

Abstract
Extensive microbial gene flows affect how we understand virology, microbiology, medical sciences, genetic modification, and evolutionary biology. Phylogenies only provide a narrow view of these gene flows: plasmids and viruses, lacking core genes, cannot be attached to cellular life on phylogenetic trees. Yet viruses and plasmids have a major impact on cellular evolution, affecting both the gene content and the dynamics of microbial communities. Using bipartite graphs that connect up to 149,000 clusters of homologous genes with 8,217 related and unrelated genomes, we can in particular show patterns of gene sharing that do not map neatly with the organismal phylogeny. Homologous genes are recycled by lateral gene transfer, and multiple copies of homologous genes are carried by otherwise completely unrelated (and possibly nested) genomes, that is, viruses, plasmids and prokaryotes. When a homologous gene is present on at least one plasmid or virus and at least one chromosome, a process of “gene externalization,” affected by a postprocessed selected functional bias, takes place, especially in Bacteria. Bipartite graphs give us a view of vertical and horizontal gene flow beyond classic taxonomy on a single very large, analytically tractable, graph that goes beyond the cellular Web of Life.
Keywords: microbial evolution, bipartite graph, virus, network
Introduction
A major problem for biology is to understand short and long taxonomical range sharing of genes, be they acquired by vertical descent or introgression (Beiko et al. 2005; Kunin et al. 2005; Puigbo et al. 2010; Andam and Gogarten 2011; Kloesges et al. 2011; Popa et al. 2011; Smillie et al. 2011; Cong et al. 2017). Another problem is to acknowledge and evaluate the role of viruses (and other mobile genetic elements) in the evolution of cells (Simmonds et al. 2017). Considered from a genetic perspective, mobile elements impact cellular evolution first by moving genes into cells and this often results in a kind of paralogy within microbial communities, when a gene copy is carried on the genome of a mobile element as well as on a chromosome. This process can create new phenotypes (Busby et al. 2013), opportunities for the coming of additional genes in genomes (Roux et al. 2015), and contribute to the resilience of microbial communities by dispersing physiologically important genes on multiple vectors (Sullivan et al. 2010; Biller et al. 2015). Moreover, the number of prophages inserted in prokaryotic genomes (and their biological impact on their hosts) is likely underestimated (Roux et al. 2015). However, we have a poor framework to study dynamics of gene flow between mobile elements and cells.
Here, we exploited a novel approach to study gene sharing in the microbial world, which expands over the numerous approaches currently being developed. These latter display some limits when it comes to study together gene sharing between mobile genetic elements and cells. Tracking the multitude of transfer paths (be they vertical or horizontal) is difficult but important, especially since sets of genes can be transferred together for functional reasons (Jain et al. 1999). On the one hand, phylogenetic methods such as the one using highways of gene sharings (Kunin et al. 2005; MacLeod et al. 2005; Beiko et al. 2006; Dagan and Martin 2006) can be used to investigate these genes movements, yet phylogenetic approaches are limited to related entities, that is, related genomes diverged from a last common ancestral genome, which belong to a monophyletic group. The simultaneous study of viruses, plasmids, plasmids and viruses, viruses and cells, plasmids and cells together remains therefore difficult. On the other hand, binary matrices (Nelson-Sathi et al. 2015), similarity networks, such as genome networks (Fondi and Fani 2010; Halary et al. 2010; Tamminen et al. 2012) and bipartite gene–genome networks analyzed with heuristic community detection method (Iranzo, Koonin, et al. 2016; Iranzo, Krupovic, et al. 2016), have provided exciting results. Such networks have been used to test hypotheses about the phylogenetic or environmental drivers of genomic diversity (Kloesges et al. 2011; Cheng et al. 2014; Forster et al. 2015) and the selective advantages of introgressed genes (Bapteste 2014). These approaches can in principle include all key evolutionary players in a common framework (although so far the vast majority of genome networks, binary matrices, and bipartite graphs have been limited to either chromosomes; Kloesges et al. 2011) or genomes of mobile elements (Lima-Mendez et al. 2008; Desnues et al. 2012; Yutin et al. 2013). These binary matrices are mathematically equivalent to bipartite graphs; however, it is more natural to speak in terms of graph when using tools from graph theory.
Bipartite graphs consist of nodes of two fundamentally different kinds. These nodes are connected by edges such that two nodes on either end of an edge are never of the same kind. Bipartite graphs have been successfully used to explore gene–disease relationships (Hwang et al. 2008), the evolution of malaria parasites (Larremore et al. 2013), recipe ingredient data sets (Ahn et al. 2011), and social media networks (Murata 2009). In the context of gene flow, cluster of homologous genes (CHG)–genome networks (fig. 1, panel A) can reveal, like binary matrices, which exact groups of homologous genes are shared exclusively by certain groups of genomes (a pattern called “twins” in graph theory, formally defined as sets of CHG nodes that have identical connectivity to genome nodes). Network analyses can also reveal “articulation points,” that is, CHG nodes that are connected to parts of the graph that otherwise share no CHG nodes.
Fig. 1.
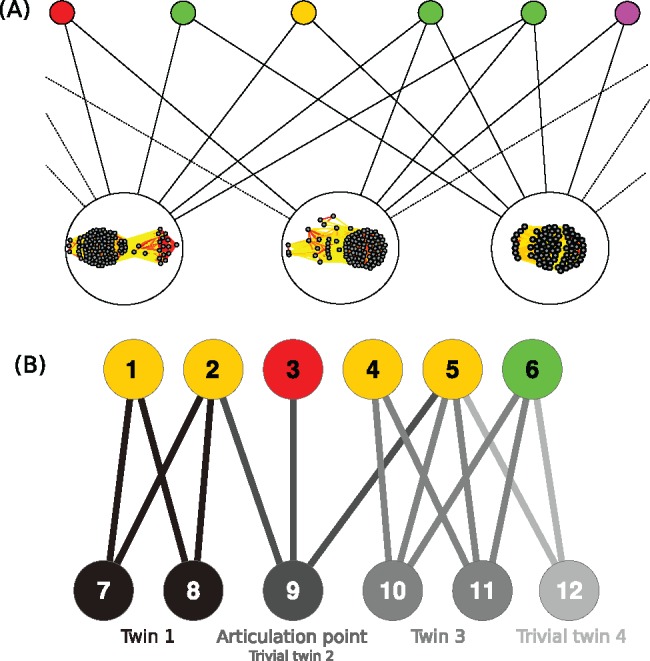
Construction of the bipartite graphs and identification of the twins and articulation points. (A) Construction of the genome-CHG bipartite graphs. Top nodes represent genomes of cells and mobile genetic elements. Bottom nodes represent CHG: we display the corresponding connected component of the sequence similarity network (see Materials and Methods), edge color (from yellow to red) indicates increasing % ID. (B) Bottom twins and articulation points: bottom nodes forming a twin class and their incident edges are drawn in the same shade of gray. Nodes 7 and 8 have the same neighbors (nodes 1 and 2) thus form twin class 1. Twins 2 and 4 are trivial since they contain only one node. Node 9 is an articulation point since its removal disconnects the graph.
Specifically, bipartite graphs can be used to track sets of genes that have been laterally transferred together, genes that unite genomes that otherwise have no homologs in common at a given threshold, and also to uncover biases in transfer and/or retention of genes between mobile elements and cells. Importantly, bipartite graphs can be analyzed in two complementary ways (Barber 2007; Alzahrani and Horadam 2016; Iranzo, Krupovic, et al. 2016; Jaffe et al. 2016). As in (Iranzo, Koonin, et al. 2016), gene–genome networks can be partitioned using heuristic methods of community detections. They can also be decomposed exactly as in (Jaffe et al. 2016), avoiding a heuristic treatment of the graph. In this paper, we looked at twins and at articulation points (fig. 1, panel B). These patterns have indeed the interesting property to be uniquely defined (unlike communities that can strongly depend on the clustering algorithm that has been used), whereas uncovering already interesting biological phenomena.
We applied this approach to 8,214 genomes, thereby extending a former analysis of gene sharing between cells and mobile elements, that identified genetic worlds (Halary et al. 2010). On the one hand, the resulting bipartite web of life effectively generalizes conclusions from microbial evolution. It shows that the web of life is composed of phylogenetically distinct elements, which are genetically intertwined, according to detectable rules: genes are primarily shared between groups of closely related genomes (i.e., taxonomically consistent groups) and between groups of genomes with the same type (i.e., typologically consistent groups, for example, phages with phages and plasmids with plasmids). It also shows that transposases navigate across the branches of the web. The greatest influence on gene sharing was host type with many prokaryotic (cellular) and MGE (acellular) kinds characterized by exclusive gene contents. Moreover, using graph compression, we analyzed “gene externalization” (Corel et al. 2016), a situation which occurs when a CHG is present on at least one extrachromosomal element and at least one chromosome. This observation is different from lateral gene transfer between two cells, because gene externalization occurs between otherwise completely unrelated genomes, that is, viruses, plasmids, and prokaryotes, which do not show a single last common ancestor. We unraveled that gene externalization was especially significant among Bacteria, and mainly driven by gene function, illustrating strong biases in the kind of genes that persist on multiple vectors in this kind of prokaryotes.
