


Abstract
High-throughput sequencing data are increasingly being made available to the research community for secondary analyses, providing new opportunities for large-scale association studies. However, heterogeneity in target capture and sequencing technologies often introduce strong technological stratification biases that overwhelm subtle signals of association in studies of complex traits. Here, we introduce the Cross-Platform Association Toolkit, XPAT, which provides a suite of tools designed to support and conduct large-scale association studies with heterogeneous sequencing datasets. XPAT includes tools to support cross-platform aware variant calling, quality control filtering, gene-based association testing and rare variant effect size estimation. To evaluate the performance of XPAT, we conducted case-control association studies for three diseases, including 783 breast cancer cases, 272 ovarian cancer cases, 205 Crohn disease cases and 3507 shared controls (including 1722 females) using sequencing data from multiple sources. XPAT greatly reduced Type I error inflation in the case-control analyses, while replicating many previously identified disease–gene associations. We also show that association tests conducted with XPAT using cross-platform data have comparable performance to tests using matched platform data. XPAT enables new association studies that combine existing sequencing datasets to identify genetic loci associated with common diseases and other complex traits.
INTRODUCTION
The rapid development pace of high-throughput sequencing technology is enabling large-sequencing studies at a scale that was previously only obtainable for genome-wide association studies (GWAS) based on high-density Single Nucleotide Polymorphisms (SNP) arrays (1). The resulting sequencing datasets from these primary studies are increasingly being deposited into public repositories available to the research community. As a result, there is a broad interest in combined association analyses that merge data from multiple studies, which involve a variety of sequencing platforms and laboratory protocols. Meta-analyses, which combine summary statistics from individual association studies, are used regularly in SNP-based GWAS and provide an effective means of controlling for technological heterogeneity between studies (2,3). However, for a variety of reasons, pooled analyses or ‘mega-analyses’ are often inherently preferable in sequence-based association studies, resulting in increased signal for low-frequency and rare variants (4), particularly when combined with rare variant association tests (5). Pooled analyses have been employed extensively in SNP-based GWAS (6–9). However, analysis of cross-platform sequencing data is far more challenging than for SNP-based GWAS due to technological stratification biases caused by differences in sample preparation, target capture, sequencing platforms and various bioinformatics pipelines. Although pooled cross-platform sequence-based association studies have been conducted (10,11), due to technological stratification, these studies have required balanced case and control proportions across each sequencing platform and laboratory. Consequently, such studies are generally unable to incorporate datasets that have been sequenced outside of the context of a case-control study specific to the disease of interest. This limitation is particularly consequential given the inability to leverage public repositories to increase control sample sizes and thus statistical power. The lack of effective tools to control for cross-platform heterogeneity have therefore greatly limited the impact of cross-platform sequencing association studies.
To address the need for improved support for cross-platform sequencing studies, we have developed the Cross-Platform Association Toolkit (XPAT) (Figure 1; Supplementary Figures S1 and 2). XPAT includes a suite of tools designed to support and conduct sequencing association studies involving technologically heterogeneous datasets. We demonstrate the utility of XPAT by conducting whole-exome case–control association studies in breast and ovarian cancer involving 272 ovarian cancer cases, 873 breast cancer cases and 1722 female controls. We consider the performance of XPAT using single marker tests and four gene-based tests, SKAT-O, variable threshold (VT), weighted sum statistic (WSS) and VAAST 2, evaluating control of Type I error and statistical power to detect known associations. To demonstrate the applicability of XPAT to other human diseases, we also conducted an additional sequence-based case-control analysis involving 205 Crohn Disease cases and 3507 controls.
Figure 1.

Diagram of the components of XPAT. The four modules in XPAT are shown in blue boxes. The input, output and intermediate files are shown in black.
MATERIALS AND METHODS
XPAT is comprised of four major modules designed to support and conduct cross-platform sequencing studies (Figure 1). The features supported include data alignment and variant calling, quality control (QC), association testing, and rare variant effect size estimation. The inputs, outputs and runtime of XPAT’s components are summarized in Figure 1; Supplementary Tables S1 and 2. Users specify the platform for each sample using a PED input file; platform is then treated as a categorical variable by XPAT throughout the analysis. Here, platform can refer to any detail of sample processing which could produce technological stratification bias, which includes but is not limited to input DNA type or quality, target capture designs or protocols, sequencing technologies or protocols, experiment batches and other laboratory protocols.
Sequencing reads alignment and variant genotype calling
The data alignment and variant calling module in XPAT provides automated parallel computing workflows that interface with the Burrows-Wheeler Aligner (BWA) (12) (v0.7.9a), the Genome Analysis Toolkit (GATK) (13) (v3.3), and other tools to filter low quality sequencing reads and to align cleaned reads to a reference genome (14,15). For each individual, XPAT uses BWA to align the sequencing reads onto a reference genome, generating a SAM file for each individual. XPAT uses Picard tools (http://broadinstitute.github.io/picard) to clean SAM files by soft-clipping beyond-end-of-reference alignments and setting MAPQ to 0 for unmapped reads. XPAT calls Samtools (16) to convert the cleaned SAM files into BAM files and sorts the BAM files. XPAT calls Picard's MarkDuplicates module to de-duplicate redundant reads from PCR amplification or non-random genome fragmentation. XPAT performs local realignment, minimizing the number of mismatched bases across reads using GATK’s IndelRealigner module. XPAT recalibrates the base score quality using GATK’s BaseRecalibrator module to generate the final clean BAM file for each individual for genotype calling.
XPAT calls GATK HaplotypeCaller to conduct variant genotyping, either by calling each sample individually or jointly calling all samples together. Joint variant genotyping is particularly beneficial in cross-platform association studies, as demonstrated in other cohort and pedigree studies (17,18) and our analyses (Supplementary Figures S3 and 4). To optimize the runtime speed of joint calling, XPAT combines per-sample gVCF files produced by HaplotypeCaller into a multi-sample gVCF file using CombineGVCFs by cohort and then performs joint genotype calling on the combined gVCF files. To conduct cross-platform aware variant genotyping, XPAT interfaces with GATK to perform Variant Quality Score Recalibration (VQSR) with variants located in the union (default) or intersection of genomic regions targeted by each platform.
