

Abstract
Introduction:
Transforming growth factor-β (TGF-β) and connective tissue growth factor (CTGF) are often described as the initial pro-fibrotic mediators upregulated early in fibrosis models dependent on angiotensin II (Ang-II). In the present study, we explore the mechanistic link between TGF-β and CTGF expression by using a novel TGF-β trap.
Materials and methods:
NIH/3T3 fibroblasts were subjected to TGF-β with or without TGF-β trap or 1D11 antibody, CTGF or CTGF plus TGF-β for six or 24 hours, and then used for quantitative real-time polymerase chain reaction (qRT-PCR) or immunocytochemistry. Male C57BL/6 mice were infused with Ang-II and randomly assigned TGF-β trap for six or 24 hours. Hearts were harvested for histological analyses, qRT-PCR and western blotting.
Results:
Exogenous TGF-β-induced fibroblasts resulted in significant upregulation of CTGF, TGF-β and type I collagen transcript levels in vitro. Additionally, TGF-β promoted the differentiation of fibroblasts into α-SMA+ myofibroblasts. CTGF expression was reduced by the addition of TGF-β trap or neutralizing antibody, confirming that its expression is dependent on TGF-β signaling. In contrast, exogenous CTGF did not appear to have an effect on fibroblast production of pro-fibrotic transcripts or fibroblast differentiation. Ang-II infusion in vivo led to a significant increase in TGF-β and CTGF mRNA expression at six and 24 hours with corresponding changes in Smad2 phosphorylation (pSmad2), indicative of increased TGF-β signaling. Ang-II animals that received the TGF-β trap demonstrated reduced CTGF mRNA levels and pSmad2 at six hours, suggesting that early CTGF expression is dependent on TGF-β signaling.
Conclusions:
We demonstrated that CTGF expression is dependent on TGF-β signaling both in vitro and in vivo in a model of myocardial fibrosis. This also suggests that early myocardial CTGF mRNA expression (six hours) after Ang-II exposure is likely dependent on latent TGF-β activation via the canonical Smad-dependent pathway in resident cardiac cells.
Keywords: Cardiac, TGF-β, CTGF, inflammation, myocardial fibrosis, fibroblasts, angiotensin-II, TGF-β trap
Introduction
Myocardial fibrosis is a common pathophysiological process that is commonly associated with cardiovascular conditions such as hypertension and is believed to partake in the final pathway towards diastolic heart failure.1 It is often characterized by overexpression of the renin angiotensin system (RAS), as well as excess production and deposition of extracellular matrix (ECM) proteins, thus resulting in increased scar tissue formation.2,3 This accumulation of ECM proteins renders the myocardium dysfunctional due to increased wall stiffness and impaired relaxation.4 Despite well-established therapies targeting the RAS, our ability to address the burden of diastolic heart failure remains limited.5 This highlights the importance of understanding the underlying mechanisms associated with myocardial fibrosis in order to bridge this knowledge gap.
Transforming growth factor-β (TGF-β) is a pleiotropic cytokine that has been well-established as a key mediator of fibrogenesis and is found upregulated and activated in numerous models of fibrotic diseases, including models with upregulated components of the RAS.6–8 The importance of TGF-β in cardiac remodeling has been implicated in numerous animal studies. In an aging model of myocardial fibrosis, TGF-β-deficient heterozygous mice (TGF-β+/-) exhibited a decrease in myocardial fibrosis and greater compliance compared to age-matched controls.9 Additionally, transgenic mice overexpressing TGF-β demonstrated significant myocardial hypertrophy and fibrosis compared to non-transgenic controls.10 However, several studies involving TGF-β blockade have demonstrated opposing results, thus emphasizing the complexity of the role of TGF-β in cardiac remodeling.11–14 Thus, there has been a shift towards focusing on downstream mediators of TGF-β in order to identify biomarkers and develop antifibrotic therapies, such as targeting Smad proteins directly downstream of TGF-β activation, as well as associated pro-fibrotic mediators, such as connective tissue growth factor (CTGF).15,16 Specifically, TGF-β signaling has been shown to upregulate CTGF production via the Smad pathway, which was also shown to correlate with fibrosis progression.17,18
Cardiac fibroblasts are one of the most abundant non-cardiomyocyte cell types in the myocardium, and are thought to be the main source and effector cells of ECM protein production and cardiac remodeling after injury.19,20 As such, fibroblasts are ideal cell types to study pro-fibrotic cytokine regulation in vitro. In animal models, our group has focused our attention on the initial few days post-angiotensin II (Ang-II) infusion and demonstrated that significant inflammation and fibrosis development were associated with increased expression of TGF-β and CTGF in the myocardium.18,21–23 Highlighting the importance of time-dependent events leading up to fibrosis, our previous work also demonstrated a marked elevation in CTGF gene expression prior to significant TGF-β production that remains to be fully characterized.18
In the present study, we used an in vitro system of fibroblast monocultures to examine the mechanistic link between TGF-β and CTGF on fibroblast activation and differentiation. We also used a novel soluble TGF-β trap designed based on TGF-β type II receptor (TβRII) ectodomains to explore this link.24 The TGF-β trap is designed to yield a peptide that is approximately threefold smaller in size compared to antibodies and is able to sequester active TGF-β. Finally, our findings were reflected in vivo using a well-established model of hypertension and myocardial fibrosis secondary to Ang-II infusion.
Materials and methods
In vitro fibroblast monoculture
NIH/3T3 (ATCC, Manassas, VA, USA) fibroblasts were used and cultured as previously described.21 In brief, NIH/3T3 fibroblasts were maintained in Dulbecco’s modified Eagle media (DMEM; Invitrogen, Carlsbad, CA, USA) complete (DMEM-C), which contained DMEM, 10% heat-inactivated fetal bovine serum (FBS), 2 mmol/L L-glutamine, 100 mg/ml streptomycin and 100 U/ml penicillin. NIH/3T3 fibroblasts were washed with Dulbecco’s phosphate-buffered saline (DPBS), lifted using 0.25% trypsin-EDTA (Invitrogen), counted, and re-plated at 1.0 × 105 cells/well on 0.1% gelatin-coated Corning 6-well tissue culture plates in DMEM-C with or without 0.1% gelatin-coated coverslips.
NIH/3T3 fibroblasts were incubated overnight in a humidified chamber at 37°C with 5% CO2 and then starved in 1% FBS/DMEM (DMEM, L-glutamine, 100 mg/ml streptomycin, 100 U/ml penicillin and 1% FBS) overnight before being treated with either sterile-filtered full-length recombinant human CTGF produced in HEK293 cells (125 ng/ml; CS363B, Cell Sciences, Canton, MA), sterile-filtered full-length recombinant active human TGF-β1 (10 ng/ml; ab50036, Abcam, Cambridge, UK), CTGF plus TGF-β1, or media (1% FBS/DMEM) only for six and/or 24 hours. Some cultures were treated with TGF-β1 plus TGF-β trap (400 nM), TGF-β1 plus pan-TGF-β neutralizing antibody (1D11; 15 μg/ml; MAB1835, R&D Systems, Minneapolis, MN, USA) or TGF-β trap only for six hours.