Results and Discussion
We initially constructed bipartite graphs from a data set of 382 prokaryotic genomes and 7,832 mobile element genomes. This family-based data set was carefully selected to avoid the sequencing bias in microbial genomics toward Bacteria. In addition, the resulting data set size is amenable to BLAST-based sequence-similarity analyses, and extends over a former study of gene sharing between cells and mobile elements (see supplementary fig. S1, Supplementary Material online for a genome network updated with respect to Halary et al. 2010). In these bipartite graphs, the “top” nodes correspond to the genomes and the “bottom” nodes correspond to CHG, defined at various stringencies (fig. 1, panel A). The stringency parameters of minimum percentage of identity in sequences allowed us to focus, for example, on recent gene family transmissions (i.e., when two sequences could be aligned over ≥ 80% of their mutual length, and were ≥ 95% identical in sequence; ≥95% ID for short). Varying stringency parameters allowed us to consider a variety of evolutionary time scales (see Materials and Methods). Still, the criterion of ≥ 80% mutual cover, critical to identify homologous genes, typically filtered out information about partial similarity, that is, between recombinant gene forms, such as fused genes or remodeled genes (Jachiet et al. 2014; Méheust et al. 2016). This means that our estimates of genetic sharing and the proportion of genes externalization presented below are conservative, restricted to the identification of full-sized (externalized) genes.
An undirected edge connecting a top and a bottom node indicated that a member of a CHG was found in a genome. Thus, these graphs include simultaneously several levels of organization (showing genes and genomes), and several agents (showing chromosomes and plasmids or viruses). Hence, they provide information about the distributions of CHGs across a broad range of genomes. As in any inference of comparative genomics however, the distribution of homologs across taxa only approximates actual gene exchanges, possibly because of the size of our sample, but also since intermediate unknown players are likely (see, for instance, those discovered lately in Hug et al. 2016). Despite these limits, the structure of these graphs is already very informative.
The network is amenable to standard structural analyses. The CHG sizes follow a power-law-like distribution (supplementary fig. S2-1, Supplementary Material online). The node degree distribution in these graphs differs according to the node type, consistently with (Iranzo, Krupovic, et al. 2016). The degree of a genome represents the part of the genome that is shared at the given similarity level. The degree of a CHG represents the number of genomes in which a given CHG is found. The degree distribution of CHGs seems to display a power law behavior, whereas genomes show a different degree distribution. Viruses and plasmids display a subpower law with far less small degree nodes, and a two-mode bump likely inherited from the bimodal size distribution of both types of MGEs. This last feature can also be observed in the results reported by (Iranzo, Krupovic, et al. 2016). This trend is moreover stable with the size of the data set (supplementary fig. S2-2, Supplementary Material online). Beyond these topological features, the network is also amenable to analyses that help understanding microbial evolution.
Connected Component Analysis Reveals Groups of Genomes with Exclusive Gene Pools
For each network at a given stringency threshold, we first enumerated all its connected components (CCs), that is, all sets of nodes for which there is always an interconnecting path. These CCs represent groups of genomes associated with an exclusive pool of CHGs, that is, a CHG found in a CC is by definition absent in any other CC. The robust recovery of multiple CCs (522 in the graph at ≥ 95% ID and 156 in the graph at ≥ 30% ID, also see supplementary fig. S1, Supplementary Material online) is consistent with the genetic worlds identified in (Halary et al. 2010), albeit with a now much larger data set and a different network approach (supplementary fig. S1, Supplementary Material online). The discrete nature of this graph suggests a discontinuity in vertical and horizontal transmission of full-sized genes between genomes belonging to different CCs. This barrier may reflect phylogenetic isolation, ecological isolation, the use of an alternative genetic code or quite simply the nonexhaustive data set of genes and genomes at our disposal. For example, the CC in supplementary figure S2, Supplementary Material online, illustrates the case of the Spiroplasma phages, which are characterized by the alternative use of the codon UGA to encode Tryptophan instead of “STOP” (i.e., the Mycoplasma/Spiroplasma code). The taxonomic homogeneity of this CC suggests that these phages have been exclusively sharing a unique pool of genes, in effect privatized by their own lineage (McInerney et al. 2011). Note that, conversely, members of a given CC do not necessarily directly share a CHG, meaning that even genomes belonging to the same CC are not necessarily connected by vertical or horizontal gene transmission. Indeed, our bipartite graphs also display a giant CC (gCC), encompassing 6,362 (i.e., 80.1%) genomes and 80,136 (99%) CHGs (at ≥ 30% ID) (supplementary table S1 and fig. S4-1, Supplementary Material online). This single gCC include genomes that have no homologous genes in common, yet participate in a giant network of gene sharing. The ability to reconstruct such a pattern is a significant advantage associated with the use of bipartite graphs.
Twin CHG Are Likely Genetic Public Goods
To understand the coinheritance of CHGs, and also to provide us with a tool for the study of phenotype evolution when phenotypes are not associated with a single monophyletic taxonomic group, we analyzed each CC, including the gCC, at a more fine-grained level, by enumerating all the twins of bottom nodes (BT) within these connected components (fig. 1, panel A). Twins are nodes with identical sets of neighbors in a graph. BTs represent CHGs that are exclusively present in exactly the same set of genomes. Therefore, a group of genes that are cotransmitted, vertically and/or horizontally (from a common ancestor or via LGT) within a club of genomes, will be detectable as a BT (above a sufficient similarity level, see supplementary fig. S3, Supplementary Material online and below). We verified that the compositions of our BTs were significantly different from the ones expected from random networks (empirical adjusted p value = 4.76 × 10−3, on 1,165 simulations with permuted genome attributions for genes, see Materials and Methods) and robust (see proportions for different subsets of data in supplementary fig. S4, Supplementary Material online). This observation confirmed that there is an evolutionary structure in the network, but it cannot be used to determine the respective strengths of the vertical and horizontal modes of inheritance.
Detecting individual or sets of CHGs shared by many genomes with otherwise totally distinct gene contents (at a given similarity threshold) is essential to track long-distance horizontal gene transmission across the web of life. We demonstrated that this detection can be achieved via exact graph compression. We reduced the bipartite graph by grouping together BT nodes into bottom metanodes, and simplified it further by removing all BTs that were present in only one genome (see Materials and Methods). We did not further exploit here the information regarding the number of paralogs within the CHG contributing to the metanodes. Notably, the result of this graph reduction is unique and robust, that is, it does not depend from the order in which twins are merged. This merging produces a quotient, BT-free, bipartite graph with no loss of information due to this compression. It is then trivial to enumerate all bottom articulation points (BAPs) in such reduced graphs, that is, all nodes whose removal would increase the number of connected components. Although strictly topological, the notion of BAPs could in principle help to detect public genetic goods (McInerney et al. 2011), that is, genetic material that is being shared by taxonomically distant genomes, which possibly benefit from the properties these shared genes confer, for some other reason than genealogy (i.e., genes coding for environmental adaptation or others hitch-hiking with them…). Around 16% of the articulation points in the network at ≥ 30% ID (and up to 71% at ≥ 90% ID) were proposed as horizontally transferred according to our horizontality test with majority as decision rule (Materials and Methods).
We report for instance the case of the 3′-phosphoadenosine 5′-phosphosulfate sulfotransferase (PAPS reductase)/FAD synthetase CHG, shared by the Gram-positive bacterium Ruminococcus bromii and the Gram-negative bacterium Fibrobacter succinogenes, forming a BAP in our graph at ≥ 90% ID. This CHG encodes an enzyme with the rare ability to store two electrons without the need for cofactor or prosthetic groups, which likely enhances the success rate of transfer for this CHG in the rumen (supplementary fig. S5, Supplementary Material online). At a stringency of ≥ 90% ID, 56 BAP nodes (out of 811 BAP nodes) encompass transposases which, as the graph suggests (supplementary tables S3 and S4, Supplementary Material online), possess the capability to move across distantly related genomes (Hooper et al. 2009).
The Topology of the Web of Life Shows Patterns of Gene Transmission
Simple graph patterns in a CHG-genome network subjected to exact decomposition are already sufficient to provide abundant biological information. Detecting recurrent patterns in the compressed bipartite graphs of this data set of 382 prokaryotic genomes and 7,832 mobile elements (3,613 viruses and 4,219 plasmids) confirmed prior knowledge about vertical or horizontal gene transmission, whereas extending these conclusions to a more comprehensive data set. More precisely, a single analysis of CC and BT analyses (supplementary figs. S6-1 and S6-2, Supplementary Material online, also supplementary tables S1–S7, Supplementary Material online) generalize observations about several rules of gene transmission, that had been made on separate studies (Beiko et al. 2005; Kunin et al. 2005; MacLeod et al. 2005;Hooper et al. 2009; Halary et al. 2010; Puigbo et al. 2010; Kloesges et al. 2011; Schliep et al. 2011; Smillie et al. 2011; Desnues et al. 2012; Tamminen et al. 2012; Busby et al. 2013; Yutin et al. 2013; Iranzo, Koonin, et al. 2016; Iranzo, Krupovic, et al. 2016; Jaffe et al. 2016; Popa et al. 2017).
