

Abstract
Hearing loss is a major public health concern with no pharmaceutical intervention for hearing protection or restoration. Using zebrafish neuromast hair cells—a robust model for mammalian auditory and vestibular hair cells— we identified a urea-thiophene carboxamide, 1 (ORC-001) as protective against aminoglycoside antibiotic (AGA)-induced hair cell death. The 50% protection (HC50) concentration conferred by 1 is 3.2 μM with protection against 200μM neomycin approaching 100%. 1 was sufficiently safe and drug-like to validate otoprotection in an in vivo rat hearing loss model. We explored the structure activity relationship (SAR) of this compound series to improve otoprotective potency, improve pharmacokinetic properties and eliminate off-target activity. We present the optimization of 1 to yield 90 (ORC-13661). 90 protects mechanosensory hair cells with HC50 of 120 nM and demonstrates 100% protection in the zebrafish assay, superior physiochemical and pharmacokinetic and toxicologic properties as well as complete in vivo protection in rats.
Graphical Abstract
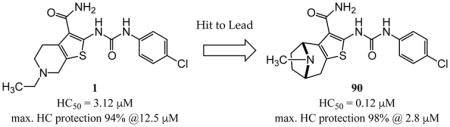
Introduction
Acquired hearing loss can be caused by a variety of environmental and organic factors ranging from noise exposure and pharmaceutical agents to infections, late onset genetic disorders and aging.1, 2 Hair cells of the inner ear are the mechanosensors that transduce a physical stimulus - vibrations transmitted by the fluid-filled compartment of the cochlea and vestibule - into nerve impulses that are converted in the central nervous system into the perception of sound and vestibular reflexes. Most forms of hearing loss are sensorineural in nature, meaning they result from death or injury of the cochlear hair cells or other cell types in the organ of Corti of the cochlea or the neurons connecting the hair cells to the brain.3 Unlike aquatic vertebrates and birds, mammals lack the ability to regenerate hair cells making hearing loss irreversible.4, 5 The mechanisms by which hair cells are killed are complex and overlapping. Elements of programmed cell death, or apoptosis, as well as necrotic signatures have been identified in damaged hair cells in vitro and in vivo.5–8 Antioxidants including aspirin, N-acetyl cysteine (NAC) and selenium compounds9 have been reported to offer some degree of protection in model organisms,3, 10 but have yielded disappointing results in human clinical trials.11–13 Mechanistically, stress pathways, including the c-Jun N-terminal kinase (JNK), mixed lineage kinase (MLK) and p38 kinase pathways have been implicated in drug-induced ototoxicity and small molecule inhibitors of these pathways have been proposed as protective agents.8, 14–17 However, clinical trials have again been disappointing, possibly because the protection is not robust and falls off at higher doses of the ototoxic agent.18 These clinical findings indicate a need for better therapeutic agents effective at higher doses of ototoxic agent.
Aminoglycoside antibiotics (AGAs) constitute a class of natural product antibiotics that are effective against a variety of clinically important gram-negative bacteria.11 However, because of side effects that include irreversible hearing and balance problems (ototoxicity) and kidney damage (nephrotoxicity), their use is limited in North America and Western Europe to combating life-threatening infections.19–21 Exceptions are the use of systemic AGAs for controlling flare-ups of chronic Pseudomonas aeruginosa infections in cystic fibrosis patients and multi-drug resistant tuberculosis.22–25 AGAs exert their antimicrobial effect partly by binding to ribosomal RNA and altering or inhibiting ribosomal protein synthesis.26–29 A more controversial model of toxicity in bacterial and mammalian cells through direct induction of reactive oxygen species (ROS) has been proposed but remains hotly contested.30–32 Additionally, proteins that bind AGAs have been identified and proposed as relevant mediators of in vivo ototoxicity.33
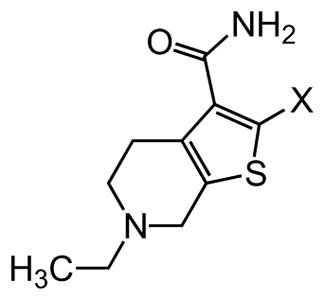
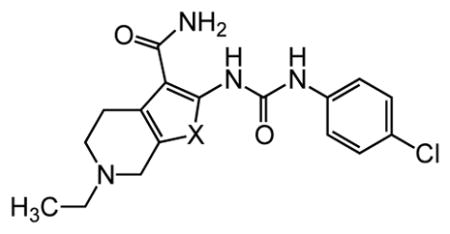
In an unbiased approach, we used the lateral line neuromast hair cells in free-swimming larval zebrafish (Danio rerio) to screen for compounds that protect these mechanosensory hair cells against AGA-induced cell death.34 Like mammalian auditory and vestibular hair cells, lateral line hair cells transduce a mechanical stimulus of changing liquid pressure to nerve impulses and like their auditory counterparts, are exquisitely sensitive to damage or death due to AGA exposure. Neuromast hair cell toxicity can be used as a highly reliable and versatile screening assay to identify modifiers, either enhancers or suppressors, of ototoxic agents.34 The assay is equally effective in identifying genetic and pharmacological modifiers. In a screen of structurally diverse, small molecule, drug-like compounds, we identified a thiophene-urea-carboxamide 1 (ORC-001, PROTO-1, Figure 1),20 as an agent that provided dose-dependent protective activity (otoprotection) against a range of AGA concentrations. Further experiments revealed that 1 was sufficiently drug-like to allow proof-of-concept testing in a rat model of AGA-induced hearing loss (see below). However, 1 exhibited pharmacokinetic and toxicological liabilities including only adequate solubility, moderate oral bioavailability, short in vivo half-life and moderate inhibition of the human Ether-à-go-go-Related Gene (hERG, aka KV11.1) potassium ion channel. Inhibition of hERG by a diverse array of pharmaceuticals has been linked to cardiac arrhythmias through QT interval prolongation.35–38 Because of this, hERG inhibition is generally viewed as a liability in drug development. Finally, as a hit in a phenotypic screen, 1 was uncharacterized in terms of its molecular target or mechanism of action.39, 40 To overcome these limitations, we carried out a systematic evaluation of analogs using a phenotypic hair cell protection assay in zebrafish to generate a structure activity relationship for compounds that protect against aminoglycoside antibiotic-induced hearing loss. The results of this evaluation and validation of hearing protection in mammals by one of the final lead compounds are the subjects of the current manuscript.
Figure 1.

Structure of 1.
Results
Synthesis of 1 and 3-aryl urea derivatives
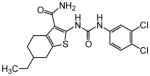
In a phenotypic screen using zebrafish neuromast hair cells we identified 1 as highly protective against aminoglycoside antibiotic-induced hair cell death. Early ADME characterization revealed several promising properties as a lead molecule (adequate aqueous solubility and membrane permeability, reasonable plasma protein binding, no issues with CYP stability or inhibition and an acceptable clearance profile), although we identified several pharmacokinetic limitations and a hERG liability (hERG IC50 3.26 μM). However, 1 was sufficiently safe and drug-like to be used to validate otoprotection in an in vivo rat model of aminoglycoside-induced hearing loss as a measure of proof of concept (vide infra). We then explored the structure activity relationships of this compound series with three goals in mind: improve otoprotective potency, improve pharmacokinetic properties and eliminate/minimize off-target activity.
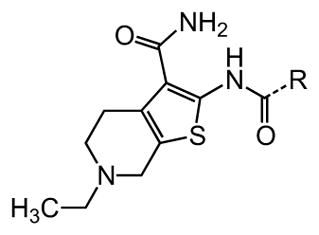
The preparation of 1 and N3′-aryl derivatives began with the Gewald condensation41, 42 of 4-ethyl-4-piperidone with cyanoacetamide in the presence of elemental sulfur and morpholine to produce 2-amino-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxamide 28 as shown in Scheme 1. Subsequent reaction of 28 with aryl isocyanates produced the urea derivatives 1–15 in moderate to good yields. Formation of HCl salts stabilized compounds containing a basic nitrogen in the tetrahydropyridine ring and related structures, which otherwise slowly underwent aereal oxidation to generate fluorescent conjugated products that interfered in the primary bioassay. In all, over 400 analogues including amines and quaternary ammonium salts were prepared and evaluated in the zebrafish neuromast hair cell protection assay.
Scheme 1.

Gewald reaction to form 2-amino-thiophene carboxamides.
In vivo screen of 1 analogs in zebrafish
New compounds were tested in the zebrafish assay as previously described.34, 43 In summary, newly hatched free-swimming AB zebrafish larvae were raised at 28.5°C in petri dishes and transferred to cell culture baskets placed in 6-well culture plates in groups of ten fish per basket. Fish were pre-treated with test compound for 1 hour at five three-fold dilutions followed by treatment with 200 μM neomycin. Following neomycin exposure, fish were rinsed briefly with a staining agent (DASPEI), anesthetized and mounted on the stage of an epifluorescence dissecting microscope. Hair cell staining of ten neuromasts on one side of each animal was evaluated visually each neuromast was scored for presence of a normal compliment of hair cells, with reduced or absent DASPEI staining indicating a reduction in hair cell number. Composite scores were calculated for animals in each treatment group, normalized to the control group and expressed as % hair cell survival. If hair cell survival was at least 50%, the HC50 was calculated. These data are shown in Tables 1–8 and representative dose response relationship experiments for several analogues in the zebrafish hair cell protection assay are shown in Figures 2 and 3. All zebrafish protocols were approved by the University of Washington IACUC.
Table 1. Urea substituents.
Protective activity of urea variants in the zebrafish hair cell protection assay. NA – not active (HC50 > 25 μM).
|
R | HC50 (μM) |
|---|---|---|
| 1 |
|
3.12 |
| 2 |
|
3.13 |
| 3 |
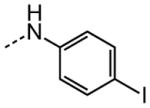
|
1.62 |
| 4 |
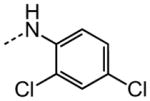
|
11.5 |
| 5 |
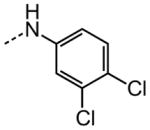
|
17.4 |
| 6 |
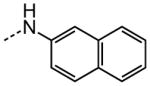
|
1.78 |
| 7 |
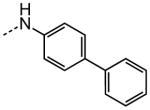
|
3.39 |
| 8 |
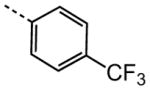
|
2.91 |
| 9 |
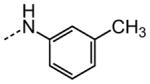
|
13.0 |
| 10 |
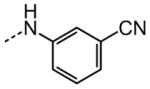
|
10.3 |
| 11 |
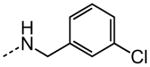
|
NA |
| 12 |
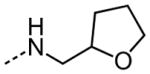
|
NA |
| 13 |
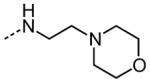
|
NA |
| 14 |
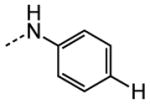
|
NA |
| 15 |
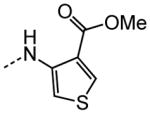
|
NA |
Figure 2.

