

Abstract
A coordinated, multidisciplinary approach to care is essential for optimum management of the primary manifestations and secondary complications of Duchenne muscular dystrophy (DMD). Contemporary care has been shaped by the availability of more sensitive diagnostic techniques and the earlier use of therapeutic interventions, which have the potential to improve patients’ duration and quality of life. In part 2 of this update of the DMD care considerations, we present the latest recommendations for respiratory, cardiac, bone health and osteoporosis, and orthopaedic and surgical management for boys and men with DMD. Additionally, we provide guidance on cardiac management for female carriers of a disease-causing mutation. The new care considerations acknowledge the effects of long-term glucocorticoid use on the natural history of DMD, and the need for care guidance across the lifespan as patients live longer. The management of DMD looks set to change substantially as new genetic and molecular therapies become available.
Introduction
The 2010 care considerations for Duchenne muscular dystrophy (DMD)1,2 advocated a multidisciplinary approach to the management of this severe, progressive neuromuscular disease. This three-part update was necessitated by a number of themes that characterise contemporary DMD care: the increasing complexity of subspecialty care and the need for a multidisciplinary clinical team; the use of more sensitive diagnostic techniques and earlier therapeutic interventions; the expectation of prolonged survival, prompting the need for care guidance across the lifespan; and the recognition that the natural history of DMD has been altered by the long-term use of glucocorticoids.3 The new care considerations have also been shaped by the expectation that emerging genetic and molecular therapies will substantially change the nature of DMD management in the near future.
In 2014, the DMD Care Considerations Working Group steering committee, comprising experts from a wide range of disciplines, identified 11 topics to be included in this update. Part 2 contains the latest care considerations for respiratory, cardiac, bone health and osteoporosis, and orthopaedic and surgical management. Large-scale, randomised controlled trials (RCTs) are rare in this field, so guidance was developed using a method that queries a group of experts on the appropriateness and necessity of specific assessments and interventions, using clinical scenarios. This methodology was designed to produce an essential toolkit for DMD care; only assessments and interventions that have been deemed both appropriate and necessary are recommended. A complete description of the methods is provided in part 1 and the appendix.
Figure 1 in part 1 of this Review provides a brief overview of assessments and interventions across all topics, organised by stage of disease. It is intended to serve as a pocket guide to overall disease management.
Figure 1. Assessments and interventions for respiratory care of patients with Duchenne muscular dystrophy by stage of disease.

DMD=Duchenne muscular dystrophy. FVC=forced vital capacity. MEP=maximum expiratory pressure. MIP=maximum inspiratory pressure. PCF=peak cough flow. petCO2=end-tidal partial pressure of CO2. ptcCO2=transcutaneous partial pressure of CO2. SpO2=blood oxygen saturation by pulse oximetry. *See text for definitions of sleep study results. †All specified threshold values of PCF, MEP, and MIP apply to older teenage and adult patients. ‡Fatigue, dyspnoea, morning or continuous headaches, frequent nocturnal awakenings or difficult arousal, hypersomnolence, difficulty concentrating, awakenings with dyspnoea and tachycardia, or frequent nightmares. §We strongly endorse the use of non-invasive methods of assisted ventilation instead of tracheostomy to optimise patient quality of life; indications for tracheostomy include patient preference, inability of patient to use non-invasive ventilation successfully, three failed extubation attempts during a critical illness despite optimum use of non-invasive ventilation and mechanically assisted coughing, or failure of non-invasive methods of cough assistance to prevent aspiration of secretions into the lungs due to weak bulbar muscles.
Respiratory management
Respiratory complications are a major cause of morbidity and mortality in people with DMD. Complications include respiratory muscle fatigue, mucus plugging, atelectasis, pneumonia, and respiratory failure. If left untreated, patients are at risk of severe dyspnoea, lengthy hospital admissions due to atelectasis or pneumonia, and death due to respiratory arrest or respiratory-induced cardiac arrhythmias.4-6
An anticipatory approach to management includes monitoring of respiratory muscle function and the timely use of lung volume recruitment, assisted coughing, nocturnally assisted ventilation, and subsequent daytime ventilation. These core therapies can decrease respiratory complications, improve quality of life, and prolong survival.4,7-10 Patients should typically be using most or all of these core therapies by the age of 18–21 years, before their transition from paediatric to adult respiratory care providers.
Implementation of respiratory care considerations and guidelines1,2,11-15 requires a multidisciplinary team— including physicians, respiratory therapists (or physical therapists in some health-care systems), and home caregivers—to perform pulmonary function testing and sleep studies and to initiate and manage lung volume recruitment,16 manual and mechanically assisted coughing,5,17 non-invasive ventilation, and invasive ventilation via tracheostomy. Decisions for optimum respiratory management need to be made with awareness of the patient’s other body systems, especially the cardiac system.10,18
In this update, we endorse higher pulmonary function thresholds (ie, milder levels of respiratory impairment) for initiation of assisted coughing and assisted ventilation than were recommended in the 2010 care considerations. The new criteria are intended to result in more anticipatory use of these interventions, with the possibility that therapy will be initiated in slightly younger patients.
