 | 8.16 | 0 | 11.22 | 0 |
| Heterocapsa triquetra | 4.17 | 50.00 | 4.17 | 0 | 0 | 25.00 | 0 | 0 | 12.50 | 0 | 4.17 | 0 |
| Karenia mikimotoi | 0 | 30.77 | 14.80 | 0.11 | 0.35 | 34.27 | 7.11 | 0 | 8.86 | 0.82 | 2.79 | 0.12 |
| Karlodinium veneficum | 0.55 | 52.44 | 1.93 | 0.18 | 0.83 | 34.31 | 5.52 | 0 | 0.83 | 0.09 | 3.04 | 0.28 |
| Lingulodinium polyedrum | 9.09 | 27.27 | 0 | 9.09 | 9.09 | 0 | 0 | 0 | 0 | 0 | 45.45 | 0 |
| Pyrocystis lunula | 0 | 38.29 | 0.54 | 0.54 | 0.54 | 37.62 | 12.18 | 0.40 | 0.80 | 0.27 | 8.84 | 0 |
| Symbiodinium minutum | 0.26 | 55.27 | 2.31 | 0 | 0.26 | 13.62 | 6.94 | 0 | 6.68 | 0 | 14.65 | 0 |
Fig. 2.
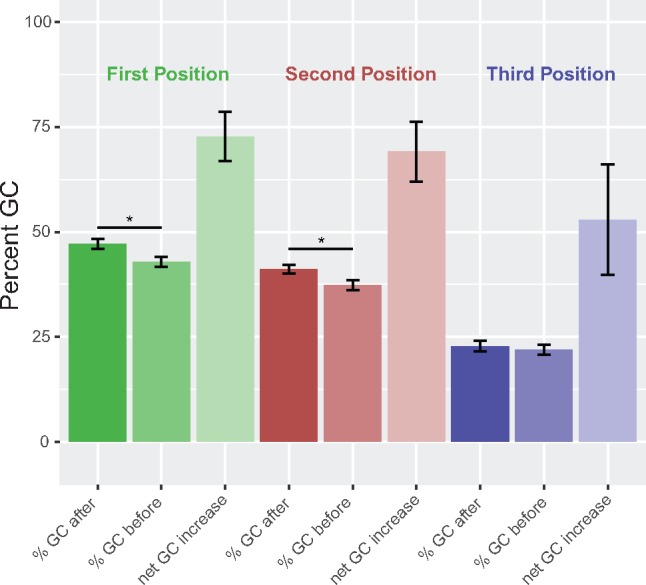
—Effect of editing on GC content. This figure shows the inherent GC content bias among codon positions in dinoflagellates, with higher GC content in positions one and two, and the effect of editing to increase GC content significantly in positions one and two, but not three. Bars are color-coded by codon position. Net GC increase defined as the difference between GC-enriching (i.e., A or T to G or C) and GC-depleting edits. Error bars denote 95% confidence intervals.
A recent study suggested the presence of motifs directing a subset of editing events in the S. microadriaticum nuclear genome (Liew et al. 2017). To test this in our data set, we extracted flanking regions for each editing event and searched for sets of motifs that direct editing events across lineages (Materials and Methods). Though we could identify motifs enriched in these flanking regions relative to controls, these were not conserved across all lineages. Additionally, searches using those motifs previously identified scored poorly compared with ab initio predictions (typically, P value of e-05 compared with e-20), including in the related S. minutum. Thus, it does not appear that sequence elements within a 200-nucleotide window show consistent signals to direct these events across dinoflagellates.
Editing Events Are Clustered along Genes
Previous studies have suggested clustering of editing events in both dinoflagellate mitochondria and plastid sequences (Lin et al. 2007; Zhang et al. 2008; Richardson et al. 2014). We developed an automated analysis to measure quantitatively clustering of editing events across genes by comparing our results to simulated data (Materials and Methods). We found that editing is clustered in all genes of C. horridum, Kare. mikimotoi, L. polyedrum, and P. lunula, and is clustered in the majority (>70%) of genes from other taxa (supplementary table S6, Supplementary Material online). We also asked whether editing events occurred more frequently in the same, or adjacent, codon to other editing events, and found this to be highly significant (fig. 3). Thus, editing is clustered along the length of genes, and this clustering is unlikely under models of randomly distributed events.
Fig. 3.
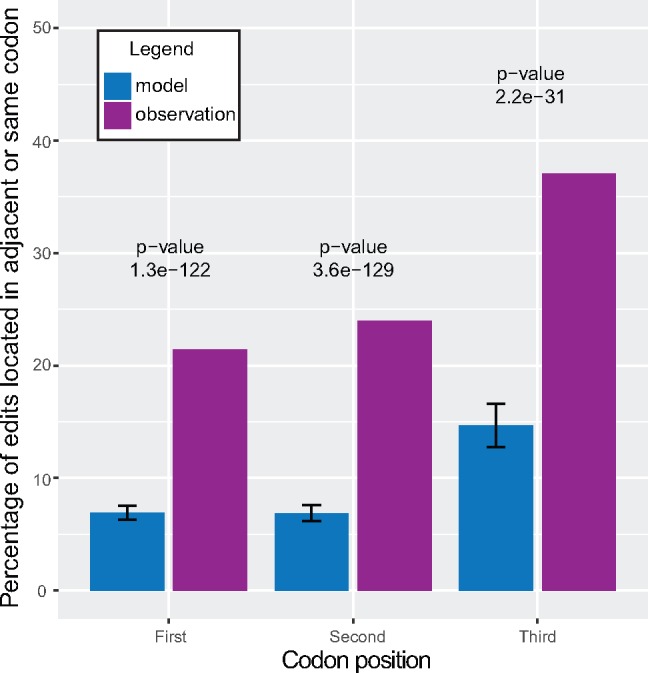
—Editing events occur in close proximity to each other. This figure shows the significant propensity of edits to occur in the same, or adjacent, codon as other edits. For each codon position, the number of edits expected to occur in the same or adjacent codon was determined based on a random distribution of edits with the same codon position preferences as observed in real data. Comparison of the actual distribution to the expected distribution showed highly significant clustering when considering edits in all three positions. Error bars represent SD of the simulation (n=100).
Functional Consequences of Editing
We next aimed to discern functional trends in editing. Here, we focus on S. minutum and P. lunula from the peridinin dinoflagellates and Kare. mikimotoi and Karl. veneficum from the fucoxanthin dinoflagellates, as these represent our most complete plastid editing data sets.