Dose response functions for zebrafish neuromast hair cells treated with neomycin (200 μM) plus 1, 53, 76, 53, 87 or neomycin alone.
Figure 3.

Dose response functions for zebrafish neuromast hair cells treated with neomycin (200 μM) plus 1, 67, 90, 99 or neomycin alone.
Structure Activity Relationship (SAR)
For the purposes of the determination of structure activity relationships, 1 was conceptualized as composed of six structural units: 1) N3-aryl substituents on the urea; 2) the 1,3-di-substituted urea; 3) thiophene core; 4) primary carboxamide C2 substitution; 5) tetrahydropyridine core, and; 6) N-ethyl group. The goal of the SAR-determining studies described here was three-fold: first, to determine the contribution of each structural element described above to the observed biological activity; second, to improve the protective activity of the compound series, and; third, to improve the pharmacokinetic and toxicological properties of the lead compounds. A systematic variation of each structural element and its effect on protective activity is described.
The examination of the effects of N3-substitution to replace the p-chlorophenyl group relied on the reaction of readily synthesized isocyanates or their equivalents with 2-amino-thiophene-3-carboxamide 28. A variety of aromatic and non-aromatic isocyanates gave rise to a panel of substituted ureas as shown in Table 1. The pattern of activity was indicative of a narrow range of allowable substituents with p-chlorophenyl being close to optimal. Several aryl ureas had comparable protective activity to 1 (HC50 = 3.12 μM), such as the classic isosteric replacements at the C4 position (2, 3, 7 and 8, HC50 = 1.6–3.4 μM range). Interestingly, the unsubstituted analog 14 was inactive, and the bioisosteric 3,4-disubstituted pair 5 and 6 differed in potency by 10-fold (HC50 = 1.78 for 6 versus 17.4 μM for 5), reinforcing the narrow requirement for allowable substitution. The ‘extended’ benzyl analogues represented by 11 were inactive, as were N-alkyl derivatives, represented by 12 and 13, clearly lying out of the tolerable substitution range. Heterocyclic derivative 15 was similarly inactive, as were the majority of the 2-substituted aromatic analogues (data not shown).
As shown in Table 2, replacement of the primary carboxamide with a carboxylic acid 16, methyl ester 17, N-methyl amide 18, carbonitrile 19 and tertiary amide 20 greatly reduces the activity, suggesting that both protons on the primary amide are required for activity. Similarly, carboxamide bioisosteric compounds 1,2,5-oxadiazole 21 and 1,3,4-oxadiazole 22 are inactive. Only the 2-hydroxyethyl carboxamide 24 and 2-(N-morpholino)ethyl carboxamide 25 retain modest activity (both HC50 = 9.4 μM), suggesting the possibility that an internal hydrogen bond with the proximal urea proton may provide a pre-organization of molecular structure which contributes to compound potency.
Table 2. Carboxamide variants.
Protective activity of carboxamide variants in the zebrafish hair cell protection assay. NA – not active (HC50 > 25 μM).

|
X | HC50 (μM) |
|---|---|---|
| 1 |
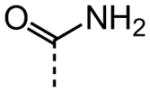
|
3.12 |
| 16 |
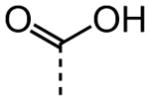
|
NA |
| 17 |
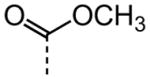
|
NA |
| 18 |
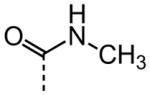
|
NA |
| 19 |
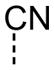
|
NA |
| 20 |
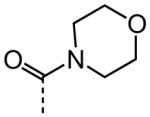
|
NA |
| 21 |
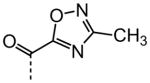
|
NA |
| 22 |
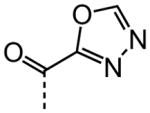
|
NA |
| 23 |
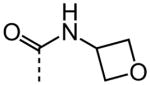
|
NA |
| 24 |
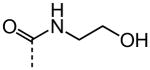
|
9.40 |
| 25 |
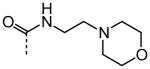
|
9.38 |
As shown in Table 3, efforts to replace the di-substituted urea moiety were largely unsuccessful, although the di-substituted thiourea 26 did provide modest potency, albeit two-fold less (HC50 = 6.57 versus. 3.12 μM). Replacement of urea with several types of amide structures resulted in no protective activity in 27, 29–30 or 32–33, and unsurprisingly the core compound 28 without a disubstituted urea functional group was inactive. Cyclopropyl malonamide 31 displayed a lack of potency, and likewise substitution at either the urea N1 or N3 position was also not tolerated (data not shown). Replacement of the di-substituted urea with a peptide bond or as a squarate di-amide also completely abolished protective activity (data not shown).
Table 3. Urea variants.
Protective activity of 2-aminothiophene variants in the zebrafish hair cell protection assay. NA – not active (HC50 > 25 μM).
|
X | HC50 (μM) |
|---|---|---|
| 1 |
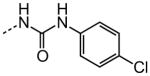
|
3.12 |
| 26 |
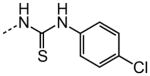
|
6.57 |
| 27 |
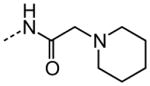
|
NA |
| 28 |
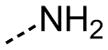
|
NA |
| 29 |
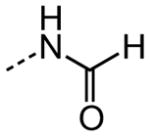
|
NA |
| 30 |
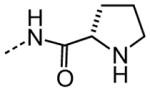
|
NA |
| 31 |
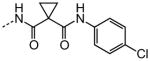
|
NA |
| 32 |
|
NA |
| 33 |

|
NA |
The thiophene portion of the core fused bicyclic ring is also important for activity (Table 4). Several 5- and 6-membered ring thiophene variants were prepared from commercial building blocks to give 34–37, none of which provided protective activity. The bioisosteric tetrahydroisoquinoline 38 was also prepared, but likewise displayed no potency in the bioassay.
Table 4. Thiophene variants.
Protective activity of thiophene variants in the zebrafish hair cell protection assay. NA – not active (HC50 > 25 μM).
|
X | HC50 (μM) |
|---|---|---|
| 1 |
|
3.12 |
| 34 |
|
NA |
| 35 |
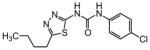
|
NA |
| 36 |
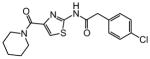
|
NA |
| 37 |
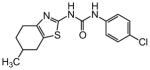
|
NA |
| 38 |
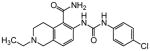
|
NA |
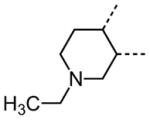
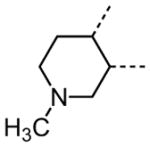
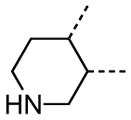
The variation of the parent tetrahydrothieno[2,3-c]pyridine ring proved feasible without eliminating biological activity, with protective activity remaining in the 1–5 μM range (Table 5). Movement of the nitrogen to produce tetrahydrothieno[3,2-c]pyridine 41 was tolerated (HC50 = 4.61 μM), as was ring-contraction to the dihydrothieno[2,3-c]pyrrole 42 (HC50 = 4.34 μM) and ring-expansion to the tetrahydrothienoazepines 43 and 44 (HC50 = 3.98 and 5.80 μM, respectively). However, replacement of the tetrahydropyridine ring with the carbocyclic cyclohexanes 46–48 completely abolished protective activity. Breaking up the fused bicyclic core to provide the tertiary aminomethyl tetra-substituted thiophene 45, provided a compound of moderate but measurable activity (HC50 = 6.92 μM).
Table 5. Tetrahydropyridine variants.
Protective activity of tetrahydropyridine variants in the zebrafish hair cell protection assay. NA – not active (HC50 > 25 μM).
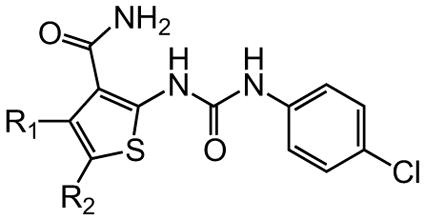
|
R1/R2 | HC50 (μM) |
|---|---|---|
| 1 |
|
3.12 |
| 39 |
|
4.70 |
| 40 |
|
3.26 |
| 41 |
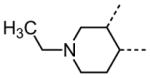
|
4.61 |
| 42 |
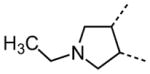
|
4.34 |
| 43 |
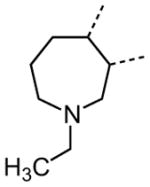
|
3.98 |
| 44 |

|
5.80 |
| 45 |
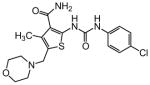
|
6.92 |
| 46 |
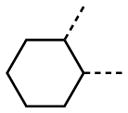
|
NA |
| 47 |
|
NA |
| 48 |
|
NA |
Interestingly, removal of the N-ethyl substituent as in 40 had only a modest effect on activity (HC50 = 3.26 μM for 40 versus 3.12 μM for 1), leading to an exploration of the effect of N-substitution on potency, as shown in Table 6. A series of N-alkyl hydrocarbon analogues 49–58 were prepared to provide a simple exploration of changes in length and steric bulk. While the increase in bulk at the terminal end of the linear case reduced activity (N-ethyl 1, HC50 = 3.12 μM, to N-isobutyl 50, HC50 = 4.51 μM, to N-neopentyl 51, HC50 = 5.56 μM, to N-isopropyl 52, HC50 = 1.49 μM), a few alkyl substituents bearing terminal cyclic motifs led to improved additional improvements in protective activity [N-(cyclopropyl)ethyl 53, HC50 = 1.22 μM, to N-(cyclobutyl)ethyl 54, HC50 = 1.36 μM, to N-(cyclopentyl)ethyl 55, HC50 = 1.83 μM, to N-(cyclohexyl)methyl 56, HC50 = 1.89 μM, versus 3.12 μM for 1]. Unfortunately, with along with increases in hearing protection potency, a number of compounds in this same set also exhibited increases in hERG activity as determined by the patch clamp assay (hERG IC50 = 2.67 μM for 53 and 0.73 μM for 54 versus 3.26 μM for 1). To test the effect amine basicity on potency, carbamate 59 and aminoethyl ester 60 were prepared, with the result that protective activity was completely suppressed. Interestingly, as the hydrocarbon chain connecting the trialkylamine to the ester functionality was increased to two carbons to give 61, potency was increased (HC50 = 1.10 μM for 61 versus 3.12 μM for 1). When the length was increased between three to five methylene units in 62–64, the potency was improved to less than a micromolar activity (HC50 = 0.6–0.7 μM range). Unlike the hydrocarbon substituent series, however, the increases in potency were not necessarily paralleled by their hERG inhibition, (hERG IC50 = 3.84 μM for 62 versus 3.26 μM for 1). Compounds incorporating an amide functionality distal to the tetrahydropyridine nitrogen as in 65–67 also retained improved protective activity, providing some of the most potent compounds prepared in the N-alkyl SAR series (HC50 = 0.1–0.5 μM range).
