

Abstract
β-guanidinopropionic acid (β-GPA) has been used as a nutritional supplement for increasing physical strength and endurance with positive and predictable results. In muscles, it works as a nonadaptive stimulator of mitochondria biogenesis; it also increases lipid metabolism. There are data indicating that β-GPA can be also neuroprotective, but its mechanisms of action in the brain are less understood. We studied the effects of β-GPA on animal behavior and mitochondrial biogenesis in the cortex and midbrain of mid-age healthy mice. We found that even short-term 3-week-long β-GPA treatment increased the mitochondrial DNA (mtDNA) copy number in the cortex and ventral midbrain, as well as the expression of several key antioxidant and metabolic enzymes—indicators of mitochondria proliferation and the activation of Nrf2/ARE signaling cascade. At the same time, β-GPA downregulated the expression of the β-oxidation genes. Administration of β-GPA in mice for 3 weeks improved the animals’ physical strength and endurance health, ie, increased their physical strength and endurance and alleviated anxiety. Thus, β-GPA might be considered an adaptogene affecting both the muscle and brain metabolism in mammals.
Keywords: β-guanidinopropionic acid, brain, mitochondrial biogenesis, Nrf2/ARE signaling cascade, β-oxidation, behavior
Introduction
Mitochondrial biogenesis plays a key role in mitochondrial homeostasis essential for the normal functioning of brain cells.1 Impairments in the regulation of mitochondrial biogenesis is one of the causes of metabolism dysregulation in brain cells. Mitochondrial damages result in the development of various neurodegenerative disorders, such as Alzheimer’s disease,2 Parkinson’s disease,3 multiple sclerosis,4 and amyotrophic lateral sclerosis.5 It was reported that the mitochondrial content decreases with age,6 which together with the activation of oxidative stress could be the cause of aging.7
The interaction between transcription factors (TFAM, NRF1, PPARα, etc) and regulatory coactivators, mainly PGC-1α, is the critical stage in mitochondrial proliferation.8 However, numerous recent studies demonstrated that the key role in the activation of mitochondrial biogenesis is due to the Nrf2/ARE signaling cascade that upregulates expression of nuclear genes through the interaction of the Nrf2 transcription factor (NF-E2 p45-related factor 2) with antioxidant response elements (AREs).9 The Nrf2/ARE cascade is activated by various xenobiotics and oxidants, such as superoxide, H2O2, hydroxyl radicals, and nitric oxide,10 that stabilize free Nrf2.11 It is then translocated to the nucleus, where it binds to AREs in the promoters of genes mostly responsible for the antioxidant defense and mitochondrial biogenesis.12 Therefore, the Nrf2/ARE cascade could be considered to be an example of mitohormesis. The essence of this concept is that mild stress activates mitochondrial mechanisms that promote organism survival.13
One of the ways to induce mild stress is a competitive inhibition of creatine kinase with the creatine analog β-guanidinopropionic acid (β-GPA). Regular consumption of β-GPA mimics endurance training by inducing the chronic depletion of intracellular phosphocreatine and adenosine triphosphate (ATP).14 This process has been extensively studied in skeletal muscles, but the mechanism of β-GPA action on the nervous system is poorly understood, especially regarding cognitive changes and mitochondria biogenesis. In this study, we investigated the effects of a short-term β-GPA treatment on selected cognitive functions, mitochondria biogenesis, and the activation of the Nrf2/ARE signaling cascade in different brain regions.
Materials and Methods
Animals
All experimental procedures with animal, such as maintenance, injections, and sacrifice, were performed in accordance with the rules set by Voronezh State University Ethical Committee on Biomedical Research (Section of Animal Care and Use). C57Bl/6 mice, 12-month-old males, were obtained from Stolbovaya (Russia). All animals were maintained under controlled conditions of 12 hour light/12 hour dark and received water ad libitum and a standard laboratory diet (Ssniff-Spezialdiäten GmbH, Germany).
The following animal groups were used for the experiment: (1) mice that were not subjected to injections (n = 10)—this group was used only for physiological tests and were not killed after the experiment; (2) saline-treated mice (n = 8)—the volume of injections was 200 μL; and (3) β-GPA-treated mice (n = 8)—concentration of β-GPA (Sigma-Aldrich, USA) was 1 mg/kg/day. The volume of injections was 200 μL.
