

Abstract
Prolonged seizure activity or status epilepticus (SE) is one of the most critical manifestations of organophosphate exposure. Previous studies in our laboratory have demonstrated that oxidative stress is a critical mediator of SE-induced neuronal injury. The goal of this study was to determine if diisopropylflurorphoshate (DFP) exposure in rats resulted in oxidative stress and whether scavenging reactive oxygen species attenuated DFP-induced neurotoxicity. DFP treatment increased indices of oxidative stress in a time- and region- dependent manner. Neuronal loss measured by Fluoro-Jade B staining was significantly increased in the hippocampus, piriform cortex and amygdala following DFP. Similarly, levels of the proinflammatory cytokines, particularly TNF-α, IL-6, and KC/GRO were significantly increased in the piriform cortex and in the hippocampus following DFP treatment. The catalytic antioxidant AEOL10150, when treatment was initiated 5 min after DFP-induced SE, significantly attenuated indices of oxidative stress, neuroinflammation and neuronal damage. This study suggests that catalytic antioxidant treatment may be useful as a novel therapy to attenuate secondary neuronal injury following organophosphate exposure.
Keywords: seizures, antioxidant, oxidative stress, neuroinflammation, diisopropylflurorphoshate, pharmacokinetic analysis
Organophosphorus compounds (OP) are highly toxic chemicals that exert their effects mainly through irreversible inhibition of acetylcholinesterase (AChE). This activity has made OP compounds effective pesticides however these compounds carry the inherent risk of human toxicity and have been used in military combat and by terrorists (Jett, 2007, 2010). Accidental or intentional exposure to OP compounds including nerve agents necessitates the investigation of novel treatment paradigms to prevent mortality and secondary effects from exposure. The CounterACT program is a transNIH initiative whose primary goal is to promote the development of medical countermeasures to prevent mortality and secondary effects caused by exposure to OP compounds and nerve agents (Jett, 2010). The most critical manifestation of OP exposure and resultant inhibition of AChE is prolonged seizure activity known as status epilepticus (SE). SE results in high mortality rates, neuronal loss, and neuroinflammation in those that survive. Current therapeutics such as oximes and benzodiazepines can inhibit SE and reduce mortality; however, they are associated with serious side effects and must be administered within minutes of exposure (de Araujo Furtado et al., 2012; Jett, 2007; Shih et al., 2003). There is therefore a great need for novel neuroprotective countermeasures.
Oxidative stress is a phenomenon associated with SE and a known contributor to neuronal injury and neuroinflammation. Treatments targeting oxidative stress may therefore hold promise as novel and efficacious neuroprotective countermeasures against OP toxicity. We have previously shown that oxidative stress contributes to neuronal loss, cognitive impairment and mortality caused by the nerve agent surrogate, pilocarpine (Pearson et al., 2015; Pearson-Smith et al., 2017). However, whether an authentic OP, such as Diisopropylflurorphoshate (DFP) also results in increased indices of oxidative stress and neuroinflammation is relatively poorly understood (Flannery et al., 2016; Zaja-Milatovic et al., 2009). Furthermore, whether pharmacological targeting of DFP-induced oxidative stress confers neuroprotection remains to be determined.
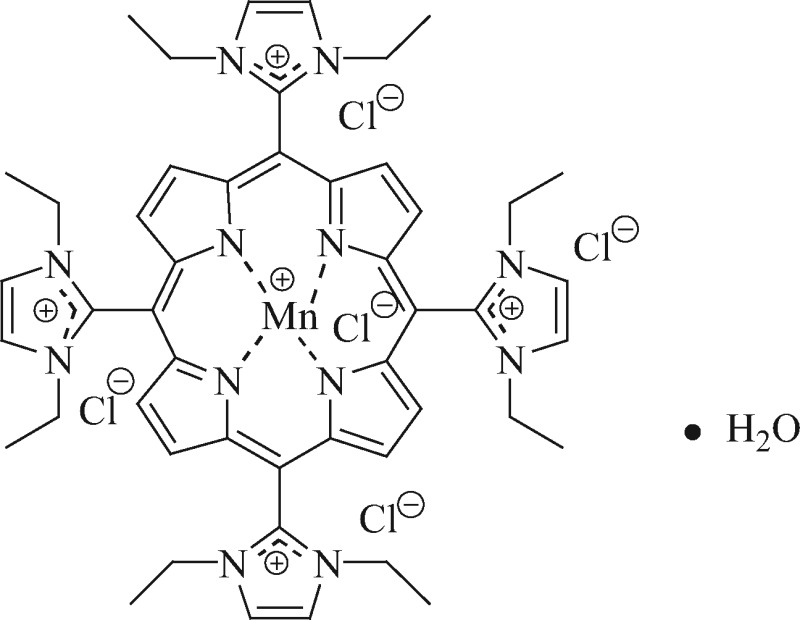
The metalloporphyrin, AEOL10150, is a broad spectrum catalytic antioxidant and a superoxide dismutase mimetic capable of scavenging multiple forms of reactive oxygen species (ROS) and nitrogen species including superoxide, hydrogen peroxide, lipid peroxides and peroxynitrite (Table 1). We have previously demonstrated efficacy of AEOL10150 against kainic acid and pilocarpine-induced oxidative stress, mitochondrial dysfunction and neuronal loss (Pearson et al., 2015; Rowley et al., 2015).
Table 1.
Structure and Antioxidant Properties of AEOL10150
| ROS | Superoxide | Hydrogen peroxide | Lipid Peroxides | Peroxynitrite | |
|---|---|---|---|---|---|
 |
Activity | kcat() = 6.78 × 107 M−1 s−1 | k(H2O2) = 2.2 min−1 | F2-IP IC50 = 0.1 µM | k(ONOO−) = 1.01 × 107 M−1 s −1 |
Structure of metalloporphyrin catalytic antioxidant AEOL10150. The effect of AEOL10150 to destroy superoxide (as measured by pulse radiolysis), hydrogen peroxide (Clark oxygen electrode), peroxynitrite (stop-flow) and inhibit lipid peroxidation (F2-isoprostanes) (Kachadourian et al., 2004).
The goal of this study was to determine if treatment with AEOL10150 could attenuate indices of oxidative stress, neuroinflammation and neurodegeneration associated with exposure to DFP. Particularly, we sought to determine if post-treatment with AEOL10150 after DFP injection was sufficient to exert neuroprotection so as to evaluate the therapeutic potential of AEOL10150 as a medical countermeasure agent. Pharmacokinetic data in control and DFP-treated rats weres used to determine the ideal dosing paradigm while a therapeutic window of the efficacy of AEOL10150 treatment against indices of oxidative stress was established. Our findings indicate that suppression of oxidative stress by AEOL10150 can attenuate DFP-induced release of proinflammatory cytokines and neurodegeneration. These results suggest that targeting of oxidative stress in a postexposure treatment paradigm may provide a novel therapeutic avenue to improve secondary neuronal injury after OP exposure.
MATERIALS AND METHODS
Reagents
DFP, pyridostigmine, atropine methyl nitrate, pyridine-2-aldoxime methochloride (2-pam) were purchased from Sigma Aldrich. Midazolam was purchased from Hopira Inc. (Lake Forest, Illinois). DFP was stored at −20 °C and freshly diluted with saline (from 1060 to 4.5 mg/ml). Manganese (III) meso-tetrakis (di-N-ethylimidazole) porphyrin designated as AEOL10150 (also known in the literature MnIIITDE-2-ImP5+) was pharmaceutical grade and obtained from Aeolus Pharmaceuticals.