XPAT’s QC metrics
The automated QC procedures in XPAT involve a series of analyses to identify and filter problematic samples and variants due to cross-platform biases. For sample level QC, XPAT infers gender information based on the ratio of the homozygote and heterozygote counts on chromosome X for each individual and reports possible misidentification of gender. XPAT also identifies low quality samples based primarily on NC90 scores. NC90 is a sample-level platform-specific missing genotype rate, defined as the proportion of missing genotypes in a sample among all variants with call rates of 0.9 or greater among all samples from that platform. By default, XPAT excludes individuals with NC90 > 25%. For variant level QC, XPAT provides platform-aware QC metrics to control for cross-platform biases. XPAT will mask a variant if it meets any of the following criteria: (i) VQSR tranche sensitivity score >99.9 for SNP and >98.0 for INDEL, (ii) genotype quality score <5, (iii) fraction of reads supporting minor alleles <20%, (iv) P < 10−6 in a Hardy-Weinberg equilibrium test (19) in the control population, (v) platform-wide missing-genotype rate >10%, (vi) P < 0.05 in a differential missing-genotype rate test across platforms and (vii) P < 0.05 in a differential allele frequency test across platforms. The cross-platform missing-genotype rate and differential allele frequency tests are conducted using χ2 tests. XPAT conducts missing-genotype rate across all platforms involved in the study including both cases and controls. XPAT conducts differential allele frequency tests across case platforms or across control platforms, separately, avoiding comparisons between cases and controls. For quantitative traits, XPAT first defines groups by quantiles of the trait of interest and then conducts within group cross-platform tests.
Population and technological stratification
The detection and characterization of population and technological stratification in XPAT relies on two consecutive steps of Principle Component Analysis (PCA) (20): an external step and an internal step. XPAT first performs an external PCA to select cases and controls with matched genetic ancestry, a procedure termed ‘PCA projection’ (21,22). XPAT uses the 1000 Genomes project (phase3-20130502) (23) as the default reference panel, including 427 samples from 8 population groups (i.e. CEU, CHS, KHV, LWK, PEL, PJL, PUR and YRI). XPAT constructs a reference Principal Component (PC) space with common variants (Minor Allele Frequency, MAF > 20%) from the reference panel, and then projects cases and controls onto the PC space. This PCA enables the identification and exclusion of population outliers that are poorly matched to the case and control groups with respect to genetic ancestry. In our study, we manually selected samples clustering with CEU from the 1000 Genomes project, according to the results from the external PCA. XPAT then excludes the external reference panel, conducting an internal PCA with selected cases and controls, using well-behaved markers that are selected based on the allele frequency in an external reference database. By default, XPAT includes variants with MAF > 10% in non-Finnish Europeans reported in Exome Aggregation Consortium (ExAC) (24). XPAT performs linkage disequilibrium pruning to obtain a set of unlinked markers (i.e. approximately statistically independent) markers with r2 < 0.2 to conduct the PCA, which is an approach that has been employed frequently in previous studies (25,26). The internal PCA is more sensitive to population and technological stratification compared the external PCA, enabling better control of residual cross-platform biases or sub-population stratification. We included the first two PCs from the internal PCA as covariates in each association test. Note that the allele frequency filters described above apply only to the PCA steps; lower allele frequency filters are typically applied to gene-based association tests, as described below.
Gene-based association tests
After completing QC and calculating PCs, XPAT can conduct one or more association tests throughout the sequenced regions. Currently, XPAT supports single marker tests using linear and logistic regression as well as 29 rare variant association tests. In this study, we evaluated VAAST 2 (27), SKAT-O (28), VT (29) and WSS (30) (full list in Supplementary Table S3). One of the most common applications of a rare variant association test is a gene-based test of rare, protein-coding variants. To support this application, XPAT annotates all variants using the variant annotation tool (VAT) in VAAST 2 to identify variants that may impact protein function. By default, XPAT tests all missense, nonsense, splicing, and coding INDEL variants in each gene (in VAAST 2, rare variants with sufficiently low conservation-controlled amino acid substitution matrix scores (CASM scores) may also be excluded from the test, as previously described (27)). XPAT calculates P-values using a covariate adjusted permutation test (31), which enables the analysis to control for PCs and other covariate information, even if the test statistic cannot incorporate covariates (e.g. WSS and VAAST 2). XPAT also supports analytical calculation of P-values for tests that can incorporate covariates without a permutation test (e.g. SKAT-O). Optionally, XPAT can incorporate variant prioritization scores in association tests that support variant weighting. In XPAT, the default variant prioritization method in VAAST 2 analysis is CASM (27). For SKAT-O (32) and VT (29), XPAT can use either PolyPhen-2 (PP2) (33) or transformed CASM scores (34) as weights. XPAT uses the R package ‘SKAT’ to implement SKAT-O. XPAT integrates VT Test Software (http://genetics.bwh.harvard.edu/rare_variants/) to implement VT. XPAT uses the R package ‘AssotesteR’ (https://cran.r-project.org/web/packages/AssotesteR/index.html) to implement WSS and 24 other test statistics. XPAT uses the software ‘RAML’ (http://ccge.medschl.cam.ac.uk/software/raml/) to implement the rare admixture maximum likelihood test (35). In our study, we conducted gene-based association tests using rare variant with MAF < 0.5% in each case-control dataset.
XPAT uses a permutation-based test for genes with multiple isoforms, called the Multiple Gene Isoform Test (MGIT), which can be applied using any gene-based association test (Supplementary Figure S2). MGIT simultaneously tests all known isoforms in a gene and conducts a permutation procedure to calculate a single gene-level P-value. MGIT first calculates the test statistic, Si (defined by the gene-based test statistic), for each transcript Ti of a given gene using any of the 30 supported rare variant association tests. The significance level of Si, denoted as Pi, is assessed with individual permutation tests, using BiasedUrn sampling (31), which incorporates the covariate matrix from the internal PCA. During permutation, MGIT calculates statistics sij, where i indexes the transcript and j indexes the permutation, then transforms sij into its empirical p value pij. For the jth permutation, MGIT selects the minimum pij among all transcripts of a given gene, denoted as min-pj. The minimum Pi among all transcripts is compared with the distribution of min-pj, to generate the overall gene-level p value. All XPAT gene-based results from this study were generated using MGIT, except where specifically noted.