Injections were performed 3 times a week for 3 weeks. The mice’s weight and the total amount of consumed food and water were measured daily. Once a week, a string test was performed. Measurements of the oxygen consumption rates of live animals and the open field test were performed after the course of injections. After 3 weeks, the mice were killed; brain dissection was performed to extract the cortex and the ventral midbrain (VMB) for quantitative real-time polymerase chain reaction (qRT-PCR) analysis. We evaluated the cortex because its role in anxiety15 and midbrain because of its obvious role in motor functions16: the two major behavioral changes we see in GPA-treated animals. The timeline of the experiment is presented in Figure 1.
Figure 1.

The timeline of experiments and the results of string test. (A) The timeline of treatments and experiments. (B) The results of string test. β-GPA-treated mice already exhibited increased strength and endurance during the second week of injections (*P < .01). The test was performed and scored as described in “Materials and Methods” section.
β-GPA, β-guanidinopropionic acid.
Physiological tests
For the estimation of physical strength and endurance, the string test was performed. A mouse was placed by its front paws on a string 50 cm long and 42 cm above a padded surface for 1 minute. The test scoring was performed as described by Cardozo-Pelaez et al.17
For the estimation of anxiety and exploratory behavior, the open field test was used. A mouse was placed in the corner of an open field (60 × 60 × 40 cm) with randomly placed holes (0.5 cm diameter). The recording time was 5 minutes. We evaluated the number of occurrences in the center of area, time spent in the center, the horizontal activity, number of rearing acts, and number of hole poking for studying exploratory behavior. We estimated the number of grooming acts for studying the anxiety behavior.18
Measurements of oxygen consumption and carbon dioxide emission in live animals were performed with a gas-phase oximeter (“Vernier,” USA). The animals were placed in an air-tight vessel. The O2 consumption and the CO2 emission were recorded for 8 minutes. The rates were expressed in mol O2(CO2)/min/g body weight. The respiratory quotient (RQ) was calculated as the ratio of the rate of carbon dioxide emission/rate of oxygen consumption.
Quantitative real-time polymerase chain reaction
Total RNA was isolated using an ExtractRNA kit (Evrogen, Russia) according to protocol. Quantitative assay of total RNA was performed with a Qubit 2.0 (Thermo Fisher Scientific, USA). An amount of 500 μg of RNA was used for making complementary DNA (cDNA) using an MMLV RT kit (Evrogen, Russia). An amount of 10 μg of cDNA was used for quantitative real-time PCR with a qPCRmix-HS SYBR+LowROX (Evrogen). Primer sequences for PCR were as follows:
Cox1—F: 5′-TCGCAATTCCTACCGGTCTC-3′;
R: 5′-CGTGTAGGGTTGCAAGTCAGC-3′;
Tfam—F: 5′-ATTCCGAAGTGTTTTTCCAGCA-3′;
R: 5′-TCTGAAAGTTTTCGATCTGGGT-3′;
Nrf1—F: 5′-AGCACGGAGTGACCCAAA-3′;
R: 5′-TGTACGTGGCTACATGGACCT-3′;
Ho-1—F: 5′-CACGCATATACCCGCTACCT-3′;
R: 5′-CCAGAGTGTTCATTCGAGCA-3′;
Prdx3—F: 5′-GGTTGCTCGTCATGCAAGTG-3′;
R: 5′-CCACAGTATGTCTGTCAAACA-3′;
Prdx5—F: 5′-GGTTGCTCGTCATGCAAGTG-3′;
R: 5′-CCACAGTATGTCTGTCAAACA-3′;
Sod2—F: 5′-CAGACCTGCCTTACGACTATGG-3′;
R: 5′-CTCGGTGGCGTTGAGATTGTT-3′;
Gclc—F: 5′-GCAGCTTTGGGTCGCAAGTAG-3′;
R: 5′-TGGGTCTCTTCCCAGCTCAGT-3′;
Gpx—F: 5′-AGTCCACCGTGTATGCCTTC-3′;
R: 5′-GTGTCCGAACTGATTGCACG-3′;
Pgc-1α—F: 5′-ATGTGTCGCCTTCTTGCTCT-3′;
R: 5′-CACGACCTGTGTCGAGAAAA-3′;
Pparα—F: 5′-AGAGCCCCATCTGTCCTCTC-3′;
R: 5′-ACTGGTAGTCTGCAAAACCAAA-3′;
Acadm—F: 5′-AGGGTTTAGTTTTGAGTTGACGG-3′;
R: 5′-CCCCGCTTTTGTCATATTCCG-3′;
Acox1—F: 5′-TAACTTCCTCACTCGAAGCCA-3′,
R: 5′-AGTTCCATGACCCATCTCTGCC-3′;
Cpt1—F: 5′-CTCCGCCTGAGCCATGAAG-3′;
R: 5′-CACCAGTGATGATGCCATTCT-3′;
Actβ—F: 5′-GGCTGTATTCCCCTCCATCG-3′;
R: 5′-CCAGTTGGTAACAATGCCATGT-3′;
Gapdh—F: 5′-GGCTCCCTAGGCCCCTCCTG-3′;
R: 5′-TCCCAACTCGGCCCCCAACA-3′.