Effect of Editing on Protein Size and Hydrophobicity
It was previously reported that editing in S. minutum resulted in decreased molecular weight but increased hydrophobicity of proteins (Mungpakdee et al. 2014), as measured by GRAVY score (Kyte and Doolittle 1982). We calculated molecular weight and hydrophobicity across our data set (supplementary table S7, Supplementary Material online) and found negligible correlation between them (fucoxanthin lineages rho = 0.075, P value = 0.49, peridinin lineages rho = 0.19, P value = 0.39, fig. 4A). Restricting these calculations to all members of gene families universally plastid-encoded in both peridinin and fucoxanthin lineages (atp, psa, psb, and pet) did not improve the correlation (fucoxanthin lineages rho = −0.079, P value = 0.68, peridinin lineages rho = 0.19, P value = 0.39, supplementary fig. S7, Supplementary Material online), nor did restricting the calculations to transmembrane proteins (fucoxanthin lineages rho = 0.050, P value = 0.81, peridinin lineages rho = 0.19, P value = 0.44). We did note strong correlations in specific organisms: for example, consistent with the results of Mungpakdee et al. (2014), decreased molecular weight was strongly correlated to increased protein hydrophobicity following editing in S. minutum (rho = −0.66, P value = 0.019, fig. 4B). However, a significant but contrasting correlation was observed in P. lunula, in which decreased molecular weight was associated with decreased hydrophobicity (rho = 0.79, P value = 0.0038, fig. 4B). Hence, although editing does significantly decrease molecular weight in all lineages (P value < 0.05, t-test), we conclude that there is no consistent trend of editing to change both molecular weight and protein hydrophobicity.
Fig. 4.
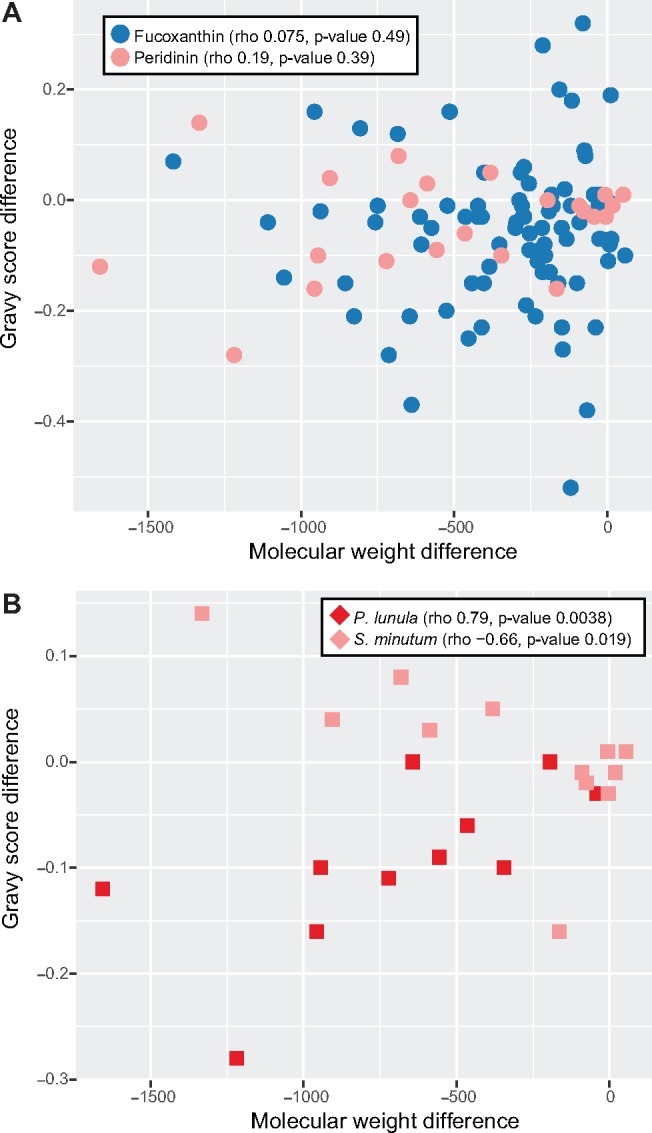
—Effect of editing on protein size and hydrophobicity. This figure shows scatter plots between molecular weight difference and GRAVY score before and after editing for fucoxanthin and peridinin dinoflagellate plastid sequences (A), and peridinin dinoflagellate data sets, Pyrocystis lunula and Symbiodinium minutum (B). Spearman’s rank correlation tests (rho) are reported along with their significance. The trend previously reported for S. minutum, that editing results in proteins that have lower molecular weight and are more hydrophobic (Mungpakdee et al. 2014), is observed here, but not in any other data set.
Editing Is Associated with Divergent Sequence Regions
We extended the sliding window analysis presented in Richardson et al. (2014) on the genes tufA and psaA to all genes across our data set (Materials and Methods). For each gene, this analysis involved comparisons of editing rate between the dinoflagellate genomic and transcript sequences to the similarity between the dinoflagellate genomic sequence and a reference sequence across the length of the gene, with the similarity between the two sequences calculated using both nucleotide identity and amino acid similarity (fig. 5A, Materials and Methods). To reduce the possibility of artefactual results based on choice of reference sequence, we compared our sequences to five taxa: the basally divergent dinoflagellate Amphidinium carterae the haptophyte Emiliania huxleyi, the stramenopile Phaeodactylum tricornutum, and the chromerid Vitrella brassicaformis, in which widespread plastid transcript editing is not observed (Barbrook et al. 2012; Dorrell and Howe 2012; Dorrell et al. 2014; fig. 1), and the haptophyte Chrysochromulina tobin, which has not yet been analyzed for editing, although is anticipated not to possess plastid RNA editing events given its broad absence from other studied haptophyte lineages (Fujiwara et al. 1993; Dorrell and Howe 2012) (Materials and Methods; supplementary table S1, Supplementary Material online).
Fig. 5.