Animals
All studies were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23). All procedures were approved by the Institute Animal Care and Use Committee of the University of Colorado Denver, which is fully accredited by the American Association for the Accreditation of Laboratory Animal Care. Male Sprague-Dawley (SD) rats (approximately 300 g) were purchased from Harlan Laboratories (Indianapolis, Indiana) and used for all experiments. Upon arrival, rats were group housed on a 14/10 light/dark cycle with ad libitum access to both food (Harlan rat chow) and filtered water. Rats were allowed 1 week of acclimation before any experiments were performed. A total of 206 rats were used for the entirety of the study and the number of rats in each group per experiment is detailed in the results section. Of the 206 rats, 144 received DFP with or without AEOL10150.
DFP and AEOL10150 administration
Male SD rats were injected with DFP (4.5 mg/kg subcutaneous s.c.), which is equivalent to 0.9 × LD50 along with the standard therapy consisting of pyridostigmine (0.1 mg/kg, i.m.) 30 min prior to DFP, atropine methyl nitrate (2 mg/kg, i.m.) and pyridine-2-aldoxime (2-PAM, 25 mg/kg, i.m.) 1 min after DFP. Midazolam (2 mg/kg, i.m.) was administrated at 1, 5, or 15 min after SE initiation. This group of rats will be referred to as DFP + standard therapy. The DFP + standard therapy protocol was performed following methods previously described in Pouliot et al. (2016) and Sisó et al. (2017). This method has been demonstrated to mimic the electrographic and pathological characteristics in human and has been approved by the CounterACT program. Control rats received all compounds except DFP which was replaced by saline, thus controls will be referred to as saline + standard therapy. In half of the rats receiving DFP + standard therapy, AEOL10150 (5 mg or 7 mg/kg s.c.) treatment was initiated at 1 min (5 mg/kg s.c), 5 min (7 mg/kg s.c) or 15 min (5 mg/kg s.c.) after SE onset and continued (5 mg/kg s.c.) every 4 h until sacrificed. Midazolam (2 mg/kg i.m.) was given at the same time point (1, 5, or 15 min) as AEOL10150. This group will be referred to as DFP + standard therapy + AEOL10150. All rats were visually monitored during SE and behavioral seizures were scored using a modified Racine scale (Racine, 1972). The seizure activities were scored on the following scale: P0—normal; P1—immobilization and staring, occasional “wet-dog shakes”; P2—head nodding, unilateral forelimb clonus, frequent “wet dog shakes”; P3—rearing, salivation, bilateral forelimb clonus; P4—generalized limbic seizures with falling, running and rearing, and P5—continuous generalized seizures with tonic limbic extension, death. SE onset was defined as the appearance of P3 or higher seizures followed by a period of continuous seizure activity
AEOL10150 administration for PK analysis
Rats with jugular vein catheter cannulation (Harlan Laboratories) received AEOL10150 (5 mg/kg dissolved in sterile saline) administrated via catheter (i.v.) or (s.c.) route. Blood samples (approximately 0.1 ml) from rat’s jugular catheter were collected 1, 3, 6, 12, 24, and 48 h after AEOL10150 administration. Plasma (50 µl) was obtained after whole blood samples were centrifuged at 13 000 rpm for 10 min. 48 h after AEOL10150 treatment, the rats were deeply anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and perfused transcardially with 80–100 ml saline and paraformaldehyde. Hippocampus was rapidly dissected and frozen with dry ice. Brain and plasma samples were stored at −80 °C for pharmacokinetic analysis.
Determination of AEOL10150 pharmacokinetics
The concentration of AEOL10150 in brain and plasma of saline + standard therapy + AEOL10150 and DFP + standard therapy + AEOL10150 rats was measured after extraction and precipitation by 50% methanol and 0.05 N perchloric acid centrifuged at 16 000 × g for 20 min. The resulting supernatant was filtered through a 0.22-μm filter before injection into the HPLC set at 466 nm. The HPLC was equipped with a spectrophotometric detector (Elite LaChrom System; Hitachi) and a YMC-Pack ODS-A column (4.6 × 150 mm, 3 μm) as previously described (Kachadourian et al., 2002). Mobile phase consisted of 20 mM trimethylamine, 20 mM trifluoroacetic acid, pH 2.7 and 70% acetonitrile. Flow rate was 1 ml/min. Recovery of metalloporphyrins from plasma samples was determined to be >95%.
Measurement of oxidative markers
Glutathione (GSH), glutathione disulfide (GSSG), tyrosine and 3-nitrotyrosine (3-NT) assays were performed with an ESA (Chelmsford, Massachusetts) 5600 CoulArray HPLC equipped with 8 electrochemical cells following the company instruction (ESA Application Note 70-3993) with small modification (Liang et al., 2007). The potentials of the electrochemical cells were set at 400/450/500/570/630/690/810/860 mV versus Pd. Analyte separation was conducted on a TOSOHAAS (Montgomeryville, PA) reverse-phase ODS 80-TM C-18 analytical column (4.6 × 250 mm; 5 μm).
Measurement of thiols and disulfides in plasma
The levels of cysteine (Cys), cystine (Cyss), GSH, and GSSG in plasma were measured with an ESA (Chelmsford, Massachusetts) 5600 CoulArray HPLC equipped with 8 electrochemical cells following the company instruction (Dionex Application Brief 131) and methods previously described (Liang and Patel, 2016). Potentials of the electrochemical cells were set at 400/500/600/650/700/750/800/850 mV versus Pd. Analyte separation was conducted on YMC-Pack ODS-A C-18 analytical column (Waters Inc., 4.6 × 250 mm; 5 μm particle size). A 2-component gradient elution system was used with component A of the mobile phase composed of 50 mM NaH2PO4 pH 2.7, 1.0 mM 1-octanesulfonic acid and component B composed of 50 mM NaH2PO4 pH 2.7, 1.0 mM 1-octanesulfonic acid and 50% methanol. 100% A is run at 0.4 ml/min from 0 to 10 min at initial time and a linear gradient to 100% A 0.6 ml/min is run over the period from 10 to 18 min. From 18 to 20 min, the conditions are maintained at 100% A 0.6 ml/min then is run a linear gradient to 92% A, 8% B 0.6 ml/min from 20 to 28 min and returned back 100% A 0.4 ml/min from 28 to 32 min. Equilibration time for the next run is 8 min. The sample tubes containing 1:1 ratio plasma and 2% PCA/0.2 M boric acid were centrifuged at 13 000 g, 4°C for 10 min to pellet protein. An aliquot (20 μl) of the supernatant was injected into the HPLC.
Fluoro-Jade B analysis
Paraffin-embedded brain sections (20 µm) were cut coronally and stained with Fluoro-Jade B (FJB; Histo-Chem) as previously described in Schmued and Hopkins (2000). FJB is an anionic fluorescein derivative that selectively stains neurons undergoing degeneration. The number of FJB positive cells were quantified in the hippocampus (CA1 and CA3), piriform cortex and amygdala using Image J software (NIH) following the method as described previously in Vargas et al. (2013). Briefly, the FJB positive cells in a given area (10× axis) were counted in 6 sections 100 μm apart from both hemispheres of each animal. The average of positive cells from 6 sections/rat was expressed as positive cell number/HP (high power, 10× axis). Images were captured using a Nikon Eclipse TE2000-U microscope.