Quantitative real-time polymerase chain reaction was performed with Bio-Rad CFX-96 (Bio-Rad, USA). The reaction conditions were as follows: 5 minutes at 95°С, followed by 35 cycles of 20 seconds at 95°С, 30 seconds at 59°С, and 30 seconds at 72°С. Actβ and Gapdh were used as controls in the comparative cycle threshold method.
Evaluation of mtDNA copy number and lesions
DNA extraction was performed using a Genome DNA extraction kit (Biosilica, Russia). The amount of mtDNA was estimated by the qPCR of mtDNA fragment by using the ΔΔCq approach. Primer sequences for mtDNA were as follows:
F: 5′-ACGAGGGTCCAACTGTCTCTTA-3′;
R: 5′-AGCTCCATAGGGTCTTCTCGT-3′.
The nucleus-encoded Gapdh gene was used as a reference. Primer sequences were as follows:
F: 5′-GGCTCCCTAGGCCCCTCCTG-3′;
R: 5′-TCCCAACTCGGCCCCCAACA-3′.
Evaluation of the number of mtDNA lesions was performed with the original method that was previously described.19 Primer sequences for mtDNA were as follows:
First fragment—F1: 5′-TAAATTTCGTGCCAGCCACC-3′;
R1 (short): 5′-GTTGACACGTTTTACGCCGA-3′;
R1 (long): 5′-ATGCTACCTTTGCACGGTCA-5′;
Second fragment—F2: 5′-ACGAGGGTCCAACTGTCTCTTA-3′;
R2 (short): 5′-AGCTCCATAGGGTCTTCTCGT-3′;
R2 (long): 5′-CCGGCTGCGTATTCTACGTT-5′;
Third fragment—F3: 5′-AAGAAGGAGCTACTCCCCACC-3′;
R3 (short): 5′-AGCTTATATGCTTGGGGAAAATAGT-3′;
R3 (long): 5′-GTTGACACGTTTTACGCCGA-5′.
The number of mtDNA lesions was calculated per 10 kb of mtDNA using the following formula:
Statistical analysis
Statistical analysis was performed using statistical variance methods (Microsoft Excel software package). The results are presented as mean ± standard error of the mean (SEM). The significance of differences between groups was estimated with the Student’s t-test; only statistically significant differences (P < .05) are discussed in this article. In the open field test, the differences among the experimental groups were analyzed using a parametric one-way analysis of variance (ANOVA).
Results
Behavioral and physiological tests
β-guanidinopropionic acid injections for 3 weeks did not cause any changes in the body weight or the amounts of food and water consumed (data not presented). However, the string test showed that by the second week, β-GPA treatment had increased the physical strength and endurance of the animals as compared with the mice receiving injections of physiological saline (Figure 1B, P < .01). The maximum (approximately, 3-fold) increase in the physical strength was observed by the end of third week of injections (P < .01). There were no statistically significant differences between mice receiving injections of physiological saline and control mice receiving no injections.
In the open field test, mice receiving β-GPA injections entered the center of the field twice as often as the mice injected with physiological saline (Figure 2A, F1,18 = 2.15; P < .05) and spent more time in the center of the field (Figure 2B, F1,18 = 4.42; P < .05), but showed no difference with this group in the number of rearing events. Mice receiving no injections displayed twice as much rearing events than the injected mice (Figure 2C). Injections of saline and β-GPA did not affect the mobility of mice, which manifested in the total time of their movement, ie, horizontal activity (Figure 2D). Injections decreased the frequency of hole poking; mice injected with β-GPA did not differ from the mice injected with saline in the number of hole-poking events (Figure 2E). These data indicate a trend toward an increase in the exploratory behavior in β-GPA-treated group compared with mice injected with saline. Injection with saline increased grooming by 64% as compared with mice receiving no injections; however, grooming was more than two times less often in mice injected with β-GPA than in those injected with saline (Figure 2F, F1,18 = 4.54; P < .01). These data showed increasing anxiety caused by injection, but β-GPA reduced this fear.
Figure 2.