Table 6. Tetrahydroprydine N-substituent variants.
Protective activity of tetrahydropyridine N-alkyl variants in the zebrafish hair cell protection assay. NA – not active (HC50 > 25 μM).
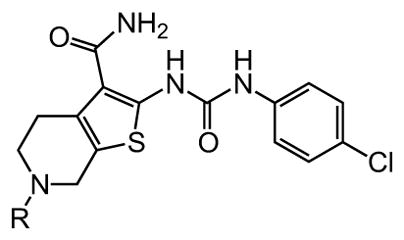
|
R | HC50 (μM) |
|---|---|---|
| 1 |
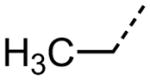
|
3.12 |
| 49 |
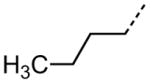
|
3.50 |
| 50 |
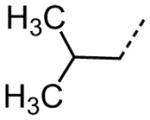
|
4.51 |
| 51 |
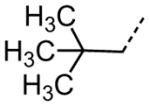
|
5.56 |
| 52 |
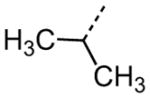
|
1.49 |
| 53 |
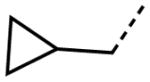
|
1.22 |
| 54 |
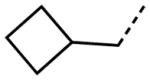
|
1.36 |
| 55 |
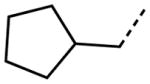
|
1.83 |
| 56 |
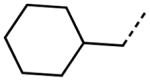
|
1.89 |
| 57 |
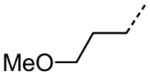
|
2.40 |
| 58 |
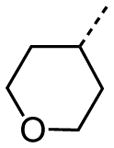
|
2.83 |
| 59 |
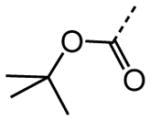
|
NA |
| 60 |
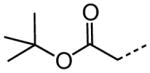
|
NA |
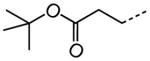
| 61 |
|
1.10 |
| 62 |
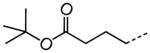
|
0.56 |
| 63 |
|
0.71 |
| 64 |
|
0.62 |
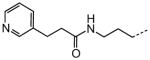
| 65 |
|
0.47 |
| 66 |
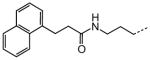
|
0.11 |
| 67 |
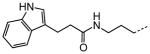
|
0.10 |
In order to more fully explore the requirement for a basic nitrogen in the tetrahydropyridine ring, we synthesized a panel of N,N-dialkyl quaternary ammonium salts (Table 7). This provides a permanent positive charge within the ring, whereas the previous compounds were assumed to be protonated at physiological pH. The initial N-methyl analogue 68 was found to be slightly more potent than the parent compound (HC50 = 1.96 μM for 68 versus 3.12 μM for 1), but interestingly it exhibited the same level of hERG inhibition (hERG IC50 = 3.38 μM for 68 versus 3.26 μM for 1). We were initially pleased to find that increasing steric bulk around the nitrogen yielded compounds with increasing activity (N-ethyl 68, HC50 = 1.96 μM, to N-isobutyl 71, HC50 = 0.38 μM, to N-neopentyl 72, HC50 = 0.29 μM), although a limit was reached with N-isopropyl compound 70 (HC50 = 0.96 μM). Similarly, increasing the size of a cyclic moiety on the terminal end of the N-substituent led to increasingly more potent compounds [N-(cyclopropyl)ethyl 73, HC50 = 0.36 μM, to N-(cyclobutyl)ethyl 74, HC50 = 0.21 μM, to N-(cyclopentyl)ethyl 75, HC50 = 0.20 μM, to N-(cyclohexyl)methyl 76, HC50 = 0.10 μM]. It was therefore a pleasant discovery that the hERG activity decreased inversely from that observed in the N-alkyl series. For instance, interaction with the hERG channel decreased as the zebrafish potency increased for the size increase from the N-ethyl compound 68 (HC50 = 1.96 μM, hERG IC50 = 3.38 μM) to N-isobutyl compound 71 (HC50 = 0.38 μM, hERG IC50 = 9.75 μM) to N-(cyclohexyl)methyl compound 76 (HC50 = 0.10 μM, hERG IC50 > 30 μM). As with the basic N-substituted variants in Table 6, quaternary ammonium salts containing methyl ether and ester functional groups tethered by increasing length aliphatic linkers yielded highly active compounds 78–81 shown in Table 7.
Table 7. Quaternary salts.
Protective activity of quaternary ammonium salt derivatives in the zebrafish hair cell protection assay.
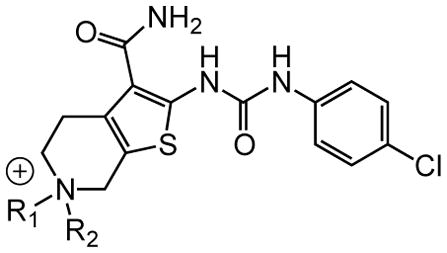
|
R1 | R2 | HC50 (μM) |
|---|---|---|---|
| 1 |
|
n/a | 3.12 |
| 68 |
|
|
1.96 |
| 69 |
|
|
0.45 |
| 70 |
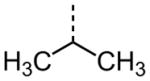
|
|
0.96 |
| 71 |

|
|
0.38 |
| 72 |
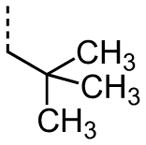
|
|
0.29 |
| 73 |
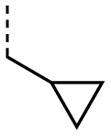
|
|
0.36 |
| 74 |
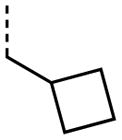
|
|
0.21 |
| 75 |
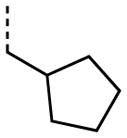
|
|
0.20 |
| 76 |
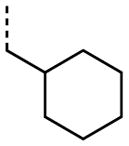
|
|
0.10 |
| 77 |
|
|
0.10 |
| 78 |
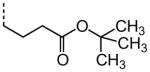
|
|
0.25 |
| 79 |
|
|
0.14 |
| 80 |
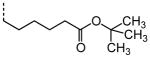
|
|
0.11 |
| 81 |
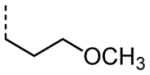
|
|
0.48 |
| 82 |
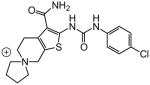
|
0.41 | |
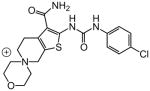
| 83 |
|
1.21 | |
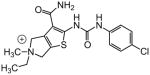
| 84 |
|
3.10 | |
| 85 |
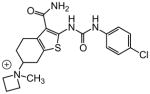
|
3.92 | |
| 86 |
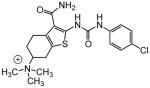
|
5.87 | |
A number of other quaternary ammonium congeners were prepared as more significant scaffold modifications (Table 7). Spirocyclic pyrrolidine-dihydrothieno[2,3-c]pyridinium compound 82 and spirocyclic morpholine-dihydrothieno[2,3-c]pyridinium compound 83 were both more potent than the N-ethyl-N-methyl parent compound (HC50 = 0.41 μM for 82 and HC50 = 1.21 μM for 83 versus HC50 = 1.96 μM for 68), but the former displayed the same level of hERG inhibition (IC50 = 3.51 μM for 82 versus IC50 = 3.38 μM for 68), whereas the latter compound displayed only moderate hERG activity (IC50 = 12.4 μM for 83). The quaternary dihydrothieno[2,3-c]pyrrole analogue 84 was similar to the parent tetrahydrothieno[2,3-c]pyridine 68 both in terms of hearing protection (HC50 = 3.10 μM for 84) and hERG inhibition (IC50 = 3.29 μM for 84). Two compounds in which the quaternary ammonium functional group was outside of the fused bicyclic ring system were prepared, but both were less active (HC50 = 3.92 μM for 85 and HC50 = 5.87 μM for 86).
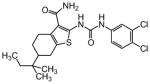
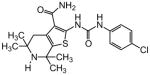
The steric environment around the basic tetrahydropyridine nitrogen was further explored by the synthesis of the 2,2,6,6-tetramethyl tetrahydropyridine derivative 87 (Table 8), which was prepared from tetramethyl piperidone through the route depicted in Scheme 1. This compound was more active than the parent compound (HC50 = 1.24 μM for 87 versus HC50 = 3.12 μM for 1), but more importantly it was devoid of hERG liability (IC50 > 30 μM for 87 versus IC50 = 3.26 μM for 1). In vitro ADME profiling of 87 demonstrated that it had excellent metabolic stability, no issues with CYP inhibition, but suffered from poor aqueous solubility (13 μM) and modestly increased plasma protein binding (96.5% in human and 95.8% in rat plasma).
Table 8. Tropane variants.
Protective activity of bicyclic tetrahydropyridine (tropane) analogues in the zebrafish hair cell protection assay.
| Compound | R | HC50 (μM) |
|---|---|---|
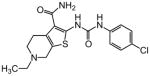
| 1 |
|
3.12 |
| 87 |
|
1.24 |
| 89 |
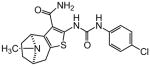
|
2.25 |
| 90 |
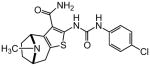
|
0.12 |
| 92 |
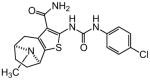
|
7.21 |
| 93 |
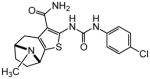
|
0.27 |
| 95 |
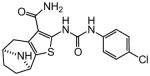
|
3.92 |
| 96 |
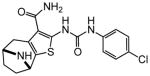
|
0.27 |
| 98 |
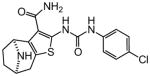
|
5.00 |
| 99 |
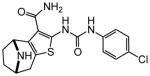
|
0.03 |
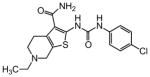
In an effort to evaluate the importance of conformational flexibility of the tetrahydropyridine unit, we set out to synthesize the bicyclic, tropane-like compound 91 as a racemate (Figure 5). The standard Gewald sequence using commercially available tropinone to react with cyanoacetamide and elemental sulfur in the presence of base yielded a mixture with two predominant products, each having the correct molecular weight of the desired product as well as 1H NMR spectra consistent with the expected product structure. An admixture of the two yielded distinct but partially overlapping NMR spectra suggesting that the two did not interconvert under these conditions. The 2-aminothiophene materials were difficult to purify and fully characterize and instead were reacted as a mixture with p-chlorophenyl isocyanate to give a mixture of racemic regioisomers 88 and 91 (Figure 5). Again, both compounds exhibited mass and 1H-NMR spectra consistent with the expected product. Both compounds had good protective activity with 88 slightly more active than 91 [HC50 = 0.36 μM for 88 versus HC50 = 0.83 μM for 91]. The expected product contains two asymmetric centers at the bridgehead carbon atoms and would be produced as a racemate. The more active of the two racemic compounds 88 was resolved by chiral HPLC chromatography to yield the individual enantiomers as 89 and 90 (ORC-13661).44
Figure 5.