Animals
Male wild-type (WT) C57BL/6 mice (6–8 weeks old) were purchased from Jackson Laboratory (Bar Harbor, ME) and were housed within the Carleton Animal Care Facility at Dalhousie University. Mice were provided with food and water ad libitum for 1 week prior to experimentation.
Saline/Ang-II infusion and TGF-β trap intraperitoneal injection
Mice were randomly assigned to either saline (vehicle) or Ang-II (2.8 mg/kg/d; Sigma-Aldrich, Oakville, ON, Canada) through the use of subcutaneously implanted Alzet osmotic minipumps (Alzet Corp., Palo Alto, CA) as previously described.21,22 In brief, isoflurane with oxygen was used to anesthetize mice. A mid-scapular skin incision (approximately 1–2 cm) was made to implant the osmotic minipump subcutaneously. Immediately prior to implantation, mice were also randomly assigned to either phosphate-buffered saline (PBS, as a vehicle control) or TGF-β trap (T22d35, also known as (TβRII)2; 10 mg/kg)24 and administered via intraperitoneal injection, giving a total of four groups (saline/vehicle, saline/trap, Ang-II/vehicle and Ang-II/trap).
The animals were recovered for six or 24 hours while provided with food and water ad libitum, and observed for pain and morbidity. All animal experiments were performed in accordance with the Canadian Council on Animal Care guidelines and approved by the Dalhousie University Committee on Laboratory Animals (reference number 14-022).
Hemodynamic measurements
Mean arterial blood pressure was measured using a non-invasive tail cuff system (Kent Scientific, Torrington, CT, USA). Mice were trained for blood pressure measurements 7 days prior to the first day of experimentation for acclimatization as previously described.22,23 Blood pressures were measured immediately prior to osmotic pump implantation for baseline measurements and at 24 hours after Ang-II exposure for a minimum of five consecutive measurements.
Tissue processing and histology
All mice hearts were harvested as previously described.18,21,23 In brief, isolated hearts were flushed with saline, weighed and then divided into three sections along the vertical axis. The base was processed for histological analyses and the remaining two sections were immediately snap frozen for molecular analyses.
Isolated mice hearts were processed for histological analyses by fixing in 10% formalin for 24 hours. Formalin-fixed tissues were paraffin-embedded and serially sectioned on a 5-μm microtome. Gross myocardial histological structures and cellular infiltration were analyzed using hematoxylin and eosin (H&E)-stained heart cross-sections. Light microscopy was performed using the Zeiss Axiovision 4.6 digital image analysis program (Carl Zeiss, Toronto, ON, Canada) on slides, visualized with an AxioCam HRc camera (Carl Zeiss) and analyzed using Adobe Photoshop CS6. An established standardized methodology was used to quantitatively assess histological images, as previously described.18,21,23 In brief, a 500 × 500 pixel grid was overlaid on a heart cross-section captured at 5× magnification. A blinded observer counted grids containing cellular infiltrates as well as the total number of grids encompassing the heart. The percent of affected grids was calculated over the total number of grids encompassing the heart.
Immunohistochemistry and immunofluorescence
Immunohistochemistry was performed on paraffin-embedded heart cross-sections for CD11b (Abcam) as previously described.18,23 Sections were deparaffinized, subjected to degrading alcohol gradient and treated for antigen retrieval before staining. Briefly, 3% hydrogen peroxide was used for endogenous peroxidase quenching, non-specific staining was blocked using 10% normal goat serum and primary antibody was incubated overnight at 4°C. Following primary antibody incubation, a specific biotin-conjugated secondary antibody was incubated for 1 hour at room temperature. The antibody complexes were conjugated to avidin–biotin complex (Vectastain ABC kit; Vector Laboratories, Inc., Burlington, CA) and developed using 3,3’diaminobenzidine as the chromagen (DAB; Dako-Cytomation, Mississauga, ON, Canada). Light microscopy was performed using the Zeiss Axiovision 4.6 digital image analysis program (Carl Zeiss) on slides, visualized with an AxioCam HRc camera (Carl Zeiss) and analyzed using Adobe Photoshop CS6.
Immunofluorescence was performed on 4% paraformaldehyde-fixed NIH/3T3 fibroblast monocultures for α-SMA as previously described.21 In brief, fixed cells were washed with PBS, blocked with 5% bovine serum albumin (BSA)/PBS, and incubated with primary antibody overnight at 4°C. Next, fixed cells were incubated with AlexaFluor488 (Invitrogen) secondary antibody for 1 hour and nuclei were counterstained with Hoechst stain (Sigma-Aldrich). Six-well plates were read for fluorescence by using an Infinite M200 Pro (Tecan, Mannedorf, Germany) and α-SMA expression was standardized to Hoechst fluorescence intensity. Slides were visualized and captured using a Zeiss Axioplan II MOT (Carl Zeiss GmbH, Gottingen, Germany) with an AxioCam HRC color camera.
Western blotting
Western blot analysis was performed on isolated snap-frozen heart samples as previously described.18 Heart sections were homogenized in radioimmunoprecipitation assay buffer (RIPA; 150 mmol/L NaCl, 50 mmol/L Tris-HCl base, 0.1% sodium dodecyl sulfate (SDS), 0.1% Triton X-100 and 0.5% deoxycholic acid) with protease inhibitor cocktail added (Roche Diagnostics, Indianapolis, IN, USA). Samples were denatured using Laemmli sample buffer with β-mercaptoethanol (Bio-Rad Laboratories, Hercules, CA, USA) and boiled at 95°C for 5 minutes. Protein was separated on a 12% SDS-polyacrylamide gel electrophoresis gel and transferred to an Immobilon polyvinylidene difluoride (PVDF; Millipore Corp., Bedford, MA) membrane. PVDF membranes were incubated with either 5% skimmed milk or 5% BSA (Sigma-Aldrich) in Tris-buffered saline with 0.1% Tween-20 (TBST; Sigma-Aldrich) for 1 hour and then incubated with anti-Smad2/3(Cell Signaling Technology, Beverly, MA), phosphoSmad2 (Cell Signaling Technology) or actin (loading control; Sigma-Aldrich) antibody overnight at 4°C. The following day, membranes were incubated with horseradish peroxidase-linked goat anti-rabbit immunoglobulin G (Vector Laboratories) and developed using an Amersham enhanced chemiluminescence kit (GE Healthcare UK, Ltd, Buckinghamshire, UK). Images were taken using the ChemiDocTM Touch Imaging System (Bio-Rad Laboratories) and analyzed using Image LabTM software (Bio-Rad Laboratories).