Multiplex proinflammatory cytokine measurement
Levels of the proinflammatory cytokines, TNF-α, IL-1β, IL-6, and KC/GRO, were measured using a rat multiplex proinflammatory cytokine array kit from Mesoscale Discovery (MSD) according to the manufacturer’s instructions and method described previously in McElroy et al. (2017). One side of the hippocampus or piriform cortex from each rat was lysed in MSD Tris Lysis buffer supplemented with protease and phosphatase inhibitors in a 1:10 ratio. The lysates were then centrifuged at 13 000 rpm for 10 min and supernatants were collected. Protein concentrations were determined in the supernatants using a Bradford protein assay with 250 µg of protein loaded per well for each sample in duplicate. Calibration curves were prepared in the supplied diluent with a range of 40 000 pg/ml to 2.45 pg/ml. Wells were blocked for 30 min, after which samples were added and incubated with shaking (300–1000 rpm) for 2 h. The plate was then washed with PBS + 0.05% Tween 20 and 25 µl of detection antibody was added and incubated for an additional 2 h. Finally, the plate was washed, MSD read buffer was added to the wells and the plate was read using Sector Imager 2400. Concentrations of analytes were determined by measuring the intensity of light emitted at 620 nm and Softmax Pro software using curve-fit models.
AChE activity assay
To assess AChE (EC 3.1.1.7) activity, the AChE activity assay kit (Sigma, MAK119) was used and experiments were performed as previously described with minor modifications per company instruction (Ellman et al., 1961). Briefly, brain tissue (10% w/v) was sonicated in 0.1 M phosphate buffer, pH 7.5, followed by centrifugation at 14 000 rpm at 4 °C for 5 min and cleared supernatants were used for assay. In total 10 µl of brain tissue supernatants or plasma was added in duplicate to a 96-well plate. Then, 190 µl of the freshly prepared working reagent was added to all sample wells and the plate was tapped briefly to mix. Samples were incubated at room temperature for 2 min and the initial absorbance at 412 nm was read followed 10 min later by another reading of absorbance at 412 nm. The calibrator (200 U/l) provided by the Kit was used as a standard. One unit of AChE is the amount of enzyme that catalyzes the production of 1.0 µmol of thiocholine per minute at pH 7.5 at room temperature.
Statistical analysis
All data are expressed as mean ± SEM. Pharmacokinetic data was obtained using a 1 compartment model with first order input and first order output (PK analyst, model No. 3, MicroMath, Salt Lake City, Utah). The time course of oxidative stress indices and determination of the therapeutic window of AEOL10150 were analyzed using a 2-way ANOVA. Significant main effects and interactions were probed using Bonferroni posttests and effect size was calculated using Eta squared (η2). Cytokine multiplex data were analyzed by 1-way ANOVA with post hoc multiple comparison test corrected for multiple comparisons (Tukey test) within a given brain region. Effect size was calculated using Eta squared. FluoroJade-B data within a given brain region were analyzed by unpaired t-test and Cohen’s d for determination of effect size. p values < .05 were considered statistically significant. Analyses were performed using Prism 5 software (GraphPad Software, San Diego, California).
RESULTS
AEOL10150 Pharmacokinetic Analysis
To determine the pharmacokinetic profile of AEOL10150 and whether this profile is altered in DFP-treated rats, AEOL10150 (5 mg/kg, 5 min after SE initiation) was administered by i.v. and s.c. routes and its concentration in plasma and hippocampus were evaluated at various time points. Concentration-time data of AEOL10150 by s.c. route best fit a 1 compartment PK model with first-order input and output. The plasma half-life (T½) of AEOL10150 (s.c., 5 mg/kg) was not significantly different between groups (n = 3 rats/group) and was estimated to be 1.5 ± 0.1 h in the saline + standard therapy group and 0.9 ± 0.3 h in the DFP + standard therapy group (Table 2, Figs. 1A and 1B). The time to achieve maximum plasma concentration (Tmax) of AEOL10150 was faster in the DFP + standard therapy group at 1.5 ± 0.3 h compared with 2.3 ± 0.1 h in the saline + standard therapy group (p = .06). The maximum plasma concentration (Cmax) was estimated to be 4397 ± 174 ng/ml in saline + standard therapy group and 6194 ± 255 ng/ml in the DFP + standard therapy group, representing a significant difference between groups (p > .01; Table 2, Figs. 1A and 1B). Area under the curve (AUC) for AEOL10150 by s.c. route was not altered by DFP treatment and was estimated to be 26 675 ± 2079 h × ml/ng in the saline + standard therapy group and 23 416 ± 5216 h × ml/ng in the DFP + standard therapy group (Table 2). Bioavailability (calculated with AUC s.c./AUC i.v. %) was not statistically different between groups and was 43% ± 3% in the saline + standard treatment group compared with 30% ± 7% in the DFP + standard therapy treatment group (Table 2). Also unchanged upon treatment with DFP was the concentration of the compound in hippocampus 48 h after s.c. or i.v. AEOL10150 treatment (Figure 1C).
Table 2.
Comparison of Plasma Pharmacokinetic Profilesa
| AEOL10150 (5 mg/kg, s.c). | Saline | DFP | p value |
|---|---|---|---|
| T1/2 (h) | 1.5 ± 0.1 | 0.9 ± 0.3 | .08 |
| Tmax (h) | 2.3 + 0.1 | 1.5 + 0.3 | .066 |
| Cmax (ng/ml) | 4397 ± 174 | 6194 ± 255 | <.01* |
| AUC (h × ml/ng) | 26 675 ± 2079 | 23 416 ± 5215 | .59 |
| % Bioavailability | 43 ± 3 | 30 ± 7 | .15 |
Data are means ± SEM, un-paired t-test. Pharmacokinetic data were obtained using a 1 compartment model with first order input and output (PK analyst, model No. 3, MicroMath, Salt Lake City, Utah).
Figure 1.

Pharmacokinetic analyses of AEOL10150. The concentration of AEOL10150 in plasma at 1, 3, 6, 12, 24, and 48 h by s.c. (A) or by i.v (B) and in hippocampus at 48 h (C) after AEOL10150 5 mg/kg, s.c. or i.v. with saline + standard therapy or DFP + standard therapy treatment (n = 3/group).
DFP Exposure Induces SE
Within 5–10 min of exposure to 0.9 × LD50 DFP (4.5 mg/kg) all rats exhibited signs of OP toxicity including hypersalivation, lacrimation and diarrhea. In all rats exposed to DFP, this was followed with tremors, wet dog shakes and limbic seizures with rearing and rolling. Each animal exhibited at least 1 P3 or greater seizure before rapidly progressing to SE. All of the animals (100%) achieved SE with this dose DFP exposure. Mortalities were 0%, 14.8%, 18.8%, and >40% when midazolam was injected at 1, 5, 15 min and with no injection after SE onset at 24 h with DFP + standard therapy, respectively.