First, the vast majority of CCs and BTs were composed of genomes consistent by type of hosts, for all stringency thresholds. For example, at ≥ 90% ID, 94.26% of the CCs, and 92.01% of BTs showed gene sharing between genomes of the same type (i.e., either exclusively cellular, exclusively viral or exclusively plasmid, see fig. 2). In addition, not only were the vast majority of CCs and BTs consistent with genome type but the constituent genomes were also generally taxonomically consistent (supplementary fig. S6-1 and tables S1 and S2, Supplementary Material online), for all stringency thresholds. For example, at ≥ 90% ID, 78.5% of the CCs, and 99% of the BTs that contain prokaryotes (i.e., 19% of all BTs) showed gene sharing among members of the same phylum (as defined independently by the NCBI taxonomy). Overall, this strong taxonomic signal reflects the fact that distantly related genomes have rather different gene contents, which is consistent with the relatively independent evolution of various kinds of cellular organisms in the web of life (Halary et al. 2010). We confirmed this disconnection of the living world (in terms of gene content) by plotting the distribution of CHGs across taxa using a heatmap, constructed on the sole criterion of ≥ 80% mutual coverage (see supplementary fig. S11, Supplementary Material online), in order not to increase the differences between taxa, which could artifactually happen at high stringency thresholds when one considers that two genomes display different gene contents, whereas their seemingly different CHGs are simply divergent groups of related sequences belonging to common ancient gene families. This approach showed that genomes from different lineages use genuinely different sets of genes, and that very few CHGs are shared widely between prokaryotes (in agreement with Ku et al. 2015). This approach was also used for the identification of Exclusively Shared CHGs (ESCHG), see below).
Fig. 2.

Support composition of twins and CHGs. The taxonomy of the carriers of the genes in a CHG is called its support composition. For every similarity threshold (on the x-axis), we report how many CHGs (on the right) or twins of CHGs (on the left) are exclusively found in Archaea, in Bacteria, in both kinds of prokaryotes (“mixed prokaryotes”), in viruses only, in plasmids only, in both kinds of MGE (“mixed MGE”), or in both cellular and mobile elements (“externalized”).
Second, within these major taxonomic lineages, genomic evolution is highly reticulated. We verified this by computing, for each group of closely related genomes in this data set, the number of CHGs that are shared by members of this group and exclusively by them. The size of these ESCHG amounts to the percentage of BTs (i.e., sets of exclusive CHGs) found exclusively in genomes belonging to the taxonomic group. This notion differs from that of core genome (at the given similarity level), since ESCHGs are not present outside this taxonomic group and the core genome can include CHGs that are also present in other groups from different lineages (e.g., housekeeping genes). Notably, categories featuring substantial relative amounts of core gene families are environmentally (Thermococci, Halobacteria) or metabolically (Methanococci, Cyanobacteria) specific lineages. The fraction of these ESCHG that are core is typically small (fig. 3): at most ∼18% for the 15 Methanococci and the 15 Thermococci, and <5% for the 16 Cyanobacteria, and 25 Halobacteria contained in our data set.
Fig. 3.

ESCHG and core. The “shared pangenome” of a lineage is composed of all CHGs that are shared by at least two genomes and contain at least one member of the lineage. (A) For each taxonomic group, we report the percentage of CHGs forming the shared pangenome that are core exclusive, exclusive but noncore and not exclusive. (B) For each taxonomic group, we report the percentage of the shared pangenome that is comprised of core exclusive, and core but not exclusive CHGs. The total height of the bar represents the core itself.
Thus, most BTs are not associated with all genomes of a taxonomic group, consistent with a high turnover of CHGs, at least of lineage-specific genes, within prokaryotic genomes. Jackknife analyses show that these trends are robust with respect to the size of the data set (supplementary fig. S4 and tables S5–S7, Supplementary Material online).
We also implemented a conservative horizontality test, which exploits the network information to determine which BTs have likely been horizontally transferred between cells (see Materials and Methods). About 80% of the taxonomically consistent BTs and 61% of the taxonomically inconsistent BTs at ≥70% ID were considered as laterally inherited for the majority decision rule (supplementary fig. S7, Supplementary Material online). These numbers represent a minimum estimate of horizontal gene transfer, since the absence of evidence for transfer for the other BTs does not mean that these other BTs were necessarily vertically inherited. They could also have been transferred between close relatives.
Our estimates of transferred genes/genomes are consistent with some published estimates found in the literature, although possibly a bit more conservative. Overall, our analysis supports the generally admitted notion that horizontal gene transfer deeply impact microbial evolution (Raymond et al. 2003; Zhaxybayeva et al. 2006; Kloesges et al. 2011; Popa et al. 2011; Koonin 2016), with rates that vary across genomes (Koonin et al. 2001; Dagan et al. 2008; Kloesges et al. 2011). Thresholding our networks allow us to compare our results with reports on both recent HGTs, as well as on cumulative HGTs. For example, Kloesges et al. (2011) reported that 9.6% of the genes within a prokaryotic genome were recently acquired, Lawrence and Ochman (2002) reported 18% of genes recently acquired by HGT in E. coli, or Hernández-López et al. (2013) proposed that up to 25% of core genes were recently transferred in Rickettsiales. Consistently, we proposed that up to 15% of the genes of the tested genomes have been recently transferred (i.e., showing ≥ 90% similarity between donor and hosts genomes, supplementary fig. S7-1, Supplementary Material online). At ≥ 30% similarity, cumulative effects of HGT become noticeable (supplementary fig. S7-1, Supplementary Material online), affecting in average 41.73% of the genomes and up to 68.34% of a genome (Starkeya novella DSM 506). These values are in the same range that the ones suggested by other publications, that is, that on an average, at least 81% ± 15% of the genes in each prokaryotic genome were involved in HGT during their history (Dagan et al. 2008), and see also (Kloesges et al. 2011), who reported that 75% of the genes of a genome were on an average affected by one HGT, and (Koonin 2015), who suggested that 60% of the information flux between prokaryotic genomes is not tree-like. Our values are however a bit higher than analyses by (Snel et al. 2002; Beiko et al. 2005; Kunin et al. 2005), that reported HGT rates varying from 20% to 39% of the prokaryotic gene families analyzed by phylogeny.
We verified with an expanded data set (Materials and Methods) that the trends described earlier were robust. Namely, we observed the following: 1) taxonomically homogeneous CCs (only ≥90% ID) and taxonomically homogeneous BTs (33–66% of all BTs, depending on the identity threshold); 2) taxonomically heterogeneous CCs (the gCC and up to 19 CCs at ≥95% ID) and taxonomically heterogeneous BTs (7–38% of all BTs); 3) typologically homogeneous CCs (from 455 at ≥95% ID to 151 at ≥30% ID) and typologically homogeneous BTs (70–83% of all BTs); 4) typologically heterogeneous CCs (the gCC and up to 42 at ≥95% ID) and typologically heterogeneous BTs (16–29% of all BTs). We also detected a broad range of externalized genes in all these prokaryotic genomes (supplementary fig. S5-3 and S5-4, Supplementary Material online). However, detailed analysis of this broader data set was out of the scope of the present paper.
Compressing Bipartite Graphs Detects Novel Instances of the Mobilization of Public Genetic Goods
Within the likely transferred BTs, we focused next on some with potential adaptive content (Karcagi et al. 2016). In the graph at ≥ 90% ID, our very discrete sampling of genomes contained 20 BTs distributed on genomes from different phyla. In the graph at ≥ 30% ID, including more ancient sharing events, there were 12,864 BTs (i.e., 30.9% of all BTs) grouping genomes from different phyla. Such taxonomically heterogeneous BTs point to candidate genetic public goods, transferred over large phylogenetic distances, that is, since these sequences are used by phylogenetically heterogeneous hosts, which was confirmed by our horizontality test (supplementary fig. S8, Supplementary Material online). For example, Twin 7227 is a CHG involved in cell wall—peptidoglycan—lysis. The protein is found in viruses and bacteria and is important in degrading the cell wall—either for the purposes of infecting a bacterium or for cell division. This kind of “cell puncturing device” is likely to enhance horizontal transfer. Twin 3034 is the LexA protein, which in purified form acts as a repressor of RecA and itself. This protein can function to reduce the level of recombination and SOS-mediated response from an organism (Pant et al. 2016). The SOS response is triggered by DNA damage, as is RecA. Therefore the function of this twin seems to be to repress recombination and to stop DNA repair processes which might prevent the integration of a sequence into a genome. Other interesting examples stem from these analyses. Twin 7401 at ≥ 90% ID corresponds to a particular prokaryotic compartment involved in the carbon fixation from atmospheric CO2 called the carboxysome (Yeates et al. 2008), shared by taxonomically divergent bacteria (two Cyanobacteria and two Gammaproteobacteria). The carboxysome is also present as twin 69 (under a sufficiently divergent form as to make a different CHG): this time it is even an articulation point linking one Bacteroidetes, one Chloroflexi, and one Actinobacterium. We also find conspicuous plant nodule associated genes: twin 1436 is a nitrogenase subunit NifH forming a twin for a club of three nodule associated Alphaproteobacteria, and twin 7710 is an articulation point, with a dehydrogenase function, between one Acidobacterium (Candidatus Solibacter usitatus) and two nodule Alphaproteobacteria (Methylocella silvestris and Mesorhizobium australicum). The removal of the articulation point neatly separates the three according to taxonomy, and seems ecologically driven, since all three are soil-dwellers (Chen et al. 2010; Challacombe et al. 2011). This hypothesis is supported by information on the isolation sites, retrieved from the GOLD database (Mukherjee et al. 2017), that is, SE Australia for Candidatus Solibacter usitatus, W Australia for M. australicum, and Europe (Germany) for M. silvestris. This Acidobacterium has moreover a large number of genes associated with MGEs (Challacombe and Kuske 2012; Fondi et al. 2016), and Mesorhizobium australicum harbors a laterally acquired 455.7-kb genomic island, indicating that these genomes are prone to acquire genes. Public goods however are not the only genes that can be shared so broadly. Twin 13016 is a toxin–antitoxin system, a famous “addiction” system (here shared between three plasmids and one phage). Both genes are needed in the genome in order to function. In general, the toxin is long-lived and the antitoxin is short-lived, and keeps the cell safe from the toxin by binding to it. When the genes are removed from the cell, then the short-lived antitoxin breaks down, leaving the toxin to kill the cell. This mechanism removes cells that have been cured of the toxin–antitoxin system, providing an advantage to those cells that have both genes (Gerdes et al. 2005; Otsuka 2016). Maximum likelihood trees reconstructed a posteriori for each of these twins confirmed that the genes discussed here were likely involved in LGT, in agreement with our test of horizontality transfer (supplementary fig. S8, Supplementary Material online).