Odds ratio estimation
Once a significant gene association is identified, XPAT can estimate odds ratios (ORs) for particular classes of variants in a gene, including likely gene disrupting (LGD) variants (e.g. stop gain, splice donor/acceptor and frame shift INDELs), missense variants, rare variants with a specified MAF, damaging variants (as measured by amino acid substitution (AAS) score (27), PP2, or SIFT (36)) and variants belonging to specific functional domains (according to annotation from InterPro database (37)). XPAT conducts variant annotation using the refGene, ljb23_pp2hvar, ljb23_sift and dbnsfp31a_interpro (for protein domain information) databases from ANNOVAR (38) (version: 2015Nov02). XPAT calculates ORs using logistic regression, accounting for population stratification and cross-platform biases by incorporating PCs and other potential covariates, using the following formula:
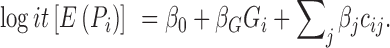 |
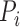 is an indicator variable for the phenotype (disease affected status) of an individual i, with 0 for control and 1 for case.
is an indicator variable for the phenotype (disease affected status) of an individual i, with 0 for control and 1 for case. 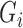 indicates whether individual i carries at least one variant of interest (Gi = 1) or not (Gi = 0).
indicates whether individual i carries at least one variant of interest (Gi = 1) or not (Gi = 0). 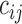 is the j-th covariate within the i-th individual.
is the j-th covariate within the i-th individual. 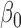 is the intercept;
is the intercept; 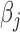 is the coefficient of the j-th covariate;
is the coefficient of the j-th covariate; 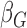 is the coefficient for the genetic variant. The OR is calculated as the exponential of the estimated
is the coefficient for the genetic variant. The OR is calculated as the exponential of the estimated 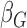 . For functional categories with zero variant counts in either cases or controls, XPAT uses a Fisher's exact test to estimate ORs and confidence intervals. In our study, we estimated ORs using rare variant with MAF < 0.5% in each case-control dataset.
. For functional categories with zero variant counts in either cases or controls, XPAT uses a Fisher's exact test to estimate ORs and confidence intervals. In our study, we estimated ORs using rare variant with MAF < 0.5% in each case-control dataset.
Benchmark QC
We compared the XPAT’s QC with benchmark QC metrics adopted from a recent whole-exome association study (10). Both QC metrics were applied to the genotypes generated from the same jointly called multi-sample VCF files. The benchmark QC metrics included the following variant level QC filters: (i) VQSR tranche scores >99.75 for SNPs and >99.50 for INDELs, (ii) genotype quality scores <30 for SNPs and <90 for INDELs, (iii) Fewer than 20 or 25% of reads supporting the minor allele for SNPs and INDELs, respectively (iv) alternate allele read depth <2 for both SNPs and INDELs, (v) study-wide missing-genotype call rates >20%, (vi) P < 10−8 in a Hardy-Weinberg equilibrium test within the control population (19), (vii) locating within low-complexity regions predicted by mdust (http://compbio.dfci.harvard.edu/tgi/) and (viii) INDELs with more than two alternate alleles or within three base pairs of another INDEL. A detailed comparison between the XPAT and benchmark QC procedures is provided in Supplementary Table S4.
Ovarian and breast cancer analyses
We downloaded the whole-exome sequencing data of 395 samples with ovarian serous cystadenocarcinoma (39,40) and 1100 samples with breast invasive carcinoma (41) from the Cancer Genome Atlas (TCGA) project through the Cancer Genomics Hub (https://cghub.ucsc.edu/). We downloaded the whole-exome sequencing data of a shared control set of 4677 samples in the Simons Simplex Collection (42) from the National Database for Autism Research (NDAR) (43). All samples were sequenced from blood-derived normal samples. We excluded all self-reported Hispanic or Latino samples, and related individuals reported by KING (44). Using PCA in XPAT, we selected female cases and controls of European ancestry, including 272 ovarian cancer cases, 873 breast cancer case and 1722 controls unaffected mothers of offspring with Autism Spectrum Disorder (ASD) from NDAR. We conducted read alignment, variant calling and QC using XPAT with default parameters.
Crohn disease analysis
We designed a target sequencing panel, consisting of 101 genes in genomic regions with established associations with Crohn disease from GWAS. We used Agilent SureDesign to design probes for targeted enrichment using the HaloPlex Target Enrichment System. The targeted regions constituted 1076 targets and 17 912 amplicons for a total of 497.3 kb of coding sequence. We sequenced 205 cases with early disease onset (≤18 years). DNA was obtained from whole blood using standard procedures. Target sequencing libraries were constructed per the manufacturer’s protocol, and sequenced using an Illumina HiSeq 2500 with a depth of 300× on average. We conducted read alignment, variant calling and QC using the procedures described above for the TCGA data, with two exceptions: variants outside of the targeted region were excluded (capture intersection), and VQSR filtering was not applied to INDELs due to their limited number. We used 3507 NDAR samples of European ancestry as controls in this case-control analysis, and performed gene-based tests for the genes in target regions with VAAST2-MGIT using the first two internal PCs as covariates.
RESULTS
We evaluated the performance of XPAT by conducting two whole-exome case-control analyses involving 272 women with ovarian cancer and 783 women with breast cancer, and a shared control set of 1722 women. The controls from NDAR were unaffected mothers of offspring with ASD. Other than ASD status, phenotype information was unavailable for the controls and thus a small proportion may have been misclassified, although the potential misclassification bias would have only a modest effect on power and effect size estimates (45). The cross-platform heterogeneity within and between datasets includes differences in sequence centers, target capture designs and sequencing coverages (Supplementary Table S5).