DFP Exposure Increases Indices of Oxidative Stress in Brain
To determine the time course and brain region distribution of oxidative/nitrative stress markers, GSH, GSSG, and 3-NT were measured in the hippocampus, piriform cortex and frontal cortex at 6, 12, 24, and 48 h after DFP + standard therapy. The levels of GSH were significantly decreased at 24 and 48 h, but not at 6 or 12 h in both hippocampus (Figure 2A, treatment F[1, 40] =86.03, p < .0001, η2 = 0.37; time F[3, 40] = 19.32, p < .0001, η2 = 0.25; interaction F[3, 40] = 16.01, p < .0001, η2 = 0.21, n = 6 rats/group) and piriform cortex (Figure 2E treatment F[1, 40] = 104.1, p < .0001, η2 = 0.38; time F[3, 40] = 20.91, p < .0001, η2 = 0.23; interaction F[3, 40] = 21.5, p < .0001, η2 = 0.24, n = 6 rats/group). Accumulation of its disulfide, GSSG was significantly increased at 24 and 48 h, but not at 6 or 12 h, in the hippocampus (Figure 2B, treatment F[1, 40] = 32.39, p < .01, η2 = 0.28; time F[3, 40] = 5.99, p < .01, η2 = 0.16; interaction F[3, 40] = 8.03, p < .001, η2 = 0.21, n = 6 rats/group) and piriform cortex, (Figure 2F, treatment F[1, 40] = 50.29, p < .0001, η2 = 0.38; time F[3, 40] = 7.08, p < .001, η2 = 0.16; interaction F[3, 40] = 7.49, p < .001, η2 = 0.17, n = 6 rats/group), resulting in an overall depletion of the ratio of GSH: GSSG in both hippocampus and piriform cortex (Figure 2C, treatment F[1, 40] = 31.02, p < .0001, η2 = 0.31; time F[3, 40] = 4.55, p < .01, η2 = 0.14; interaction F[3, 40] = 4.97, p < .01, η2 = 0.15, n = 6 rats/group; Figure 2G, treatment F[1, 40] = 63.06, p < .0001, η2 = 0.41; time F[3, 40] = 7.31, p < .001, η2 = 0.14; interaction F[3, 40] = 9.3, p < .0001, η2 = 0.18, n = 6 rats/group, respectively). The ratio of 3-NT/tyrosine followed a similar pattern and was significantly increased at 24 and 48 h, but not at 6 or 12 h, in the hippocampus (Figure 2D, treatment F[1, 40] = 34.12, p < .0001, η2 = 0.29; time F[3, 40] = 8.39, p < .001, η2 = 0.21; interaction F(3, 40) = 6.03, p < .01,η2 = 0.15, n = 6 rats/group) and piriform cortex (Figure 2H, treatment F[1, 40] = 68.57, p < .0001, η2 = 0.38; time F[3, 40] = 14.62, p < .0001, η2 = 0.24; interaction F[3, 40] = 9.26, p < .001, η2 = 0.15, n = 6 rats/group). DFP + standard therapy did not alter oxidative stress markers in the frontal cortex (Figs. 2I–L). Based on these results, the 24 h time point was chosen to test the effects of AEOL10150 on indices of oxidative stress and neuronal injury.
Figure 2.

Oxidative makers in the hippocampus, piriform cortex and frontal cortex at different time points after DFP + standard therapy treatment. The levels of GSH, GSSG, GSH/GSSG and 3-NT/tyrosine of rats at 6, 12, 24, and 48 h after DFP with standard therapy in the hippocampus (A–D), piriform cortex (E–H), and frontal cortex (I–L). Midazolam 2 mg/kg i.m. 5 min after SE initiation. Bars represent mean + SEM, *p < .01 versus saline, 2-way ANOVA, n = 6 per group.
AEOL10150 Treatment Does Not Attenuate the DFP-Induced Inhibition of AChE Activity
To ensure that any observed effects of AEOL10150 were due to its antioxidant activity and not direct effects on AChE, activity of AChE was measured 24 h after DFP + standard therapy. The AChE activities in the plasma, hippocampus and piriform cortex of the rats with saline+ standard therapy were 0.359 ± 0.005 U/ml, 5.103 ± 0.114 and 4.709 ± 0.199 U/g tissue and inhibited by 76.8%, 80.0%, and 84.1% at 24 h after DFP + standard therapy, respectively (Figs. 3A and F[2, 15] = 1254, p < .0001, η2 = 0.99; 3B, F[2, 15] = 946.9, p < .0001, η2 = 0.99; 3 C, F[2, 15] = 337.8, p < .0001, η2 = 0.98, n = 6 rats/group). Treatment with AEOL10150 starting 5 min after DFP-induced SE and continuing every 4 h; thereafter, had no effect on DFP-induced inhibition of AChE activity (Figure 3). This suggests that AEOL10150 does not act as a direct scavenger of DFP.
Figure 3.

Activity of AChE with or without AEOL10150 treatment. The activity of AChE in the plasma (A), hippocampus (B) and piriform cortex (C) of rats 24 h after DFP with standard therapy alone or with AEOL10150. Midazolam and AEOL10150 was given 5 min after SE initiation and AEOL10150 5 mg/kg s.c. or saline every 4 h thereafter. Bars represent mean + SEM, *p < .01 versus saline + standard therapy, 1-way ANOVA, n = 6 per group.