Synthesis and resolution of chiral tropane analogues. a). NCCH2CO2Et, S8, morpholine, EtOH; b). p-ClC6H4N=C=O, TEA, THF; c). chiral HPLC separation.
The structural ambiguity of the products derived from the original tropinone - Gewald reaction was eventually resolved by determining the single crystal X-ray diffraction structure of 90 HCl salt as shown in Figure 4 (Supporting Information, Figure S2, S3 and Table S2). Surprisingly, the synthesis of 90 resulted from a previously undescribed alternate reaction pathway in the Gewald condensation, resulting in a reversed regiochemistry of the thiophene relative to the bicyclic tropinone starting material.
Figure 4.

Crystal structure of 90.
The crystal structure unambiguously proves the absolute structure of 90 as (1R,8S)-4-{[(4-chlorophenyl)carbamoyl]amino}-11-methyl-5-thia-11-azatricyclo[6.2.1.02,6]undeca-2(6),3-diene-3-carboxamide hydrochloride. Based on this the crystal structure, we assigned the structures of 89, 90, 92 and 93 as shown in Figure 5, with 91, 92 and 93 representing the “expected” Gewald products. The mechanism of the “unexpected” Gewald reaction to yield 88, 89 and 90 is under investigation. An alternate synthetic approach starting from chiral (+) 2-tropinone was developed subsequently and provided 90 that was identical by mass spectrometry, 1H-NMR spectroscopy and chiral HPLC as the material derived from achiral 3-tropinone. The asymmetric synthesis of 90 will be the subject of a subsequent publication.
Protection assays in zebrafish revealed that 90 is the more potent enantiomer (HC50 = 0.12 μM) while 89 is considerably less effective (HC50 > 2.25 μM). The eudismic ratio (ER) of 18.8 suggests that 90 interacts with a chiral target. The less potent racemate 91 was also resolved by chiral HPLC to provide the individual enantiomers as 92 and 93. The levorotatory enantiomer 93 is more potent (HC50 = 0.27 μM) while the dextrorotatory enantiomer 92 is considerably less active (HC50 = 7.21 μM, ER 26.7). Reaction of the N-BOC tropinone under standard Gewald conditions followed by p-chlorophenyl-urea formation yielded a single product 94 (Figure 5), albeit in poor yield, that was carried through N-BOC removal and chiral resolution to yield the N-H compounds 95 and 96. Based on 1H NMR chemical shift of the diagnostic C-6 (X-ray structure numbering) methine proton, we assigned the structures of 94, 95 and 96 as shown (Figure 5). As with the N-methyl compounds 92 and 93, the levorotatory N-H enantiomer 96 was more potent (HC50 = 0.27 μM) while the dextrorotatory enantiomer 95 was less active (HC50 = 3.92 μM, ER 14.7), lending additional support to our structural assignments. Completing the N-H and N-methyl regiochemical tropane-fused thiophene sets, we reacted racemic N-BOC 2-tropinone under the Gewald conditions to afford racemic 97. Chiral HPLC separation of 97 could only be accomplished prior to N-BOC group deprotection. Subsequent removal of the N-BOC group yielded enantiomers 98 and 99.
Examination of the structural relationship among the active enantiomers of the two thiophene orientation Gewald products for the N-methyl 90 and 93, and N-H compounds 96 and 99 reveals a similar relationship to that seen with the regio-isomeric tetrahydropyridines 1 and 41 (Table 5). The altering the position of the basic nitrogen relative to the thiophene ring while maintaining the overall scaffold geometry yields compounds with similar biological activity. The tropinone compounds 90, 93, 96 and 99 exhibit superior protective activity with HC50 in the 33 – 270 nM range while their corresponding enantiomers 89, 92, 95 and 98 have significantly reduced activity. Based on activity and accessibility, indole-containing N-alkyl derivative 67 (Table 6), 2,2,6,6-tetramethyl tetrahydropyridine derivative 87 (Table 8) and 90 were selected for early pre-clinical characterization along with 1 (1).
Pre-clinical characterization of 1, 67, 87 and 90
Although the hit-to-lead phase of the campaign resulted in several very potent and efficacious derivatives of 1 1, the triage of compounds using the early ADME results was the determining factor in deciding which analogues to take forward for early pharmacokinetic evaluation. As a number of the quaternary ammonium salt compounds were not orally bioavailable and also found to inhibit the hERG channel, we elected to focus on the tertiary amine derivatives. As a result, we chose to profile three compounds, in addition to 1 (1, HC50 = 3.12 μM): the terminal indole-containing N-alkyl compound 67 (Table 6, HC50 = 0.10 μM), the 2,2,6,6-tetramethyl tetrahydropyridine derivative 87 (Table 8, HC50 = 1.24 μM) and the bicyclic tropane-like compound 90, Table 8, HC50 = 0.12 μM). Therefore, the pharmacokinetic, metabolic and in-vitro toxicological properties for these candidates were determined. The compounds have moderate to good aqueous solubility [85 μM for 1, 86 μM for 67, 83 μM for 87, and 76 μM for 90 for the free base forms], acceptable plasma protein binding profiles [95.9% (human) and 94.1% (rat) 1, 99.9% (human) and 99.9% (rat) for 67, 96.5% (human) and 95.8% (rat) for 87, and 90.7% (human) and 90.1% (rat) for 90], and elimination kinetics with half-lives of 2.1 hours for 1, 7.1 hours for 67, 6.2 hours for 87 and 7.8 hours for 90. Compounds 1, 87 and 90 exhibited no in vitro toxicity against the HepG2 cell line (IC50 >300 μM), whereas compound 67 inhibited growth of HepG2 cells is IC50 17μM. All four compounds were well tolerated in an acute toxicity assay in rats at 50 mg/kg i.v. As noted, hit compound 1 inhibited the hERG ion channel (hERG IC50 = 3.26 μM) giving a ratio of 1.2 (hERG/HC50). On the other hand, compounds 67, 87 and 90 were less potent hERG inhibitors with IC50 values of 20.1 μM, >30 μM and 5.6 μM, respectively, and with hERG/HC50 ratios of 201, >24 and 47, respectively. In addition to hERG inhibition, 1 was found to gradually undergo partial air oxidation to yield an inactive, highly fluorescent byproduct. Compound 67 was shown to have marginal oral bioavailability (>1%), while 1, 87 and 90 had acceptable to good oral bioavailability (46%, 21% and >50%). Compound 87 and 90 are resistant to the oxidation pathway observed for 1 (as well as 67) degradation by virtue of geminal dimethyl substitution in 87 and the bicyclic tropane framework of 90.
Compounds 1 and 90 were tested for in vitro and in vivo interference with aminoglycoside antimicrobial activity. There was no interference by either drug with neomycin minimal inhibitory concentration (MIC) or minimal bactericidal concentration (MBC) against E. coli ATCC25922. Additionally, 90 exhibited no interference in the MIC for tobramycin, amikacin, kanamycin and neomycin against five strains of P. aeruginosa or amikacin MIC against M. tuberculosis. Finally, 90 showed no interference in an in vivo mouse pulmonary tuberculosis model with amikacin, kanamycin and moxifloxacin (see Supporting Information, Table S4-1 and S4-2).
In vivo testing of ORC compounds in rats
On the basis of early pre-clinical evaluation, compounds 1 and 90 were tested in an in vivo rat model of aminoglycoside-induced hearing loss. The auditory brain stem response (ABR) assay is a widely used, non-invasive method for quantification of auditory function in vertebrates, including humans. Male Sprague Dawley rats 5–7 weeks old were purchased from Harlan Laboratories. Briefly, rats are anesthetized, placed in a sound-attenuated enclosure on a heating pad to maintain body temperature at 37.5 ºC. Electrical responses from the ear and CNS are detected with subcutaneous needle electrodes. Positive and negative electrodes are inserted at the left temporal bone above the pinna and at the vertex of the skull, with the ground electrode in the thigh. Free-field pure-tone stimuli are generated by custom software, and presented as short (5 ms) acoustic pulses with 1-ms rise/fall times, at a repetition rate of 19/s. In addition, broadband clicks are presented at the beginning and end of each session to assess any changes in the animal’s condition. Neural responses are amplified, bandpass filtered (100–3000 Hz), digitized using custom software and displayed on-line as an average response to the repeated acoustic stimulus. A sequence of pure tone stimuli of varying frequencies (2, 4, 8,16 and 32 kHz) and intensities (usually 15 – 90 dB, SPL) is presented and the resulting waveforms are recorded. All individual responses and averaged responses were saved. Examination of the averaged sound-induced waveforms by a trained experimenter yields the hearing thresholds—minimum sound level required to induce a reliable waveform at each frequency tested.
In preliminary experiments, we showed that 1 injected intravenously (i.v.) was non-toxic to rats at doses up to 25 mg/kg. This dose also yielded a plasma half-life of 1.7 hours. Based on literature precedent, we carried out an in vivo hearing protection experiments using kanamycin (500 mg/kg/day s.c.). The results of these experiments are shown in Figure 6. After initial ABR testing, male Sprague-Dawley rats 50–60 days old were treated with kanamycin 500 mg/kg/day (s.c.) for 14 days and then allowed to recover without treatment for an additional two weeks before final ABR testing. A second group was subjected to the same treatment with the addition of exposure to 1 at 25 mg/kg/day by intraperitoneal injection. Control groups included animals receiving 1 alone, the vehicle for 1 (Cremaphor EL:DMSO:EtOH:PBS) alone or no treatment. ABR testing (Figure 6) reveals a highly significant hearing loss at 8–32 kHz, that is greatest at the higher frequencies in animals treated with kanamycin alone. This hearing loss was completely prevented by 1 at 8 and 16 kHz and mostly resolved at the highest frequency tested (32 kHz). All rat protocols were approved by the University of Washington IACUC.
Figure 6.