Transposases Flood the Web of Life but Do Not Persist
We also observed the diffusion of other so-called “selfish” genes. In general, transposases were broadly distributed over MGE and chromosomes, as expected according to, for example, Aziz et al. (2010). Notably, although transposases are not limited to prophages, and all prophages do not encode a transposase, we verified that the number of transposases in prokaryotic genomes did not correlate with the number of inserted phages, confirming that these mobile elements had decoupled dynamics of chromosomal invasion (adjusted R2 coefficient = −0.001), and that chromosomes in our data set are not prone to a general inclusion of these diverse types of MGEs. In the overall graph at ≥ 90% ID, 4.78% of the CCs and 8.03% of the BTs were annotated as containing a transposase, respectively. Interestingly, transposases were overrepresented in BTs mixing different types of genomes (supplementary table S4, Supplementary Material online), because some transposases travel across different host genomes (Hooper et al. 2009). Homologous transposases are indeed known to be found and functional in different hosts, eventually from different domains of life (i.e., the piggyBac transposable element, isolated from a virus, operates in a diversity of eukaryotes; Johnson and Dowd 2014). Likewise, various unrelated studies of genomics have reported the presence of transposases on plasmids (Jones-Dias et al. 2016; Dias et al. 2018) and viruses (Sun et al. 2015; Wilson et al. 2017), occasionally with adaptive hitch-hiking genes (Ahmad et al. 2015; Manageiro et al. 2015; Sabat et al. 2015; Aleksandrzak-Piekarczyk et al. 2016; Ageevets et al. 2017; Sun et al. 2017), disseminating the view that transposases are commonly found on these types of mobile genetic elements. Other works have highlighted the evolutionary interplay between transposases and different types of mobile elements (transpovirons, Koonin and Krupovic 2017; casposons, Krupovic et al. 2017; and retroviruses, Skala 2014). Thus our results, offering a systematic survey of the distribution of transposases across mobile elements, are compatible with this background knowledge.
In our family-based data set (see supplementary tables S3 and S4, Supplementary Material online), transposases were found in 7.21% (888 out of 12,321) of the BTs joining the same type of genomes, and in 17.94% (192 out of 1,070) of the BTs joining different types of genomes (e.g., any combination of virus, plasmids, or chromosomes). Likewise, transposases were found in 3.04% (14 out of 460) of the CCs joining the same type of genomes, and in 21.43% (6 out 28) of the CCs that joined different types of genomes. In about 1/6 of these BTs with heterogeneous phyla, other CHG possibly hitch-hiked with these transposases. This suggests that transposases are actively travelling across the web of life, possibly leveraging over the mobility of other MGEs, but they do not organize the web of life. Indeed, removing annotated transposases from the analyses does not substantially change the topology of the bipartite graph. We also tested that the persistence of the network structure was not due to the CHG that have hitch-hiked with the transposases by removing all the CHG that were associated with transposases (i.e., that share the same BT than transposases, in order to reduce the impact of genes hitch-hiking with transposases). This protocol did not affect the topology of the bipartite graphs (supplementary fig. S6-1 and S6-2 bottom rows, Supplementary Material online), consistent with our claim that transposases do not organize the web of life.
Introgressive evolution has by contrast shaped mobile genetic elements—as can be seen for example in the sharing of very similar genes between viruses and plasmids within the mobilome network (i.e., 8 CCs [out of 488] and 107 BTs [out of 13,391] mixing viruses and plasmids at ≥ 90% ID).
Gene Externalization Levels Are Remarkably High in the Microbial World
Since our networks encode exact information about which genomes share which CHG, we were able to quantify the extent of “gene externalization,” that is, of sharing between chromosomes and extrachromosomal elements (e.g., when a given CHG is connected to two genomes of different kinds). The idea is that externalized genes are copied on different supports (i.e., chromosomes, plasmids, or virus). In that sense, copies of the same gene are encoded in different media (without necessary be lost from their original support). To continue this analogy, externalized genes can also be viewed as “remastered gene copies.” Gene externalization differs from LGT between chromosomes, although it can contribute to LGT when a gene from a chromosome is copied to a MGE and from that MGE to another chromosome (or to the same genome, in the case of autologs; Popa et al. 2017). The difference between gene externalization and LGT means that rules relating to gene externalization may differ from rules relating to LGT. In particular, gene externalization may be random and at a high rate, which would not be visible from LGT analyses, if the host recipient cell selects against the residency of some of the externalized genes (i.e., for example, informational genes may be more externalized than transferred). We observed an impressive proportion of externalized genes in the web of life (fig. 4 and supplementary table S8, Supplementary Material online).
Fig. 4.

Percentage of gene externalization at ≥ 30% ID for the 382 prokaryotes in our data set. The proportion of externalized genes from Bacteria (green triangles) is significantly higher (Student’s t-test, p value < 10−16) than for Archaea (yellow dots), to the notable exception of Haloarchaea (red dots). It is moreover largely uncorrelated with genome size (regression lines with shaded confidence interval at 95%).
More precisely, for our data set, Bacteria generally have higher externalization than Archaea (significant t-test for ≥ 30%, 40%, 50%, and 60% ID, see supplementary table S8, Supplementary Material online). A notable exception to this rule is the Haloarchaea, which is likely explained by their chimeric nature (Nelson-Sathi et al. 2012).
Careful analyses of the genomes with highest externalization proportions (> 60% at ≥ 30% ID, see fig. 4) did not identify structural, ecological nor taxonomical commonalities between these genomes. They were all of high quality (with usually ≥ 11-fold sequence coverage) and their gene content had been carefully investigated (Dunfield et al. 2003; Nandasena et al. 2006; Munk et al. 2011; Huo et al. 2012; Kappler et al. 2012). Some of these genomes have interesting metabolic or physiological properties, like a high trophic versatility in the sulfur-oxidizing α-proteobacterium Starkeya novella DSM 506 (Kappler et al. 2001, 2012; Wang et al. 2016), suggestive of LGT affecting that genome. Likewise, 18 putative horizontally transferred regions only had been described in the endophytic β-proteobacterium Herbaspirillum seropedicae SmR1 (Pedrosa et al. 2011), as well as a plasmid-carried photosynthetic ability in the α-proteobacterium Rhodospirillum rubrum ATC 11170 (Kuhl et al. 1984; Munk et al. 2011). The largest set of detected transferred genes concerned a genomic island of 455.7 kb in the root nodule α-proteobacterium Mesorhizobium australicum WSM2073, a symbiosis island from the original inoculant strain from the host legume (Nandasena et al. 2006). This genomic island only amounts to 7.3% of the genome of Mesorhizobium australicum WSM 2073 (Reeve et al. 2013). Thus, LGT, and some bacteriophages, transposases, genomic islands, and extra chromosomal small-sized plasmids had been described in some (but not all) of the genomes with high externalization proportion and their relatives. However, all these MGE inserted in chromosomes were found in considerably lower proportion than the proportion of externalized genes (supplementary fig. S9-1, Supplementary Material online). Indeed, there was almost no correlation between the externalization proportion and the proportion of inserted phages in these genomes (adjusted R2 coefficient = 0.1392, for the number of externalized genes vs. the number of inserted MGEs), and in prokaryotic genomes in general (adjusted R2 coefficient = 0.11, for the number of externalized genes vs. the number of inserted MGEs). Likewise, there was no correlation between the externalization proportion and the proportion of transposases present in the prokaryotic genomes (adjusted R2 coefficient = 0.07, for the number of transposases vs. the number of externalized genes). Furthermore, externalized genes were scattered across the genomes of their carriers (supplementary fig. S9-2, Supplementary Material online). Therefore, the detection of high externalization proportions describes a substantial novel general phenomenon, which can affect very different genomes.
Gene Externalization Is not random with Respect to Gene Function
We also followed the dynamics of gene externalization for networks of decreasing stringency (assuming a molecular clock, from the most recent to the most ancient externalization events) in order to identify some of externalization rules and to test whether gene externalization is random with respect to gene function. The distributions of the COG categories associated with externalized genes were markedly different, and these differences persisted at different similarity thresholds (fig. 5, and also supplementary fig. S10, Supplementary Material online). The “L” category was abundant among recently externalized genes, suggesting that transposases were among the recent CHG that have moved across different types of host genome. This finding is consistent with the reports of implications of transposons in the evolution of chimeric molecular structures (such as transpovirons, Desnues et al. 2012 and casposons, Béguin et al. 2016). However, full-sized genes from that “L” category do not tend to accumulate in their externalized form in genomes, as evidenced by the way their proportion dropped in graphs with lower % ID. This indicates that these transposases do not persist in their host genomes. By contrast, the proportion of externalized genes from the “M” (membrane biogenesis) and “T” (Signal transduction) categories was smaller for recent events than for events considered over a longer time period (i.e., genes from these categories tended to accumulate progressively in genomes as externalized). Other functional categories, such as “E” (Amino-acid metabolism and transport) and “P” (Inorganic ion metabolism and transport) presented more complex distributions.