AEOL10150 Treatment Attenuates DFP-Induced Oxidative/Nitrative Markers in Brain
To determine if treatment with AEOL10150 was able to attenuate DFP-induced oxidative and nitrative stress, GSH, GSSG and 3-NT were measured in hippocampus and piriform cortex 24 h after DFP exposure. To establish the therapeutic window for AEOL10150, the compound was given 1, 5, or 15 min after seizure initiation and continuing thereafter every 4 h. In the hippocampus, relative to the saline + standard therapy group, in rats treated with DFP + standard therapy the levels of GSH decreased by 14.5%, 23.7%, and 24.7%, after midazolam (2 mg/kg i.m.) administration at 1, 5, and 15 min after seizure initiation, respectively (Figure 4A). The levels of GSSG increased by 53.7%, 63.2%, and 90.7% in the DFP + standard therapy group compared with the saline + standard therapy group after midazolam (2 mg/kg i.m.) administration at 1, 5, and 15 min after seizure initiation, respectively (Figure 4B). The levels of 3-NT increased by 75.9%, 125%, and 153.8% in the DFP + standard therapy group compared with the saline + standard therapy group after midazolam (2 mg/kg i.m.) administration at 1, 5, and 15 min after seizure initiation, respectively (Figure 4D). In the piriform cortex, the levels of GSH were decreased by 27.6%, 47.2%, and 48.6%, the levels of GSSG were increased by 81.1%, 109.7%, and 178.8% and the levels of 3-NT were increased by 151.2%, 241.9%, and 267.6% in rats treated with DFP + standard therapy compared with the saline + standard therapy group after midazolam 2 mg/kg i.m. administration at 1, 5, and 15 min after seizures initiation, respectively (Figs. 4E, 4F, and 4H). This suggests that the longer SE is allowed to progress, the higher the levels of oxidative stress as the 15-min time point corresponded to the largest difference in oxidative stress markers. Additionally, the levels of oxidative/nitrative markers appear to be higher in the piriform cortex than in the hippocampus at all of time points observed. In the hippocampus, treatment with AEOL10150 did not attenuate the DFP-induced decrease in GSH levels (Figure 4A). However, the DFP-induced increase in its disulfide, GSSG was attenuated by treatment with AEOL10150 when given at either 5 or 15 min after seizure initiation (Figure 4B, treatment F[2, 45] = 26.52, p < .0001, η2 = 0.52; time F[2, 45] = 0.78, p > .05, η2 = 0.02; interaction F[4, 45] = 0.75, p > 0.05, η2 = 0.03, n = 6 rats/group). In the hippocampus, AEOL10150 was able to significantly attenuate the DFP-induced depletion of overall GSH: GSSG when given at the 5 min time point (Figure 4C, treatment F[2, 45] = 45.67, p < .0001, η2 = 0.65; time F[2, 45] = 0.77, p > .05, η2 = 0.01; interaction F[4, 45] = 0.5, p > .05, η2 = 0.01, n = 6 rats/group) and attenuate 3-NT levels when given at 5 or 15 min (Figure 4D, treatment F[2, 45] = 55.54, p < .0001, η2 = 0.63; time F[2, 45] = 4.88, p < .05, η2 = 0.06; interaction F[4, 45] = 2.41, p < .05, η2 = 0.05, n = 6 rats/group). In the piriform cortex, treatment with AEOL10150 at 1 and 5 min was able to attenuate the DFP-induced decrease in GSH levels (Figure 4E, treatment F[2, 45] = 129.37, p < .0001, η2 = 0.65; time F[2, 45] = 25.61, p < .0001, η2 = 0.13; interaction F[4, 45] = 11.17, p < .0001, η2 = 0.11, n = 6 rats/group), while only the 5 min time point was able to attenuate GSSG levels (Figure 4F, treatment F[2, 45] = 39.85, p < .0001, η2 = 0.45; time F[2, 45] = 15.93, p < .0001, η2 = 0.18; interaction F[4, 45] = 4.69, p < .01, η2 = 0.11, n = 6 rats/group). Treatment starting at both 1 and 5 min after seizure initiation improved the overall ratio of GSH: GSSG (Figure 4G, treatment F[2, 45] = 129.07, p < .0001, η2 = 0.75; time F[2, 45] = 10.7, p < .001, η2 = 0.06; interaction F[4, 45] = 4.98, p < .01, η2 = 0.06, n = 6 rats/group) while only the 5 min time point attenuated 3-NT levels (Figure 4H, treatment F[2, 45] = 50.95, p < .0001, η2 = 0.60; time F[2, 45] = 6.75, p < .01, η2 = 0.08; interaction F[4, 45] = 2.37, p > .05, η2 = 0.06, n = 6 rats/group). Taken together, the data suggest that treatment with AEOL10150 is effective at attenuating oxidative/nitrative stress with optimal results being achieved when given 5 min after DFP-induced seizure initiation.
Figure 4.

Determination of therapeutic window of AEOL10150 against oxidative stress markers in brain. The levels of GSH, GSSG and GSH/GSSG and 3-NT/tyrosine in the hippocampus (A–D) and piriform cortex (E–H) of rats 24 h after DFP with standard therapy alone or with AEOL10150. Midazolam 2 mg/kg i.m. treated at same time point with AEOL10150 when AEOL10150 is given 1 min (5 mg/kg, s.c.), 5 min (7 mg/kg, s.c.) or 15 min (5 mg/kg, s.c.) after SE initiation and 5 mg/kg s.c. or saline every 4 h thereafter. Bars represent mean + SEM, * p < .01 versus saline + standard therapy, #p < .05 versus DFP + standard therapy + saline, 2-way ANOVA, n = 6 per group.
AEOL10150 Treatment Attenuates DFP-Induced Oxidative Markers in Plasma
Previous studies suggest that decreased Cys and alterations to the ratio of Cys: Cyss in plasma can serve as sensitive biomarkers for oxidative damage in the kainic acid and pilocarpine models of TLE (Liang and Patel, 2016). Therefore, the levels of thiols and disulfides in plasma were measured by HPLC. The levels of Cys were significantly decreased by DFP compared with control and this decrease was significantly attenuated by AEOL10150 when given either 5 or 15 min after seizure initiation (Figure 5A, treatment F[2, 30] = 20.40, p < .0001, η2 = 0.56; time F[1, 30] = 1.28, p > .05, η2 = 0.02; interaction F[2, 30] = 0.37, p > 0.05, η2 = 0.01, n = 6 rats/group), to a level not significantly different from saline + standard therapy group. Cyss levels in the DFP + standard therapy group did not increase relative to saline + standard therapy group until 15 min after seizure initiation and this was significantly attenuated in the DFP+ standard therapy + AEOL10150 group (Figure 5B, treatment F[2, 30] = 8.1, p < .01, η2 = 0.27; time F[1, 30] = 6.11, p < .05, η2 = 0.10; interaction F[2, 30] = 3.51, p < .05, η2 = 0.12, n = 6 rats/group). The DFP-induced depletion in Cys: Cyss was significantly attenuated by AEOL10150 treatment at either time point (Figure 5C, treatment F[2, 30] = 30.11, p < .0001, η2 = 0.63; time F[1, 30] = 3.13, p > .05, η2 = 0.03; interaction F[2, 30] = 0.89, p > .05, η2 = 0.02, n = 6 rats/group). Plasma GSH, GSSG, and the ratio were unchanged by DFP exposure (Figs. 5D–F, p > .05, n = 6 rats/group). These experiments suggest that treatment with AEOL10150 can attenuate DFP-induced increased oxidative stress markers in plasma.
Figure 5.

Determination of therapeutic window of AEOL10150 against oxidative stress markers in plasma. The levels of (A) Cys, (B) Cyss, (C) Cys/Cyss, (D) GSH, (E) GSSG, and (F) GSH/GSSG in plasma of rats 24 h after DFP with standard therapy alone or with AEOL10150. Midazolam 2 mg/kg i.m. treated at same time point with AEOL10150 when AEOL10150 is given 5 min (7 mg/kg, s.c.) or 15 min (5 mg/kg, s.c.) after SE initiation and 5 mg/kg s.c. or saline every 4 h thereafter. Bars represent mean + SEM, *p < .01 versus saline + standard therapy, #p < .05 versus DFP + standard therapy + saline, 2-way ANOVA, n = 6 per group.