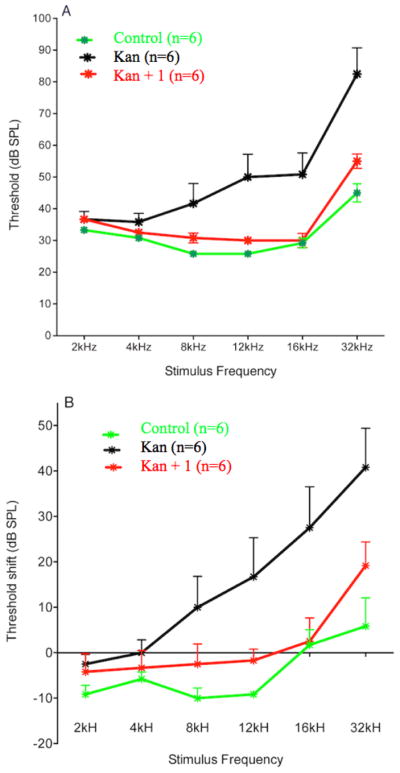
Protection of hearing in Sprague-Dawley rats treated with kanamycin for 14 days (500 mg/kg/day s.c.) by concurrent i.p. administration of 1 (25 mg/kg/day). A. Mean (+1 SEM) ABR thresholds in rats recorded an additional 14 days after the 14-day drug treatment with 1 alone, kanamycin, or co-administration with kanamycin and 1. B. Mean (+1 SEM) threshold shifts in hearing from pre-treatment levels 14 days after the treatment for each group. Positive values indicate increasing levels of hearing loss.
We selected compound 90 as our lead compound for full pre-clinical evaluation based on its superior protective activity in the zebrafish assay, decreased hERG-inhibitory activity and superior pharmacokinetic properties (Figure 7 and Supporting Information, Table S1) and efficiency of large scale synthesis. Compound 90 was tested for in vivo protection using amikacin, a more widely employed aminoglycoside antibiotic than kanamycin in the clinic. For testing the in vivo mammalian efficacy of 90, we used Fischer 344 rats 7–9 weeks old at the time of testing were purchased from Taconic. Rats were exposed to amikacin alone at 320 mg/kg/day (s.c.) for 12 days or the same amikacin exposure plus 90 (5 mg/kg/day, p.o.) for the same period. Additional rats were treated with 90 alone (5 mg/kg/day [n=3], or 25 mg/kg/day [n=3], p.o.) for 12 days. In all experiements 90 was delivered by oral gavage. We find that exposure to amikacin compared to kanamycin causes significantly more reproducible hearing loss in animals treated with AGA alone. For oral gavage the vehicle for 90 was PEG300:DMA:EtOH:H20. ABR thresholds were determined for each rat prior to drug treatment. Rats were then treated with amikacin alone or amikacin plus 90 daily for 12 days and then allowed to recover for 2 weeks before final ABR testing. As shown in Figure 8, Fischer 344 rats treated with amikacin (320 mg/kg/day s.c.) alone for 12 days exhibited moderate to severe hearing loss at all frequencies tested. Co-administration of amikacin (320 mg/kg/day s.c.) and 90 (5 mg/kg/day p.o.) for 12 days resulted in highly significant hearing protection across all frequencies tested. Furthermore, the resulting average hearing thresholds of this group are well within the normal range, and 6 of the 8 rats tested in this group showed complete protection from hearing loss due to the aminoglycoside exposure. None of the rats treated with 90 alone showed reliable changes in hearing thresholds (data not shown). In support of the audiological findings, immunohistochemical examination of the inner and outer hair cells of the organ of Corti from experimental animals showed significant disruption of normal tissue architecture (Figure 9, panel A) in Fischer 344 rats treated with amikacin alone (Figure 9, panel B). The amikacin-induced hair cell death in the organ of Corti was suppressed by co-treatment with 90.
Figure 7.

ADME Profiling for Compound 90.
Figure 8.

Protection of hearing in Fischer 344 rats treated with amikacin (320mg/kg/day s.c.) for 12 days with or without concurrent oral administration of 90 (5 mg/kg/day). Threshold shift from pre-treatment hearing levels at 2-weeks following treatment period.
Figure 9.

Confocal images of the organ of Corti basal turn: representative examples from each treatment group. A. Control: saline treated, s.c. daily for 12 days. B. Amikacin treated, s.c. daily, 320 mg/kg/day for 12 days. C. Amikacin (Ami) + 90: Amikacin s.c. daily, 320 mg/kg/day + 90 p.o. daily, 5mg/kg for 12 days. Cochleae from animals were fixed with 4% paraformaldehyde and dissected. Organs of Corti were incubated with cell-type specific antibodies: anti-myosin 7A antibody (hair cells: green), anti-Sox2 antibody (supporting cell nuclei: red) and antineurofilament (NF) antibody (neuronal processes: blue). All animals were treated with drug for 12 days. Final post-treatment ABR testing was done after an additional 12 day period. Animals euthanized after ABR testing for histological examination.
Discussion
Protection of hearing through pharmacological intervention against the myriad insults that are capable of causing hearing loss has been a long sought-after goal.45 We describe the development and preclinical characterization of a family of urea-thiophene carboxamides culminating in 90, an orally active and well tolerated agent with excellent pharmacokinetic and metabolic properties. Compound 90 protects rat hearing against amikacin-induced ototoxicity while showing no effect on in vitro or in vivo antibacterial activity of several clinically relevant AGAs. Our structure activity relationship (SAR) for this compound series is based on over 300 analogs interrogating all elements of the core structure of the screening hit 1.34 Our studies identified structural elements that are absolutely required for protective activity as well as those that can be substantially altered without disrupting the biological activity. The p-chlorophenyl urea substituent appears to be optimal as a wide variety of substituted aryl urea compounds exhibit comparable activity at best. Significant changes of the aryl substituent nature or position lead to loss of activity. The bis-mono-substituted urea is absolutely required for activity as even modest change such as replacement by an amide, cyclopropyl malonamide or N-methylation abolishes activity. Likewise, the thiophene-3-carboxamide is critical for protective activity as replacement of either thiophene or carboxamide with even conservative structural variants leads to dramatic loss of activity.
The N-ethyl tetrahydropyridine moiety of 1 provided the richest ground for structural variation. A basic nitrogen or quaternary ammonium salt is needed for activity, presumably existing in a mostly, or completely cationic state at physiological pH for favorable electrostatic interactions with its target. The positive charge can however reside at the tetrahydropyridine 2-position (as in 1) or 3-position (as in 41, Table 5), in a 5-membered ring (as in 42, Table 5) or 7-membered ring (as in 43 and 44, respectively, Table 5) or as part of a rigid bicyclo-[3.2.1]-aza-octane (tropane) ring system (as in 90 and 93, Table 8). Extending the N-alkyl substituent to 2-naphthyl- or indole-containing amides (as in 66 and 67, Table 6) yielded the most active compounds in the zebrafish assays but at the cost of physicochemical properties. Compound 67 has the high potency value (HC50 = 70 nM), but lacks solubility and oral bioavailability making it unsuitable for in vivo testing in the rat. Based on the high potency of aryl amide analogs like 67, we investigated the possibility of appending functional groups that would be useful for mechanistic and target identification studies. We discovered that a wide range of substituents are tolerated when connected to the tetrahydropyridine nitrogen through a 7- to 17-atom linker. Analogs containing a fluorescent probe (i.e. BODIPY) as well as an affinity tag (i.e. biotin) have been synthesized based on the existing SAR and are currently being used in mechanistic and target identification efforts. Among the most active ORC compounds in this series are 90, HC50 = 120 nM), 93 (HC50 = 270 nM), 96 HC50 = 270 nM) and 99 HC50 = 33 nM), which vary only in Nsubstitution on the 8-azabicyclo[3.2.1] ring system and position of the 2-carbon bridge relative to the thiophene ring (Figure 5). Because of it superior potency in the zebrafish hair cell protection assay and improved pharmacokinetic properties 90 was advanced through IND-enabling studies under the auspices of Oricula Therapeutics.
Conclusion
In the absence of a molecular target, elaboration of our original screening hit 1, into a full SAR program was made possible using the phenotypic zebrafish neuromast hair cell viability assay to predict in vivo activity in a mammalian model. This effort yielded 90 as a promising clinical candidate for the prevention of aminoglycoside induced hearing loss in patients. Clinical adoption of such an agent would reinvigorate the safe use of this underused broad class of effective antibiotics and thus expand their utility in the treatment of serious and life-threatening bacterial infections.
Experimental Section
Zebrafish neuromast hair cell protection assay
Adult AB zebrafish were bred and newly fertilized embryos were collected the week prior to testing and raised at 28.5°C in petri dishes containing embryo medium (1 mM MgSO4, 0.15 mM KH2PO4, 50 μM Na2HPO4, 1 mM CaCl2, 500 μM KCl, 115 mM NaCl, 700 μM NaHCO3). Newly hatched free-swimming larvae were fed paramecium at 4 days post fertilization (dpf). All procedures were approved by the University of Washington Animal Care and Use Committee. For initial efficacy testing of each new experimental compound, fish 5–7 dpf were transferred to cell culture baskets placed in 6-well culture plates containing 7 mL of embryo medium. Typically, tests are done with ten fish per basket but work well with up to 50 fish. All treatment and wash volumes are 7 mL. Fish were pre-treated with test compound for 1 hour at five initial concentrations (1.56, 3.125, 6.25, 12.5, and 25 μM). Following this exposure to the test compound alone, fish were moved to a new well containing the test compound plus 200 μM neomycin (neomycin sulfate, Sigma, St. Louis, MO, catalog # N1142) for 1 hr. If significant hair cell protection was observed at the lowest concentration the dosages were reduced by half until hair cell protection was eliminated. All experimental compounds were tested alongside the following controls groups: fish treated with neomycin (200 μM) alone; fish treated with test compound (25 μM) alone); fish treated with 1 at the same concentration range as test compound; and untreated fish using the same procedures.
Following neomycin exposure, fish were rinsed briefly 4 times in embryo medium and 700 μl of 0.05% DASPEI (2-{4-(dimethylamino)styryl}-N-ethylpyridinium iodide, Molecular Probes, Eugene, OR) was added and allowed to stain for 15 minutes. DASPEI brightly labels the hair cells of each neuromast due to their robust packing in mitochondria.46 Following this labelling and two rinses in embryo medium, the fish were anesthetized with 350 μl MS222 (0.55 μg/mL final concentration, 3-aminobenzoic acid ethyl ester, methansulfonate salt, Sigma, St. Louis, MO).