The results of the open field test. A. The number of entering to the center of the field; B. Time spent in the center of the field; C. Number of rearing acts; D. Total time of movement; E. Number of hole pocking acts; F. Number of grooming acts. The test was performed and scored as described in “Materials and Methods” section and according to the timeline presented in Figure 1A (*P < .05; **P < .01).
β-GPA, β-guanidinopropionic acid.
β-guanidinopropionic acid injections changed the respiration parameters (Table 1) in the experimental animals: the rate of CO2 emission in the β-GPA-injected mice decreased by 7% (P < .05), whereas no difference in the oxygen consumption rate was observed between the groups. These changes resulted in a decrease in the RQ of the GPA-injected mice from 0.96 ± 0.01 to 0.9 ± 0.02 (P < .05). No difference was found between saline-injected mice and mice receiving no injections.
Table 1.
Respiratory analysis of mouse groups.
| w/o | Saline | GPA | |
|---|---|---|---|
| CO2 | 4.49 ± 0.24 | 4.41 ± 0.16 | 4.11 ± 0.12* |
| O2 | 4.61 ± 0.32 | 4.56 ± 0.36 | 4.54 ± 0.18 |
| RQ | 0.97 ± 0.02 | 0.96 ± 0.01 | 0.9 ± 0.02* |
GPA, guanidinopropionic acid; RQ, respiratory quotient; SEM, standard error of the mean.
Values are mean ± SEM.
P < .05.
Changes in gene expression
Expression of the mitochondrial Cox1 gene increased 1.5 times in the cortex and 1.8 times in the midbrain in the β-GPA-injected mice. β-guanidinopropionic acid also upregulated the expression of Tfam and Nrf1 genes involved in the regulation of mitochondrial biogenesis. The expression levels of the Ho-1 gene were increased in both brain regions (Figure 3A and B).
Figure 3.

β-GPA-induced changes in the expression of selected enzymes—markers of the activation of Nrf2/ARE cascade or PGC-1α/PPARα cascade. (A) Brain cortex. (B) Ventral midbrain (*P < .05; **P < .01).
β-GPA, β-guanidinopropionic acid.
Expression of several antioxidant genes was upregulated in β-GPA-treated mice. Thus, expression levels of Sod2 in the cortex and midbrain and of Prdx3 in the cortex were significantly increased. At the same time, expression of Prdx5, Gclc, and Gpx was not affected in both studied brain regions.
We also observed an increase (although statistically insignificant, P = .09) in the Pgc-1α expression in the cortex only. At the same time, in the VMB, no changes in the expression of Pgc-1α and PGC-1α-targeted genes involved in regulation of fatty acid oxidation (Pparα, Acadm, Acox1, and Cpt1) were found. However, in the cortex, we observed an approximate 2-fold decrease in the expression levels of genes responsible for hydrolysis and transmembrane transport of fatty acids (Figure 3).
mtDNA copy number and lesions
Injections of β-GPA increased the mtDNA copy number 1.7 times in the cortex (P < .01) and 1.82 ± 0.35 times in the midbrain (P < .05), as compared with saline-injected mice (Figure 4). There were no differences in the number of mtDNA lesions in the studied mtDNA fragments in both the cortex and the midbrain (Table 2).
Figure 4.
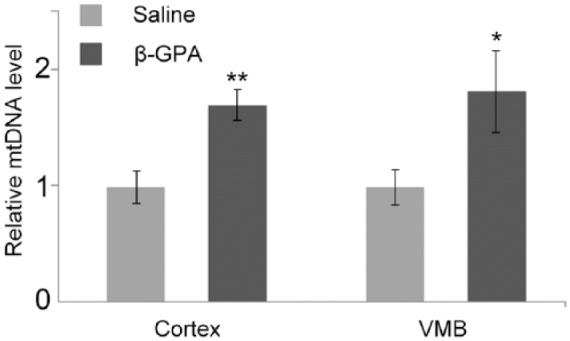
mtDNA copy numbers in brain cortex and VMB are increased by β-GPA-treatment.
Cortex, brain cortex; VMB, ventral midbrain.
*P < .05; **P < .01.
Table 2.
Number of excessive mtDNA damage induced by GPA-injection in cortex and VMB.
| First fragment | Second fragment | Third fragment | |
|---|---|---|---|
| Cortex | 0.12 ± 0.25 | −0.08 ± 0.18 | 0.06 ± 0.68 |
| VMB | 0.04 ± 0.32 | −0.12 ± 0.34 | 0.14 ± 0.62 |
GPA, guanidinopropionic acid; mtDNA, mitochondrial DNA; SEM, standard error of mean; VMB, ventral midbrain.
Values are mean ± SEM.