We performed sequencing reads alignment, joint variant calling, QC and case–control association testing using the XPAT workflow described in Figure 1. For each cancer type, we conducted single marker logistic regression analysis and four rare variant association tests, both with and without variant prioritization information. For comparison, we mirrored each XPAT case-control analysis with an analysis based on QC metrics and procedures from a recent whole-exome association study (10). In this study, the authors conducted a case-control mega-analysis using exome sequencing data involving multiple capture technologies, sequencing platforms and laboratories. The study applied stringent thresholds for QC, balancing sensitivity and specificity by training on several datasets (10). The association results exhibited no inflation or deflation of Type I error, with the caveat that the numbers of cases and controls were balanced in each platform. The QC metrics used in this study (see ‘Materials and Methods’ section) provide a benchmark that is representative of state-of-the-art cross-platform sequencing association studies.
The quantile–quantile (Q–Q) plots in Figure 2 demonstrate that the association results generated with benchmark QC metrics were characterized by high levels of Type I error inflation. For example, with an α level of 0.001, the number of significant genes exceeded the expected number under the null hypothesis by between 3- and 26-fold (Figure 3 and Supplementary Table S6). In comparison, the association results generated with XPAT were largely consistent with theoretical expectations under the null (red dots in Figure 2 and Supplementary Figure S5). XPAT reduced the proportion of significant associations in all association tests for both ovarian and breast cancer at α levels of 0.001, 0.01 and 0.05, and the proportions of significant associations were substantially closer to the nominal α levels. We then compared association test results using internal PCs or external PCs as covariates, observing inconsistent control of Type I error using external PCs in contrast to stable performance with internal PCs (Supplementary Figures S6 and 7), suggesting that internal PCAs are more sensitive to technological stratification. We also extended covariates to the first ten internal PCs and observed equivalent control of Type I error (see Supplementary Figure S7E and F).
Figure 2.

Q–Q plots of observed versus expected gene-based P-values in VAAST 2 for genes with eight or more tested variants. Cases and controls include (A) 272 ovarian cancer cases and 1722 shared controls, and (B) 783 breast cancer cases and 1722 shared controls. Blue dots: benchmark QC (see ‘Materials and Methods’ section). Red dots: XPAT. The gray band represents a 95% (pointwise) confidence region. The Q–Q plots were generated with R package ‘Haplin’.
Figure 3.

Observed proportion of significant associations at different α levels. We conducted association tests for ovarian (A–C) and breast (D–F) cancer, using eight association tests supported in XPAT. We calculated the proportions of significant associations at α levels of 0.001, 0.01 and 0.05 (dashed lines in each sub panel), and compared the performance of XPAT’s QC metrics versus benchmark QC metrics for each method and dataset.
To evaluate XPAT’s influence on statistical power, we examined the difference in P-values and genome-wide rankings of 16 well-established breast and ovarian cancer susceptibility genes (Supplementary Table S7) for association results generated with and without XPAT. Established susceptibility gene associations that replicated at P < 0.05 in one or more tests include BRCA1 (46), BRCA2 (46), RAD51C (47), RAD51D (48) and BRIP1 (49) in ovarian cancer, and BRCA1 (46), BRCA2 (46), RAD51B (50), CHEK2 (51) and ATM (52) in breast cancer (Figure 4 and Supplementary Table S7). In general, tests that incorporated variant prioritization weights (VAAST 2, SKAT-O with PP2, and VT with PP2) exhibited higher power than those without variant prioritization (VAAST 2 without CASM, SKAT-O, VT and WSS). In the ovarian cancer analysis, all tests replicated the association between ovarian cancer and BRCA1, which reached exome-wide significance (P = 2.4 × 10−6) in VAAST 2. PALB2, which has been proposed as an ovarian cancer susceptibility gene (53), showed nominal association (P = 0.044) with ovarian cancer risk in our VAAST 2 analysis (Supplementary Table S7).
Figure 4.

Observed gene-based P-values for known cancer–gene associations. The heatmap depicts the P-values of known cancer–gene associations in ovarian cancer and breast cancer for genes with P < 0.05 in one or more association tests.
We then compared our cross-platform XPAT results with platform-matched ovarian cancer case-control sequencing datasets with 1089 ovarian cases and 1133 controls from the studies of Epidemiology and Risk Factors in Cancer Heredity (SEARCH) (50,54) to assess whether the additional QC in XPAT resulted in a loss of statistical power. These datasets provide high coverage case-control sequencing data for four established ovarian cancer susceptibility genes, BRCA1, BRCA2, RAD51C and RAD51D (50,54). We performed variant calling and QC following the procedures in their original papers. To ensure that sample sizes were identical in the cross-platform and matched platform datasets, we repeatedly sampled 250 cases and 1000 controls with replacement to generate 1000 bootstrap replicates. We then conducted association tests using VAAST 2 on each bootstrap replicate. We estimated statistical power from the proportion of significant tests among all 1000 bootstrapped datasets for a given α level. As shown in Figure 5, the power from these two datasets was comparable for α levels between 1 × 10−5 and 0.1, demonstrating that the additional QC in XPAT did not substantially compromise power. The P-value distributions and OR estimates from the two datasets were also highly similar (Supplementary Figures S5 and 8).
Figure 5.

Power estimation for association tests using XPAT. The lines depict the power comparisons with VAAST 2 analysis with XPAT using TCGA ovarian cancer cases and NDAR controls (solid lines) and using platform-matched ovarian cancer cases and controls (dashed lines), for four genes: BRCA1 (red), BRCA2 (orange), RAD51D (blue), RAD51C (green). The x-axis shows the α level and the y-axis shows the statistical power. The power was calculated based on 1000 bootstraps. For each bootstrap, we sampled 250 cases and 1000 controls with replacement from each dataset.