Fig. 5.

Distribution of functional categories among externalized genes. Color bars represent the percentage of a given COG category among externalized genes above a given identity threshold (according to the color code on the right side of the figure). On the upper bar, COG categories are grouped by large functional groups (including “Poorly characterized,” which includes COG categories R and S). The “No”/0 class (on the left) refers to the genes for which no COG category was attributed). Note the very conspicuous peak for the “L” category at thresholds ≥ 90% and 95% ID.
Due to gene externalization, the web of life appears, in its prokaryotic parts, as a collection of largely disconnected, isolated, prokaryotic strands, affected by introgression between close relatives, doubled by spider-webs of mobile elements (supplementary fig. S1, Supplementary Material online). One should therefore not be misled by the taxonomical consistency of CCs and BTs. Phylogenetically consistent prokaryotic groups are typically subjected to and sustained by processes that are not simple vertical descent with modification. Gene externalization, from cells to mobile genetic elements, and from mobile genetic elements to cells, may contribute to the high turnover of genes in genomes and their patchy distribution in prokaryotic lineages. The high levels of gene externalization that we report indicate that, collectively, genomes of MGE contain most (possibly all) CHGs from several individual bacterial genomes, which are present, dispersed through fragmented copies, in the unrelated genomes of viruses and plasmids. We predict that as more MGE genomes are sequenced, the percentage of externalized genes per prokaryotic genome will increase, albeit at different rates for different biological functions.
Conclusion
Our work complements the findings of other recent studies using networks. Cong et al. (2017), for example, analyzed genetic exchange communities, by decomposing a k-mer based LGT network using notions of graph theory. They inferred functionally biased LGT between cells from various prokaryotic data sets, with up to 144 chromosomes. Our strategy was similar to this inspiring work, but aimed at a different goal. In contrast with (Cong et al. 2017), we represented a diversity of sharings (vertically transmitted genes, horizontally acquired genes, or simply shared genes when the mode of inheritance could not be determined), between a diversity of hosts (between cells, between cells and MGE, and between MGE). In that regard, we did not attempt to reconstruct genetic exchange communities of prokaryotes, united by LGT, but rather to offer a broader picture of CHG distribution across microbial genomes. This was achieved by two means: first, we used a broader data set than Cong et al. (2017), since a total of 8,214 genomes (7,832 MGEs and 382 chromosomes) were included in the network. Second, we used a different type of network, that is, a genome-CHG bipartite graph. In our analyses, we further focused on genes shared between extrachromosomal elements and chromosomes (externalized genes), because these types of sharings would not be necessarily detected by the identification of LGT between cells, whereas our work shows that the sharing of genes between chromosomal and extrachromosomal elements constitutes an important process of microbial communities, which is also functionally biased. Namely, homologous genes which are present, after selection, on both chromosomal and extrachromosomal elements have biased functional profiles, in particular a high turn-over of genes associated with the L category, because genes from this COG category, whereas overrepresented in the pools of conserved externalized genes are less abundant in the pools of divergent externalized genes than genes with, for example, the “M” and “T” functions. Thus, genes that are recently externalized are not enriched in metabolism, regulation, and transport, contrasting with the genes that Cong et al. reported as overrepresented in LGT within genetic exchange communities. Therefore, our work underscores that two distinct introgressive processes affect the evolution of microbial communities, that is, gene externalization and LGT.
Our findings also strongly echoe with the remarkable study by Popa et al. (2017). These authors used a bipartite CHG-genome network (in their case, directed between donors and hosts), with 3,982 prokaryotic genomes and phages genomes (including prophages of prokaryotic chromosomes, individualized as nodes in their network) to investigate lateral gene transfer by transduction. These authors report that transduction is mostly restricted between closely related donors and recipients, consistent with experimental observations and with our observation that viral genomes are generally peripheral on the genome network, that is, associated with specific cellular host lineages (Halary et al. 2010), which is also confirmed in our present work with an expanded data set. Interestingly, Popa et al. accurately decomposed transduction events in two phases: the uptake of a gene from a donor chromosome into a phage, and the acquisition of a gene from a prophage into a recipient chromosome. According to our own terminology, each of these phases corresponds to an externalization event (one from a cell to a virus, and another from a virus to a cell, respectively). For that reason, the edges in their network correspond to polarized externalization events between prokaryotes and phages. Interestingly, Popa et al. (2017) stress that 9% of the transduction events they detected are autologs, that is, duplication of genes mediated by mobile DNA vectors. This autology is consistent with our claim that externalization introduces genetic redundancy (and possibly resilience) within microbial communities. Of note, our work further underscores that the process of externalization (i.e., the sharing of genes on different types of supports, extrachromomal, and chromosomal elements) is very general. We report that externalized genes amount to even higher proportions of the chromosomes (i.e., >60% in Starkeya novella, which are not due to inserted prophages) than the proportion of transferred genes detected by Popa et al. (who inferred 15,298 edges between 2,573 bacteria and 4,650 phages). This is logical, because only events of gene externalization 1) beginning in a chromosome and ending in a chromosome, 2) mediated by a phage, were described in their directed bipartite networks of chromosomes and phages (Popa et al. 2017). By contrast, in our analysis, the first constraint is alleviated, and plasmids, rather than phages, appear as the main carriers of externalized genes (85–99% of externalized genes match on plasmids at ≥30% ID, see supplementary fig. S9-3, Supplementary Material online).
To summarize, we introduced an analysis of gene sharing between chromosomal and extrachromosomal elements from the microbial world based on a bipartite graph. This strategy allowed us to independently recover major trends of microbial evolution (i.e., the taxonomical and typological biases in the pattern of gene sharings, the existence of genetic worlds, in particular in networks built at low stringency thresholds, the promiscuity of transposases), to propose some novel HGT candidates, and to bring forward the general process of gene externalization. We thus show that the web of life is disconnected, with major prokaryotic kinds, largely, but not absolutely isolated from one another in terms of gene sharing, surrounded by spider-webs of mobile genetic elements, with which chromosomes share externalized genes. Moreover, transposases have recently run across this web of life. Consequently, bipartite graphs appear as a powerful way to study the processes of microbial life beyond classic taxonomy and customary genomic analyses, and we propose that beyond coding CHG, the use of bipartite graphs could be further generalized to small RNA families-genome networks and gene families-metagenome networks, and applied to even larger data sets to keep up with the impressive accumulation of molecular sequences. Finally, gene families-genome-metagenome tripartite graphs constitute an exciting horizon for expanded multilevel analyses.
Materials and Methods
Data Collection
We downloaded all complete genomes for viruses, plasmids, and Archaea available as of Nov. 2013 from the NCBI, as well as one complete genome from each bacterial family, in order to compensate for the sampling bias toward Bacteria in the available genomic databases (family-based data set). In this way, we obtained 230 Bacteria, 152 Archaea, 4,219 plasmids, and 3,613 viruses (see supplementary files S1–S4, Supplementary Material online). We verified that these MGE were not inserted in these chromosomes, that is, that the MGEs were extrachromosomal elements. We also used PHASTER (Arndt et al. 2016) to detect inserted phages. When detected, we considered such inserted phages as part of the prokaryotic genomes, since we see chromosomes as aggregates of genes from various origins, consistently, for example, with the conception of Cong et al. (2017), but see Popa et al. (2017) for an alternative treatment of inserted phages in network analysis. We also constituted an expanded genus-based data set comprising all available complete archaeal genomes (235) and one genome per bacterial genus (799) available at the NCBI in Nov. 2016. This second data set was only used to confirm the trends discovered in the family-based data set.
Sequence Analysis
We ran a BLAST all-against-all (blastp version 26, E-value 10−5) on all the 1,578,351 protein coding sequences (1,151,256 prokaryotic, 262,544 plasmidic, and 164,551 viral sequences) from the family-based data set. Because of the large size of the genus-based data set (3,834,026 sequences), we used DIAMOND v9.6.107 (Buchfink et al. 2015) with parameters “more sensitive,” E-value 10−5, no soft-masking, with tabulated BLAST-style output. DIAMOND with these parameters returns very similar results to BLAST, as we carefully assessed on the family-based data set. For both data sets, we filtered the returned hits by keeping only the best reciprocal hit between two sequences, whenever the corresponding matching length covered at least 80% of both sequences We also filtered the BLAST output of the family-based data set with an E-value cutoff of 10−11 to account for the effect of multiple testing on the size of the query sequence set (i.e., 106).
Clusters of Homologous Sequences
For a given % ID, we constructed clusters of homologous sequences (CHG) as the connected components of the following graph: nodes are protein-coding sequences, and an edge is drawn between two nodes if the sequences have ≥ 80% best reciprocal cover and the returned identity percentage is at least the required % ID (fig. 1, panel A). A CHG is called nontrivial if its cardinality is at least 2.
Heatmap Representation of Bipartite Graphs
We have graphically shown the distribution of the CHGs in the chromosomes by a heatmap representation of the adjacency matrix of the CHG-genome bipartite graph. More specifically, in this heatmap, each row represents a chromosome, and every column a nontrivial CHG, and the cell is colored by the cardinality of the CHG (supplementary fig. S11, Supplementary Material online).