For assessment of the dose-response protective functions, the fish in each experimental and control group were transferred to separate wide depression slides in embryo medium with MS222. Slides were mounted on the stage of an epifluorescence dissecting microscope equipped with a DAPSEI filter set (excitation 450–490 nM and barrier 515 nM, Chroma Technologies, Brattleboro, VT). The hair cell staining of ten neuromasts (SO1, SO2, IO1, IO2, IO3, IO4, M2, MI1, MI2 and O2) on one side of each animal was visually evaluated.47 Each neuromast was scored for presence of a normal compliment of hair cells (score=2), reduced DASPEI staining indicating a reduction in hair cell number (score=1), or absence of DASPEI staining (score=0). The total score for each animal was tabulated, to give a composite score that ranged from 0 to 20 for each fish. Average scores and standard deviations were calculated for animals in each treatment group. Scores were normalized to the control group (vehicle only, no drug, no neomycin) and expressed as % hair cell survival. If hair cell survival was at least 50%, the 50% effective concentration (HC50) was calculated as a linear extrapolation from the nearest concentrations of protective drug that produced hair cell survival below and above 50%.
Rat Auditory Brain Stem Response (ABR) assay for hearing protection
Male Fischer 344 rats were purchased from Harlan Laboratories (Livermore CA) at 40–50 days postnatal. Auditory brainstem responses (ABRs) were measured from each rat twice, once 1–2 weeks prior to drug treatments and again at 2 weeks after the termination of drug treatment. Rats were anesthetized with isoflurane, placed on a heating pad to maintain body temperature near 37°C, and placed in a sound-attenuating chamber. ABRs responses were recorded using standard subcutaneous needle electrodes with the positive and negative electrodes at the left temporal bone above the pinna and the vertex of the skull, and the ground electrode in the thigh. Free-field pure tone stimuli were generated, and ABR recordings were digitized using custom software. Tone pips were 5 ms in duration with 1-ms rise/fall times, presented at a repetition rate of 19/s. In addition, broadband clicks were presented at the beginning and end of each session to assess for any changes in the animal’s condition. All stimuli were calibrated on-line at the beginning of each experiment with a calibrated probe microphone. Neural responses were pre-amplified (100×; A-M Systems amplifier Model 3000), sent through an MA3 amplifier with an additional 20 dB post-preamp gain (Tucker Davis Technologies), bandpass filtered (100–3000 Hz; Krohn-Hite filter model 3550), and digitized at 24.4 kHz. We sampled responses in a 15-ms window (with a 5ms stimulus onset delay). The threshold was defined as the lowest sound pressure level (SPL) in which a recognizable waveform was present and repeatable. Thresholds were determined at 2, 4, 8, 16, and 32 kHz and for a broad-band click. Stimuli were presented 500 times from 80 to 20 dB SPL in steps of 10 and then 1000 repetitions in steps of 5 dB SPL when approaching threshold. Near threshold each series was repeated to determine the reliability of the waveform at the estimated threshold, 5 dB above and 10 dB below the estimated threshold. When animals appeared to be deaf at a particular frequency, the stimulus was presented at the maximum intensity generated by our system (90–100 dB SPL at least twice at 1000 repetitions to be assured of a complete hearing loss. The threshold was then arbitrarily set at 100 dB SPL. All averaged responses were examined on line and thresholds were estimated. Following the experimental session all responses were printed and rescored twice, once by the experimenter and once by an ABR expert who was blinded with regard to the experimental condition. Threshold estimates were within 5 dB in over 97 % of the response traces, and the few that differed by >10dB were re-examined by the two evaluators to come to a consensus. Data were analyzed as raw thresholds at each test frequency and as “Threshold Shift”, comparing pre-test threshold with post-drug administration threshold, where a positive number indicates a hearing loss (in dB) due to the drug treatment.
Chemistry
All reactions were performed under a dry atmosphere of nitrogen unless otherwise specified. Indicated reaction temperatures refer to the reaction bath, while room temperature (rt) is noted as 25 ºC. Commercial grade reagents and anhydrous solvents were used as received from vendors and no attempts were made to purify or dry these components further. Removal of solvents under reduced pressure was accomplished with a Buchi rotary evaporator at approximately 28 mm Hg pressure using a Teflon-linked KNF vacuum pump. Thin layer chromatography was performed using either 1” × 3” AnalTech No. 02521 or Merck 60 F254 silica gel plates with fluorescent indicator using appropriate solvent mixtures. Visualization of TLC plates was made by observation with either short wave UV light (254 nm lamp), 10% phosphomolybdic acid in ethanol or in iodine vapors. Medium pressure Flash column chromatography was carried out using either a Teledyne Isco CombiFlash Companion Unit with RediSep Rf silica gel columns or a Biotage Isolera with SiliCycle HP cartridges. Proton NMR spectra were obtained either on 300 MHz Bruker Nuclear Magnetic Resonance Spectrometer or 500 MHz Bruker Nuclear Magnetic Resonance Spectrometer and chemical shifts (δ) are reported in parts per million (ppm) and coupling constant (J) values are given in Hz, with the following spectral pattern designations: s, singlet; d, doublet; t, triplet, q, quartet; dd, doublet of doublets; m, multiplet; br, broad singlet; sym, symmetrical. Tetramethylsilane (TMS) was used as an internal reference. Melting points are uncorrected and were obtained using a MEL-TEMP Electrothermal melting point apparatus. Mass spectroscopic analyses were performed either using positive mode electron spray ionization (ESI) on a Varian ProStar LC-MS with a 1200L quadrupole mass spectrometer or using positive mode atmospheric pressure chemical ionization (APCI) on a Shimadzu LC-MS system. High performance liquid chromatography (HPLC) purity analysis was conducted using a Varian Pro Star HPLC system with a binary solvent system A and B using a gradient elution [A, H2O with 0.1% trifluoroacetic acid (TFA); B, CH3CN with 0.1% TFA] and flow rate = 1 mL/min, with UV detection at 254 nm. All final compounds were purified to ≥95% purity and these purity levels were measured by a Varian Pro Star HPLC system. Three different Varian Pro Star HPLC methods were used to establish compound purity. HPLC Method A: Phenomenex Luna C18(2) column (4.6 × 250 mm); mobile phase, A = H2O with 0.1% TFA and B = CH3CN with 0.1% TFA; gradient 10–95% B (0.0–10 min; hold for 6 min); UV detection at 254 nm. HPLC Method B: SunFire C18 column (4.6 × 250 mm); mobile phase, A = H2O with 0.1% TFA and B = CH3CN with 0.1% TFA; gradient: 10–100% B (0.0–20 min; hold for 5 min); UV detection at 254 nm. HPLC Method C: SunFire C18 column (4.6 × 250 mm); mobile phase, A = H2O with 0.1% TFA and B = CH3CN with 0.1% TFA; gradient: 0–100% B (0.0–15 min; hold for 5 min)); UV detection at 254 nm.
2-Amino-6-ethyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxamide. A stirred mixture of N-ethyl-4-pyrrolidinone (22.7 g, 178 mmol), 2-cyanoacetamide (16.5 g, 197 mmol), sulphur (6.87 g, 215 mmol) and morpholine (31.5 g, 362 mmol) in ethanol (350 mL) was heated to reflux under nitrogen for 5 h. After this time, the reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was mixed with saturated aqueous sodium bicarbonate (200 mL), water (200 mL). The aqueous mixture was extracted with methylene chloride (5 × 200 mL). The combined organic extracts were dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was triturated with cold methanol (30 mL) and filtered. The filter cake was washed with cold methanol (2 × 10 mL) and then dried under reduced pressure to provide the product as a yellow solid (21.7 g, 54%): 1H NMR (300 MHz, DMSO-d6) δ 6.98 (s, 2H), 6.52 (bs, 2H), 3.29 (s, 2H), 2.66 (d, J = 4.8 Hz, 2H), 2.60 (d, J = 4.8 Hz, 2H), 2.46 (q, J = 7.2 Hz, 2H), 1.04 (t, J = 7.2 Hz, 3H); LRMS m/z 226 [M+H]+.
2-[3-(4-Chlorophenyl)ureido]-6-ethyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxamide (1). To the stirred solution of 2-amino-6-ethyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxamide (450 mg, 2.00 mmol) in anhydrous tetrahydrofuran (10 mL) at room temperature under nitrogen was added a solution of 4-chlorophenyl isocyanate (400 mg, 2.40 mmol) in anhydrous methylene chloride (6 mL) dropwise over 3 min. Then the reaction mixture was stirred overnight at room temperature. The reaction mixture was filtered. The filter cake was washed with methylene chloride (5 mL) and then dried under reduced pressure to provide the product 1 (free base) as a white solid (301 mg, 38%): 1H NMR (300 MHz, DMSO-d6) δ 10.94 (s, 1H), 10.12 (s, 1H), 7.52–7.45 (m, 4H), 7.34 (d, J = 9.0 Hz, 2H), 3.45 (s, 2H), 2.77 (d, J = 4.8 Hz, 2H), 2.65 (d, J = 4.8 Hz, 2H), 2.55–2.45 (m, 2H), 1.07 (t, J = 6.6 Hz, 3H); LRMS m/z 379 [M+H]+.
2-[3-(4-Chlorophenyl)ureido]-6-ethyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxamide Hydrochloride (1 HCl). To the stirred mixture of compound 1 (free base) (157 mg, 0.400 mmol) in methylene chloride (50 mL) at room temperature was added hydrochloric acid (2 M in diethyl ether, 0.300 mL, 0.600 mmol). After addition, the mixture was concentrated under reduced pressure. The resulting solid was triturated with methylene chloride and filtered to afford compound (1 HCl) as pale yellow solid: 1H NMR (500 MHz, DMSO-d6) δ 10.94 (s, 1H), 10.84 (bs, 1H), 10.22 (s, 1H), 7.75–6.90 (m, 6H), 4.49 (d, J = 14.5 Hz, 1H), 4.22–4.16 (m, 1H), 3.65– 3.62 (m, 1H), 3.38–3.05 (m, 5H), 1.32 (t, J = 7.2 Hz, 3H); HRMS (ESI) m/z calculated for C17H19ClN4O2S [M+H]+ 379.09899, found 379.10085.
(+/−)-2-Amino-9-methyl-5,6,7,8-tetrahydro-4H-5,8-epiminocyclohepta[b]-thiophene-3-carboxamide. The stirred mixture of tropinone (3.00 g, 21.6 mmol), 2-cyanoacetamide (1.99 g, 23.7 mmol), sulphur (830 mg, 25.9 mmol), and morpholine (3.75 g, 43.0 mmol) in ethanol (80 mL) was heated to reflux under nitrogen for 4 h. After this time, the reaction mixture was cooled to room temperature and concentrated under reduced pressure. The resulting residue was diluted with saturated aqueous sodium bicarbonate (60 mL) and extracted with methylene chloride (3 × 150 mL). The combined organic extracts were dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel eluting with methanol/methylene chloride (1:9) to provide compound (+/−)-2-Amino-9-methyl-5,6,7,8-tetrahydro-4H-5,8-epiminocyclohepta[b]-thiophene-3-carboxamide as a brown solid (396 mg, 8%): LRMS m/z 238 [M+H]+.