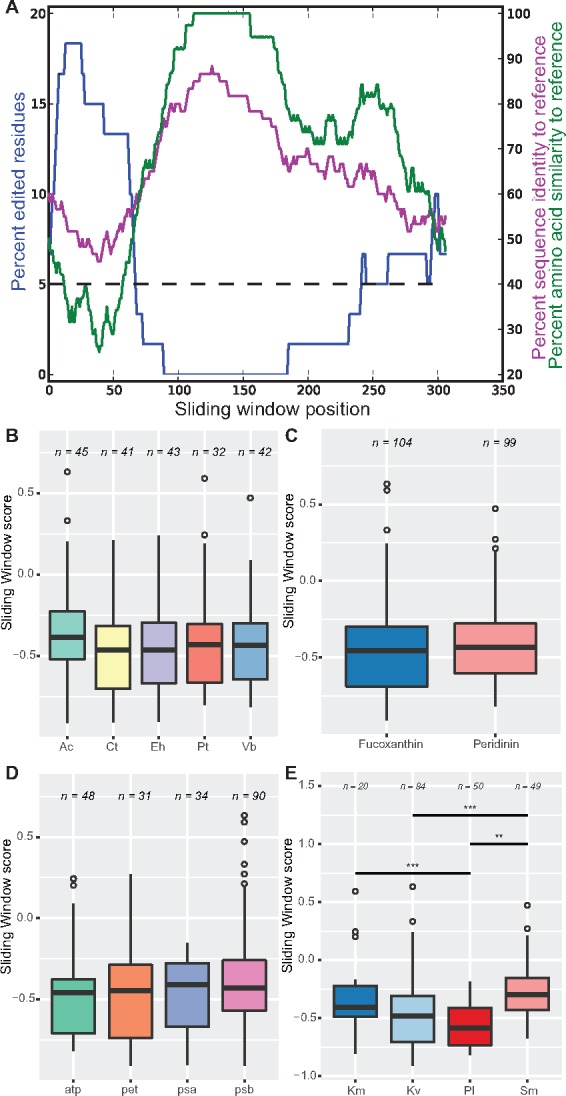
—Sliding window editing analysis and protein sequence conservation. This figure outlines the rationale behind correlating local editing rate to sequence conservation with reference sequences, and the overall effect that editing associates with more divergent regions of genes. (A) Exemplar graph showing the result of correlative sliding window analysis in the Karlodinium veneficum psaB gene. The x axis denotes the position along the gene, whereas the left hand axis denotes the percentage of edited residues in a given window, and is indicated by a blue line. The right hand axis denotes the percentage identity to a reference sequence (Emiliania huxleyi) for both the amino acid sequence (green line) and the nucleotide sequence (magenta line) in a given window. The horizontal dotted line denotes the average editing rate across the entire gene sequence. (B–E) Boxplots quantifying sliding window analysis, as in (A), as the Pearson’s correlation coefficient between per window amino acid similarity and editing rate across the whole sequence. Separate plots illustrate these values between reference sequences (B), between plastid types (C), by gene family (D), and by organism (E). Correlations with P value >0.05 were not included; n denotes sample size (number of genes). Significance is denoted: *P value<0.05, **P value<0.01, ***P value<0.001. Abbreviations: Ac, Amphidinium spp.; Ct, Chrysochromulina tobin; Eh, Emiliania huxleyi; Pt, Phaeodactylum tricornutum; Vb, Vitrella brassicaformis; Km, Karenia mikimotoi; Kv, Karlodinium veneficum; Pl, Pyrocystis lunula; Sm, Symbiodinium minutum.
Our initial studies demonstrated negative correlations between editing and sequence divergence from references in some but not all organism/reference pairs. Cross-referencing to relevant alignments revealed that large indels are present in some genes that affected global correlation calculations (supplementary fig. S8, Supplementary Material online). Hence, we developed an automated method to trim out indels without altering sequence reading frames (Materials and Methods) and applied this to our data sets. Repeating the analysis on trimmed data sets revealed an overall negative correlation between editing of any given sequence and sequence similarity to reference sequences, regardless of nucleotide/amino acid comparison, or the number of edits applied to the gene (fig. 5A and supplementary fig. S9A and table S8, Supplementary Material online).
We focused on all members of the atp, pet, psa, and psb gene families for further analysis, as these genes are located in the plastid genomes of both peridinin and fucoxanthin-containing dinoflagellates (Gabrielsen et al. 2011; Mungpakdee et al. 2014; Dorrell et al. 2017). We excluded any correlation values that were not significant (excluded 19/229 nucleotide; 26/229 amino acid values; P value > 0.05, supplementary table S8, Supplementary Material online). Regardless of the reference used, the overall correlations obtained were typically around −0.5 between sequence conservation and amino acid similarity following editing (fig. 5B), and were similar between fucoxanthin and peridinin lineages (fig. 5C andsupplementary fig. S9B, Supplementary Material online). There was no difference among genes regardless of plastid affiliation (fig. 5D). Repeating these analyses considering only nonsynonymous editing events, which actually change the resulting amino acid sequence, or considering nucleotide sequences, gave similar results (supplementary table S8, Supplementary Material online). Comparison of individual taxa revealed a weaker correlation in both Kare. mikimotoi and S. minutum (fig. 5E), and these values were significantly (P value < 0.05) different when compared with both Karl. veneficum and P. lunula (S. minutum) or P. lunula only (K. mikimotoi).
To further investigate this phenomenon, we compiled larger reference data sets of between 27 and 30 reference sequences from the majority of plastid lineages, for each gene of eleven genes conserved across our core dinoflagellate data set (supplementary table S1, Supplementary Material online). We compared each of these to the corresponding amino acid translations of each dinoflagellate genomic/transcript pair to determine the diversity of residues, measured by a modified Shannon’s entropy score (Sander and Schneider 1991; Valdar 2002) that we term “positional entropy” (Materials and Methods), among reference sequences at edited positions. We calculated positional entropy by taking into account the presence of indels and normalizing so that the final score ranged between zero (maximum entropy) and one (complete conservation). We did not score sites at which 50% or more of the reference sequences contained a gap, as these could correspond to indels or poorly aligned regions.
We noted that the distribution of positional entropy scores was similar between peridinin and fucoxanthin lineages, and among organisms, with a high frequency of conserved positions tapering off at lower values of positional entropy (fig. 6A and B;supplementary table S9, Supplementary Material online). This is consistent with conservation of plastid orthologues among distantly related taxa. Next, we compared the distribution of positional entropy scores between edited and nonedited residues, and found that editing occurs in residues with a significantly lower positional entropy score (P value < 0.001, fig. 6C). Thus, editing is generally associated with regions capable of tolerating a higher level of variability in dinoflagellate plastids.
Fig. 6.

—Positional entropy analysis of sequences. This figure shows that, although orthologues from all lineages under study are overall well-conserved compared with plastid sequences without editing, amino acids changed by RNA editing are present in significantly less well conserved positions than those unchanged by RNA editing. Separate boxplots show positional entropy scores of all sites between plastid types (A) and by organism (B), and between edited and nonedited sites (C). Positions for which residues were absent in more than half of reference sequences were not scored; n denotes sample size (number of positions). Significance is denoted: *P value<0.05, **P value<0.01, ***P value<0.001. Abbreviations: Km, Karenia mikimotoi; Kv, Karlodinium veneficum; Pl, Pyrocystis lunula; Sm, Symbiodinium minutum.
Finally, we investigated the biological underpinning of this effect by comparing editing status of individual amino acid positions with their known functions in protein and cofactor interactions and metabolic functions, based on previous annotations (Dorrell et al. 2017; supplementary table S10, Supplementary Material online). In proteins with more than one biochemical environment, we observed no significant difference in editing event distribution between these environments (lumen 428/4,294, stroma 231/2,164, transmembrane 306/2,734 positions edited, P value = 0.29, χ2 test). We did however observe significantly fewer edits in residues with functional annotations compared with those without known functions (140/2,388 positions vs. 1,044/9,034, P value = 3.9e-06, t-test). Thus, the majority of editing events cluster in regions of low sequence similarity to orthologous sequences from lineages that do not perform plastid transcript editing, and are depleted in residues with known functional roles, in both peridinin and fucoxanthin dinoflagellates.
Editing Is Primarily Corrective
Editing has the potential to alleviate otherwise deleterious mutations in the underlying genomic sequence prior to their having phenotypic consequences, which we henceforth term “corrective editing.” One example of this in dinoflagellate plastid sequences is the removal of premature STOP codons (Dorrell and Howe 2012; Jackson et al. 2013; Richardson et al. 2014), which we confirm occurs in C. horridum (1), H. triquetra (1), Kare. mikimotoi (4), Karl. veneficum (10), P. lunula (7), and S. minutum (2), through alteration of usually one or two, but in rare cases three, bases (supplementary table S3, Supplementary Material online).