Functional Annotation of Genes
The genes were clustered according to the COG categories using the software RPSBlast (Marchler-Bauer et al. 2015) with parameters (E-value ≤10−5). For the family-based data set, 934,742 genes were attributed at least one COG category (70.6% of prokaryotic genes, 38.6% of plasmid genes, and 12.5% of viral genes).
Bipartite Graphs Generation
We constructed the CHG-genome bipartite graphs by taking as top nodes the genomes and as bottom nodes the nontrivial CHGs, an edge connecting a genome node to a CHG node if the genome contained at least one member of the CHG (fig. 1, panel B). Since we focus on gene sharing, we further simplified this bipartite graph by removing all bottom nodes having degree 1, that is, genes that have no detectable similarity with any other at that threshold (“singletons”) as well as nontrivial CHGs that were only found in one genome (“specific paralogs”). We detected BTs, quotiented the graph, and detected BAPs using custom Python computer code (MultiTwin package, submitted for publication).
Simulation Protocol
In order to assess the stability of our findings, we have performed two types of simulation. We have first redone all the previous analyses on random samples comprising 20%, 50%, and 75% of the genomes in the family-based data set. We have found similar trends on the distribution of CHG and twins among the different kingdoms (see supplementary fig. S4 and tables S5–S7, Supplementary Material online). Second, we have also constructed permuted versions of the bipartite graphs, by changing the name of the genome containing a given gene. This simple randomization process means that genome labels are assigned randomly to sequences, which allows to test for the contribution of taxonomy, since taxonomical relationships are “broken” in the random network. We have performed 1,165 such random label permutations on the different bipartite graphs at all % ID. In this way, the identity of the top and bottom nodes was unchanged, and their degree distribution only marginally altered. All reported statistics on the CC and BT distributions were far outside the range of the simulated ones (p value = 4.76 × 10−3 corrected for multiple testing with a FDR of 5%; Benjamini and Yekutieli 2001).
Assignment of Putative Lateral Gene Transfer Events
In order to determine which edges in our sequence similarity graphs could be considered as candidates to lateral transfer, we implemented the following protocol, inspired by (Brilli et al. 2008; Fondi and Fani 2010). For every edge, we compared its similarity weight (i.e., the percentage of identity between the sequences it connects) to the empirical distribution of percentages of identity between the two corresponding taxa, restricted to the sequences satisfying the condition of reciprocal cover ≥80%. To avoid pushing the distribution artifactually down, due to the presence of paralogs, we only kept the largest similarity value whenever the corresponding pair of taxa appeared several times in a given CHG. We have determined a confidence interval for the 95% quantile of this distribution by a bootstrap procedure (10,000 samples with replacement), and reported a percentage of identity to be significantly higher than expected when it is higher than the upper bound of this confidence interval. To declare a cluster of genes as candidate for LGT, we have used three increasingly stringent criteria on the proportion of edges, significantly more similar than expected, that are required to declare. With the minority criterion, one significant edge is sufficient, whereas the majority (resp. unanimity) criteria require that the majority of edges (respectively all) be significantly more similar than expected. Additionally, we constructed phylogenies for twins highlighted as likely public goods in Compressing Bipartite Graphs Detects Novel Instances of the Mobilization of Public Genetic Goods (supplementary fig. S8-1–S8-7, Supplementary Material online). For every such twin, we have lowered the similarity threshold as much as we could do so that the sequences forming the corresponding twin could reasonably be aligned. The resulting sequences were aligned with muscle (v. 3.8.31) (Edgar 2004), trimmed with Gblocks (Castresana 2000), and ML trees were constructed (under the WAG model) with Seaview (Gouy et al. 2010). The visualization and annotation was carried out with iTOL (Letunic and Bork 2016). Alternative trees (see SI, description of additional archive http://www.evol-net.fr/index.php/fr/downloads/MBE_Corel_LGT_Trees.tar.bz2; last accessed January 18, 2018.) were constructed by aligning sequences with mafft (Katoh et al. 2005), trimming the alignment with trimal (Capella-Gutiérrez et al. 2009) and using ultrafast bootstrap approximation to infer an ML tree with iqtree (Minh et al. 2013; Nguyen et al. 2015).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Author contributions
E.C., P.L., and E.B. designed the study, R.M. collected and processed the data, E.C. and J.O.M. performed the analyses, E.B. wrote the paper. All authors discussed the results and commented on the manuscript.
Supplementary Material
Acknowledgments
This work was supported by the ERC (grant FP7/2007-2013 Grant Agreement #615274 to E.B. and E.C.). We thank D. Bhattacharya and H. Le Guyader and four anonymous reviewers for reading the manuscript and critical comments.
References
- Ageevets V, Julia S, Irina L, Maya M, Elena I, Elena K, Vladislav B.. 2017. Genetic environment of the blaKPC-2 gene in a Klebsiella pneumoniae isolate that may have been imported to Russia from Southeast Asia. Antimicrob Agents Chemother. 612:e01856–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad N, Chong TM, Hashim R, Shukor S, Yin W-F, Chan K-G.. 2015. Draft genome of multidrug-resistant Klebsiella pneumoniae 223/14 carrying KPC-6, isolated from a general hospital in Malaysia. J Genomics 3:97–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn Y-Y, Ahnert SE, Bagrow JP, Barabási A-L.. 2011. Flavor network and the principles of food pairing. Sci Rep. 11:196.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksandrzak-Piekarczyk T, Koryszewska-Bagińska A, Grynberg M, Nowak A, Cukrowska B, Kozakova H, Bardowski J.. 2016. Genomic and functional characterization of the unusual pLOCK 0919 plasmid harboring the spaCBA Pili cluster in Lactobacillus casei LOCK 0919. Genome Biol Evol. 81:202–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzahrani T, Horadam KJ.. 2016. Complex systems and networks In: Lü J, Yu X, Chen G, Yu W, editors. Understanding complex systems. Vol. 73 Berlin, Heidelberg: Springer Berlin Heidelberg. [Google Scholar]
- Andam CP, Gogarten JP.. 2011. Biased gene transfer in microbial evolution. Nat Rev Microbiol. 97:543–555. 10.1038/nrmicro2593 [DOI] [PubMed] [Google Scholar]
- Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS.. 2016. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44(W1):W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz RK, Breitbart M, Edwards RA.. 2010. Transposases are the most abundant, most ubiquitous genes in nature. Nucleic Acids Res. 3813:4207–4217. 10.1093/nar/gkq140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapteste E. 2014. The origins of microbial adaptations: how introgressive descent, Egalitarian evolutionary transitions and expanded kin selection shape the network of life. Front Microbiol. 5(Mar):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber MJ. 2007. Modularity and community detection in bipartite networks. Phys Rev E Stat Nonlin Soft Matter Phys. 766:66102. 10.1103/PhysRevE.76.066102 [DOI] [PubMed] [Google Scholar]
- Béguin P, Charpin N, Koonin EV, Forterre P, Krupovic M.. 2016. Casposon integration shows strong target site preference and recapitulates protospacer integration by CRISPR-Cas systems. Nucleic Acids Res. 4421:gkw821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beiko RG, Harlow TJ, Ragan MA.. 2005. Highways of gene sharing in prokaryotes. Proc Natl Acad Sci U S A. 10240:14332–14337. National Academy of Sciences: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beiko RG, Keith JM, Harlow TJ, Ragan MA.. 2006. Searching for convergence in phylogenetic Markov Chain Monte Carlo. Syst Biol. 554:553–565. 10.1080/10635150600812544 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Yekutieli D.. 2001. The control of the false discovery rate in multiple testing under dependency. Ann Stat. 294:1165–1188. [Google Scholar]
- Biller SJ, Berube PM, Lindell D, Chisholm SW.. 2015. Prochlorococcus: the structure and function of collective diversity. Nat Rev Microbiol. 131:13–27. [DOI] [PubMed] [Google Scholar]
- Brilli M, Mengoni A, Fondi M, Bazzicalupo M, Liò P, Fani R.. 2008. Analysis of plasmid genes by phylogenetic profiling and visualization of homology relationships using Blast2Network. BMC Bioinformatics 91:551.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, Huson DH.. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods 121:59–60. 10.1038/nmeth.3176 [DOI] [PubMed] [Google Scholar]
- Busby B, Kristensen DM, Koonin EV.. 2013. Contribution of phage-derived genomic islands to the virulence of facultative bacterial pathogens. Environ Microbiol. 152:307–312. 10.1111/j.1462-2920.2012.02886.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T.. 2009. trimAl: a Tool For Automated Alignment Trimming In Large-Scale Phylogenetic Analyses. Bioinformatics Appl Note 2515:1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 174:540–552. 10.1093/oxfordjournals.molbev.a026334 [DOI] [PubMed] [Google Scholar]
- Challacombe J, Kuske C.. 2012. Mobile genetic elements in the bacterial phylum Acidobacteria. Mobile Genet Elem. 24:179–183. 10.4161/mge.21943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challacombe JF, Eichorst SA, Hauser L, Land M, Xie G, Kuske CR, Steinke D.. 2011. Biological consequences of ancient gene acquisition and duplication in the large genome of Candidatus solibacter Usitatus Ellin6076. PLoS One 69:e24882–e24882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Crombie A, Rahman MT, Dedysh SN, Liesack W, Stott MB, Alam M, Theisen AR, Murrell JC, Dunfield PF.. 2010. Complete genome sequence of the aerobic facultative methanotroph Methylocella silvestris BL2. J Bacteriol. 19214:3840–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Karkar S, Bapteste E, Yee N, Falkowski P, Bhattacharya D.. 2014. Sequence similarity network reveals the imprints of major diversification events in the evolution of microbial life. Front Ecol Evol. 2(Nov):1–13. [Google Scholar]