AEOL10150 Treatment Attenuates DFP-Induced Proinflammatory Cytokines
To determine if treatment with AEOL10150 could attenuate neuroinflammation, proinflammatory cytokines including TNF-α, IL-1β, IL-6, and KC/GRO were measured in the hippocampus and piriform cortex 24 h after DFP-induced SE. The control values (mean ± SEM) in the hippocampus were 1.26 ± 0.17, 33.33 ± 1.25, 190.4 ± 8.76, and 3.35 ± 0.34 pg/ml (per 250 µg protein) and in the piriform cortex were 1.34 ± 0.16, 32.35 ± 3.32, 201.3 ± 7.48, and 3.21 ± 0.36 pg/ml (per 250 µg protein) for TNF-α, IL-1β, IL-6, and KC/GRO, respectively. The levels of TNF-α, IL-6, and KC/GRO were significantly increased 4.8-, 43.0-, and 95.9-fold in the hippocampus and the levels of TNF-α, IL-6, IL-1β, and KC/GRO were significantly increased 32.1-, 0.92-, 173.5-, and 82.9-fold in the piriform cortex at 24 h after DFP-induced SE, respectively (Figs. 6A–D;p < .05). Treatment with AEOL10150 + standard therapy was able to significantly attenuate elevated TNF-α and IL-6 but not IL-1β or KC/GRO in the hippocampus relative to DFP + standard therapy (Figure 6A, F[2, 16] = 16.92, p < .0001, η2 = 0.68, n = 6–7 rats/group; 6 C, F[2, 15] = 18.72, p < .0001, η2 = 0.71, n = 5–7 rats/group; 6B, F[2, 16] =1.25, p > .05, η2 = 0.14, n = 6–7 rats/group; 6 D, F[2, 15] = 14.90, p > .05, n = 6 rats/group, respectively). In the piriform cortex, treatment with AEOL10150 + standard therapy was able to significantly attenuate elevated levels of all proinflammatory cytokines measured compared with the group receiving DFP + standard therapy (Figure 6A, F[2, 16] = 14.29, p < .001, η2 = 0.64, n = 6–7 rats/group; 6B, F2, 16] = 16.12, p < .0001, η2 = 0.67, n = 6–7 rats/group; 6 C, F[2, 15] = 14.40, p < .001, η2 = 0.66, n = 6–7 rats/group; 6 D, F[2, 16] = 14.21, p < .001, η2 = 0.64, n = 6–7 rats/group).
Figure 6.

The brain levels of cytokines in rats 24 h after DFP and AEOL10150 treatment. The level of (A) TNF-α, (B) IL-β, (C) IL-6, or (D) KC/GRO in the hippocampus and piriform cortex of rats 24 h after DFP with standard therapy alone or with AEOL10150. Midazolam 2 mg/kg i.m. and AEOL10150 7 mg/kg, s.c. treated 5 min after SE initiation and AEOL10150 5 mg/kg s.c. or saline every 4 h thereafter. Bars represent mean + SEM, *p < .01 versus saline + standard therapy, #p < .05 versus DFP + standard therapy + saline, 1-way ANOVA within given brain region, n = 5–7 per group.
AEOL10150 Treatment Attenuates DFP-Induced Neuronal Damage
To investigate whether treatment with AEOL10150 could attenuate DFP-induced neuronal damage, FJB staining was performed and quantified in the hippocampus, amygdala and piriform cortex. No FJB positive cells were found in any brain area of rats treated with saline or AEOL10150 alone. The averages of cells positively stained with FJB from 6 sections/rat of 6 rats were 328.2 ± 29.8; 383.7 ± 37.2; 182.7 ± 16.7; 200.2 ± 21.4 positive cells/high power (10 × axis) in the piriform cortex, amygdala, CA3 and CA1 of rats receiving DFP + standard therapy + saline, respectively. DFP-induced neuronal damage in the piriform cortex including amygdala was significant higher than in the CA3 and CA1 which is similar to the pattern of increased DFP-induced oxidative/nitrative markers in the piriform cortex relative to the hippocampus. In the hippocampus (Figure 7A), treatment with AEOL10150 significantly attenuated FJB staining in the CA1 (Figs. 7B and 7G, F[1, 32] = 3.39; p < .01, d = 1.1, n = 6 rats/group) and CA3 regions (Figs. 7C and 7G, F[1, 34] = 1.81; p < .05, d = 0.72, n = 6 rats/group). This represents a change of 38.8% and 24.7%, respectively. Treatment with AEOL10150 also significantly attenuated FJB staining by 26.8% in the piriform cortex (Figs. 7D, 7E, and 7G, F[1, 33] = 1.07; p < .05, d = 0.71, n = 6 rats/group) and by 28.7% in the amygdala (Figs. 7D, 7F, and 7G, F[1, 21] = 2.87; p < .05, d = 1.0, n = 6 rats/group).
Figure 7.

Neuronal damage as assessed by FluoroJade B staining. Representative FluoroJade B staining images of (A) whole hippocampi, (B) CA1, (C) CA3, (D) temporal lobe, (E) piriform cortex, and (F) amygdala 24 h after DFP with standard therapy alone or with AEOL10150. Midazolam 2 mg/kg i.m. and AEOL10150 7 mg/kg, s.c. treated 5 min after SE initiation and AEOL10150 5 mg/kg s.c. or saline every 4 h thereafter. (B) and (C) are the enlarged images from the white rectangle insets of (A) and (C) and (D) are the enlarged images from the white rectangle insets of (E). (G) Quantification of average number of FJB stained neurons. The average of positive cells from 6 sections/rat was expressed as positive cell number/HP (high power, 10× axis) *p < .01 versus DFP + standard therapy + saline, t-test within given brain region, n = 6 rats per group.
DISCUSSION
Here, we report that exposure to the OP and surrogate nerve agent, DFP results in increased oxidative stress, neuroinflammation and neuronal death. We show that a small molecular weight catalytic antioxidant, AEOL10150 can cross the blood-brain barrier and attenuate oxidative stress in the brain and plasma. Additionally, this compound significantly decreased proinflammatory cytokines and neuronal death without attenuating the DFP-induced inhibition of AChE activity. Taken together, the data demonstrate the ability of AEOL10150 to serve as an adjunct therapy for OP and/or nerve agent exposure and highlight the importance of oxidative stress as a therapeutic target after OP exposure. In this study, we provide a novel target for OP intoxication and therapeutic agent for ameliorating its downstream deleterious consequences.
Some pharmacokinetic analyses of AEOL10150 have been done in previous studies in rats exposed to sulfur mustard or chlorine gas (McGovern et al., 2011; O’Neill et al., 2010). Additionally, data from this laboratory have demonstrated that AEOL10150 penetrates the blood-brain barrier and maintains a relatively stable concentration in the brain of rats treated with pilocarpine (Pearson et al., 2015). Here, we extend the literature on this compound to show that it can access the blood-brain barrier and maintain relatively stable levels in brain after either intravenous or s.c. injection in DFP-treated rats. The levels of AEOL10150 in DFP-treated rats were similar to those in saline-treated rats. The time to reach maximum plasma levels (Tmax) and plasma half-life (T1/2) in DFP-treated rats was used to establish the dosing regimen of 5 mg/kg (s.c.) every 4 h. It was interesting to observe that DFP exposure had some effects on AEOL10150 pharmacokinetic profile including a shortened T1/2 and Tmax and a greater Cmax. These data suggest that DFP treatment may be affecting the rate of AEOL10150 uptake from the s.c. injection site. The observation that AEOL10150 can accumulates in target tissues after exposure to chemical agents and nerve agent surrogates make the compound potentially useful in circumstances of intentional or accidental exposure.