To investigate the contribution of different functional categories of variants to cancer risk in specific genes, we conducted association tests with a subset of the variants by excluding all LGD variants for TCGA datasets. Notably, though we observed attenuation of the signal after excluding LGD variants, five gene–cancer associations remained nominally significant (P < 0.05): BRCA1, BRCA2, RAD51B and CHEK2 in breast cancer and BRCA1 in ovarian cancer, (Figure 4 and Supplementary Table S8). We then applied XPAT to estimate ORs for various functional categories of rare variants in each of these genes (Figure 6). We used XPAT to investigate effect sizes of the following four types of variants: LGD variants, rare (MAF < 0.5%) predicted non-damaging missense (CASM score < 2) variants, rare predicted damaging missense variants (CASM score ≥ 2) and rare damaging missense variants belonging to functional domains. The OR estimates for LGD variants in BRCA1, BRCA2 and RAD51B for breast cancer were 5.50 (Confidence Intervals, CI: 1.01 to 29.76), 5.27 (CI: 1.76 to 15.80) and ∞ (CI: 0.41 to ∞), respectively, supporting the findings from previous studies (40,55,56) (Supplementary Table S9).
Figure 6.

OR estimates for ovarian cancer susceptibility genes. We estimated the ORs with TCGA-NDAR data and platform-matched data. Dotted lines indicate null value (OR = 1.0). Each sub panel contains the OR estimates for four categories of variants on cancer risk: LGD (black), missense and non-damaging (green), missense and damaging (blue), and missense and damaging and domain region variants (red). For variant categories with zero counts in either cases or controls, we estimated the OR using a Fisher’s exact test (indicated by star).
Alternative splicing can generate multiple transcripts of the same gene, which code protein products with various functions. In the transcript-based association tests, we observed that the effect sizes of missense variants from two small BRCA1 transcripts were larger than those from the three large transcripts, especially for missense variants predicted to be damaging (Supplementary Table S10). We also observed that rare missense variants in the RING and BRCT protein domains of BRCA1 exhibited effect sizes comparable with LGD variants, although with wide confidence intervals (OR = 16.30, 95% C.I, 1.47 to 180.97 for ovarian cancer and OR = 11.70, 95% C.I, 1.34 to 102.10 for breast cancer) (Supplementary Table S11).
To demonstrate the applicability of XPAT to other human diseases, we conducted an additional sequence-based case-control analysis involving 205 Crohn Disease cases and 3507 controls from NDAR. The case data were generated from a targeted sequencing panel of 101 genes near Crohn disease susceptibility loci previously identified in GWAS, while the control dataset was generated from whole-exome capture and sequencing. XPAT was able to successfully eliminate Type I error rate inflation in this study, while replicating a well-established Crohn Disease susceptibility gene, NOD2 (57,58), with P = 1.0 × 10−6 (Supplementary Figure S9).
DISCUSSION
Given the sample sizes typically needed to identify new rare variant associations, the appeal of mega-analysis efforts that combine sequencing data from multiple sources is clear. However, in practice, the increase in power gained by merging sequencing datasets can easily be overwhelmed by the increase in false positives resulting from batch effects between datasets. In this study, we consistently observed major batch effect biases when using QC metrics and procedures typically applied in sequencing association studies, regardless of the test statistic used (blue lines in Figure 2 and Supplementary Figure S5). XPAT addresses this problem with a highly automated platform aware QC pipeline designed to identify and control for cross-platform biases, with support for parallelized sequencing data alignment, cross-platform aware joint variant genotype calling, case–control association testing and gene-based effect size estimation. The inputs and outputs of each module in XPAT are summarized in Figure 1 and Supplementary Table S1. After applying XPAT, we observed very little evidence of cross-platform bias with any of the tests we evaluated (VAAST 2, SKAT-O, VT, WSS and single marker logistic regression, see Figures 2 and 3; Supplementary Figure S5).
Throughout our analyses, successful control of Type I error relied critically on jointly called genotypes. With individually called genotypes, we observed near complete separation of cases and controls in the internal PCA (Supplementary Figures S3 and 4). This separation is indicative of high levels of technological stratification, and is in sharp contrast to the internal PCAs generated from jointly called genotypes (Supplementary Figure S6). We attribute the reduction in technological stratification resulting from joint calling to two major factors: first, because joint calling improves variant detection sensitivity and specificity (18), the approach can reduce differences in variant sensitivity and specificity between platforms. Second, joint calling produces missing genotype data for all variants detected by any platform, which can then be used to identify variants with systematic biases between platforms. In some situations, it may be logistically difficult or computationally infeasible to obtain and process all raw sequencing data. If gVCF files are available for each sample, an alternative approach is to conduct joint calling directly from the initial gVCF files, skipping the data realignment step. This approach is only feasible if the gVCF files were generated using the same genome reference version, and the process may introduce additional cross-platform artifacts due to potential parameter differences in the GATK pipeline. However, when appropriate, joint calling from pre-generated gVCF files results in a one-to-two order of magnitude reduction in runtime. Supplementary Table S2 estimates the runtime, in CPU hours, for each step in XPAT, with sample sizes ranging from 1000 to 100,000 samples. In all cases, the QC, association testing and effect size estimation procedures should consume only a fraction of the CPU hours required for data alignment and variant calling. All results presented in this study were generated from jointly-called genotypes, except where specifically noted.
To characterize the behavior of the individual QC steps in XPAT, we compared the numbers of variants masked by each of XPAT’s QC criteria and the benchmark QC criteria used by Singh et al. (10) (Supplementary Figure S10). Although XPAT was far more effective at eliminating cross-platform artifacts, XPAT actually filtered out far fewer SNVs than the benchmark QC pipeline, due to the relatively relaxed VQSR thresholds for SNVs (99.9 in XPAT versus 99.75 in the benchmark). In total, 689,844 and 956,437 coding SNVs passed all QC steps in XPAT compared to 244,843 and 252,251 SNVs with benchmark QC, for ovarian cancer and breast cancer, respectively. These observations suggest that, for many heterogeneous sequencing studies, cross-platform biases cannot be avoided by restricting the analysis to high confidence variants based solely on VQSR or other variant quality metrics. However, by incorporating the cross-platform QC metrics used in XPAT (e.g. differential missing genotype rate test and differential allele frequency test among platforms), most cross-platform biases can be eliminated, even with relatively relaxed baseline SNV QC metrics. In contrast, the VQSR threshold for INDELs was more stringent in XPAT compared to the benchmark pipeline (98.0 versus 99.5), which was necessary to fully control for Type I error inflation (Supplementary Figure S11). However, due to using relaxed thresholds for INDEL genotype quality, sequencing depth and allelic balance, XPAT still filtered out fewer INDELs than the benchmark QC pipeline. In total, 10,594 and 14,414 coding INDELs passed all QC steps in XPAT compared to 4,547 and 4,570 INDELs with the benchmark QC pipeline, for ovarian cancer and breast cancer, respectively. The additional INDELs and SNVs included in the XPAT pipeline resulted in a 25% to 170% increase in the number of testable genes (Supplementary Figure S12).