In order to determine if other corrective editing events outside of premature STOP codon removal occur we quantified the relative biochemical consequence of each edit with an “editing score.” This was equivalent to the difference obtained from comparison of both genomic and transcript amino acids to the homologous amino acid in a reference sequence using the Blosum62 substitution matrix (Materials and Methods). Positive scores therefore indicate an increase in biochemical similarity to the amino acid in the reference sequence, whereas negative scores indicate a decrease in biochemical similarity. For consistency, we chose the same reduced gene set and reference organisms as for our sliding window correlation analysis. To test if the overall corrective effect of editing could be explained simply by the biases we identified in terms of both codon position and base conversion (table 2 and supplementary table S3, Supplementary Material online), we compared the editing scores observed to patterns of editing found on simulated sequences based on our observed data, ensuring that the proportion of editing events associated with each codon position and base conversion were not significantly different from those observed in real data (P value > 0.05, χ2 test, Materials and Methods).
Scatterplots of editing score frequency indicate overall positive scores and moderate negative values falling between 0 and −2 (fig. 7A and supplementary table S11, Supplementary Material online). In each species, the values were found to be significantly higher than would be expected by chance in the majority of cases (Karl. veneficum 48%, K. mikimotoi 67%, S. minutum 83%, and P. lunula 91%), and in all cases the observed score was higher than the simulated score (supplementary table S6, Supplementary Material online). No significant differences were found between the reference sequences used and any of the results obtained (fig. 7B). Thus, the majority of editing events in dinoflagellate plastids have an overall corrective effect.
Fig. 7.

—Summary of corrective editing. This figure highlights the overall corrective effect of editing to result in amino acids that are more similar to their homologues in orthologues from organisms that do not undergo RNA editing than those originally encoded in the genomic sequence. (A) Scatter plot of the relationship between the number of editing events and the average editing score for each gene. (B) Boxplots showing that the corrective effect is independent of choice of reference organism; n denotes sample size (number of events). Abbreviations: Ac, Amphidinium spp.; Ct, Chrysochromulina tobin; Eh, Emiliania huxleyi; Pt, Phaeodactylum tricornutum; Vb, Vitrella brassicaformis; Km, Karenia mikimotoi; Kv, Karlodinium veneficum; Pl, Pyrocystis lunula; Sm, Symbiodinium minutum.
Editing Functions Vary between Lineages
Similar to the sliding window correlation analysis (fig. 5D), we observed no significant difference in editing score between genes (fig. 8A), in all lineages (supplementary fig. S10A, Supplementary Material online). We did however note a negative correlation between editing rate and score, that is, that editing in genes with low editing rates is primarily corrective (PC = −0.26, P value = 0.0039). We did not find any significant correlation between the sliding window correlations to assess the magnitude of editing in divergent sequence regions, with average editing score, indicating that editing functions are conserved regardless of whether editing primarily occurs in conserved or variable regions of genes (supplementary fig. S10B, Supplementary Material online). Nevertheless we noted that peridinin lineages have significantly higher editing scores than fucoxanthin lineages, that is, editing has a stronger corrective effect in peridinin dinoflagellates (P value < 0.001, Tukey’s test, fig. 8B). This result was borne out by comparison of the individual taxa (fig. 8C). Although peridinin lineages almost always had positive scores, there was a significantly greater spread of editing scores in fucoxanthin lineages (F = 1.20, P value = 1.9e-6, F-test), including a substantial number of editing events with negative scores.
Fig. 8.

—Editing functions vary across genes and lineages. This figure shows how, despite that editing is corrective overall, this corrective effect varies across organisms and that noncorrective events occur in variable and conserved positions. Separate boxplots show editing scores of all sites between genes (A), plastid types (B), and by organism (C); n denotes sample size (number of events). (D) Scatter plot showing the relationship between positional entropy of edited positions and the effect of editing events, as assessed by average editing score. Lines represent density of values for clarity. Note not only that the plot is skewed toward positive editing score values but also that a number of noncorrective events exist, including in highly (i.e., entropy > 0.90) conserved positions. (E) Plot as in (D), but focussing on individual organisms. Significance is denoted: *P value < 0.05, **P value < 0.01, ***P value < 0.001. Abbreviations: Km, Karenia mikimotoi; Kv, Karlodinium veneficum; Pl, Pyrocystis lunula; Sm, Symbiodinium minutum.
As very detrimental editing events would likely be short-lived in a population, we reasoned that noncorrective editing changes may occur in variable positions within protein sequences. To understand better the potential relationship between editing and conservation, we calculated an average editing score for each edited position. This involved the same editing score calculated previously, but averaged the values obtained across all reference sequences for each site and hence serves as a rough measure of the overall corrective effect of editing for a given position, when considering orthologues from diverse plastids. Editing scores were in general skewed toward positive values, consistent with our previous analyses; however, we found that many of the positions with negative editing scores had low positional entropies, that is, strong conservation in reference sequences, including in invariant sites, for both fucoxanthin and peridinin lineages (fig. 8D), suggesting the existence of a population of noncorrective editing events across lineages that cannot be explained through relaxed sequence constraints alone.
To investigate this further, we compared the occurrence of noncorrective edits to functional annotation data (Dorrell et al. 2017; supplementary table S9, Supplementary Material online). Although functional positions were generally associated with corrective edits, we found several instances where noncorrective events occurred in presumably important functional residues in fucoxanthin, but not peridinin, lineages. This includes, for example, the edited removal of otherwise conserved residues implicated in cofactor binding and intersubunit interactions within the C-terminal region of K. veneficum psbD transcripts (supplementary fig. S11, Supplementary Material online). Thus, there are some differences in the functions of editing in peridinin and fucoxanthin lineages, which may relate to editing on individual fucoxanthin plastid genes having noncorrective functions.
Discussion
In this study, we have presented the first systematic analysis of transcript editing across dinoflagellate plastids. We have employed novel bioinformatic methods to investigate the dynamics of editing across multiple species, including novel data for the fucoxanthin dinoflagellate Kare. mikimotoi and the peridinin dinoflagellate P. lunula. Overall, we found that editing occurs frequently, with an average rate of ∼5% (table 1), though we observed rates as high as 14.33% (Karl. veneficum psbD) and confirmed the complete absence of editing from psbA and psaB in Heterocapsa triquetra (Dang and Green 2009) (also for psbB when not considering the poly-U tail; supplementary table S3, Supplementary Material online). We did not observe clear trends in editing associated with gene families or protein functions, though some gene families appear to be more highly edited than others; for example, the pet genes are edited at an average of 5.44%, whereas the psb genes are edited at an average of 2.38%.