Discussion
During the last few decades, β-GPA has been used as a nutritional supplement for the nonadaptive increase of physical strength and endurance. This compound is known to exert a noticeable effect on the energy metabolism in skeletal muscles and other tissues as well.20 In line with this, we observed that mice demonstrated an increase in the physical strength in the string test after just 2 weeks of β-GPA injections (Figure 1). Therefore, the fast effect of the drug is explained by the fast substitution of the creatine level in skeletal muscle. It was shown earlier that the creatine level decreased by 50% after 2 weeks of β-GPA treatment21 and decreased by more than 2 times after 3 weeks of β-GPA treatment.22 Notably, the effect of the β-GPA injections on nonmotor behavior parameters was also significant. We observed statistically significant changes in several parameters, such as an increased number of occurrences in the center, increased time spent in the center of area, and decreased number of grooming acts that demonstrated a decrease of anxiety in the mice, which may be partly caused by the series of repeated intraperitoneal injections. However, we did not find that the β-GPA had an effect on an increased number of acts of rearing, horizontal activity, and number of hole-poking, which could be markers of the enhanced exploratory behavior. Although interpreting some of these data might vary and certainly require a more extensive array of tests, we think that the overall pattern agrees well with the anxiolytic action of β-GPA injections.
The mechanism of the β-GPA action on the fatty acid metabolism is still poorly understood. It was found that β-GPA intensifies the lipid metabolism through upregulating expression of fatty acid transporters in different types of muscles23 and through lowering the mitochondrial respiratory control index.24 Our results on basal (resting) oxygen consumption demonstrate a small but significant decrease in RQ in the β-GPA-treated mice (Table 1). In principle, this does not contradict with an increased contribution of fatty acid β-oxidation to the body’s metabolism. However, it is unlikely due to the increased oxidation of fatty acids in the brain of the treated animals, because we did not observe any activation of the expression of β-oxidation genes in the brain. On the contrary, the amounts of messenger RNAs (mRNAs) for acetyl-CoA dehydrogenases (Acadm), acetyl-CoA oxidase (Acox1), and carnitine palmitoyl transporter (Cpt1) were slightly decreased in the cortex. Significant changes in the mRNA level of these genes were not shown in the VMB. It may be connected with different physiological roles of the creatine cycle in different brain compartments. A lower level of creatine kinase was shown in the cortex compared with the midbrain and the brain as a whole.25 Probably, low doses of β-GPA have a more pronounced effect on gene expression in the cortex. In addition, this may be due to the transport of β-GPA at the blood–brain barrier. The transport of guanidino compounds at the blood–brain barrier plays important roles in energy homeostasis in the brain.26 Apparently, there are differences in the transport of β-GPA in the VMB and cortex. This important point needs further careful study.
The absence of an increase in β-oxidation gene expression correlates well with the results of earlier β-GPA studies that revealed no changes in the blood concentration of triglycerides.27,28 Therefore, we cannot assert that β-GPA produces no effect on the fatty acid metabolism in the organism, but our data suggest that these changes take places in tissues other than those of the brain, eg, brown fat.29
However, the data are interesting regarding the question which signaling cascades are initiated in the brain by β-GPA. Fatty acid oxidation is tightly controlled by transcription factors, in particular PGC-1α, which is a coactivator for the transcriptional factors of the PPAR family. It also plays a key role in the activation of mitochondrial biogenesis.8 Creatine analogs induce a mild ATP deficiency in cells, thereby activating adenosine monophosphate–activated protein kinase (AMPK), which, in its turn, activates PGC-1α, as it has been convincingly demonstrated in skeletal muscles14,23,28,30,31. However, in the brain, we found no expression upregulation for the Pgc-1α, Pparα, and a number of genes involved in fatty acid oxidation. These results are surprising, especially in the light of data obtained by Horvath et al,24 who demonstrated that β-GPA treatment affected mitochondrial biogenesis through AMPK and PGC-1α. This discrepancy might be due to the fact that the our studies were performed in healthy mid-age mice, whereas Horvath et al studies were performed in mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a toxin used to chemically model Parkinson’s disease.