Relative real-time quantitative polymerase chain reaction
TRIzol (Life Technologies, Carlsbad, CA) was used according to the manufacturer’s protocol for total RNA isolation from snap-frozen heart sections and NIH/3T3 fibroblast monocultures. First-strand cDNA was synthesized from extracted RNA using an iScriptTM Advanced cDNA Synthesis Kit (Bio-Rad Laboratories). Real-time quantitative polymerase chain reaction (qRT-PCR) was performed using iQ SYBR Green Supermix (Bio-Rad Laboratories), detected using Rotor-GeneTM 6000 (Corbett Life Science, San Francisco, CA, USA) and analyzed with Rotor-Gene Q Series software (QIAGEN Sciences, Germantown, MD, USA). Efficiency curves and no-template controls accompanied each run and melt curves were run after cycling for target specificity. Primers were designed against mRNA sequences of CTGF (forward, 5′-TCAACCTCAGACACTGGTTTCG-3′; reverse, 5′-TAGAGCAGGTCTGTCTGCAAGC-3′), TGF-β (forward, 5′-GCCTGAGTGGCTGTCTTTTG-3′; reverse, 5′-CTGTATTCCGTCTCCTTGGTTC-3′), collagen-IαI (forward, 5′-CAACAGTCGCTTCACCTACAGC-3′; reverse, 5′-GTGGAGGGAGTTTACACGAAGC-3′), 18S ribosomal RNA (reference gene; forward, 5′-TCAACTTTCGATGGTAGTCGCCGT-3′; reverse, 5′-TCCTTGGATGTGGTAGCCGTTTCT-3′) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (reference gene; forward, 5′-CCTTCCGTGTTCCTACCCC-3′; reverse, 5′-GCCCAAGATGCCCTTCAGT-3′). Relative expression levels were normalized to reference genes GAPDH and/or the 18S ribosomal gene, and further normalized to controls using the Pfaffl method.
Statistical analyses
Data are presented as means ± SEM. One-way analysis of variance (ANOVA) was performed using the Bonferroni post-test for multiple comparisons to compare experimental groups with control groups. All direct comparisons between two groups were evaluated using the two-tailed Student’s t-test for changes in relative expression. All statistical calculations were computed using GraphPad PRISM software version 6 (GraphPad Software, Inc., La Jolla, CA, USA) and significance was determined if p ≤ 0.05.
Results
Expression of fibroblast fibrogenic mediators and differentiation into α-SMA myofibroblasts is dependent on TGF-β signaling in vitro
Using a monoculture of NIH/3T3 fibroblasts, the addition of exogenous TGF-β resulted in a significant increase in CTGF mRNA expression after six hours of exposure (Figure 1(a); 7.7-fold ± 0.7 vs. media control, p < 0.001). This increase was significantly reduced with 1D11, an anti-TGF-β neutralizing antibody (Figure 1(a); 2.6-fold ± 1.0 vs. media control, 66% reduction, p < 0.01). The addition of the TGF-β trap reduced CTGF expression but this failed to reach significance (Figure 1(a); 4.8-fold ± 0.7 vs. media control, 38% reduction, not significant). Exogenous TGF-β stimulation of NIH/3T3 fibroblasts also resulted in a significant increase of TGF-β transcript (Figure 1(b); 4.1-fold ± 1.1 vs. media control, p < 0.05), suggestive of an autoregulatory mechanism promoting TGF-β expression. Additionally, exogenous TGF-β stimulation of NIH/3T3 fibroblasts increased type I collagen production, as indicated by an elevation of collagen-IαI transcript levels six hours after treatment (Figure 1(c); 1.6-fold ± 0.1 vs. media control, p < 0.05). Treatment with TGF-β trap or 1D11 antibody resulted in reductions of TGF-β transcript levels (Figure 1(b); 24 and 56% reduction, respectively) and collagen-IαI transcript levels (Figure 1C; 44 and 56% reduction, respectively). Taken together, the expression of fibrogenic mediators by fibroblasts in vitro is largely dependent on TGF-β signaling.
Figure 1.

Effect of TGF-β trap on exogenous TGF-β-induced upregulation of fibrogenic mediators CTGF, TGF-β and collagen type I transcript levels in NIH/3T3 fibroblasts in vitro.
NIH/3T3 fibroblasts were incubated with exogenous TGF-β only, TGF-β trap only, TGF-β plus TGF-β trap, or TGF-β plus 1D11 antibody for six hours. Transcript levels of (a) CTGF, (b) TGF-β and (c) collagen type I were obtained using qPCR analyses. Transcript levels are reported relative to housekeeping genes ribosomal 18S and GAPDH.
Data are represented as means ± SEM and expressed relative to media control.
n = 3 independent experiments in triplicate.
*p < 0.05, **p ≤ 0.01 and ***p ≤ 0.001, compared to media control.
CTGF: connective tissue growth factor; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; qPCR: quantitative polymerase chain reaction; TGF-β: transforming growth factor-β; 1D11: pan-TGF-β neutralizing antibody.
NIH/3T3 fibroblasts grown in monoculture and exposed to exogenous CTGF did not result in changes in CTGF, TGF-β or type I collagen transcript alone. Furthermore, the addition of CTGF to TGF-β did not provide any synergism (Figure 2(a) to (c)). This contrasts with exogenous TGF-β stimulation, which resulted in significant upregulation of CTGF mRNA in NIH/3T3 fibroblasts (Figure 2(a); 7.3-fold ± 0.5 vs. media control, p ≤ 0.01), as well as a trending increase in TGF-β and type I collagen transcript levels (Figure 2(b) to (c)).
Figure 2.

Exogenous CTGF incubation does not appear to have an effect on NIH/3T3 fibroblast production of fibrogenic CTGF, TGF-β or collagen type I transcripts independently or synergistically with exogenous TGF-β incubation in vitro. NIH/3T3 fibroblasts were incubated with exogenous CTGF only, TGF-β only, or CTGF plus TGF-β for six hours. Transcript levels of (a) CTGF, (b) TGF-β and (c) collagen type I were obtained using qPCR analyses. Transcript levels are reported relative to housekeeping genes ribosomal 18S and GAPDH.
Data are represented as means ± SEM and expressed relative to media control.
n = 3 independent experiments in triplicate.
*p<0.05, **p ≤ 0.01, compared to media control.
CTGF: connective tissue growth factor; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; qPCR: quantitative polymerase chain reaction; TGF-β: transforming growth factor-β.
We next assessed the differentiation of fibroblasts into α-SMA+ myofibroblasts, which are the effector cells of fibrosis development. The addition of TGF-β for 24 hours resulted in significant upregulation of α-SMA expression, indicative of increased fibroblast differentiation into myofibroblasts, compared to media control (Figure 3(a), (c) and (e); 1.9-fold ± 0.2 vs. media control, p ≤ 0.01). Alternatively, CTGF had no effect on fibroblast α-SMA expression independently or in combination with TGF-β, suggesting that CTGF does not play an important early role in fibroblast differentiation into myofibroblasts (Figure 3(a), (b), (d) and (e)).