GSH is the most abundant thiol containing nonenzymatic antioxidant in tissues including the brain and plays an important role in preventing oxidative damage (Meister and Anderson, 1983). The levels of GSH, GSSG and ratio of this redox couple, GSH/GSSG, serve as major intracellular indicators of redox imbalance and oxidative stress markers in cells and in vivo (Reed and Saage, 1995). 3-NT is an indicator of nitration to tyrosine residues which is induced by the highly reactive nitrogen species peroxynitrite (ONOO−) (Sawa et al., 2000). Increased 3-NT has been well established as a biomarker of oxidative/nitrative protein damage and reported to accumulate in brain tissue isolated from Parkinson’s disease (PD) patients and in rodent models of PD, cerebral ischemia and temporal lobe epilepsy (Coeroli et al., 1998; Giasson et al., 2000; Pennathur et al., 1999; Ryan et al., 2014). In this study, we show a time and region-dependent increase in these oxidative stress markers after DFP exposure. Specifically, markers of oxidative stress were detected at 24 and 48 h, but not at 6 or 12 h, in the hippocampus and piriform cortex but not in the frontal cortex. This time course of cellular redox status disruption is similar to what has been previously reported in the kainate model of TLE (Liang and Patel, 2006; Ryan et al., 2014). We have previously shown the efficacy of AEOL10150 against oxidative/nitrative stress and neurodegeneration induced by chemoconvulsant agents, kainate and pilocarpine when administered at a 5 mg/kg s.c. every 4 h dosing paradigm (Liang et al., 2016; Pearson et al., 2015). This same dosing paradigm resulted in significant protection against pilocarpine-induced mortality (Pearson-Smith et al., 2017). This, in conjunction with the pharmacokinetic data detailed above lead us to determine the therapeutic window of AEOL10150 against DFP-induced oxidative stress markers. Indices of oxidative stress and altered cellular redox status were measured 24 h after DFP in the hippocampus, piriform cortex and plasma when AEOL10150 was given at various times after SE initiation. When treatment with AEOL10150 was initiated 5 min after DFP-induced SE onset, indices of oxidative stress in the hippocampus, piriform cortex, and plasma were significantly attenuated while mortality seemed relatively unaffected, establishing this as the ideal time point to ascertain the role of oxidative stress in contributing to DFP-induced neuronal loss and neuroinflammation.
Previous studies have demonstrated that SE induced by OP agents or chemoconvulsant results in activation of glial cells and release of proinflammatory cytokines (Flannery, et al., 2016; Li et al., 2015; Ravizza et al., 2008; Vezzani and Granata, 2005). This neuroinflammation and associated neurodegeneration extends beyond the acute DFP intoxication stage and can persist for weeks after the initial exposure, contributing to poor neurological outcomes (Flannery et al., 2016). Oxidative stress is known to contribute to both neuroinflammation and neurodegeneration and targeting of this process in a post-OP exposure paradigm is a novel therapeutic avenue. DFP induced a robust cytokine release, particularly TNF-α, IL-6, and KC/GRO which was greater in the piriform cortex than in the hippocampus, consistent with oxidative/nitrative makers and the observed pattern of neurodegeneration. The DFP-induced increase of TNF-α, and IL-6 in the hippocampus and the increased TNF-α, IL-1β, IL-6, and KC/GRO in the piriform cortex was significantly attenuated by AEOL10150 treatment. Additionally, treatment with the catalytic antioxidant significantly attenuated DFP-induced neurodegeneration in piriform cortex, amygdala and hippocampus. These results suggest that oxidative processes may contribute to DFP-induced neuroinflammation and neurodegeneration and clearly indicate that antioxidant treatment can exert neuroprotection against DFP-induced toxicity.
In conclusion, the results reported here suggest targeting of oxidative stress in the DFP model of OP intoxication can improve neuroinflammation and neurodegeneration. Although neuroinflammation is positively associated with seizure severity, oxidative stress has been observed in humans and animals exposed to subconvulsive doses of DFP (Lukaszewicz-Hussain, 2008; Muniz et al., 2008; Ranjbar et al., 2002; Trevisan et al., 2008; Zaja-Milatovic, et al., 2009). Thus, targeting of oxidative stress may provide benefits under a wide variety of exposure conditions.
FUNDING
This research work was supported by the National Institutes of Health (1U01NS083422-02 to M.P). Dr Day and Dr Patel are consultants for Aeolus Pharmaceuticals which develops catalytic antioxidants for human diseases including the compound used in this work. Dr Day holds equity in Aeolus Pharmaceuticals.
REFERENCES
- Coeroli L., Renolleau S., Arnaud S., Plotkine D., Cachin N., Plotkine M., Ben-Ari Y., Charriaut-Marlangue C. (2002). Nitric oxide production and perivascular tyrosine nitration following focal ischemia in neonatal rat. J. Neurochem. 70, 2516–2525. [DOI] [PubMed] [Google Scholar]
- de Araujo Furtado M., Rossetti F., Chanda S., Yourick D. (2012). Exposure to nerve agents: From status epilepticus to neuroinflammation, brain damage, neurogenesis and epilepsy. Neurotoxicology 33, 1476–1490. [DOI] [PubMed] [Google Scholar]
- Ellman G. L., Courtney K. D., Andres V. Jr, Feather-Stone R. M. (1961). A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95. [DOI] [PubMed] [Google Scholar]
- Flannery B. M., Bruun D. A., Rowland D. J., Banks C. N., Austin A. T., Kukis D. L., Li Y., Ford B. D., Tancredi D. J., Silverman J. L. et al. , (2016). Persistent neuroinflammation and cognitive impairment in a rat model of acute diisopropylfluorophosphate intoxication. J. Neuroinflammation 13, 267.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson B. I., Duda J. E., Murray I. V., Chen Q., Souza J. M., Hurtig H. I., Ischiropoulos H., Trojanowski J. Q., Lee V. M. (2000). Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290, 985–989. [DOI] [PubMed] [Google Scholar]
- Jett D. A. (2010). Finding new cures for neurological disorders: A possible fringe benefit of biodefense research? Sci. Transl. Med. 2, 23ps12–23ps12. 10.1126/scitranslmed.3000752). [DOI] [PubMed] [Google Scholar]
- Jett D. A. (2007). Neurological aspects of chemical terrorism. Ann. Neurol. 61, 9–13.http://dx.doi.org/10.1002/ana.21072 [DOI] [PubMed] [Google Scholar]
- Kachadourian R., Johnson C. A., Min E., Spasojevic I., Day B. J. (2004). Flavin-dependent antioxidant properties of a new series of meso-n, n′-dialkyl-imidazolium substituted manganese(iii) porphyrins. Biochem. Pharmacol. 67, 77–85. [DOI] [PubMed] [Google Scholar]
- Kachadourian R., Menzeleev R., Agha B., Bocckino S. B., Day B. J. (2002). High-performance liquid chromatography with spectrophotometric and electrochemical detection of a series of manganese(iii) cationic porphyrins. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 767, 61–67. [DOI] [PubMed] [Google Scholar]