Ambulatory stage
Figure 1 shows respiratory diagnostic tests and therapies for individuals with DMD, by stage of disease. Spirometry should be initiated when the patient is 5–6 years of age. Serial monitoring of pulmonary function is critical for respiratory management. Typically, forced vital capacity (FVC) rises with growth, until an individual becomes non-ambulatory. FVC reaches a peak, followed by a plateau, and then deteriorates over time.19-21 Deteriorating FVC can occur in the absence of dyspnoea and remain unrecognised unless pulmonary function is measured regularly. In a large cohort study in boys who had not been treated with corticosteroids, the age at loss of ambulation was predictive of the age at which peak FVC was realised, the absolute peak FVC, and the rate of subsequent decline.19 For example, earlier loss of ambulation was associated with an earlier and lower peak FVC as well as a more rapid decline in FVC than was later loss of ambulation. However, because the rate of change in FVC over time can vary greatly among individuals, serial measurement of FVC is necessary to characterise each individual’s respiratory phenotype or trajectory.
Sleep studies with capnography might be necessary during the ambulatory stage, especially for individuals with weight gain due to glucocorticoid therapy and for individuals with symptoms of sleep-disordered breathing. Sleep studies can also be used as an alternative method to monitor respiratory status among individuals who cannot cooperate with pulmonary function testing.
Individuals with DMD should receive yearly immunisation with the inactivated influenza vaccine (ie, the injectable vaccine, not the live, attenuated nasal vaccine) and pneumococcal vaccines (including PCV13 and PPSV23), according to guidelines available from the US Centers for Disease Control and Prevention,22 other public health entities such as the Immunization Action Coalition,23 and Parent Project Muscular Dystrophy.24
Patients and their caregivers should be educated about respiratory complications during the ambulatory stage of DMD to prepare them for future medical complications and therapies.
Early non-ambulatory stage
The need for respiratory interventions occurs mainly after the loss of ambulation (figure 1). Seated FVC (expressed both as an absolute value and as a percentage predicted on the basis of arm span or ulnar length), maximum inspiratory and expiratory pressures, peak cough flow, and blood oxygen saturation by pulse oximetry (SpO2) should be measured at least every 6 months in all non-ambulatory individuals. Additionally, end-tidal or transcutaneous partial pressure of carbon dioxide in the blood (petCO2 or ptcCO2, respectively) should be measured every 6 months or any time SpO2 is 95% or lower on room air, when the necessary equipment is available.
As their vital capacity decreases, patients with DMD develop stiff, non-compliant chest walls and lung volume restriction. To preserve lung compliance, lung volume recruitment is indicated when FVC is 60% predicted or less, achieved with a self-inflating manual ventilation bag or mechanical insufflation– exsufflation device to provide deep lung inflation once or twice daily.25–27
During the early non-ambulatory stage, some individuals with DMD need surgery for progressive scoliosis.28 Previously published guidelines address respiratory management of patients undergoing surgery, including indications for preoperative training in the use of assisted cough devices and non-invasive ventilators.13 For patients who are cognitively impaired and unable to reliably perform pulmonary function testing, preoperative polysomnography might be helpful.
Late non-ambulatory stage
As they progress through the non-ambulatory stage, individuals with DMD develop weak cough efforts, placing them at risk of atelectasis, pneumonia, ventilation–perfusion mismatch, and progression to respiratory failure, especially during respiratory tract infections. Treatment consists of manual and mechanically assisted coughing, which are indicated when FVC is less than 50% predicted, when peak cough flow is less than 270 L/min, or when maximum expiratory pressure is less than 60 cm H2O (figure 1).29–31
We advise having a home pulse oximeter for individuals treated with assisted coughing during respiratory infections. When SpO2 is less than 95% on room air, the frequency of assisted coughing should be increased to prevent and treat mucus plugging, atelectasis, and pneumonia. We also recommend initiation of antibiotic therapy during acute respiratory illnesses when individuals have three of the following five signs of pneumonia: fever, elevated white blood count or C-reactive protein concentration, sputum production, a pulmonary infiltrate on chest radiograph, or hypoxaemia or respiratory distress.