Figure 3.

Exogenous CTGF incubation does not appear to affect differentiation of NIH/3T3 fibroblasts into α-SMA+ myofibroblasts independently or synergistically with exogenous TGF-β incubation in vitro. NIH/3T3 fibroblasts were incubated with exogenous CTGF only, TGF-β only or CTGF plus TGF-β for 24 hours. Representative fields of view stained for α-SMA, Hoechst and phase-contrast are shown for fibroblasts in (a) media control (1% FBS/DMEM), (b) exogenous CTGF, (c) exogenous TGF-β and (d) exogenous CTGF plus TGF-β. (e) Graphical representation of α-SMA expression as measured by fluorescence intensity relative to Hoechst fluorescence intensity and represented as relative to media controls.
Data are represented as means ± SEM.
n = 3 independent experiments in triplicates.
*p < 0.05, **p ≤ 0.01, compared to media control.
α-SMA: alpha-smooth muscle actin; AU: arbitrary units; CTGF: connective tissue growth factor; DMEM: Dulbecco’s modified Eagle media; FBS: fetal bovine serum; TGF-β: transforming growth factor-β.
Taken together, these findings confirm that CTGF expression is dependent on TGF-β activation but suggest that CTGF does not promote the pro-fibrotic effects of TGF-β based on collagen expression or α-SMA differentiation.
Early CTGF expression is partially dependent on latent TGF-β activation in vivo
Previous published work by us and others has suggested that the predominant molecular mediators involved in the early fibrotic response to Ang-II exposure are TGF-β and CTGF.17,18,21,25. In this study, we demonstrated that CTGF transcript levels were significantly elevated as early as six hours after Ang-II infusion (Figure 4(a); 19.3-fold ± 5.4 vs. saline controls, p < 0.05) and remained elevated after 24 hours (Figure 4(a); 17.0-fold ± 3.1 vs. saline controls, p < 0.05). TGF-β transcript levels were also elevated at six hours and remained elevated at 24 hours (Figure 4(b); 2.5-fold ± 0.2, p < 0.01, and 3.2-fold ± 0.4, p < 0.001, respectively, vs. saline controls), although not to the same extent as CTGF levels.
Figure 4.

Early upregulation of myocardial CTGF and TGF-β transcript levels appeared as early as six hours after Ang-II exposure. Myocardial (a) CTGF and (b) TGF-β transcript levels after six or 24 hours of Ang-II infusion relative to saline controls. Transcript levels are reported relative to housekeeping gene ribosomal 18S.
Data are expressed as means ± SEM.
n = 5–6.
*p < 0.05, **p ≤ 0.01 and ***p ≤ 0.001, compared to saline controls.
Ang-II: Angiotensin II; CTGF: connective tissue growth factor; TGF-β: transforming growth factor-β.
Histological myocardial cross-sections were stained with H&E and assessed based on the percentage of grids containing cellular infiltration over the total number of grids containing the myocardial cross-section. The rationale for assessing myocardial-infiltrating cells is based on a body of evidence demonstrating early inflammatory cell accumulation secondary to Ang-II exposure.21,23 There were no significant differences in cellular infiltration six hours of after Ang-II exposure compared to saline controls (Figure 5(a) and (b)). After 24 hours of Ang-II exposure, we demonstrated a significant increase in mononuclear cell infiltration compared to saline controls (Figure 5(c) to (e); 18.3% ± 1.4 vs. 0.9% ± 0.1, respectively, p < 0.0001). There were little to no polymorphonuclear cells, suggesting little role for granulocytes in this model (Figure 5(f)). Using immunohistochemistry staining for CD11b, our findings were confirmed by demonstrating that a significant number of infiltrated cells were positive for CD11b (Figure 5(g) and (h)). Taken together, our results show that initiation of CTGF expression at six hours occurred prior to significant mononuclear cell infiltration, suggesting that resident cardiac cells, such as cardiac fibroblasts and endothelial cells rather than infiltrating cells, must be the predominant source of these factors early after Ang-II exposure.
Figure 5.

Mononuclear cellular infiltration in the myocardium was present after 24 hours of Ang-II infusion. Representative images of myocardial cross-sections stained with H&E from mice that received ((a) and (c)) saline or ((b), (d) and (f)) Ang-II. (e) H&E sections were semi-quantified for cellular infiltration in Ang-II animals relative to saline controls. Immunohistochemistry was used to characterize mononuclear cell infiltrates as CD11b+. Representative myocardial cross-sections from animals treated with (g) saline or (h) Ang-II.
Data are expressed as means ± SEM.
n = 5.
****p < 0.0001, compared with saline control. Original magnification: ×20 (a) to (d); ×40 (f).
Ang-II: Angiotensin II; H&E: hematoxylin and eosin.
It has been well documented that in the myocardium, TGF-β activity occurs via the canonical Smad-dependent signaling pathway, which upregulates downstream pro-fibrotic mediators in fibrosis development.4,17,26 We performed western blotting to identify phosphorylation of Smad2 relative to total Smad2/3 protein levels at six and 24 hours and assess the activation of the TGF-β-Smad-dependent pathway. Ang-II-exposed hearts demonstrated significant upregulation of Smad2 phosphorylation (pSmad2) levels relative to reference protein actin (Figure 6(a) and (b); 5.7-fold ± 1.2, p < 0.05) compared to saline control, whereas total Smad2/3 protein levels were not affected (Figure 6(a)). Thus, the ratio between pSmad2 levels relative to total Smad2/3 levels is elevated upon Ang-II exposure, suggestive of increased TGF-β activity (Figure 6(b), 5.7-fold ± 1.3, p < 0.05). Similarly, 24 hours of Ang-II infusion resulted in an increasing trend in pSmad2 levels relative to reference protein actin (Figure 6(c) and (d), 3.1-fold ± 0.9, p = 0.0686), while total Smad2/3 levels remain unchanged (Figure 6(c)). This resulted in a trending increase in pSmad2 levels relative to total Smad2/3 levels (Figure 6(d), 4.1 ± 1.3, p = 0.0722), suggestive of a maintained increase in TGF-β activity in the myocardium following Ang-II exposure.
Figure 6.

Levels of Smad2 phosphorylation were upregulated after Ang-II exposure, indicative of TGF-β activity. (a) Representative western blot images and semi-quantification using densitometry of pSmad2 and total Smad2/3 protein levels relative to actin at six hours. (b) Densitometry was used for semi-quantification of pSmad2 levels relative to total Smad2/3 protein levels. (c) Representative western blot images and semi-quantification using densitometry of pSmad2 and total Smad2/3 protein levels relative to actin at 24 hours. (d) Densitometry was used for semi-quantification of pSmad2 levels relative to total Smad2/3 protein levels.