We considered whether specific editing events between lineages are conserved across multiple species, or whether they represent independent applications within groups or lineages. Previous studies have suggested phylogenetic correlation between editing events and taxonomic distribution in the cob and cox1 genes of dinoflagellate mitochondria (Zhang et al. 2008). However, in plant plastids, where more data are available, it does not appear that specific editing events are structured in a phylogenetic context (Takenaka et al. 2013, inter alia); for example, several editing events are conserved in Nicotiana tabacum and N. sylvestris ndhB and ndhD, but absent from the closely related N. tomentosiformis (Sasaki et al. 2003). Certain plastid transcript editing appears to be conserved between closely related dinoflagellate species, as inferred from detailed inspections of either psaA (Mungpakdee et al. 2014) or psbA editotypes (supplementary fig. S3, Supplementary Material online). A lack of conservation between the evolutionarily unrelated plastids of peridinin and fucoxanthin dinoflagellates is not unexpected, but we show this trend holds within each lineage (peridinin or fucoxanthin dinoflagellates) as well (supplementary fig. S3, Supplementary Material online). This may reflect that editing is an extremely dynamic feature, that is, specific editing events may originate and then be secondarily lost in individual dinoflagellate plastids, or that the extremely fast sequence evolution in dinoflagellate plastid genomes (Janouškovec et al. 2010; Dorrell et al. 2017), which may compensate for previous mutation events, or render individual editing events functionally redundant, precludes the establishment of evolutionarily stable editing sites in multiple species.
In contrast with the highly dynamic evolution of individual editing sites within dinoflagellates, we found many examples of conserved patterns within editing. In both fucoxanthin and peridinin species, the majority (>85%) of editing events were nonsynonymous, and occurred in either the first or second position of codons; editing sites are clustered and biased toward highly divergent regions of individual plastid genes; and editing events have principally corrective functions (table 1;figs. 3, 5, and7). We additionally find evidence of editing trends that are found in nearly all species studied or in highly unrelated species, suggesting that they can potentially occur globally across both peridinin and fucoxanthin plastid lineages. These include the removal of premature STOP codons from plastid sequences in all species except the lone gene from L. polyedrum; cases of editing generating STOP codons initially absent in the genomic sequence, in Kare. mikimotoi psbX and in P. lunula atpB and psbD (supplementary table S3, Supplementary Material online); and all twelve editing interconversions in both peridinin and fucoxanthin plastid lineages, barring C-to-G events and G-to-U events, which appear restricted to fucoxanthin plastids and P. lunula, respectively (fig. 1).
The wide range of functions conserved between the editing machinery of peridinin and fucoxanthin plastids is highly consistent with a common origin. This could result if the incoming fucoxanthin plastid acquired an editing machinery retained from an ancestral peridinin plastid, or if the fucoxanthin and peridinin plastids independently acquired their editing machinery from the same source. It has been suggested that machinery mediating editing events may translocate between genomes within a single cell, increasing the likelihood that multiple genomic contexts within a lineage may adopt editing (Smith and Keeling 2015). Mitochondrial transcript editing has been recognized in dinoflagellates for over a decade (Lin et al. 2002), and a recent report in S. microadriaticum suggests extensive nuclear transcript editing as well (Liew et al. 2017). Many of the editing trends observed in dinoflagellate plastids (e.g., clustering of editing events, bias toward specific codon positions and nonsynonymous substitutions, and use of an expanded or complete repertoire of editing interconversions) are also found in dinoflagellate mitochondrial and nuclear editosomes (Jackson et al. 2007; Zhang et al. 2008; Liew et al. 2017), consistent with a common ultimate origin of editing in all three organelles.
Understanding how the plastid RNA editing systems in dinoflagellates originated rests on identifying the underlying effector proteins, which remain poorly understood. Transcript editing is known to have evolved independently in multiple organelles in distantly related eukaryotic lineages (Gray 2012; Smith and Keeling 2015), involving distinct machinery in each case. Though the full editing machinery is not currently known in plant plastids, an array of PPR, MORF, and OZ proteins are known to be critical components (Sun et al. 2016). Of these, PPR proteins are known to bind sequence motifs upstream of editing events, and direct editing site specificity (Shikanai 2015). Though PPR proteins have been identified in dinoflagellates, and some putative sequence elements defined (Liew et al. 2017), it is unknown whether these PPR proteins are targeted to, or function in, dinoflagellate plastids, and we were unable to determine universal motifs in a 200-nucleotide window surrounding each event, suggesting that relevant sequence features may be specific to small numbers of events. It is also possible that multiple systems of RNA editing machinery are present in a single organelle; discussing this possibility in dinoflagellates, Lin et al. (2007) note that the slime mould Physarum polycephalum has both substitutional and insertional transcript editing within the mitochondria. Previous studies, including Lin et al. (2007) and Mungpakdee et al. (2014), have proposed models of sequential acquisition of plastid editing machinery during dinoflagellate evolution, allowing the possibility of more than one type of editing event. However, our data show a much larger variety of edit types present in both fucoxanthin and peridinin dinoflagellates than have been previously reported, and support a single—or relatively limited—number of evolutionary transitions in the amount of edit types available within the plastid.
A related question is why transcript editing evolved and is maintained in extant lineages, including application to the replacement fucoxanthin plastid. Our results show that editing sites are broadly distributed over plastid genomes and favour divergent or less functionally critical regions, suggesting that their application may be promiscuous, similar to some A-to-I editing in mammals (Nishikura 2010). Our observation that editing is higher in regions of divergent sequence (fig. 5A), suggests these represent either mutational hotspots within the plastid genome, or encode protein segments more tolerant of amino acid changes. However, our observations regarding codon position bias and nonsynonymous editing, combined with our simulation studies, suggest transcript editing is not simply a random process. This is also supported by dN/dS calculations of editing, corrected for base and codon preferences, which suggest editing event retention is under selective control (supplementary table S12, Supplementary Material online). Therefore, retention of transcript editing is likely to be due to its functional significance, via a constructive neutral model of evolution. One hypothesis, which has already been proposed for fucoxanthin dinoflagellates, is that editing serves to correct deleterious changes in the genomic sequence prior to translation (Dorrell and Howe 2012; Richardson et al. 2014). In this view, editing allowed for the fixation of genomic mutations, which subsequently increased dependency on editing machinery through the decreasing probability of correction of multiple mutations by spontaneous reversion. Evolutionary “ratchet” mechanisms have previously been proposed to explain RNA editing systems across eukaryotes, and, on a general level, may act as a driver of “irremediable complexity” in cellular systems (Lynch 2007; Gray et al. 2010; Lukeš et al. 2011).
A final unresolved question is why the editing machinery varies in terms of function between different dinoflagellate species. This variation may occur at the level of individual species: for example, a global effect of editing to decrease protein size and increase protein hydrophobicity is restricted to S. minutum (Mungpakdee et al. 2014; fig. 4B andsupplementary table S7, Supplementary Material online). More dramatically, detrimental editing events that reduce the sequence conservation of otherwise highly invariant positions are more prevalent in fucoxanthin than peridinin lineages (fig. 8 and supplementary fig. S11, Supplementary Material online). The different functional consequences of editing in individual dinoflagellate plastid lineages might reflect lineage-specific changes in the selective environments encountered, or the biochemistry and protein–protein interactions observed in individual plastids. Previous EST studies have indicated that fucoxanthin plastids utilize nuclear-encoded proteins of peridinin origin (e.g., phosphoribulokinase) to support core metabolic pathways (Patron et al. 2006; Waller et al. 2006; Dorrell and Howe 2015). Plastid-encoded proteins that interact with these proteins (e.g., the RuBisCo components RbcS and RbcL) might be placed under a different selective landscape, and develop a different editotype, than would evolve in the absence of such chimeric interactions. Understanding the physiological drivers underpinning individual dinoflagellate plastids rests on better understanding the different plastid-targeted proteins found in each lineage. The recent publication of two dinoflagellate genomes (for S. minutum and Symbiodinium kawagutii; Shoguchi et al. 2013; Lin et al. 2015), alongside high-quality transcriptome libraries for >50 further species via MMETSP (Keeling et al. 2014) and other sequence projects, will likely prove invaluable for exploring this question.