(+/−)-2-[3-(4-Chlorophenyl)ureido]-9-methyl-5,6,7,8-tetrahydro-4H-5,8-epiminocyclohepta[b]thiophene-3-carboxamide 88. To the stirred mixture of 2-Amino-9-methyl-5,6,7,8-tetrahydro-4H-5,8-epiminocyclohepta[b]-thiophene-3-carboxamide (375 mg, 1.58 mmol) in methylene chloride (10 mL) at room temperature under nitrogen was added a solution of 4-chlorophenyl isocyanate (255 mg, 1.66 mmol) in methylene chloride (10 mL). The reaction mixture was stirred overnight and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel eluting with methanol/methylene chloride (15:85) to provide compound 88 as an off-white solid (369 mg, 60%): 1H NMR (300 MHz, DMSO-d6) δ 10.70 (bs, 1H), 10.05 (bs, 1H), 7.90–6.50 (m, 6H), 4.11 (bs, 1H), 2.99 (d, J = 14.1 Hz, 1H), 2.35–1.95 (m, 7H), 1.79 (bs, 1H), 1.48 (bs, 1H); LRMS m/z 391 [M+H]+.
(−)-2-[3-(4-Chlorophenyl)ureido]-9-methyl-5,6,7,8-tetrahydro-4H-5,8-epiminocyclohepta[b]thiophene-3-carboxamide (89) and (+)-2-[3-(4-Chlorophenyl)ureido]-9-methyl-5,6,7,8-tetrahydro-4H-5,8-epiminocyclohepta[b]thiophene-3-carboxamide (90)
88 (720 mg) was separated by chiral preparative HPLC (20 μm CHIRALCEL OD, 5 cm × 50 cm, 100 mL/min flow rate, 120 mg/injection) eluting with 0.1% diethylamine in 20% ethanol/heptane to provide 89 (268 mg, 37%)as a white solid, LRMS m/z 391 [M+H]+, followed by 90 (250 mg, 35%) as a white solid. LRMS m/z 391 [M+H]+. Free base optical rotations. 89: −19.5º (c = 0.2, MeOH), T=25 ºC. 90: +18.5º (c = 0.2, MeOH), T=25 ºC.
(−)-2-[3-(4-Chlorophenyl)ureido]-9-methyl-5,6,7,8-tetrahydro-4H-5,8-epiminocyclohepta[b]thiophene-3-carboxamide Hydrochloride 89 and (+)-2-[3-(4-Chlorophenyl)ureido]-9-methyl-5,6,7,8-tetrahydro-4H-5,8-epiminocyclohepta[b]thiophene-3-carboxamide Hydrochloride (89). To the stirred mixture of compound 89 (free base) (240 mg, 0.610 mmol) in methanol (30 mL) at room temperature was added 1 M hydrochloric acid (1.25 mL, 1.25 mmol) dropwise. After addition, the mixture was stirred for 10 min. After this time, the mixture was diluted with water (10 mL) and lyophilized to provide compound 89 as a white solid (255 mg, 97%): 1H NMR (300 MHz, DMSO-d6) δ 11.24 (bs, 0.40 H), 10.34 and 10.32 (2 s, 1H), 10.19–10.16 (m, 1.60H), 7.51–7.48 (m, 4H), 7.35 (d, J = 9.0 Hz, 2H), 4.95–4.82 (m, 1H), 4.19–4.08 (m, 1H), 3.43–3.17 (m, 1H), 2.88–2.68 (m, 4H), 2.45–2.09 (m, 3H), 1.89–1.80 (m, 1H); optical rotation [α]D= −5.5º (c = 0.2, MeOH); HRMS (ESI) m/z calculated for C18H19ClN4O2S [M+H]+ 391.09899, found 391.09906.
(+)-2-[3-(4-Chlorophenyl)ureido]-9-methyl-5,6,7,8-tetrahydro-4H-5,8-epiminocyclohepta[b]thiophene-3-carboxamide Hydrochloride 89 and (+)-2-[3-(4-Chlorophenyl)ureido]-9-methyl-5,6,7,8-tetrahydro-4H-5,8-epiminocyclohepta[b]thiophene-3-carboxamide Hydrochloride (90). To the stirred mixture of compound 90 (free base)(220 mg, 0.60 mmol) in methanol (30 mL) at room temperature was added 1 M hydrochloric acid (1.25 mL, 1.25 mmol) dropwise. After addition, the mixture was stirred for 10 min. After this time, the mixture was diluted with water (10 mL) and lyophilized to provide compound 90 as a white solid (243 mg, 96%): 90: 1H NMR (300 MHz, DMSO-d6) δ 11.20 (bs, 0.34H), 10.34 and 10.31 (2 s, 1H), 10.18 (s, 1H), 10.16 (bs, 0.66H), 7.54–7.33 (m, 6H), 4.94–4.90 (m, 1H), 4.20–4.06 (m, 1H), 3.42–3.16 (m, 1H), 2.88–2.65 (m, 4H), 2.49–2.28 (m, 2H), 2.21–2.09 (m, 1H), 1.89–1.80 (m, 1H); optical rotation [α]D= +3.5º (c = 0.2, MeOH); HRMS (ESI) m/z calculated for C18H19ClN4O2S [M+H]+ 391.09899, found 391.09907.
Supplementary Material
Acknowledgments
We thank AMRI chemistry colleagues Randall Davis and Feryan Ahmed for synthetic efforts in support of this program. We also thank Dr. Brandon Mercado of the Yale University Chemical and Biophysical Instrumentation Center for X-ray crystal structure determination of 90 HCl. We are also grateful for the assistance of Kelly Owens, Allison Hixon, Divya Mehta, Heather Kinnear and Patricia Wu for carrying out the zebrafish assays, to Carol Robbins, Brianna Moore, Robin Gibson and Van Redilia for carrying out the mammalian ABR studies. Tor Linbo and Glen MacDonald provided invaluable technical assistance and David White provided excellent maintenance and breeding advice in the zebrafish facility. We gratefully acknowledge the contributions of Jill Heemskerk, Nancy Freeman, and Charles Cywin (National Institutes of Health program staff). The research described here was supported by the grants U01NS074506, RO1DC013688, R01DC05987, and RO1DC009807 from the National Institutes of Health and funds from Washington State Life Sciences Discovery Fund.
Abbreviations Used
- ADME
absorption, distribution, metabolism and excretion
- AGAs
aminoglycoside antibiotics
- dpf
days post fertilization
- hERG
human Ether-à-go-go-Related Gene
- ABR
auditory brain stem response
- DASPEI
2-{4-(dimethylamino)styryl}-N-ethylpyridinium iodide
- TEA
triethylamine
- THF
tetrahydrofuran
Footnotes
Accession code
X-ray crystallographic structure of 90 HCl has been deposited at The Cambridge Crystallographic Database (CSD) with the accession code CCDC-1557948 (http://ccdc.cam.ac.uk).
The Supporting Information is available free of charge on the ACS Publications Website at DOI: (to be inserted by publisher).
Figure S1: Reproducibility of Zebrafish Lateral Line Hair Cells Viability Assay; Table S1: Pharmacokinetic and Toxicological Properties of 90; Figure S2 & S3, Figures S2, S3 and Table S2: Compound 90 Crystal Structure Determination Tables S4 and S5: Non-interference in Antimicrobial Activity of Aminoglycoside Antibiotics; Full Experimental Procedures and Characterization of Compounds 1–99 (PDF)
SMILES strings for reported compounds (CSV)
Notes
The authors declare the following competing financial interest(s): Part of the research reported was funded by Oricula Therapeutics. G.J., V.E.G., D.W.R., E.WR and J.A.S are founders of Oricula Therapeutics.