Data are expressed as means ± SEM and normalized to actin.
n = 5–6.
*p < 0.05 and **p < 0.01, compared to saline control.
Ang-II: Angiotensin II; PBS: phosphate-buffered saline; pSMAD2: phosphorylated Smad2; Ser: serine.
Collectively, the increase in phosphorylation of Smad proteins downstream of TGF-β signaling seen at six hours is unlikely to be directly induced by newly synthesized TGF-β protein from the early (6 hours) upregulation of TGF-β mRNA given the short time course. Increased CTGF expression could be dependent on TGF-β already present in the myocardial environment in the form of latent TGF-β. Thus, we next assessed whether the early increase in CTGF expression is a result of increased TGF-β signaling. The administration of the TGF-β trap significantly reduced CTGF mRNA levels by 64.2% in Ang-II animals at six hours compared to PBS vehicle controls (Figure 7(a), left; 19.3-fold ± 5.4 vs. 6.9-fold ± 1.6, respectively, p < 0.05). These results support our in vitro observations and suggest that early CTGF expression is at least partially dependent on TGF-β signaling. In particular, it is possible that this is due to latent TGF-β activation in the myocardium rather than entirely on TGF-β-independent effects of Ang-II exposure. Interestingly, there appears to be a trend towards an increase in TGF-β expression at six hours, although it failed to reach significance and did not affect levels at 24 hours (Figure 7(a) and (b), right). Administration of the TGF-β trap reduced myocardial pSmad2 levels relative to total Smad2/3 protein in animals exposed to Ang-II compared to PBS controls (Figure 8(a) and (b); 5.7-fold ± 1.5 vs. 2.1-fold ± 0.5, respectively, compared to saline control, p < 0.05). However, the effect of the TGF-β trap was no longer maintained at 24 hours with regards to CTGF expression (Figure 7). Similarly, the levels of pSmad2 relative to total Smad2/3 proteins were not significantly different after 24 hours of Ang-II exposure (Figure 8(c) and (d)).
Figure 7.

Administration of TGF-β trap reduced levels of Ang-II-induced early upregulation of CTGF transcript levels at six hours. Myocardial CTGF and TGF-β transcript levels after (a) six hours or (b) 24 hours of Ang-II infused animals injected with TGF-β trap or PBS relative to saline controls. Transcript levels are reported relative to housekeeping gene ribosomal 18S.
Data are expressed as means ± SEM.
n = 5–6.
*p < 0.05, compared to PBS group.
Ang-II: Angiotensin II; CTGF: connective tissue growth factor; PBS: phosphate-buffered saline; Trap: TGF-β trap; TGF-β: transforming growth factor-β.
Figure 8.

TGF-β trap administration to partially reduced Ang-II-induced elevation of phosphorylated Smad2 levels at six hours. (a) Representative western blot images and semi-quantification using densitometry of pSmad2 and total Smad2/3 protein levels relative to actin at six hours. (b) Densitometry was used for semi-quantification of pSmad2 levels relative to total Smad2/3 protein levels. (c) Representative western blot images and semi-quantification using densitometry of pSmad2 and total Smad2/3 protein levels relative to actin at 24 hours. (d) Densitometry was used for semi-quantification of pSmad2 levels relative to total Smad2/3 protein levels.
Data are expressed as means ± SEM and normalized to actin.
n = 5–6.
*p < 0.05, compared to saline control.
Ang-II: Angiotensin II; PBS: phosphate-buffered saline; pSMAD2: phosphorylated Smad2; Ser: serine; Trap: TGF-β trap; TGF-β: transforming growth factor-β.
Taken together, these results confirm that CTGF production in vivo after Ang-II exposure is dependent on TGF-β signaling, likely via latent TGF-β activation in the myocardium and particularly in the first six hours. Furthermore, the elevated TGF-β activity that in part led to significant upregulation of CTGF transcript levels was through the activation of the canonical Smad-dependent pathway.
In vivo effect of TGF-β trap on fibrosis
The in vivo ability of TGF-β trap to affect early fibrotic changes was assessed by comparing three groups of animals over a 24-hour period receiving Ang-II or Ang-II and Trap compared to saline control. Ang-II exposure for 24 hours was associated with some increase in collagen deposition as assessed by histology (Sirius Red/Fast Green) and qRT-PCR, significant cellular infiltration by mononuclear cells and significant cell proliferation assessed using Ki-67 expression (Supplementary Figures 1–3). In contrast, the co-administration of the TGF-β trap with Ang-II did not limit collagen expression in vivo at 24 hours, increased cellular infiltration or the significant increase in Ki-67+ mononuclear cells within the myocardium that was seen in animals exposed to Ang-II (Supplementary Figures 1–3). Taken together, our findings suggest that in vivo the TGF-β trap did not impact initial changes normally associated with fibrosis, namely, collagen expression, mononuclear cell infiltration or cellular proliferation.
Physiological changes seen in Ang-II-exposed animals (Appendix 1)
Ang-II infusion resulted in a significant increase in mean arterial blood pressure after 24 hours compared to their WT counterparts (133.9 ± 6.5 mmHg vs. 109.3 ± 7.3 mmHg respectively, p < 0.05).
Discussion
Our understanding of how the renin-angiotensin-aldosterone system is involved in the fibrotic process is heavily dependent on our understanding of TGF-β and its downstream mediators like CTGF.4 The pleiotropic protein TGF-β is believed to play a central role in most fibrotic conditions.6,7 Active TGF-β has a high affinity for its receptor, transmembrane TβRIIs, and a small fraction of binding can generate maximal cellular response.27 TGF-β signaling via the canonical pathway requires Smad family protein phosphorylation, such as of Smad2 and Smad3, which then forms a complex with Smad4 and translocates into the nucleus to elicit downstream gene transcription.28 Additionally, CTGF (also referred to as CCN2), a matricellular protein of the CCN family, has also been suggested to be a key mediator of fibrosis downstream of TGF-β signaling.4,18,29–32 By using a NIH/3T3 in vitro fibroblast monoculture system, we demonstrate that CTGF expression is dependent on TGF-β activity. In addition to detecting changes in CTGF expression, our in vitro system was capable of detecting increases in the expression of TGF-β, type I collagen and subsequently fibroblast differentiation into α-SMA+ myofibroblasts, all of which supports their role in the fibrotic process. This increase in CTGF expression was significantly limited by inhibiting TGF-β signaling using a TGF-β trap and a pan-anti-TGF-β neutralizing antibody, confirming that CTGF expression is largely dependent on TGF-β signaling.