For cross-platform association studies involving data generated from DNA target capture, an additional QC consideration involves the potential pre-filtering of variants detected in regions of the genome that fall outside of the designed capture regions. During the development of XPAT, we evaluated three approaches to capture-aware QC: (i) inclusion of all detected variants (capture naïve), (ii) inclusion of variants covered by the capture design of every platform (capture intersection) and (iii) inclusion of variants covered by the capture design of any platform (capture union). With capture naïve filtering, we were unable to fully control for Type I error with XPAT QC. In contrast, capture intersection and union filtering both exhibited no signs of Type I error inflation with XPAT QC. However, capture intersection filtering was overly conservative, detecting 381,231 and 531,102 fewer coding variants with XPAT QC relative to the capture union filtering, for ovarian and breast cancer, respectively. This reduction in variant detection sensitivity resulted in a considerable loss of signal for known breast and ovarian cancer susceptibility genes, as shown in Supplementary Table S12. Based on these observations, we employed capture union filtering throughout this study. Capture union filtering is also the default option in XPAT, although all three options are supported. During the variant recalibration QC step, XPAT will only include variants that pass the specified capture filter. The inclusion of variants in regions that were unintentionally sequenced may initially seem counterintuitive. However, the majority of variants (98%) that passed XPAT QC and were present in the capture union but absent in the capture intersection were located within 100 bp of a capture region in every sequencing platform. These variants could be expected to achieve reliable coverage depth, assuming typical read lengths and DNA fragment sizes.
Several factors will influence the number of variants that pass cross-platform QC metrics, including the number of platforms included in the analysis, and differences in exome capture design and data quality within each platform. In general, platforms with lower data quality will have a disproportionate influence on the number of variants that fail QC. In this study, variants lost due to cross-platform QC did not noticeably decrease the power to detect known ovarian cancer–gene associations, based on comparisons with matched platform case-control data for BRCA1, BRCA2, RAD51C and RAD51D (Figure 5). We expect similar performance on other whole-exome sequencing datasets, but our findings are not fully generalizable given the dependence on platform data quality. In each new study, we recommend careful assessment of the data quality of each platform and the proportion of variants that fail QC using the reports generated by XPAT.
In evaluating the power to replicate known cancer–gene associations, we observed that tests which incorporated variant prioritization weights (i.e. VAAST 2, SKAT-O + PP2 and VT + PP2) generally outperformed their alternative versions without variant prioritization (i.e. VAAST 2 without CASM scores, SKAT-O and VT) (Figure 4). This result is consistent with previous work, which has shown that variant prioritization typically increases power to identify rare variant disease associations in sequencing association studies (27,59,60). Among the tests we evaluated, VAAST 2 had the highest power to replicate established cancer–gene associations (27). In the VAAST 2 breast cancer results, we observed nominally significant gene–cancer associations (BRCA1, BRCA2, RAD51B and CHEK2) when only considering missense variants (Figure 4). This result adds to the growing evidence of a substantial contribution of rare missense variants to cancer susceptibility (50,61).
XPAT incorporates a novel method, MGIT. Gene-based tests typically include variants from all coding exons in a gene, irrespective of gene isoform. For genes with multiple isoforms, this test is often essentially equivalent to a test of the largest isoform in the gene (see Supplementary Table S10). Because smaller isoforms tend to be enriched for the core functional domains of a gene, they may also be enriched for susceptibility variants with larger effect sizes. MGIT employs a permutation approach to test each isoform of a gene, summarizing the contribution of each transcript without the need to explicitly model correlation between transcripts, which can increase statistical power in scenarios where variant effect sizes vary between transcripts. MGIT can be applied in conjunction with any gene-based association test to assess gene level significance; XPAT offers MGIT functionality for each of the 29 gene-based tests included in the toolkit.
The examples presented in this study all involve case–control association analyses with cross-platform whole-exome sequencing data. XPAT can be applied to other sequencing datasets as well, including targeted gene panel and whole genome sequencing data. In addition to case-control study designs, XPAT also supports family-based and quantitative trait analyses. The toolkit also produces variant output in a variety of commonly used data formats, and thus can be easily incorporated into other association test frameworks.
CONCLUSION
XPAT is a toolkit designed to support cross-platform association studies with heterogeneous sequencing datasets. We have demonstrated that XPAT can greatly reduce Type I error inflation resulting from cross-platform biases without reducing power to detect true disease–gene associations. XPAT enables new studies that leverage heterogeneous sequencing datasets from public repositories to search for novel genetic loci associated with common human diseases and other complex traits.
DATA AVAILABILITY
XPAT described in this paper is freely available for academic use and can be downloaded at http://www.hufflab.org/software/xpat/.
Supplementary Material
ACKNOWLEDGEMENTS
An allocation of computer time on the University of Texas MD Anderson Research Computing High Performance Computing (HPC) facility is gratefully acknowledged.
Author contributions: C.H. conceived and directed the study. Y.Y. performed all experiments and developed the software. H.H., F.H., J.C., S.G. and M.Y. tested the software and contributed to the association studies. C.H. and J.F. contributed to the development of the software with the support of P.S. and M.H. Y.Y., R.B. and C.H. wrote the manuscript. All authors read and approved the final manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
US National Institutes of Health [R01 CA195614, R01 GM104390, R01 HG005859, R01 DK091374]. This research was supported by the Andrew Sabin Family Fellowship. RJB was supported by NCI Award Numbers R25CA057730 (PI: Shine Chang, PhD) and CA016672 (PI: Ronald Depinho, MD). Funding for open access charge: University of Texas MD Anderson Cancer Center.