- Cong Y, Chan Y-b, Phillips CA, Langston MA, Ragan MA.. 2017. Robust inference of genetic exchange communities from microbial genomes using TF-IDF. Front Microbiol. 8:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corel E, Lopez P, Méheust R, Bapteste E.. 2016. Network-thinking: graphs to analyze microbial complexity and evolution. Trends Microbiol. 243:224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagan T, Artzy-Randrup Y, Martin W.. 2008. Modular networks and cumulative impact of lateral transfer in prokaryote genome evolution. Proc Natl Acad Sci U S A. 10529:10039–10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagan T, Martin W.. 2006. The tree of one percent. Genome Biol. 710:118.. 10.1186/gb-2006-7-10-118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desnues C, La Scola B, Yutin N, Fournous G, Robert C, Azza S, Jardot P, Monteil S, Campocasso A, Koonin EV, Raoult D.. 2012. Provirophages and transpovirons as the diverse mobilome of giant viruses. Proc Natl Acad Sci U S A. 10944:18078–18083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias ACF, Cotta SR, Andreote FD, van Elsas JD. 2018. The parA region of broad-host-range PromA plasmids is a carrier of mobile genes. Microbial Ecol. 75:479–486. [DOI] [PubMed] [Google Scholar]
- Dunfield PF, Khmelenina VN, Suzina NE, Trotsenko YA, Dedysh SN.. 2003. Methylocella silvestris sp. nov., a novel methanotroph isolated from an acidic forest cambisol. Int J Syst Evol Microbiol. 535:1231–1239. [DOI] [PubMed] [Google Scholar]
- Edgar RC. 2004. {MUSCLE}: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fondi M, Fani R.. 2010. The horizontal flow of the plasmid resistome: clues from inter-generic similarity networks. Environ Microbiol. 1212:3228–3242. 10.1111/j.1462-2920.2010.02295.x [DOI] [PubMed] [Google Scholar]
- Fondi M, Karkman A, Tamminen MV, Bosi E, Virta M, Fani R, Alm E, McInerney JO.. 2016. ‘Every gene is everywhere but the environment selects’: global geolocalization of gene sharing in environmental samples through network analysis. Genome Biol Evol. 85:1388–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster D, Bittner L, Karkar S, Dunthorn M, Romac S, Audic S, Lopez P, Stoeck T, Bapteste E.. 2015. Testing ecological theories with sequence similarity networks: marine ciliates exhibit similar geographic dispersal patterns as multicellular organisms. ISME J. 131:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes K, Christensen SK, Løbner-Olesen A.. 2005. Prokaryotic toxin–antitoxin stress response loci. Nat Rev Microbiol. 35:371–382. [DOI] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O.. 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 272:221–224. 10.1093/molbev/msp259 [DOI] [PubMed] [Google Scholar]
- Halary S, Leigh JW, Cheaib B, Lopez P, Bapteste E.. 2010. Network analyses structure genetic diversity in independent genetic worlds. Proc Natl Acad Sci U S A. 1071:127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-López A, Chabrol O, Royer-Carenzi M, Merhej V, Pontarotti P, Raoult D.. 2013. To tree or not to tree? Genome-wide quantification of recombination and reticulate evolution during the diversification of strict intracellular bacteria. Genome Biol Evol. 512:2305.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper SD, Mavromatis K, Kyrpides NC.. 2009. Microbial co-habitation and lateral gene transfer: what transposases can tell us. Genome Biol. 104:R45.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, Butterfield CN, Hernsdorf AW, Amano Y, Ise K et al. , 2016. A new view of the tree of life. Nat Microbiol. 15:16048. [DOI] [PubMed] [Google Scholar]
- Huo Y-Y, Cheng H, Han X-F, Jiang X-W, Sun C, Zhang X-Q, Zhu X-F, Liu Y-F, Li P-F, Ni P-X.. 2012. Complete genome sequence of Pelagibacterium halotolerans B2T. J Bacteriol. 1941:197–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang TH, Sicotte H, Tian Z, Wu B, Kocher J-P, Wigle DA, Kumar V, Kuang R.. 2008. Robust and efficient identification of biomarkers by classifying features on graphs. Bioinformatics 2418:2023–2029. [DOI] [PubMed] [Google Scholar]
- Iranzo J, Koonin EV, Prangishvili D, Krupovic M, Sandri-Goldin RM.. 2016. Bipartite network analysis of the archaeal virosphere: evolutionary connections between viruses and capsid-less mobile elements. J Virol. 9024:11043–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iranzo J, Krupovic M, Koonin EV.. 2016. The double-stranded DNA virosphere as a modular hierarchical network of gene sharing. mBio 74:e00978-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jachiet P-AP, Colson P, Lopez P, Bapteste E.. 2014. Extensive gene remodeling in the viral world : new evidence for non-gradual evolution in the mobilome network. Genome Biol Evol. 69:2195–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AL, Corel E, Pathmanathan JS, Lopez P, Bapteste E.. 2016. Bipartite graph analyses reveal interdomain LGT involving ultrasmall prokaryotes and their divergent, membrane-related proteins. Environ Microbiol. 18:5072–5081. [DOI] [PubMed] [Google Scholar]
- Jain R, Rivera MC, Lake JA.. 1999. Horizontal gene transfer among genomes: the complexity hypothesis. Proc Natl Acad Sci U S A. 967:3801–3806. 10.1073/pnas.96.7.3801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ET, Dowd PF.. 2014. A non-autonomous insect piggyBac transposable element is mobile in tobacco. Mol Genet Genomics 2895:895–902. 10.1007/s00438-014-0860-2 [DOI] [PubMed] [Google Scholar]
- Jones-Dias D, Manageiro V, Graça R, Sampaio DA, Albuquerque T, Themudo P, Vieira L, Ferreira E, Clemente L, Caniça M.. 2016. QnrS1- and Aac(6′)-Ib-Cr-producing Escherichia coli among isolates from animals of different sources: susceptibility and genomic characterization. Front Microbiol. 7(May):671.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappler U, Davenport K, Beatson S, Lucas S, Lapidus A, Copeland A, Berry KW, Glavina Del Rio T, Hammon N, Dalin E et al. , 2012. Complete genome sequence of the facultatively chemolithoautotrophic and methylotrophic alpha proteobacterium Starkeya novella type strain (ATCC 8093T). Stand Genomic Sci. 71:44–58. 10.4056/sogs.3006378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappler U, Friedrich CG, Trüper HG, Dahl C.. 2001. Evidence for two pathways of thiosulfate oxidation in Starkeya novella (formerly Thiobacillus novellus). Archiv Microbiol. 1752:102–111. [DOI] [PubMed] [Google Scholar]
- Karcagi I, Draskovits G, Umenhoffer K, Fekete G, Kovács K, Méhi O, Balikó G.. 2016. Indispensability of horizontally transferred genes and its impact on bacterial genome streamlining. Mol Biol Evol. 335:1257–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Kuma K, Toh H, Miyata T.. 2005. {MAFFT} Version 5: improvement in Accuracy of Multiple Sequence Alignment. Nucleic Acids Res. 332:511–518. 10.1093/nar/gki198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloesges T, Popa O, Martin W, Dagan T.. 2011. Networks of gene sharing among 329 proteobacterial genomes reveal differences in lateral gene transfer frequency at different phylogenetic depths. Mol Biol Evol. 282:1057–1074. 10.1093/molbev/msq297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV. 2015. The turbulent network dynamics of microbial evolution and the statistical tree of life. J Mol Evol. 80(5–6):244–250. 10.1007/s00239-015-9679-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV. 2016. Horizontal gene transfer: essentiality and evolvability in prokaryotes, and roles in evolutionary transitions. F1000 Res. 5:1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Krupovic M.. 2017. Polintons, virophages and transpovirons: a tangled web linking viruses, transposons and immunity. Curr Opin Virol. 25(Aug):7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, Aravind L.. 2001. Horizontal gene transfer in prokaryotes: quantification and classification. Annu Rev Microbiol. 551:709.. 10.1146/annurev.micro.55.1.709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupovic M, Béguin P, Koonin EV.. 2017. Casposons: mobile genetic elements that gave rise to the CRISPR-Cas adaptation machinery. Curr Opin Microbiol. 38(Aug):36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku C, Nelson-sathi S, Roettger M, Sousa FL, Lockhart PJ, Bryant D, Hazkani-covo E, Mcinerney JO, Landan G, Martin WF.. 2015. Endosymbiotic origin and differential loss of eukaryotic genes. Nature 5247566:427–432. [DOI] [PubMed] [Google Scholar]
- Kuhl SA, Wimer LT, Yoch DC.. 1984. Plasmidless, photosynthetically incompetent mutants of Rhodospirillum rubrum. J Bacteriol. 1593:913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunin V, Goldovsky L, Darzentas N, Ouzounis CA.. 2005. The net of life: reconstructing the microbial phylogenetic network. Genome Res. 157:954–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larremore DB, Clauset A, Buckee CO, Antia R.. 2013. A network approach to analyzing highly recombinant malaria parasite genes. PLoS Comput Biol. 910:e1003268.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JG, Ochman H.. 2002. Reconciling the many faces of lateral gene transfer. Trends Microbiol. 101:1–4. 10.1016/S0966-842X(01)02282-X [DOI] [PubMed] [Google Scholar]
- Letunic I, Bork P.. 2016. Interactive Tree of Life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44(W1):W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima-Mendez G, Van Helden J, Toussaint A, Leplae R.. 2008. Reticulate representation of evolutionary and functional relationships between phage genomes. Mol Biol Evol. 254:762–777. [DOI] [PubMed] [Google Scholar]
- MacLeod D, Charlebois RL, Doolittle F, Bapteste E.. 2005. Deduction of probable events of lateral gene transfer through comparison of phylogenetic trees by recursive consolidation and rearrangement. BMC Evol Biol. 5:27.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manageiro V, Pinto M, Caniça M.. 2015. Complete sequence of a BlaOXA-48 -harboring IncL plasmid from an Enterobacter cloacae clinical isolate. Genome Announc. 35:e01076-15.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Panchenko AR, Shoemaker BA, Thiessen PA,, Geer LY,, Bryant SH.. 2015. CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 35:e01076-15–e01083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerney JO, Pisani D, Bapteste E, O’Connell MJ.. 2011. The public goods hypothesis for the evolution of life on earth. Biol Dir 61:41.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méheust R, Zelzion E, Bhattacharya D, Lopez P, Bapteste E.. 2016. Protein networks identify novel symbiogenetic genes resulting from plastid endosymbiosis. Proc Natl Acad Sci U S A. 11313:3579–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh BQ, Nguyen MAT, von Haeseler A.. 2013. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 305:1188–1195. 10.1093/molbev/mst024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Stamatis D, Bertsch J, Ovchinnikova G, Verezemska O, Isbandi M, Thomas AD, Ali R, Sharma K, Kyrpides NC, Reddy TBK.. 2017. Genomes OnLine Database (GOLD) v.6: data updates and feature enhancements. Nucleic Acids Res. 45(D1):D446–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munk AC, Copeland A, Lucas S, Lapidus A, Del Rio TG, Barry K, Detter JC, Hammon N, Israni S, Pitluck S et al. , 2011. Complete genome sequence of Rhodospirillum rubrum type strain (S1). Stand Genomic Sci. 43:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata T. 2009. Detecting Communities from Bipartite Networks Based on Bipartite Modularities. In 2009 International Conference on Computational Science and Engineering, Vancouver, BC, 4:50–57. IEEE.