In conclusion, we use novel bioinformatic methods to analyze plastid transcript editing in the dinoflagellates Kare. mikimotoi and P. lunula in the context of publicly available data for other organisms. Though specific editing events appear to be lineage-specific, we identified conserved large scale effects of editing, including changes in GC content, positional bias of editing in codons and across genes, and an overall trend for editing events to associate with divergent sequence regions and increase amino acid biochemical similarity toward consensus. These results support the model of a single origin of editing machinery within the ancestor of peridinin and fucoxanthin dinoflagellates, with the capability for all possible editing conversions. The fact that editing trends are conserved across diverse taxa, in spite of the absence of specific conserved events, suggests a scenario in which a conserved complement of editing machinery has acted independently in each lineage to produce similar overall effects. The corrective function of editing has become an essential part of dinoflagellate RNA metabolism, preventing accumulation of deleterious mutations in the fast-evolving plastid genome that could affect the encoded gene products. Transcript editing appears to be shaped predominantly through selection upon promiscuous editing sites, with the notable exception of some highly conserved positions; the exact nature and mechanism of editing function in this context is a fascinating area for future research.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
The authors wish to thank Manoj Khadka for culturing the Pyrocystis lanula used in this study, David Lea-Smith and Shilo Dickens (DNA sequencing facility, Department of Biochemistry, University of Cambridge) for assistance in generating expanded gDNA sequence libraries for Karenia mikimotoi. R.G.D. was supported by an EMBO Long-Term Fellowship (ALTF 1124-2014). C.B. additionally thanks the French Government “Investissements d’Avenir” programmes MEMO LIFE (ANR-10-LABX-54), PSL* Research University (ANR-11-IDEX-0001-02), and Oceanomics (ANR-11-BTBR-0008). Work in the Dacks Lab is supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (RES0021028). C.M.K. is funded by an Alberta Innovates Health Solutions Fulltime Studentship and a Canada Vanier Graduate Scholarship. His research has been funded in part by the generosity of the Stollery Children’s Hospital Foundation and supporters of the Lois Hole Hospital for Women through the Women and Children’s Health Research Institute. E.R. is funded by an Alberta Innovates—Technology Futures Graduate Studentship and a Vanier Canada Graduate Scholarship.
Literature Cited
- Adl SM, et al. , 2012. The revised classification of eukaryotes. J Eukaryot Microbiol. 59(5):429–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, et al. , 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25(17):3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachvaroff TR, Concepcion GT, Rogers CR, Herman EM, Delwiche CF.. 2004. Dinoflagellate expressed sequence tag data indicate massive transfer of chloroplast genes to the nuclear genome. Protist 155(1):65–78. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Elkan C.. 1994. Fitting a mixture model by expectation maximization to discover motifs in bipolymers. Proc Second Int Conf Intell Syst Mol Biol. 2:28–36. [PubMed] [Google Scholar]
- Bailey TL, Gribskov M.. 1998. Combining evidence using p-values: application to sequence homology searches. Bioinformatics 14(1):48–54. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Johnson J, Grant CE, Noble WS.. 2015. The MEME Suite. Nucleic Acids Res. 43(W1):W39–W49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbrook AC, et al. , 2012. Polyuridylylation and processing of transcripts from multiple gene minicircles in chloroplasts of the dinoflagellate Amphidinium carterae. Plant Mol Biol. 79(4–5):347–357. [DOI] [PubMed] [Google Scholar]
- Barbrook AC, Santucci N, Plenderleith LJ, Hiller RG, Howe CJ.. 2006. Comparative analysis of dinoflagellate chloroplast genomes reveals rRNA and tRNA genes. BMC Genomics 7:297.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbrook AC, Symington H, Nisbet RER, Larkum A, Howe CJ.. 2001. Organisation and expression of the plastid genome of the dinoflagellate Amphidinium operculatum. Mol Genet Genomics 266(4):632–638. [DOI] [PubMed] [Google Scholar]
- Butterfield ER, Howe CJ, Nisbet RER.. 2016. Identification of sequences encoding symbiodinium minutum mitochondrial proteins. Genome Biol Evol. 8(2):439–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoon AB, Nauss JA, Stanley CD, Qureshi A.. 2017. Deep transcriptome sequencing of two green algae, Chara vulgaris and Chlamydomonas reinhardtii, provides no evidence of organellar RNA editing. Genes (Basel) 8(12):80. [Google Scholar]
- Cock PJ, et al. , 2009. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25(11):1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costea PI, et al. , 2017. metaSNV: a tool for metagenomic strain level analysis. PLoS One 12(7):e0182392.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang Y, Green BR.. 2009. Substitutional editing of Heterocapsa triquetra chloroplast transcripts and a folding model for its divergent chloroplast 16S rRNA. Gene 442(1–2):73–80. [DOI] [PubMed] [Google Scholar]
- Dang Y, Green BR.. 2010. Long transcripts from dinoflagellate chloroplast minicircles suggest ‘rolling circle’ transcription. J Biol Chem. 285(8):5196–5203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- do Rosário Gomes H, et al. , 2014. Massive outbreaks of Noctiluca scintillans blooms in the Arabian Sea due to spread of hypoxia. Nat Commun. 5:4862.. [DOI] [PubMed] [Google Scholar]
- Dorrell RG, Drew J, Nisbet RER, Howe CJ.. 2014. Evolution of chloroplast transcript processing in plasmodium and its chromerid algal relatives. PLoS Genet. 10(1):e1004008.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell RG, et al. , 2017. Progressive and biased divergent evolution underpins the origin and diversification of peridinin dinoflagellate plastids. Mol Biol Evol. 34(2):361–379. [DOI] [PubMed] [Google Scholar]
- Dorrell RG, Hinksman GA, Howe CJ.. 2016. Diversity of transcripts and transcript processing forms in plastids of the dinoflagellate alga Karenia mikimotoi. Plant Mol Biol. 90(3):233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell RG, Howe CJ.. 2012. Functional remodeling of RNA processing in replacement chloroplasts by pathways retained from their predecessors. Proc Natl Acad Sci U S A. 109(46):18879–18884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell RG, Howe CJ.. 2015. Integration of plastids with their hosts: lessons learned from dinoflagellates. Proc Natl Acad Sci U S A. 112(33):10247–10254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espelund M, et al. , 2012. Genome fragmentation is not confined to the peridinin plastid in Dinoflagellates. PLoS One 7(6):e38809.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara S, Iwahashi H, Someya J, Nishikawa S, Minaka N.. 1993. Structure and cotranscription of the plastid-encoded rbcL and rbcS genes of Pleurochrysis carterae (Prymnesiophyta). J Phycol. 29(3):347–355. [Google Scholar]