However, we also observed an increase in the mtDNA copy number in the midbrain and the cortex together with upregulation in expression of the mitochondrial cytochrome oxidase gene Cox1 (Figures 3 and 4). We also found an increase in the expression levels of the Tfam gene required for initiation of DNA replication32 and Nrf1, which is a downstream gene for PGC-1α and encodes a transcription factor for TFAM.28 This may serve as an indirect confirmation of the mitochondrial biogenesis induction by β-GPA, as reported by Horvath et al.24
The Nrf2/ARE signaling cascade is an alternative mechanism for the activation of the mitochondrial biogenesis program. Piantadosi et al33 discovered in the Nrf1 gene promoter four AREs that could bind Nrf2 and initiate activation of mitochondrial biogenesis. Involvement of the Nrf2 cascade in the activation of mitochondrial biogenesis in the brain at different physiological conditions has been demonstrated in numerous studies during the past few years.34–40 We can hypothesize that beta-GPA-dependent increase in mitochondrial biogenesis could contribute to the improvement of behavior. Many other effects of β-GPA were described. They are associated with the inhibition of creatine kinase and some transporters, such as TauT (taurine transporter)41 and GAT3 and GAT4 (gamma-aminobutyric acid [GABA] transporters),42 which are important drug targets.43
There are no data so far on the negative effects of β-GPA, except possible myocardium hypertrophy and accompanying hypertension.44 However, insofar, as mtDNA replication is seemingly involved in β-GPA effects in the brain, it was interesting to check whether it is aggravated by an accumulation of mtDNA damage. To this end, we investigated mtDNA damage in the D-loop and ribosomal RNA (rRNA) genes, because these regions have no corresponding pseudogenes in the mouse nuclear genome, yet are highly susceptible to damage by various external factors.19 However, we observed no mtDNA damage in any of the studied fragments, thereby proving that β-GPA does not have a genotoxic effect on brain mtDNA. In conjunction with the lack of reports of negative effects of β-GPA, it can be concluded that it is safe to be used as an energy metabolism modulator.
Nrf2 was originally discovered to be a transcription factor that regulates the expression of antioxidant genes. After the specific inhibition of negative regulators (Keap1 and GSK-3β), free Nrf2 translocates from cytosol to nucleus and binds to ARE regions of target genes.12 We found that β-GPA activates the expression of the Sod2 gene in both the midbrain and the cortex and of the Prdx3 gene in the cortex. Moreover, we also observed upregulation in the cortex, and in the midbrain, the expression of Ho-1 (Figure 3), which is a key protein in the heme metabolism and serves as a classical marker of the Nrf2/ARE cascade activation.45
Conclusions
Therefore, we can assume that the creatine kinase inhibition–related decrease in the ATP content in the brain can cause the activation of the AMPK/PGC-1α and Nrf2/ARE signaling cascades. According to Navarro et al,40 both signal cascades are essential for maintaining the number of mitochondria. Dysregulation of these cascades significantly impairs mitochondrial homeostasis. The Nrf2/ARE cascade facilitates adaptation to stress induced by changes in the energy metabolism resulting from the ATP deficit caused by creatine kinase inhibition via stimulating mitochondrial biogenesis and activating the expression of cytoprotective, antioxidant, and anti-inflammatory enzymes. These biochemical changes are reflected not only in changes in muscle strength and endurance, but also in the cognitive behavioral patterns, eg, the apparent anxiolytic action of β-GPA.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Ministry of Education and Science of the Russian Federation (State Assessment N 6.4656.2017/8.9); President grant for support of leading scientific school, (Agreement 14.Z57.18.3457-NSh) and Russian Fund for Basic Research (17-29-02505 ofi_m).
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: All authors have full access to all the data reported in this manuscript and take responsibility for the integrity of the data and the accuracy of the data analysis. AAS and VNP involved in the study concept and design. APG and EAS participated in the acquisition, analysis, and interpretation of data. APG drafted the manuscript. Critical revision of the manuscript for the content was done by AAS.