In the context of myocardial fibrosis, elevated expression of TGF-β has been reported as early as 1 day after Ang-II exposure.18,23 Using the Ang-II infusion model of myocardial fibrosis, we also demonstrated a significant upregulation of TGF-β as well as CTGF gene expression as early as six hours after Ang-II exposure. However, this early increase in CTGF is unlikely the result of newly synthesized TGF-β given the short time course of six hours. Our findings suggest that early CTGF expression is likely a result of latent TGF-β activation, which was supported by our findings demonstrating increased pSmad2 protein levels as early as six hours after Ang-II exposure. Others have shown that TGF-β is abundantly stored in the ECM as latent TGF-β and requires proteolytic cleavage for bioactivity6,27 In our Ang-II model of myocardial fibrosis, the administration of the TGF-β trap in Ang-II animals resulted in a significant reduction of CTGF gene expression after six hours, supporting our in vitro observations indicating that CTGF expression was dependent on TGF-β signaling. This is further supported by our western blot findings where pSmad2 levels relative to total Smad 2/3 protein were partially reduced and thus, linking the canonical TGF-β signaling pathway (TGF-β/Smad-dependent pathway) involved in the induction of early CTGF expression.
Our findings here confirm that early molecular changes in CTGF and TGF-β are seen prior to significant inflammatory cell accumulation in the myocardium that appeared after 24 hours of Ang-II exposure. To date, much remains unknown about the exact role of CTGF and it is therefore the subject of significant work.33–35 While our study highlights a mechanism explaining how CTGF is upregulated in a TGF-β-dependent manner, we provide no clear role for CTGF in the fibrotic process. We demonstrate that while CTGF expression is clearly linked to TGF-β, it does not appear to promote classical fibrotic effects. In particular, we have shown that CTGF does not appear to promote the activation of fibroblasts in vitro either by production of pro-fibrotic mediators (CTGF, TGF-β or type I collagen) or by promoting their differentiation into α-SMA+ myofibroblasts. Similar to our study, emerging evidence has demonstrated the negligible role of CTGF in the progression of myocardial fibrosis through the use of transgenic mice with myocardial-specific gain or loss of CTGF.36,37 In the present manuscript, we did not assess other roles of CTGF that have been previously described, such as its role in cardiomyocyte hypertrophy.38 Thus, future studies regarding the exact role and importance of CTGF in fibrosis progression is warranted.
The TGF-β trap is a soluble TβRII peptide that has the ability to sequester active TGF-β in the ECM.24 Previous work using the TGF-β trap by Zwaagstra et al. has demonstrated that the administration of the TGF-β trap into established 4T1 tumors can result in TGF-β neutralization in the tumor environment.24 In our Ang-II model, we have shown that the effect of the TGF-β trap appeared limited to the first six hours in vivo as they did not elicit an effect in CTGF mRNA levels after 24 hours of Ang-II infusion. There are many potential explanations for this observation, but they are beyond the scope of the present manuscript. Here, the TGF-β trap was mainly used as a tool to study early CTGF expression rather than address its therapeutic effects. Despite this, one explanation could possibly be that Ang-II can elicit direct effects on resident cells via its receptor Ang-II type I receptor (AT1R) through a number of Ang-II signaling pathways. Iwanciw et al. have shown that Ang-II activation via AT1R induced CTGF mRNA and protein production in a human fibroblast cell line through the mitogen-activated protein kinase (MAPK) signaling pathway.39 RhoA/Rho kinase, p38 MAPK and JNK signaling, and calcineurin-dependent pathways have also been shown to induce CTGF production directly upon Ang-II stimulation.40,41
In the present study, we have demonstrated significant mononuclear cellular infiltration after 24 hours of Ang-II exposure. While not the focus of the present manuscript, work by us and others has demonstrated the temporal relationship between early mononuclear cell infiltration and subsequent collagen deposition.21,23 Furthermore, we have shown that depletion of monocytes from the circulation can significantly limit myocardial fibrosis, providing a causal relationship between immune cell infiltration and fibrosis.21 Administering the TGF-β trap did not affect cellular infiltration at 24 hours despite the reduction in CTGF expression observed at six hours, which suggests that TGF-β is unlikely to be responsible for the migration of mononuclear cells in the context of Ang-II exposure. These findings illustrate the complexity that exists between pro-fibrotic changes at the cellular and molecular levels and the concomitant inflammatory responses in the context of myocardial fibrosis secondary to hypertension. In this study, we directed our attention to the first 24 hours of Ang-II exposure specifically to explore early mechanistic changes of resident cells in the myocardium, which is particularly evident at six hours, rather than focusing on the later fibrotic changes associated downstream of Ang-II infusion.
One of the limitations in using this novel TGF-β trap in our Ang-II model is the consideration of the half-life of the TGF-β trap, which is short (< 1 hour in vivo), and its effect may not be possible to be maintained in vivo for extended periods of time, such as for 24 hours24. At the time of the study, we did not have the ability to demonstrate if the TGF-β trap remained present in tissue at the 24-hour time point. Thus, we cannot disregard the possibility that elimination of the TGF-β trap from the system has occurred, which may explain the insignificant observations at 24 hours. As demonstrated by Zwaagstra et al., this dosage and delivery method may be sufficient in the context of tumor growth but may need to be altered in order to observe notable effects in the myocardium24. As such, future experiments will need to be designed to optimize the dosage of the TGF-β trap to repeated intraperitoneal injections in order to address effects at 24 hours or beyond. These limitations of the TGF-β trap are best illustrated in our in vivo findings on fibrosis seen in the Supplementary Figures 1–3. Our findings suggest that early cellular infiltration, cellular proliferation and collagen expression seen in animals receiving Ang-II was not affected by the co-administration of the TGF-β trap. While this may mean that these early fibrotic changes are not TGF-β-dependent, one cannot conclusively make this statement based on the complexity of the system and the limitation of our approach.
As demonstrated in our in vitro monoculture, the use of the 1D11 neutralizing antibody appears to be more efficacious in inhibiting effects of TGF-β activity compared to the TGF-β trap, particularly in the case of CTGF and TGF-β transcript levels. Despite these results, we moved forward into our in vivo model using the TGF-β trap as opposed to the 1D11 antibody. In a murine model of myocardial infarction via left coronary artery ligation, Frantz et al. have demonstrated detrimental effects of TGF-β inhibition using the 1D11 antibody, likely due to altering ECM remodeling after myocardial injury.13 This highlights the importance of time-dependent changes after myocardial injury and thus care must be taken when manipulating the TGF-β signaling response. Thus, because of the short half-life of the TGF-β trap, there was the possibility that this may in fact be beneficial in terms of short-term neutralization in the context of myocardial fibrosis but could not be demonstrated by our approach.