- Li Y., Lein P. J., Ford G. D., Liu C., Stovall K. C., White T. E., Bruun D. A., Tewolde T., Gates A. S., Distel T. J. et al. , (2015). Neuregulin-1 inhibits neuroinflammatory responses in a rat model of organophosphate-nerve agent-induced delayed neuronal injury. J. Neuroinflammation 12, 64.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L.-P., Patel M. (2006). Seizure-induced changes in mitochondrial redox status. Free Radic. Biol. Med. 40, 316–322.http://dx.doi.org/10.1016/j.freeradbiomed.2005.08.026 [DOI] [PubMed] [Google Scholar]
- Liang L. P., Huang J., Fulton R., Day B. J., Patel M. (2007). An orally active catalytic metalloporphyrin protects against 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine neurotoxicity in vivo. J. Neurosci. 27, 4326–4333.http://dx.doi.org/10.1523/JNEUROSCI.0019-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L. P., Patel M. (2016). Plasma cysteine/cystine redox couple disruption in animal models of temporal lobe epilepsy. Redox. Biol. 9, 45–49.http://dx.doi.org/10.1016/j.redox.2016.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukaszewicz-Hussain A. (2008). Subchronic intoxication with chlorfenvinphos, an organophosphate insecticide, affects rat brain antioxidative enzymes and glutathione level. Food Chem. Toxicol. 46, 82–86.http://dx.doi.org/10.1016/j.fct.2007.06.038 [DOI] [PubMed] [Google Scholar]
- McElroy P. B., Liang L. P., Day B. J., Patel M. (2017). Scavenging reactive oxygen species inhibits status epilepticus-induced neuroinflammation. Exp. Neurol. 298, 13–22.http://dx.doi.org/10.1016/j.expneurol.2017.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern T., Day B. J., White C. W., Powell W. S., Martin J. G. (2011). Aeol10150: A novel therapeutic for rescue treatment after toxic gas lung injury. Free Radic. Biol. Med. 50, 602–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister A., Anderson M. E. (1983). Glutathione. Annu. Rev. Biochem. 52, 711–760.http://dx.doi.org/10.1146/annurev.bi.52.070183.003431 [DOI] [PubMed] [Google Scholar]
- Muniz J. F., McCauley L., Scherer J., Lasarev M., Koshy M., Kow Y. W., Nazar-Stewart V., Kisby G. E. (2008). Biomarkers of oxidative stress and DNA damage in agricultural workers: A pilot study. Toxicol. Appl. Pharmacol. 227, 97–107. [DOI] [PubMed] [Google Scholar]
- O'Neill H. C., White C. W., Veress L. A., Hendry-Hofer T. B., Loader J. E., Min E., Huang J., Rancourt R. C., Day B. J. (2010). Treatment with the catalytic metalloporphyrin aeol 10150 reduces inflammation and oxidative stress due to inhalation of the sulfur mustard analog 2-chloroethyl ethyl sulfide. Free Radic. Biol. Med. 48, 1188–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson J. N., Rowley S., Liang L. P., White A. M., Day B. J., Patel M. (2015). Reactive oxygen species mediate cognitive deficits in experimental temporal lobe epilepsy. Neurobiol. Dis. 82, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson-Smith J. N., Liang L. P., Rowley S. D., Day B. J., Patel M. (2017). Oxidative stress contributes to status epilepticus associated mortality. Neurochem. Res. 42, 2024–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennathur S., Jackson-Lewis V., Przedborski S., Heinecke J. W. (1999). Mass spectrometric quantification of 3-nitrotyrosine, ortho-tyrosine, and o, o'-dityrosine in brain tissue of 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-treated mice, a model of oxidative stress in parkinson's disease. J. Biol. Chem. 274, 34621–34628. [DOI] [PubMed] [Google Scholar]
- Pouliot W., Bealer S. L., Roach B., Dudek F. E. (2016). A rodent model of human organophosphate exposure producing status epilepticus and neuropathology. Neurotoxicology 56, 196–203.http://dx.doi.org/10.1016/j.neuro.2016.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine R. J. (1972). Modification of seizure activity by electrical stimulation: Ii. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32, 281–294.http://dx.doi.org/10.1016/0013-4694(72)90177-0 [DOI] [PubMed] [Google Scholar]
- Ranjbar A., Pasalar P., Abdollahi M. (2002). Induction of oxidative stress and acetylcholinesterase inhibition in organophosphorous pesticide manufacturing workers. Hum. Exp. Toxicol. 21, 179–182.http://dx.doi.org/10.1191/0960327102ht238oa [DOI] [PubMed] [Google Scholar]
- Ravizza T., Gagliardi B., Noe F., Boer K., Aronica E., Vezzani A. (2008). Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol. Dis. 29, 142–160. [DOI] [PubMed] [Google Scholar]
- Reed D. J., Savage M. K. (1995). Influence of metabolic inhibitors on mitochondrial permeability transition and glutathione status. Biochim. Biophys. Acta 1271, 43–50.http://dx.doi.org/10.1016/0925-4439(95)00008-R [DOI] [PubMed] [Google Scholar]
- Rowley S., Liang L. P., Fulton R., Shimizu T., Day B., Patel M. (2015). Mitochondrial respiration deficits driven by reactive oxygen species in experimental temporal lobe epilepsy. Neurobiol. Dis. 75, 151–158.http://dx.doi.org/10.1016/j.nbd.2014.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K., Liang L. P., Rivard C., Patel M. (2014). Temporal and spatial increase of reactive nitrogen species in the kainate model of temporal lobe epilepsy. Neurobiol. Dis.ease 64, 8–15.http://dx.doi.org/10.1016/j.nbd.2013.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa T., Akaike T., Maeda H. (2000). Tyrosine nitration by peroxynitrite formed from nitric oxide and superoxide generated by xanthine oxidase. J. Biol. Chem. 275, 32467–32474.http://dx.doi.org/10.1074/jbc.M910169199 [DOI] [PubMed] [Google Scholar]
- Schmued L. C., Hopkins K. J. (2000). Fluoro-jade b: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 874, 123–130.http://dx.doi.org/10.1016/S0006-8993(00)02513-0 [DOI] [PubMed] [Google Scholar]
- Shih T.-M., Duniho S. M., McDonough J. H. (2003). Control of nerve agent-induced seizures is critical for neuroprotection and survival. Toxicol. Appl. Pharmacol. 188, 69–80. [DOI] [PubMed] [Google Scholar]
- Sisó S., Hobson B. A., Harvey D. J., Bruun D. A., Rowland D. J., Garbow J. R., Lein P. J. (2017). Editor’s highlight: Spatiotemporal progression and remission of lesions in the rat brain following acute intoxication with diisopropylfluorophosphate. Toxicol. Sci. 157, 330–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisan R., Uliano-Silva M., Pandolfo P., Franco J. L., Brocardo P. S., Santos A. R. S., Farina M., Rodrigues A. L. S., Takahashi R. N., Dafre A. L. (2008). Antioxidant and acetylcholinesterase response to repeated malathion exposure in rat cerebral cortex and hippocampus. Basic Clin. Pharmacol. Toxicol. 102, 365–369. [DOI] [PubMed] [Google Scholar]
- Vargas J. R., Takahashi D. K., Thomson K. E., Wilcox K. S. (2013). The expression of kainate receptor subunits in hippocampal astrocytes after experimentally induced status epilepticus. J. Neuropathol. Exp. Neurol. 72, 919–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A., Granata T. (2005). Brain inflammation in epilepsy: Experimental and clinical evidence. Epilepsia 46, 1724–1743.http://dx.doi.org/10.1111/j.1528-1167.2005.00298.x [DOI] [PubMed] [Google Scholar]
- Zaja-Milatovic S., Gupta R. C., Aschner M., Milatovic D. (2009). Protection of dfp-induced oxidative damage and neurodegeneration by antioxidants and nmda receptor antagonist. Toxicol. Appl. Pharmacol. 240, 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]