Ethical Approval: All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
ORCID iD: Artem P Gureev  https://orcid.org/0000-0003-3562-5329
https://orcid.org/0000-0003-3562-5329
References
- 1. Golpich M, Amini E, Mohamed Z, et al. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: pathogenesis and treatment. CNS Neurosci Ther. 2017;23:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pedros I, Patraca I, Martinez N, et al. Molecular links between early energy metabolism alterations and Alzheimer’s disease. Front Biosci. 2016;21:8–19. [DOI] [PubMed] [Google Scholar]
- 3. Pyle A, Anugrha H, Kurzawa-Akanbi M, Yarnall A, Burn D, Hudson G. Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol Aging. 2016;38:e7–216.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blokhin A, Vyshkina T, Komoly S, Kalman B. Variations in mitochondrial DNA copy numbers in MS brains. J Mol Neurosci. 2008;35:283–287. [DOI] [PubMed] [Google Scholar]
- 5. DaCruz SD, Parone PA, Lopes VS, et al. Elevated PGC-1α activity sustains mitochondrial biogenesis and muscle function without extending survival in a mouse model of inherited ALS. Cell Metab. 2012;15:778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hartmann N, Reichwald K, Wittig I, et al. Mitochondrial DNA copy number and function decrease with age in the short-lived fish Nothobranchius furzeri. Aging Cell. 2011;10:824–831. [DOI] [PubMed] [Google Scholar]
- 7. Harman D. Origin and evolution of the free radical theory of aging: a brief personal history, 1954–2009. Biogerontology. 2009;10:773–781. [DOI] [PubMed] [Google Scholar]
- 8. Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. [DOI] [PubMed] [Google Scholar]
- 9. Tkachev VO, Menshchikova EB, Zenkov NK. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry (Mosc). 2011;76:407–422. [DOI] [PubMed] [Google Scholar]
- 10. Lyakhovich VV, Vavilin VA, Zenkov NK, Menshchikova EB. Active defense under oxidative stress. The antioxidant responsive element. Biochemistry (Mosc). 2006;71:962–974. [DOI] [PubMed] [Google Scholar]
- 11. Fourquet S, Guerois R, Biard D, Toledano MB. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J Biol Chem. 2010;285:8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. 2015;88:179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ristow M, Zarse K. How increased oxidative stress promotes longevity and metabolic health: the concept of mitochondrial hormesis (mitohormesis). Exp Gerontol. 2010;45:410–418. [DOI] [PubMed] [Google Scholar]
- 14. Zong H, Ren JM, Young LH, et al. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–15987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Davidson RJ. Anxiety and affective style: role of prefrontal cortex and amygdala. Biol Psychiatry. 2002;51:68–80. [DOI] [PubMed] [Google Scholar]
- 16. Fanselow MS. The midbrain periaqueductal gray as a coordinator of action in response to fear and anxiety. In: Fanselow MS, ed. The Midbrain Periaqueductal Gray Matter. Boston, MA: Springer; 1991:151–173. [Google Scholar]
- 17. Cardozo-Pelaez F, Song S, Parthasarathy A, Hazzi C, Naidu K, Sanchez-Ramos J. Oxidative DNA damage in the aging mouse brain. Mov Disord. 1999;14:972–980. [DOI] [PubMed] [Google Scholar]
- 18. Gureev AP, Syromyatnikov MY, Gorbacheva TM, Starkov AA, Popov VN. Methylene blue improves sensorimotor phenotype and decreases anxiety in parallel with activating brain mitochondria biogenesis in mid-age mice. Neurosci Res. 2016;113:19–27. [DOI] [PubMed] [Google Scholar]
- 19. Gureev AP, Shaforostova EA, Starkov AA, Popov VN. Simplified qPCR method for detecting excessive mtDNA damage induced by exogenous factors. Toxicology. 2017;382:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oudman I, Clark JF, Brewster LM. The effect of the creatine analogue beta-guanidinopropionic acid on energy metabolism: a systematic review. PLoS ONE. 2013;8:e52879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eijnde BO, Lebacq J, Ramaekers M, Hespel P. Effect of muscle creatine content manipulation on contractile properties in mouse muscles. Muscle Nerve. 2004;29:428–435. [DOI] [PubMed] [Google Scholar]
- 22. Robinson DM, Loiselle DS. Effect of creatine manipulation on fast-twitch skeletal muscle of the mouse. Clin Exp Pharmacol Physiol. 2002;29:1105–1111. [DOI] [PubMed] [Google Scholar]
- 23. Pandke KE, Mullen KL, Snook LA, Bonen A, Dyck DJ. Decreasing intramuscular phosphagen content simultaneously increases plasma membrane FAT/CD36 and GLUT4 transporter abundance. Am J Physiol Regul Integr Comp Physiol. 2008;295:R806–R813. [DOI] [PubMed] [Google Scholar]