- Gabrielsen TM, et al. , 2011. Genome evolution of a tertiary dinoflagellate plastid. PLoS One 6(4):e19132.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavelis GS, White RA, Suttle CA, Keeling PJ, Leander BS.. 2015. Single-cell transcriptomics using spliced leader PCR: evidence for multiple losses of photosynthesis in polykrikoid dinoflagellates. BMC Genomics 16:528.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gornik SG, et al. , 2012. Loss of nucleosomal DNA condensation coincides with appearance of a novel nuclear protein in dinoflagellates. Curr Biol. 22(24):2303–2312. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, et al. , 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29(7):644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MW. 2012. Evolutionary origin of RNA editing. Biochemistry 51(26):5235–5242. [DOI] [PubMed] [Google Scholar]
- Gray MW, Lukes J, Archibald JM, Keeling PJ, Doolittle WF.. 2010. Irremediable complexity? Science 330(6006):920–921. [DOI] [PubMed] [Google Scholar]
- Green BR. 2004. The chloroplast genome of dinoflagellates–a reduced instruction set? Protist 155(1):23–31. [DOI] [PubMed] [Google Scholar]
- Guillard RRL, Hargraves PE.. 1993. Stichochrysis immobilis is a diatom, not a chrysophyte. Phycologia 32(3):234–236. [Google Scholar]
- Hackett JD, Anderson DM, Erdner DL, Bhattacharya D.. 2004. Dinoflagellates: a remarkable evolutionary experiment. Am J Bot. 91(10):1523–1534. [DOI] [PubMed] [Google Scholar]
- Hackett JD, Yoon HS et al. , 2004. Migration of the plastid genome to the nucleus in a peridinin dinoflagellate. Curr Biol. 14(3):213–218. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG.. 1992. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 89(22):10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller RG. 2001. ‘Empty’ minicircles and petB/atpA and psbD/psbE (cytb559 alpha) genes in tandem in Amphidinium carterae plastid DNA. FEBS Lett. 505(3):449–452. [DOI] [PubMed] [Google Scholar]
- Hoppenrath M, Leander BS.. 2010. Dinoflagellate phylogeny as inferred from heat shock protein 90 and ribosomal gene sequences. PLoS One 5(10):e13220.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovde BT, et al. , 2014. The mitochondrial and chloroplast genomes of the haptophyte Chrysochromulina tobin contain unique repeat structures and gene profiles. BMC Genomics 15:604.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida S, Kobiyama A, Ogata T, Murakami A.. 2009. Identification of transcribed and persistent variants of the psbA gene carried by plastid minicircles in a dinoflagellate. Curr Genet. 55(5):583–591. [DOI] [PubMed] [Google Scholar]
- Imanian B, Pombert JF, Keeling PJ.. 2010. The complete plastid genomes of the two ‘dinotoms’ Durinskia baltica and Kryptoperidinium foliaceum. PLoS One 5(5):e10711.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CJ, et al. , 2007. Broad genomic and transcriptional analysis reveals a highly derived genome in dinoflagellate mitochondria. BMC Biol. 5:41.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CJ, Gornik SG, Waller RF.. 2012. The mitochondrial genome and transcriptome of the basal dinoflagellate hematodinium sp.: character evolution within the highly derived mitochondrial genomes of dinoflagellates. Genome Biol Evol. 4(1):59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CJ, Gornik SG, Waller RF.. 2013. A tertiary plastid gains RNA editing in its new host. Mol Biol Evol. 30(4):788–792. [DOI] [PubMed] [Google Scholar]
- Janouškovec J, et al. , 2013. Split photosystem protein, linear-mapping topology, and growth of structural complexity in the plastid genome of Chromera velia. Mol Biol Evol. 30(11):2447–2462. [DOI] [PubMed] [Google Scholar]
- Janouškovec J, et al. , 2017. Major transitions in dinoflagellate evolution unveiled by phylotranscriptomics. Proc Natl Acad Sci U S A. 114(2):E171–E180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janouškovec J, Horák A, Oborník M, Lukes J, Keeling PJ.. 2010. A common red algal origin of the apicomplexan, dinoflagellate, and heterokont plastids. Proc Natl Acad Sci U S A. 107(24):10949–10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones E, Oliphant T, Peterson P et al. , 2001. SciPy: open source scientific tools for python [cited 2015 Feb 10]. Available from: http://www.scipy.org/, last accessed March 21, 2018.
- Kamikawa R, et al. , 2015. Plastid genome-based phylogeny pinpointed the origin of the green-colored plastid in the dinoflagellate Lepidodinium chlorophorum. Genome Biol Evol. 7(4):1133–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, et al. , 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12):1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ, et al. , 2014. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS Biol. 12(6):e1001889.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp C, et al. , 2015. Subcellular investigation of photosynthesis-driven carbon and nitrogen assimilation and utilization in the symbiotic reef coral Pocillopora damicornis. mBio 6(1):e02299-14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kultima JR, et al. , 2016. MOCAT2: a metagenomic assembly, annotation and profiling framework. Bioinformatics 32(16):2520–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF.. 1982. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 157(1):105–132. [DOI] [PubMed] [Google Scholar]
- Laatsch T, Zauner S, Stoebe-Maier B, Kowallik KV, Maier UG.. 2004. Plastid-derived single gene minicircles of the dinoflagellate Ceratium horridum are localized in the nucleus. Mol Biol Evol. 21(7):1318–1322. [DOI] [PubMed] [Google Scholar]
- Laslett D, Canback B.. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32(1):11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bescot N, et al. , 2016. Global patterns of pelagic dinoflagellate diversity across protist size classes unveiled by metabarcoding. Environ Microbiol. 18(2):609–626. [DOI] [PubMed] [Google Scholar]
- Leung SK, Wong JT.. 2009. The replication of plastid minicircles involves rolling circle intermediates. Nucleic Acids Res. 37(6):1991–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew YJ, Li Y, Baumgarten S, Voolstra CR, Aranda M.. 2017. Condition-specific RNA editing in the coral symbiont Symbiodinium microadriaticum. PLoS Genet. 13(2):e1006619.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, et al. , 2015. The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 350(6261):691–694. [DOI] [PubMed] [Google Scholar]
- Lin S, Zhang H, Gray MW.. 2007. RNA editing in dinoflagellates and its implications for the evolutionary history of the editing machinery In: RNA and DNA editing: molecular mechanisms and their integration into biological systems. Hoboken, NJ, USA: John Wiley & Sons, pp. 280–309. [Google Scholar]
- Lin S, Zhang H, Spencer DF, Norman JE, Gray MW.. 2002. Widespread and extensive editing of mitochondrial mRNAS in dinoflagellates. J Mol Biol. 320(4):727–739. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR.. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25(5):955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukeš J, Archibald JM, Keeling PJ, Doolittle WF, Gray MW.. 2011. How a neutral evolutionary ratchet can build cellular complexity. IUBMB Life 63(7):528–537. [DOI] [PubMed] [Google Scholar]
- Luo R, et al. , 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1(1):18.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2007. The frailty of adaptive hypotheses for the origins of organismal complexity. Proc Natl Acad Sci U S A. 104(Suppl 1):8597–8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T.. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, 2010, pp. 1–8.