In summary, our findings highlight the mechanistic link between TGF-β and CTGF and their interactions and contributions in creating a pro-fibrotic myocardial environment early following Ang-II exposure. We demonstrate that early CTGF upregulation is dependent on TGF-β signaling, likely from the activation of preformed latent TGF-β in the myocardium on cardiac resident cells. We also demonstrate that while CTGF expression is linked to TGF-β, it does not appear to promote classical fibrotic effects such as collagen expression or myofibroblast differentiation. Finally, we have provided evidence to support the use of short-term neutralization of TGF-β by using the TGF-β trap as a molecular tool in understanding mechanisms involving the TGF-β pathway that are common to various fibrotic diseases.
Supplemental Material
Supplemental material, jraas-supp._Fig_1 for Connective tissue growth factor expression after angiotensin II exposure is dependent on transforming growth factor-β signaling via the canonical Smad-dependent pathway in hypertensive induced myocardial fibrosis by Chloe Kok Sum Wong, Alec Falkenham, Tanya Myers and Jean-Francois Légaré in Journal of the Renin-Angiotensin-Aldosterone System
Supplemental Material
Supplemental material, jraas-supp._Fig_2 for Connective tissue growth factor expression after angiotensin II exposure is dependent on transforming growth factor-β signaling via the canonical Smad-dependent pathway in hypertensive induced myocardial fibrosis by Chloe Kok Sum Wong, Alec Falkenham, Tanya Myers and Jean-Francois Légaré in Journal of the Renin-Angiotensin-Aldosterone System
Supplemental Material
Supplemental material, jraas-_Supp.Fig_3 for Connective tissue growth factor expression after angiotensin II exposure is dependent on transforming growth factor-β signaling via the canonical Smad-dependent pathway in hypertensive induced myocardial fibrosis by Chloe Kok Sum Wong, Alec Falkenham, Tanya Myers and Jean-Francois Légaré in Journal of the Renin-Angiotensin-Aldosterone System
Acknowledgments
We would like to extend a great thank you to Dr John Zwaagstra and the National Research Council in Montreal for providing us with the TGF-β trap (T22d35, aka (TβRII)2) for this study. We would also like to thank Pat Colp for her technical skills and expertise that contributed to this manuscript.
Appendix
Appendix 1.
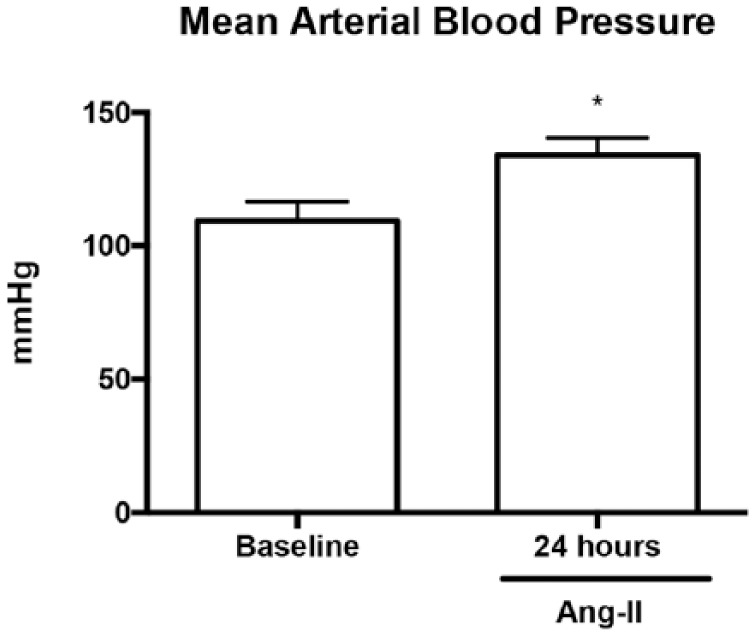
Hemodynamic parameters of Ang-II-exposed mice. Mice exposed to Ang-II for 24 hours demonstrated a significant increase in mean arterial blood pressure compared to WT counterparts.
Data expressed as means ± SEM. n = 5–8. *p < 0.05, compared with baseline.
Ang-II: angiotensin II; WT: wild-type.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funding from the Canadian Institutes of Health Research (Restitution Enhancement in Arthritis and Chronic Heart Disease REACH Program THC135230), as well as the Heart and Stroke Foundation BrightRed Graduate Research Award.
References
- 1. Díez J. Mechanisms of cardiac fibrosis in hypertension. J Clin Hypertens (Greenwich) 2007; 9: 546–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brilla CG. Renin-angiotensin-aldosterone system and myocardial fibrosis. Cardiovasc Res 2000; 47: 1–3. [DOI] [PubMed] [Google Scholar]
- 3. Li AH, Liu PP, Villarreal FJ, et al. Dynamic changes in myocardial matrix and relevance to disease: translational perspectives. Circ Res 2014; 114: 916–927. [DOI] [PubMed] [Google Scholar]
- 4. Leask A. Getting to the heart of the matter: new insights into cardiac fibrosis. Circ Res 2015; 116: 1269–1276. [DOI] [PubMed] [Google Scholar]
- 5. Shearer F, Lang CC, Struthers AD. Renin-angiotensin-aldosterone system inhibitors in heart failure. Clin Pharmacol Ther 2013; 94: 459–467. [DOI] [PubMed] [Google Scholar]
- 6. Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-beta signaling in fibrosis. Growth Factors 2011; 29: 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol 2011; 51: 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Harris WT, Kelly DR, Zhou Y, et al. Myofibroblast differentiation and enhanced TGF-B signaling in cystic fibrosis lung disease. PLoS One 2013; 8: e70196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brooks WW, Conrad CH. Myocardial fibrosis in transforming growth factor beta(1)heterozygous mice. J Mol Cell Cardiol 2000; 32: 187–195. [DOI] [PubMed] [Google Scholar]
- 10. Rosenkranz S, Flesch M, Amann K, et al. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta1. Am J Physiol Heart Circ Physiol 2002; 283: H1253–H1262. [DOI] [PubMed] [Google Scholar]
- 11. Ellmers LJ, Scott NJ, Medicherla S, et al. Transforming growth factor-beta blockade down-regulates the renin-angiotensin system and modifies cardiac remodeling after myocardial infarction. Endocrinology 2008; 149: 5828–5834. [DOI] [PubMed] [Google Scholar]
- 12. Engebretsen KV, Skardal K, Bjornstad S, et al. Attenuated development of cardiac fibrosis in left ventricular pressure overload by SM16, an orally active inhibitor of ALK5. J Mol Cell Cardiol 2014; 76: 148–157. [DOI] [PubMed] [Google Scholar]
- 13. Frantz S, Hu K, Adamek A, et al. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res Cardiol 2008; 103: 485–492. [DOI] [PubMed] [Google Scholar]
- 14. Ikeuchi M, Tsutsui H, Shiomi T, et al. Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc Res 2004; 64: 526–535. [DOI] [PubMed] [Google Scholar]
- 15. Bjornstad JL, Skrbic B, Marstein HS, et al. Inhibition of SMAD2 phosphorylation preserves cardiac function during pressure overload. Cardiovasc Res 2012; 93: 100–110. [DOI] [PubMed] [Google Scholar]
- 16. Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res 2010; 106: 1675–1680. [DOI] [PubMed] [Google Scholar]
- 17. Huang XR, Chung AC, Yang F, et al. Smad3 mediates cardiac inflammation and fibrosis in angiotensin II-induced hypertensive cardiac remodeling. Hypertension. 2010; 55: 1165–1171. [DOI] [PubMed] [Google Scholar]