- 24. Horvath TL, Erion DM, Elsworth JD, Roth RH, Shulman GI, Andrews ZB. GPA protects the nigrostriatal dopamine system by enhancing mitochondrial function. Neurobiol Dis. 2011;43:152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ilyin SE, Sonti G, Molloy G, Plata-Salamán CR. Creatine kinase-B mRNA levels in brain regions from male and female rats. Brain Res Mol Brain Res. 1996;41:50–56. [DOI] [PubMed] [Google Scholar]
- 26. Tachikawa M, Hosoya K. Transport characteristics of guanidino compounds at the blood-brain barrier and blood-cerebrospinal fluid barrier: relevance to neural disorders. Fluids Barriers CNS. 2011;8:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pelouch V, Kokif F, Khuchua ZA, et al. Cardiac phosphocreatine deficiency induced by GPA during postnatal development in rat. Mol Cell Biochem. 1996;163–164:67-76. [DOI] [PubMed] [Google Scholar]
- 28. Bergeron R, Ren JM, Cadman KS, et al. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2001;281:E1340–E1346. [DOI] [PubMed] [Google Scholar]
- 29. Yamashita H, Ohira Y, Wakatsuki T, et al. Increased growth of brown adipose tissue but its reduced thermogenic activity in creatine-depleted rats fed beta-guanidinopropionic acid. Biochim Biophys Acta. 1995;1230:69–73. [DOI] [PubMed] [Google Scholar]
- 30. Williams DB, Sutherland LN, Bomhof MR, et al. Muscle-specific differences in the response of mitochondrial proteins to beta-GPA feeding: an evaluation of potential mechanisms. Am J Physiol Endocrinol Metab. 2009;296:E1400–E1408. [DOI] [PubMed] [Google Scholar]
- 31. Chaturvedi RK, Adhihetty P, Shukla S, et al. Impaired PGC-1alpha function in muscle in Huntington’s disease. Hum Mol Genet. 2009;18:3048–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chang DD, Clayton DA. Priming of human mitochondrial DNA replication occurs at the light-strand promoter. Proc Natl Acad Sci U S A. 1985;82:351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res. 2008;103:1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hota KB, Hota SK, Chaurasia OP, Singh SB. Acetyl-L-carnitine-mediated neuroprotection during hypoxia is attributed to ERK1/2-Nrf2-regulated mitochondrial biosynthesis. Hippocampus. 2012;22:723–736. [DOI] [PubMed] [Google Scholar]
- 35. Aguiar AS, Jr, Duzzioni M, Remor AP, et al. Moderate-intensity physical exercise protects against experimental 6-hydroxydopamine-induced hemiparkinsonism through Nrf2-antioxidant response element pathway. Neurochem Res. 2015;41:64–72. [DOI] [PubMed] [Google Scholar]
- 36. Ahuja M, Kaidery NA, Yang L, et al. Distinct Nrf2 signaling mechanisms of fumaric acid esters and their role in neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced experimental Parkinson’s-like disease. J Neurosci. 2016;36:6332–6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li X, Wang H, Gao Y, et al. Protective effects of quercetin on mitochondrial biogenesis in experimental traumatic brain injury via the Nrf2 signaling pathway. PLoS ONE. 2016;11:e0164237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu KLH, Wu C-W, Chao Y-M, Hung C-Y, Chan JYH. Impaired Nrf2 regulation of mitochondrial biogenesis in rostral ventrolateral medulla on hypertension induced by systemic inflammation. Free Radic Biol Med. 2016;97:58–74. [DOI] [PubMed] [Google Scholar]
- 39. Hayashi G, Jasoliya M, Sacca F, et al. Dimethyl fumarate mediates Nrf2-dependent mitochondrial biogenesis in mice and humans. Hum Mol Genet. 2017;26:2864–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Navarro E, Gonzalez-Lafuente L, Pérez-Liébana I, et al. Heme-oxygenase I and PCG-1α regulate mitochondrial biogenesis via microglial activation of alpha7 nicotinic acetylcholine receptors using PNU282987. Antioxid Redox Signal. 2017;10:93–105. [DOI] [PubMed] [Google Scholar]
- 41. Tachikawa M, Kasai Y, Yokoyama R, et al. The blood-brain barrier transport and cerebral distribution of guanidinoacetate in rats: involvement of creatine and taurine transporters. J Neurochem. 2009;111:499–509. [DOI] [PubMed] [Google Scholar]
- 42. Liu QR, López-Corcuera B, Mandiyan S, Nelson H, Nelson N. Molecular characterization of four pharmacologically distinct gamma-aminobutyric acid transporters in mouse brain. J Biol Chem. 1993;268:2106–2112. [PubMed] [Google Scholar]
- 43. Gether U, Andersen PH, Larsson OM, Schousboe A. Neurotransmitter transporters: molecular function of important drug targets. Trends Pharmacol Sci. 2006;27:375–383. [DOI] [PubMed] [Google Scholar]
- 44. Clark JF, Khuchua Z, Kuznetsov AV, et al. Actions of the creatine analogue beta-guanidinopropionic acid on rat heart mitochondria. Biochem J. 1994;300:211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Buendia I, Michalska P, Navarro E, Gameiro I, Egea J, Leon R. Nrf2-ARE pathway: an emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol Ther. 2016;157:84–104. [DOI] [PubMed] [Google Scholar]