Conflict of interest statement. None declared.
REFERENCES
- 1. Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. et al. . Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010; 42:30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ioannidis J.P., Patsopoulos N.A., Evangelou E.. Heterogeneity in meta-analyses of genome-wide association investigations. PLoS One. 2007; 2:e841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. de Bakker P.I., Ferreira M.A., Jia X., Neale B.M., Raychaudhuri S., Voight B.F.. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum. Mol. Genet. 2008; 17:R122–R128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Evangelou E., Ioannidis J.P.. Meta-analysis methods for genome-wide association studies and beyond. Nat. Rev. Genet. 2013; 14:379–389. [DOI] [PubMed] [Google Scholar]
- 5. Liu L., Sabo A., Neale B.M., Nagaswamy U., Stevens C., Lim E., Bodea C.A., Muzny D., Reid J.G., Banks E. et al. . Analysis of rare, exonic variation amongst subjects with autism spectrum disorders and population controls. PLoS Genet. 2013; 9:e1003443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Major Depressive Disorder Working Group of the Psychiatric, G.C. Ripke S., Wray N.R., Lewis C.M., Hamilton S.P., Weissman M.M., Breen G., Byrne E.M., Blackwood D.H., Boomsma D.I. et al. . A mega-analysis of genome-wide association studies for major depressive disorder. Mol. Psychiatry. 2013; 18:497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pemov A., Sung H., Hyland P.L., Sloan J.L., Ruppert S.L., Baldwin A.M., Boland J.F., Bass S.E., Lee H.J., Jones K.M. et al. . Genetic modifiers of neurofibromatosis type 1-associated cafe-au-lait macule count identified using multi-platform analysis. PLoS Genet. 2014; 10:e1004575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schizophrenia Psychiatric Genome-Wide Association Study, C Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011; 43:969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Olgiati P., Politis A., Albani D., Rodilossi S., Polito L., Ateri E., Zisaki A., Piperi C., Liappas I., Stamouli E. et al. . Association of SORL1 alleles with late-onset Alzheimer's disease. findings from the GIGAS_LOAD study and mega-analysis. Curr. Alzheimer Res. 2012; 9:491–499. [DOI] [PubMed] [Google Scholar]
- 10. Singh T., Kurki M.I., Curtis D., Purcell S.M., Crooks L., McRae J., Suvisaari J., Chheda H., Blackwood D., Breen G. et al. . Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci. 2016; 19:571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khera A.V., Won H.H., Peloso G.M., O’Dushlaine C., Liu D., Stitziel N.O., Natarajan P., Nomura A., Emdin C.A., Gupta N. et al. . Association of rare and common variation in the lipoprotein lipase gene with coronary artery disease. Jama. 2017; 317:937–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M. et al. . The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. et al. . A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011; 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., Del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J. et al. . From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics. 2013; 43:11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. Genome Project Data Processing, S. . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nielsen R., Paul J.S., Albrechtsen A., Song Y.S.. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011; 12:443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nho K., West J.D., Li H., Henschel R., Bharthur A., Tavares M.C., Saykin A.J.. Comparison of multi-sample variant calling methods for whole genome sequencing. IEEE Int. Conf. Syst. Biol. 2014; 2014:59–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guo S.W., Thompson E.A.. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics. 1992; 48:361–372. [PubMed] [Google Scholar]
- 20. Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D.. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006; 38:904–909. [DOI] [PubMed] [Google Scholar]
- 21. McVean G. A genealogical interpretation of principal components analysis. PLoS Genet. 2009; 5:e1000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moorjani P., Patterson N., Hirschhorn J.N., Keinan A., Hao L., Atzmon G., Burns E., Ostrer H., Price A.L., Reich D.. The history of African gene flow into Southern Europeans, Levantines, and Jews. PLoS Genet. 2011; 7:e1001373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Genomes Project, C. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al. . A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B. et al. . Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hamza T.H., Zabetian C.P., Tenesa A., Laederach A., Montimurro J., Yearout D., Kay D.M., Doheny K.F., Paschall J., Pugh E. et al. . Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat. Genet. 2010; 42:781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hunter D.J., Kraft P., Jacobs K.B., Cox D.G., Yeager M., Hankinson S.E., Wacholder S., Wang Z., Welch R., Hutchinson A. et al. . A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat. Genet. 2007; 39:870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hu H., Huff C.D., Moore B., Flygare S., Reese M.G., Yandell M.. VAAST 2.0: improved variant classification and disease-gene identification using a conservation-controlled amino acid substitution matrix. Genet. Epidemiol. 2013; 37:622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee S., Emond M.J., Bamshad M.J., Barnes K.C., Rieder M.J., Nickerson D.A., Team N.G.E.S.P.-E.L.P., Christiani D.C., Wurfel M.M., Lin X.. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 2012; 91:224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Price A.L., Kryukov G.V., de Bakker P.I., Purcell S.M., Staples J., Wei L.J., Sunyaev S.R.. Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 2010; 86:832–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Madsen B.E., Browning S.R.. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009; 5:e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Epstein M.P., Duncan R., Jiang Y., Conneely K.N., Allen A.S., Satten G.A.. A permutation procedure to correct for confounders in case-control studies, including tests of rare variation. Am. J. Hum. Genet. 2012; 91:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu M.C., Lee S., Cai T., Li Y., Boehnke M., Lin X.. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011; 89:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R.. A method and server for predicting damaging missense mutations. Nat. Methods. 