References
- 1.Parham K, McKinnon BJ, Eibling D, Gates GA. Challenges and Opportunities in Presbycusis. Otolaryngol Head Neck Surg. 2011;144:491–495. doi: 10.1177/0194599810395079. [DOI] [PubMed] [Google Scholar]
- 2.Mohr PE, Feldman JJ, Dunbar JL. The Societal Costs of Severe to Profound Hearing Loss in the United States. Policy Anal Brief H Ser. 2000;2:1–4. [PubMed] [Google Scholar]
- 3.Wong AC, Ryan AF. Mechanisms of Sensorineural Cell Damage, Death and Survival in the Cochlea. Front Aging Neurosci. 2015;7:58. doi: 10.3389/fnagi.2015.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubel EW, Furrer SA, Stone JS. A Brief History of Hair Cell Regeneration Research and Speculations on the Future. Hear Res. 2013;297:42–51. doi: 10.1016/j.heares.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cunningham LL, Cheng AG, Rubel EW. Caspase Activation in Hair Cells of the Mouse Utricle Exposed to Neomycin. J Neurosci. 2002;22:8532–8540. doi: 10.1523/JNEUROSCI.22-19-08532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang H, Sha SH, Schacht J. Kanamycin Alters Cytoplasmic and Nuclear Phosphoinositide Signaling in the Organ of Corti in Vivo. J Neurochem. 2006;99:269–276. doi: 10.1111/j.1471-4159.2006.04117.x. [DOI] [PubMed] [Google Scholar]
- 7.Op de Beeck K, Schacht J, Van Camp G. Apoptosis in Acquired and Genetic Hearing Impairment: The Programmed Death of the Hair Cell. Hear Res. 2011;281:18–27. doi: 10.1016/j.heares.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J, Van De Water TR, Bonny C, de Ribaupierre F, Puel JL, Zine A. A Peptide Inhibitor of C-Jun N-Terminal Kinase Protects against Both Aminoglycoside and Acoustic Trauma-Induced Auditory Hair Cell Death and Hearing Loss. J Neurosci. 2003;23:8596–8607. doi: 10.1523/JNEUROSCI.23-24-08596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Battin EE, Brumaghim JL. Antioxidant Activity of Sulfur and Selenium: A Review of Reactive Oxygen Species Scavenging, Glutathione Peroxidase, and Metal-Binding Antioxidant Mechanisms. Cell Biochem Biophys. 2009;55:1–23. doi: 10.1007/s12013-009-9054-7. [DOI] [PubMed] [Google Scholar]
- 10.Schacht J, Talaska AE, Rybak LP. Cisplatin and Aminoglycoside Antibiotics: Hearing Loss and Its Prevention. Anat Rec. 2012;295:1837–1850. doi: 10.1002/ar.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rizzi MD, Hirose K. Aminoglycoside Ototoxicity. Curr Opin Otolaryngol Head Neck Surg. 2007;15:352–357. doi: 10.1097/MOO.0b013e3282ef772d. [DOI] [PubMed] [Google Scholar]
- 12.Le TN, Straatman LV, Lea J, Westerberg B. Current Insights in Noise-Induced Hearing Loss: A Literature Review of the Underlying Mechanism, Pathophysiology, Asymmetry, and Management Options. J Otolaryngol Head Neck Surg. 2017;46:41. doi: 10.1186/s40463-017-0219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bagger-Sjoback D, Stromback K, Hakizimana P, Plue J, Larsson C, Hultcrantz M, Papatziamos G, Smeds H, Danckwardt-Lilliestrom N, Hellstrom S, Johansson A, Tideholm B, Fridberger A. A Randomised, Double Blind Trial of N-Acetylcysteine for Hearing Protection During Stapes Surgery. PLoS One. 2015;10:e0115657. doi: 10.1371/journal.pone.0115657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pirvola U, Xing-Qun L, Virkkala J, Saarma M, Murakata C, Camoratto AM, Walton KM, Ylikoski J. Rescue of Hearing, Auditory Hair Cells, and Neurons by Cep-1347/Kt7515, an Inhibitor of C-Jun N-Terminal Kinase Activation. J Neurosci. 2000;20:43–50. doi: 10.1523/JNEUROSCI.20-01-00043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ylikoski J, Xing-Qun L, Virkkala J, Pirvola U. Blockade of C-Jun N-Terminal Kinase Pathway Attenuates Gentamicin-Induced Cochlear and Vestibular Hair Cell Death. Hear Res. 2002;163:71–81. doi: 10.1016/s0378-5955(01)00380-x. [DOI] [PubMed] [Google Scholar]
- 16.Bodmer D, Brors D, Pak K, Gloddek B, Ryan A. Rescue of Auditory Hair Cells from Aminoglycoside Toxicity by Clostridium Difficile Toxin B, an Inhibitor of the Small Gtpases Rho/Rac/Cdc42. Hear Res. 2002;172:81–86. doi: 10.1016/s0378-5955(02)00514-2. [DOI] [PubMed] [Google Scholar]
- 17.Eshraghi AA, Wang J, Adil E, He J, Zine A, Bublik M, Bonny C, Puel JL, Balkany TJ, Van De Water TR. Blocking C-Jun-N-Terminal Kinase Signaling Can Prevent Hearing Loss Induced by Both Electrode Insertion Trauma and Neomycin Ototoxicity. Hear Res. 2007;226:168–177. doi: 10.1016/j.heares.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 18.Sugahara K, Rubel EW, Cunningham LL. Jnk Signaling in Neomycin-Induced Vestibular Hair Cell Death. Hear Res. 2006;221:128–135. doi: 10.1016/j.heares.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sagwa EL, Ruswa N, Mavhunga F, Rennie T, Leufkens HG, Mantel-Teeuwisse AK. Comparing Amikacin and Kanamycin-Induced Hearing Loss in Multidrug-Resistant Tuberculosis Treatment under Programmatic Conditions in a Namibian Retrospective Cohort. BMC Pharmacol Toxicol. 2015;16:36. doi: 10.1186/s40360-015-0036-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harris T, Peer S, Fagan JJ. Audiological Monitoring for Ototoxic Tuberculosis, Human Immunodeficiency Virus and Cancer Therapies in a Developing World Setting. J Laryngol Otol. 2012;126:548–551. doi: 10.1017/S0022215112000357. [DOI] [PubMed] [Google Scholar]
- 21.Lawson L, Yassin MA, Abdurrahman ST, Parry CM, Dacombe R, Sogaolu OM, Ebisike JN, Uzoewulu GN, Lawson LO, Emenyonu N, Ouoha JO, David JS, Davies PD, Cuevas LE. Resistance to First-Line Tuberculosis Drugs in Three Cities of Nigeria. Trop Med Int Health. 2011;16:974–980. doi: 10.1111/j.1365-3156.2011.02792.x. [DOI] [PubMed] [Google Scholar]
- 22.Garinis AC, Cross CP, Srikanth P, Carroll K, Feeney MP, Keefe DH, Hunter LL, Putterman DB, Cohen DM, Gold JA, Steyger PS. The Cumulative Effects of Intravenous Antibiotic Treatments on Hearing in Patients with Cystic Fibrosis. J Cystic Fibrosis. 2017;16:401–409. doi: 10.1016/j.jcf.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skolnik K, Kirkpatrick G, Quon BS. Nontuberculous Mycobacteria in Cystic Fibrosis. Curr Treat Options Infect Dis. 2016;8:259–274. doi: 10.1007/s40506-016-0092-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee BY, Clemens DL, Silva A, Dillon BJ, Maslesa-Galic S, Nava S, Ding X, Ho CM, Horwitz MA. Drug Regimens Identified and Optimized by Output-Driven Platform Markedly Reduce Tuberculosis Treatment Time. Nat Commun. 2017;8:14183. doi: 10.1038/ncomms14183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayer-Hamblett N, Kloster M, Rosenfeld M, Gibson RL, Retsch-Bogart GZ, Emerson J, Thompson V, Ramsey BW. Impact of Sustained Eradication of New Pseudomonas Aeruginosa Infection on Long-Term Outcomes in Cystic Fibrosis. Clin Infect Dis. 2015;61:707–715. doi: 10.1093/cid/civ377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lando D, Cousin MA, Privat de Garilhe M. Misreading, a Fundamental Aspect of the Mechanism of Action of Several Aminoglycosides. Biochemistry. 1973;12:4528–4533. doi: 10.1021/bi00746a035. [DOI] [PubMed] [Google Scholar]
- 27.Le Goffic F, Capmau ML, Tangy F, Baillarge M. Mechanism of Action of Aminoglycoside Antibiotics. Binding Studies of Tobramycin and Its 6′-N-Acetyl Derivative to the Bacterial Ribosome and Its Subunits. Eur J Biochem. 1979;102:73–81. doi: 10.1111/j.1432-1033.1979.tb06264.x. [DOI] [PubMed] [Google Scholar]
- 28.Yoshizawa S, Fourmy D, Puglisi JD. Structural Origins of Gentamicin Antibiotic Action. EMBO J. 1998;17:6437–6448. doi: 10.1093/emboj/17.22.6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brodersen DE, Clemons WM, Jr, Carter AP, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. The Structural Basis for the Action of the Antibiotics Tetracycline Pactamycin and Hygromycin B on the 30s Ribosomal Subunit. Cell. 2000;103:1143–1154. doi: 10.1016/s0092-8674(00)00216-6. [DOI] [PubMed] [Google Scholar]
- 30.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A Common Mechanism of Cellular Death Induced by Bactericidal Antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 31.Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. Oxidation of the Guanine Nucleotide Pool Underlies Cell Death by Bactericidal Antibiotics. Science. 2012;336:315–319. doi: 10.1126/science.1219192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalghatgi S, Spina CS, Costello JC, Liesa M, Morones-Ramirez JR, Slomovic S, Molina A, Shirihai OS, Collins JJ. Bactericidal Antibiotics Induce Mitochondrial Dysfunction and Oxidative Damage in Mammalian Cells. Sci Transl Med. 2013;5:192ra185. doi: 10.1126/scitranslmed.3006055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karasawa T, Wang Q, David LL, Steyger PS. Climp-63 Is a Gentamicin-Binding Protein That Is Involved in Drug-Induced Cytotoxicity. Cell Death Dis. 2010;1:e102. doi: 10.1038/cddis.2010.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Owens KN, Santos F, Roberts B, Linbo T, Coffin AB, Knisely AJ, Simon JA, Rubel EW, Raible DW. Identification of Genetic and Chemical Modulators of Zebrafish Mechanosensory Hair Cell Death. PLoS Genet. 2008;4:e1000020. doi: 10.1371/journal.pgen.1000020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown AM. Herg Block, Qt Liability and Sudden Cardiac Death. Novartis Found Symp. 2005;266:118–131. doi: 10.1007/978-1-59259-884-7_4. [DOI] [PubMed] [Google Scholar]
- 36.Taboureau O, Jorgensen FS. In Silico Predictions of Herg Channel Blockers in Drug Discovery: From Ligand-Based and Target-Based Approaches to Systems Chemical Biology. Comb Chem High Throughput Screening. 2011;14:375–387. doi: 10.2174/138620711795508322. [DOI] [PubMed] [Google Scholar]
- 37.Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. Herg K(+) Channels: Structure, Function, and Clinical Significance. Physiol Rev. 2012;92:1393–1478. doi: 10.1152/physrev.00036.2011. [DOI] [PubMed] [Google Scholar]
- 38.Witchel HJ. Drug-Induced Herg Block and Long Qt Syndrome. Cardiovasc Ther. 2011;29:251–259. doi: 10.1111/j.1755-5922.2010.00154.x. [DOI] [PubMed] [Google Scholar]
- 39.Eggert US. The Why and How of Phenotypic Small-Molecule Screens. Nat Chem Biol. 2013;9:206–209. doi: 10.1038/nchembio.1206. [DOI] [PubMed] [Google Scholar]
- 40.Wagner BK, Schreiber SL. The Power of Sophisticated Phenotypic Screening and Modern Mechanism-of-Action Methods. Cell Chem Biol. 2016;23:3–9. doi: 10.1016/j.chembiol.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eller GA, Holzer W. First Synthesis of 3-Acetyl-2-Aminothiophenes Using the Gewald Reaction. Molecules. 2006;11:371–376. doi: 10.3390/11050371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang Y, Domling A. The Gewald Multicomponent Reaction. Mol Divers. 2011;15:3–33. doi: 10.1007/s11030-010-9229-6. [DOI] [PubMed] [Google Scholar]
- 43.Coffin AB, Rubel EW, Raible DW. Bax, Bcl2, and P53 Differentially Regulate Neomycin- and Gentamicin-Induced Hair Cell Death in the Zebrafish Lateral Line. J Assoc Res Otolaryngol. 2013;14:645–659. doi: 10.1007/s10162-013-0404-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simon JA, Johnson GER, Raible DW, Gonzales MD, Metzler PC, Miao W. Compound and Methods for Preventing or Treating Sensory Hair Cell Death. 9,493,482. US Patent. 2016
- 45.Cheng AG, Cunningham LL, Rubel EW. Mechanisms of Hair Cell Death and Protection. Curr Opin Otolaryngol Head Neck Surg. 2005;13:343–348. doi: 10.1097/01.moo.0000186799.45377.63. [DOI] [PubMed] [Google Scholar]
- 46.Harris JA, Cheng AG, Cunningham LL, MacDonald G, Raible DW, Rubel EW. Neomycin-Induced Hair Cell Death and Rapid Regeneration in the Lateral Line of Zebrafish (Danio Rerio) J Assoc Res Otolaryngol. 2003;4:219–234. doi: 10.1007/s10162-002-3022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raible DW, Kruse GJ. Organization of the Lateral Line System in Embryonic Zebrafish. J Comp Neurol. 2000;421:189–198. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.