- Mungpakdee S, et al. , 2014. Massive gene transfer and extensive RNA editing of a symbiotic dinoflagellate plastid genome. Genome Biol Evol. 6(6):1408–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash EA, et al. , 2007. Organization of the mitochondrial genome in the dinoflagellate Amphidinium carterae. Mol Biol Evol. 24(7):1528–1536. [DOI] [PubMed] [Google Scholar]
- Nisbet RER, Kurniawan DP, Bowers HD, Howe CJ.. 2016. Transcripts in the plasmodium apicoplast undergo cleavage at tRNAs and editing, and include antisense sequences. Protist 167(4):377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K. 2010. Funtions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 79:321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenkott B, Yamaguchi K, Tsuji-Tsukinoki S, Knie N, Knoop V.. 2014. Chloroplast RNA editing going extreme: more than 3400 events of C-to-U editing in the chloroplast transcriptome of the lycophyte Selaginella uncinata. RNA 20(10):1499–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owari S, Hayashi A, Ishida KI.. 2014. Subcellular localization of minicircle DNA in the dinoflagellate Amphidinium massartii. Phycol Res. 62(1):1–8. [Google Scholar]
- Patron NJ, Waller RF, Keeling PJ.. 2006. A tertiary plastid uses genes from two endosymbionts. J Mol Biol. 357(5):1373–1382. [DOI] [PubMed] [Google Scholar]
- Puerta MVS, Bachvaroff TR, Delwiche CF.. 2005. The complete plastid genome sequence of the haptophyte Emiliania huxleyi: a comparison to other plastid genomes. DNA Res. 12(2):151–156. [DOI] [PubMed] [Google Scholar]
- Richardson E, Dorrell RG, Howe CJ.. 2014. Genome-wide transcript profiling reveals the coevolution of plastid gene sequences and transcript processing pathways in the fucoxanthin dinoflagellate Karlodinium veneficum. Mol Biol Evol. 31(9):2376–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, et al. , 2012. Mrbayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldarriaga JF, McEwan ML, Fast NM, Taylor FJR, Keeling PJ.. 2003. Multiple protein phylogenies show that Oxyrrhis marina and Perkinsus marinus are early branches of the dinoflagellate lineage. Int J Syst Evol Microbiol. 53(1):355–365. [DOI] [PubMed] [Google Scholar]
- Sander C, Schneider R.. 1991. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins 9(1):56–68. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Yukawa Y, Miyamoto T, Obokata J, Sugiura M.. 2003. Identification of RNA editing sites in chloroplast transcripts from the maternal and paternal progenitors of tobacco (Nicotiana tabacum): comparative analysis shows the involvement of distinct trans-factors for ndhB editing. Mol Biol Evol. 20(7):1028–1035. [DOI] [PubMed] [Google Scholar]
- Ševčíková T, et al. , 2015. Updating algal evolutionary relationships through plastid genome sequencing: did alveolate plastids emerge through endosymbiosis of an ochrophyte? Sci Rep. 5:10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikanai T. 2015. RNA editing in plants: machinery and flexibility of site recognition. Biochim Biophys Acta 1847(9):779–785. [DOI] [PubMed] [Google Scholar]
- Shoguchi E, et al. , 2013. Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr Biol. 23(15):1399–1408. [DOI] [PubMed] [Google Scholar]
- Smith DR, Keeling PJ.. 2015. Mitochondrial and plastid genome architecture: reoccurring themes, but significant differences at the extremes. Proc Natl Acad Sci U S A. 112(33):10177–10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Bentolila S, Hanson MR.. 2016. The unexpected diversity of plant organelle RNA editosomes. Trends Plant Sci. 21(11):962–973. [DOI] [PubMed] [Google Scholar]
- Takenaka M, Zehrmann A, Verbitskiy D, Härtel B, Brennicke A.. 2013. RNA editing in plants and its evolution. Annu Rev Genet. 47:335–352. [DOI] [PubMed] [Google Scholar]
- Taylor FJR, Hoppenrath M, Saldarriaga JF.. 2008. Dinoflagellate diversity and distribution. Biodivers Conserv. 17(2):407–418. [Google Scholar]
- Tengs T, et al. , 2000. Phylogenetic analyses indicate that the 19’Hexanoyloxy-fucoxanthin-containing dinoflagellates have tertiary plastids of haptophyte origin. Mol Biol Evol. 17(5):718–729. [DOI] [PubMed] [Google Scholar]
- Valdar WSJ. 2002. Scoring residue conservation. Proteins Struct Funct Genet. 48(2):227–241. [DOI] [PubMed] [Google Scholar]
- Waller RF, Gornik SG, Koreny L, Pain A.. 2016. Metabolic pathway redundancy within the apicomplexan-dinoflagellate radiation argues against an ancient chromalveolate plastid. Commun Integr Biol. 9(1):e1116653.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller RF, Slamovits CH, Keeling PJ.. 2006. Lateral gene transfer of a multigene region from cyanobacteria to dinoflagellates resulting in a novel plastid-targeted fusion protein. Mol Biol Evol. 23(7):1437–1443. [DOI] [PubMed] [Google Scholar]
- Wang Y, Morse D.. 2006. Rampant polyuridylylation of plastid gene transcripts in the dinoflagellate Lingulodinium. Nucleic Acids Res. 34(2):613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernersson R. 2006. Virtual ribosome – a comprehensive DNA translation tool with support for integration of sequence feature annotation. Nucleic Acids Res. 34(Web Server issue):W385–W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada N, Sym SD, Horiguchi T.. 2017. Identification of highly-divergent diatom-derived chloroplasts in dinoflagellates, including a description of Durinskia kwazulunatalensis sp. nov. (Peridiniales, Dinophyceae). Mol Biol Evol. 34(6):1335–1351. [DOI] [PubMed] [Google Scholar]
- Zauner S, Greilinger D, Laatsch T, Kowallik KV, Maier UG.. 2004. Substitutional editing of transcripts from genes of cyanobacterial origin in the dinoflagellate Ceratium horridum. FEBS Lett. 577(3):535–538. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E.. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18(5):821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bhattacharya D, Maranda L, Lin S.. 2008. Mitochondrial cob and cox1 genes and editing of the corresponding mRNAs in Dinophysis acuminata from Narragansett Bay, with special reference to the phylogenetic position of the genus Dinophysis. Appl Environ Microbiol. 74(5):1546–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Green BR, Cavalier-Smith T.. 1999. Single gene circles in dinoflagellate chloroplast genomes. Nature 400(6740):155–159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.