- 18. Rosin NL, Falkenham A, Sopel MJ, et al. Regulation and role of connective tissue growth factor in AngII-induced myocardial fibrosis. Am J Pathol 2013; 182: 714–726. [DOI] [PubMed] [Google Scholar]
- 19. Fan D, Takawale A, Lee J, et al. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012; 5: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res 2009; 105: 1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Falkenham A, de Antueno R, Rosin N, et al. Nonclassical resident macrophages are important determinants in the development of myocardial fibrosis. Am J Pathol 2015; 185: 927–942. [DOI] [PubMed] [Google Scholar]
- 22. Rosin NL, Sopel M, Falkenham A, et al. Myocardial migration by fibroblast progenitor cells is blood pressure dependent in a model of angII myocardial fibrosis. Hypertens Res 2012; 35: 449–456. [DOI] [PubMed] [Google Scholar]
- 23. Sopel MJ, Rosin NL, Lee TD, et al. Myocardial fibrosis in response to Angiotensin II is preceded by the recruitment of mesenchymal progenitor cells. Lab Invest 2011; 91: 565–578. [DOI] [PubMed] [Google Scholar]
- 24. Zwaagstra JC, Sulea T, Baardsnes J, et al. Engineering and therapeutic application of single-chain bivalent TGF-beta family traps. Mol Cancer Ther 2012; 11: 1477–1487. [DOI] [PubMed] [Google Scholar]
- 25. Ruperez M, Lorenzo O, Blanco-Colio LM, et al. Connective tissue growth factor is a mediator of angiotensin II-induced fibrosis. Circulation 2003; 108: 1499–1505. [DOI] [PubMed] [Google Scholar]
- 26. Wei LH, Huang XR, Zhang Y, et al. Smad7 inhibits angiotensin II-induced hypertensive cardiac remodelling. Cardiovasc Res 2013; 99: 665–673. [DOI] [PubMed] [Google Scholar]
- 27. Annes JP. Making sense of latent TGFbeta activation. J Cell Sci 2003; 116: 217–224. [DOI] [PubMed] [Google Scholar]
- 28. Shi Y, Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003; 113: 685–700. [DOI] [PubMed] [Google Scholar]
- 29. Chen MM, Lam A, Abraham JA, et al. CTGF expression is induced by TGF-beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol 2000; 32: 1805–1819. [DOI] [PubMed] [Google Scholar]
- 30. Chujo S, Shirasaki F, Kawara S, et al. Connective tissue growth factor causes persistent proalpha2(I) collagen gene expression induced by transforming growth factor-beta in a mouse fibrosis model. J Cell Physiol 2005; 203: 447–456. [DOI] [PubMed] [Google Scholar]
- 31. Gravning J, Ahmed MS, von Lueder TG, et al. CCN2/CTGF attenuates myocardial hypertrophy and cardiac dysfunction upon chronic pressure-overload. Int J Cardiol 2013; 168: 2049–2056. [DOI] [PubMed] [Google Scholar]
- 32. Yang F, Chung AC, Huang XR, et al. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension 2009; 54: 877–884. [DOI] [PubMed] [Google Scholar]
- 33. Lipson KE, Wong C, Teng Y, et al. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair. 2012; 5: S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen MM, Lam A, Abraham JA, et al. CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol 2000; 32: 1805–1819. [DOI] [PubMed] [Google Scholar]
- 35. Ahmed MS, Oie E, Vinge LE, et al. Connective tissue growth factor–a novel mediator of angiotensin II-stimulated cardiac fibroblast activation in heart failure in rats. J Mol Cell Cardiol 2004; 36: 393–404. [DOI] [PubMed] [Google Scholar]
- 36. Accornero F, van Berlo JH, Correll RN, et al. Genetic analysis of connective tissue growth factor as an effector of transforming growth factor beta signaling and cardiac remodeling. Mol Cell Biol 2015; 35: 2154–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fontes MS, Kessler EL, van Stuijvenberg L, et al. CTGF knockout does not affect cardiac hypertrophy and fibrosis formation upon chronic pressure overload. J Mol Cell Cardiol 2015; 88: 82–90. [DOI] [PubMed] [Google Scholar]
- 38. Panek AN, Posch MG, Alenina N, et al. Connective tissue growth factor overexpression in cardiomyocytes promotes cardiac hypertrophy and protection against pressure overload. PLoS One. 2009; 4: e6743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Iwanciw D, Rehm M, Porst M, et al. Induction of connective tissue growth factor by angiotensin II: integration of signaling pathways. Arterioscler Thromb Vasc Biol 2003; 23: 1782–1787. [DOI] [PubMed] [Google Scholar]
- 40. Fickenberg P, Inkinen K, Ahonen J, et al. Angiotensin II induces connective tissue growth factor gene expression via calcineurin-dependent pathways. Am J Pathol 2003; 163: 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ruperez M, Rodrigues-Diez R, Blanco-Colio LM, et al. HMG-CoA reductase inhibitors decrease angiotensin II-induced vascular fibrosis: role of RhoA/ROCK and MAPK pathways. Hypertension 2007; 50: 377–383. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, jraas-supp._Fig_1 for Connective tissue growth factor expression after angiotensin II exposure is dependent on transforming growth factor-β signaling via the canonical Smad-dependent pathway in hypertensive induced myocardial fibrosis by Chloe Kok Sum Wong, Alec Falkenham, Tanya Myers and Jean-Francois Légaré in Journal of the Renin-Angiotensin-Aldosterone System
Supplemental material, jraas-supp._Fig_2 for Connective tissue growth factor expression after angiotensin II exposure is dependent on transforming growth factor-β signaling via the canonical Smad-dependent pathway in hypertensive induced myocardial fibrosis by Chloe Kok Sum Wong, Alec Falkenham, Tanya Myers and Jean-Francois Légaré in Journal of the Renin-Angiotensin-Aldosterone System
Supplemental material, jraas-_Supp.Fig_3 for Connective tissue growth factor expression after angiotensin II exposure is dependent on transforming growth factor-β signaling via the canonical Smad-dependent pathway in hypertensive induced myocardial fibrosis by Chloe Kok Sum Wong, Alec Falkenham, Tanya Myers and Jean-Francois Légaré in Journal of the Renin-Angiotensin-Aldosterone System