- Nandasena KG, O’Hara GW, Tiwari RP, Howieson JG.. 2006. Rapid in situ evolution of nodulating strains for Biserrula pelecinus L. through lateral transfer of a symbiosis island from the original mesorhizobial inoculant. Appl Environ Microbiol. 7211:7365–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson-Sathi S, Dagan T, Landan G, Janssen A, Steel M, McInerney JO, Deppenmeier U, Martin WF.. 2012. Acquisition of 1,000 eubacterial genes physiologically transformed a methanogen at the origin of Haloarchaea. Proc Natl Acad Sci U S A. 10950:20537–20542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson-Sathi S, Sousa FL, Roettger M, Lozada-Chávez N, Thiergart T, Janssen A, Bryant D, Landan G, Schönheit P, Siebers B et al. , 2015. Origins of major archaeal clades correspond to gene acquisitions from bacteria. Nature 5177532:77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ.. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 321:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka Y. 2016. Prokaryotic toxin–antitoxin systems: novel regulations of the toxins. Curr Genet. 622:379–382. 10.1007/s00294-015-0557-z [DOI] [PubMed] [Google Scholar]
- Pant A, Anbumani D, Bag S, Mehta O, Kumar P, Saxena S, Nair GB, Das B, DiRita VJ.. 2016. Effect of LexA on chromosomal integration of CTXφ in Vibrio cholerae. J Bacteriol. 1982:268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrosa FO, Monteiro RA, Wassem R, Cruz LM, Ayub RA, Colauto NB, Fernandez MA, Fungaro MHP, Grisard EC, Hungria M et al. , 2011. Genome of Herbaspirillum seropedicae strain SmR1, a specialized diazotrophic endophyte of tropical grasses. PLoS Genet. 75:e1002064.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa O, Hazkani-Covo E, Landan G, Martin W, Dagan T.. 2011. Directed networks reveal genomic barriers and DNA repair bypasses to lateral gene transfer among prokaryotes. Genome Res. 214:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa O, Landan G, Dagan T.. 2017. Phylogenomic networks reveal limited phylogenetic range of lateral gene transfer by transduction. ISME J. 112:543–554. 10.1038/ismej.2016.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigbo P, Wolf YI, Koonin EV.. 2010. The tree and net components of prokaryote evolution. Genome Biol Evol. 20:745–756. 10.1093/gbe/evq062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond J, Zhaxybayeva O, Gogarten JP, Blankenship RE.. 2003. Evolution of photosynthetic prokaryotes: a maximum-likelihood mapping approach. Philos Trans R Soc Lond B Biol Sci. 3581429:223.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve W, Nandasena K, Yates R, Tiwari R, O’Hara G, Ninawi M, Gu W, Goodwin L, Detter C, Tapia R et al. , 2013. Complete genome sequence of Mesorhizobium australicum type strain (WSM2073T). Stand Genomic Sci. 92:410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S, Hallam SJ, Woyke T, Sullivan MB.. 2015. Viral dark matter and virus–host interactions resolved from publicly available microbial genomes. eLife 4(Jul):e08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabat AJ, Pournaras S, Akkerboom V, Tsakris A, Grundmann H, Friedrich AW.. 2015. Whole-genome analysis of an oxacillin-susceptible CC80 mecA -positive Staphylococcus aureus clinical isolate: insights into the mechanisms of cryptic methicillin resistance. J Antimicrob Chemother. 7011:2956–2964. [DOI] [PubMed] [Google Scholar]
- Schliep K, Lopez P, Lapointe F-J, Bapteste E.. 2011. Harvesting evolutionary signals in a forest of prokaryotic gene trees. Mol Biol Evol. 284:1393–1405. 10.1093/molbev/msq323 [DOI] [PubMed] [Google Scholar]
- Simmonds P, Adams MJ, Benkő M, Breitbart M, Rodney Brister J, Carstens EB, Davison AJ.. 2017. Consensus statement: virus taxonomy in the age of metagenomics. Nat Rev Microbiol. 15:161–168. [DOI] [PubMed] [Google Scholar]
- Skala AM. 2014. Retroviral DNA transposition: themes and variations. Microbiol Spectr. 25:MDNA3-0005-2014. [DOI] [PubMed] [Google Scholar]
- Smillie CS, Smith MB, Friedman J, Cordero OX, David LA, Alm EJ.. 2011. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 4807376:241–244. [DOI] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen MA.. 2002. Genomes in flux: the evolution of archaeal and proteobacterial gene content. Genome Res. 121:17–25. 10.1101/gr.176501 [DOI] [PubMed] [Google Scholar]
- Sullivan MB, Huang KH, Ignacio-Espinoza JC, Berlin AM, Kelly L, Weigele PR, DeFrancesco AS, Kern SE, Thompson LR, Young S et al. , 2010. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ Microbiol. 1211:3035–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Feschotte C, Wu Z, Mueller RL.. 2015. DNA transposons have colonized the genome of the giant virus Pandoravirus salinus. BMC Biol. 131:38.. 10.1186/s12915-015-0145-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Zhang P, Qu T, Chen Y, Hua X, Shi K, Yu Y.. 2017. Identification of novel conjugative plasmids with multiple copies of fosB that confer high-level fosfomycin resistance to vancomycin-resistant enterococci. Front Microbiol. 8(Aug):1541.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamminen M, Virta M, Fani R, Fondi M.. 2012. Large-scale analysis of plasmid relationships through gene-sharing networks. Mol Biol Evol. 294:1225–1240. 10.1093/molbev/msr292 [DOI] [PubMed] [Google Scholar]
- Wang Y-N, Wang L, Tsang YF, Fu X, Hu J, Li H, Le Y.. 2016. Response of Cbb gene transcription levels of four typical sulfur-oxidizing bacteria to the CO2 concentration and its effect on their carbon fixation efficiency during sulfur oxidation. Enzyme Microbial Technol. 92(Oct):31–40. [DOI] [PubMed] [Google Scholar]
- Wilson WH, Gilg IC, Moniruzzaman M, Field EK, Koren S, LeCleir GR, Martínez Martínez J, Poulton NJ, Swan BK, Stepanauskas R et al. , 2017. Genomic exploration of individual giant ocean viruses. ISME J. 118:1736–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeates TO, Kerfeld CA, Heinhorst S, Cannon GC, Shively JM.. 2008. Protein-based organelles in bacteria: carboxysomes and related microcompartments. Nat Rev Microbiol. 69:681–691. [DOI] [PubMed] [Google Scholar]
- Yutin N, Raoult D, Koonin EV, Yutin N, Raoult D, Koonin EV.. 2013. Virophages, polintons, and transpovirons: a complex evolutionary network of diverse selfish genetic elements with different reproduction strategies. Virol J. 101:158.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhaxybayeva O, Gogarten JP, Charlebois RL, Ford Doolittle W, Thane Papke R.. 2006. Phylogenetic analyses of cyanobacterial genomes: quantification of horizontal gene transfer events. Genome Res. 169:1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.