2010; 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu H., Coon H., Li M., Yandell M., Huff C.D.. VARPRISM: incorporating variant prioritization in tests of de novo mutation association. Genome Med. 2016; 8:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tyrer J.P., Guo Q., Easton D.F., Pharoah P.D.. The admixture maximum likelihood test to test for association between rare variants and disease phenotypes. BMC Bioinformatics. 2013; 14:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ng P.C., Henikoff S.. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003; 31:3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Apweiler R., Attwood T.K., Bairoch A., Bateman A., Birney E., Biswas M., Bucher P., Cerutti L., Corpet F., Croning M.D. et al. . The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 2001; 29:37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang K., Li M., Hakonarson H.. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bell D., Berchuck A., Birrer M., Chien J., Cramer D.W., Dao F., Dhir R., DiSaia P., Gabra H., Glenn P. et al. . Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kanchi K.L., Johnson K.J., Lu C., McLellan M.D., Leiserson M.D., Wendl M.C., Zhang Q., Koboldt D.C., Xie M., Kandoth C. et al. . Integrated analysis of germline and somatic variants in ovarian cancer. Nat. Commun. 2014; 5:3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koboldt D.C., Fulton R.S., McLellan M.D., Schmidt H., Kalicki-Veizer J., McMichael J.F., Fulton L.L., Dooling D.J., Ding L., Mardis E.R. et al. . Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Iossifov I., O’Roak B.J., Sanders S.J., Ronemus M., Krumm N., Levy D., Stessman H.A., Witherspoon K.T., Vives L., Patterson K.E. et al. . The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014; 515:216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fischbach G.D., Lord C.. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010; 68:192–195. [DOI] [PubMed] [Google Scholar]
- 44. Manichaikul A., Mychaleckyj J.C., Rich S.S., Daly K., Sale M., Chen W.M.. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010; 26:2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Colhoun H.M., McKeigue P.M., Davey Smith G.. Problems of reporting genetic associations with complex outcomes. Lancet. 2003; 361:865–872. [DOI] [PubMed] [Google Scholar]
- 46. Antoniou A., Pharoah P.D., Narod S., Risch H.A., Eyfjord J.E., Hopper J.L., Loman N., Olsson H., Johannsson O., Borg A. et al. . Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am. J. Hum. Genet. 2003; 72:1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Meindl A., Hellebrand H., Wiek C., Erven V., Wappenschmidt B., Niederacher D., Freund M., Lichtner P., Hartmann L., Schaal H. et al. . Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010; 42:410–414. [DOI] [PubMed] [Google Scholar]
- 48. Loveday C., Turnbull C., Ramsay E., Hughes D., Ruark E., Frankum J.R., Bowden G., Kalmyrzaev B., Warren-Perry M., Snape K. et al. . Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011; 43:879–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rafnar T., Gudbjartsson D.F., Sulem P., Jonasdottir A., Sigurdsson A., Jonasdottir A., Besenbacher S., Lundin P., Stacey S.N., Gudmundsson J. et al. . Mutations in BRIP1 confer high risk of ovarian cancer. Nat. Genet. 2011; 43:1104–1107. [DOI] [PubMed] [Google Scholar]
- 50. Song H., Dicks E., Ramus S.J., Tyrer J.P., Intermaggio M.P., Hayward J., Edlund C.K., Conti D., Harrington P., Fraser L. et al. . Contribution of germline mutations in the RAD51B, RAD51C, and RAD51D genes to ovarian cancer in the population. J. Cin. Oncol. 2015; 33:2901–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Walsh T., Casadei S., Coats K.H., Swisher E., Stray S.M., Higgins J., Roach K.C., Mandell J., Lee M.K., Ciernikova S. et al. . Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA. 2006; 295:1379–1388. [DOI] [PubMed] [Google Scholar]
- 52. Renwick A., Thompson D., Seal S., Kelly P., Chagtai T., Ahmed M., North B., Jayatilake H., Barfoot R., Spanova K. et al. . ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006; 38:873–875. [DOI] [PubMed] [Google Scholar]
- 53. Dansonka-Mieszkowska A., Kluska A., Moes J., Dabrowska M., Nowakowska D., Niwinska A., Derlatka P., Cendrowski K., Kupryjanczyk J.. A novel germline PALB2 deletion in Polish breast and ovarian cancer patients. BMC Med. Genet. 2010; 11:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Song H., Cicek M.S., Dicks E., Harrington P., Ramus S.J., Cunningham J.M., Fridley B.L., Tyrer J.P., Alsop J., Jimenez-Linan M. et al. . The contribution of deleterious germline mutations in BRCA1, BRCA2 and the mismatch repair genes to ovarian cancer in the population. Hum. Mol. Genet. 2014; 23:4703–4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lhota F., Zemankova P., Kleiblova P., Soukupova J., Vocka M., Stranecky V., Janatova M., Hartmannova H., Hodanova K., Kmoch S. et al. . Hereditary truncating mutations of DNA repair and other genes in BRCA1/BRCA2/PALB2-negatively tested breast cancer patients. Clin. Genet. 2016; 90:324–333. [DOI] [PubMed] [Google Scholar]
- 56. Risch H.A., McLaughlin J.R., Cole D.E., Rosen B., Bradley L., Fan I., Tang J., Li S., Zhang S., Shaw P.A. et al. . Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: a kin-cohort study in Ontario, Canada. J. Natl. Cancer Inst. 2006; 98:1694–1706. [DOI] [PubMed] [Google Scholar]
- 57. Hugot J.P., Chamaillard M., Zouali H., Lesage S., Cezard J.P., Belaiche J., Almer S., Tysk C., O’Morain C.A., Gassull M. et al. . Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001; 411:599–603. [DOI] [PubMed] [Google Scholar]
- 58. Ogura Y., Bonen D.K., Inohara N., Nicolae D.L., Chen F.F., Ramos R., Britton H., Moran T., Karaliuskas R., Duerr R.H. et al. . A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001; 411:603–606. [DOI] [PubMed] [Google Scholar]
- 59. Yandell M., Huff C., Hu H., Singleton M., Moore B., Xing J., Jorde L.B., Reese M.G.. A probabilistic disease-gene finder for personal genomes. Genome Res. 2011; 21:1529–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hu H., Roach J.C., Coon H., Guthery S.L., Voelkerding K.V., Margraf R.L., Durtschi J.D., Tavtigian S.V., Shankaracharya, Wu W. et al. . A unified test of linkage analysis and rare-variant association for analysis of pedigree sequence data. Nat. Biotechnol. 2014; 32:663–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tavtigian S.V., Chenevix-Trench G.. Growing recognition of the role for rare missense substitutions in breast cancer susceptibility. Biomark. Med. 2014; 8:589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
XPAT described in this paper is freely available for academic use and can be downloaded at http://www.hufflab.org/software/xpat/.