In the late non-ambulatory stage, individuals with DMD need assisted ventilation to prolong survival.32 Ventilation devices should incorporate a back-up rate of breathing to avoid apnoea. Indications for nocturnally assisted ventilation include signs or symptoms of hypoventilation or sleep-disordered breathing, irrespective of the level of pulmonary function; relevant symptoms include fatigue, dyspnoea, morning or continuous headaches, frequent nocturnal awakenings or difficult arousal, hypersomnolence, difficulty concentrating, awakenings with dyspnoea and tachycardia, and frequent nightmares. However, some individuals remain asymptomatic despite the presence of hypoventilation.33 Thus, nocturnally assisted ventilation should be initiated when a patient’s FVC is less than 50% predicted, or when the absolute value of maximum inspiratory pressure is less than 60 cm H2O. It should also be initiated when the individual is awake and, because of daytime hypoventilation, any of the following is true: petCO2 or ptcCO2 is more than 45 mm Hg; arterial, venous, or capillary blood pCO2 is more than 45 mm Hg; or baseline SpO2 is less than 95% on room air (figure 1).33–38
Nocturnal ventilation is also indicated for individuals with abnormal sleep studies, including overnight oximetry, combination oximetry–capnography, and polysomnography with capnography. Non-ambulatory individuals with symptoms of sleep-disordered breathing should have sleep studies as often as annually, if possible. Sleep study results that indicate the need for assisted ventilation include petCO2 or ptcCO2 of more than 50 mm Hg for at least 2% of sleep time, a sleep-related increase in petCO2 or ptcCO2 of 10 mm Hg above the awake baseline for at least 2% of sleep time, an SpO2 of 88% or less for at least 2% of sleep time or for at least 5 min continuously, or an apnoea–hypopnoea index of five events per h or more.37,39 Because patients with DMD inevitably need assisted ventilation to treat hypoventilation, nocturnal non-invasively assisted ventilation (rather than continuous positive airway pressure at a constant level) is first-line therapy for individuals with DMD with obstructive sleep apnoea.
Non-invasive ventilation can also be used during and after procedures involving anaesthesia or sedation and, in conjunction with assisted coughing, to extubate individuals who are mechanically ventilated for respiratory infections.40 In DMD, hypoxaemia is usually due to hypoventilation, atelectasis, or pneumonia. Therefore, supplemental oxygen therapy should not be used alone. In conjunction with assisted ventilation and assisted coughing, oxygen therapy can be safe, especially when blood CO2 levels are monitored.
With declining pulmonary function, patients develop symptoms of hypoventilation such as dyspnoea, fatigue, and difficulty concentrating, despite their use of assisted ventilation during sleep; those with very low FVCs (<680 mL in one study41) are at particular risk. Thus, patients often self-extend their use of assisted ventilation into the daytime, ultimately up to 24 h/day. The indications for daytime-assisted ventilation are listed in figure 1. Options for continuous non-invasive ventilation include mouthpiece or so-called sip ventilation with a portable volume ventilator during the day, changing to nasal ventilation with a bi-level pressure device overnight. Alternatively, 24 h/day nasal ventilation with a bi-level pressure device can be effective and well tolerated.7,9,42 These devices should have an internal battery for safety and portability.
Whether individuals with DMD should be ventilated via tracheostomy or non-invasively is a controversial question. Some centres use time on the ventilator (eg, 16 h/day or more) as an indication for tracheostomy.43–45 However, clinical experience supports the use of non-invasively assisted ventilation for up to 24 h/day.7,42,46 We strongly endorse the use of non-invasive ventilation in most clinical situations. Our indications for tracheostomy are listed in figure 1, and include patient preference, inability to use non-invasive ventilation, three failed extubation attempts during a critical illness despite optimum use of non-invasive ventilation and mechanically assisted coughing, or failure of noninvasive methods of cough assistance to prevent aspiration of secretions into the lungs due to weak bulbar muscles. Overall, the decision is highly dependent on each individual’s preference and clinical course, the skills and usual practices of the individual’s clinicians, the local standard of care, and the availability of home resources, such as overnight nursing.47 The use of non-invasive respiratory aids is especially challenging when individuals with very advanced DMD have acute respiratory illnesses and when they have chronic difficulty swallowing their saliva.
Continuous ventilation provides life support, so a back-up ventilator and a manual resuscitator should be available in case the primary ventilator malfunctions. Batteries or a generator should be available for use during a power outage. The ventilation device and battery should attach to the individual’s wheelchair for mobility and quality of life. When practical, the presence of a night nurse can greatly decrease the risk of potentially catastrophic medical events, such as mucus plugging of the trachea.
Cardiac management
Cardiovascular complications are a leading cause of disease-related morbidity and mortality among individuals with DMD.48 Dystrophin deficiency in the heart manifests as a cardiomyopathy. As the disease progresses, the myocardium fails to meet physiological demands and clinical heart failure develops. The failing myocardium is also at risk of life-threatening rhythm abnormalities.49
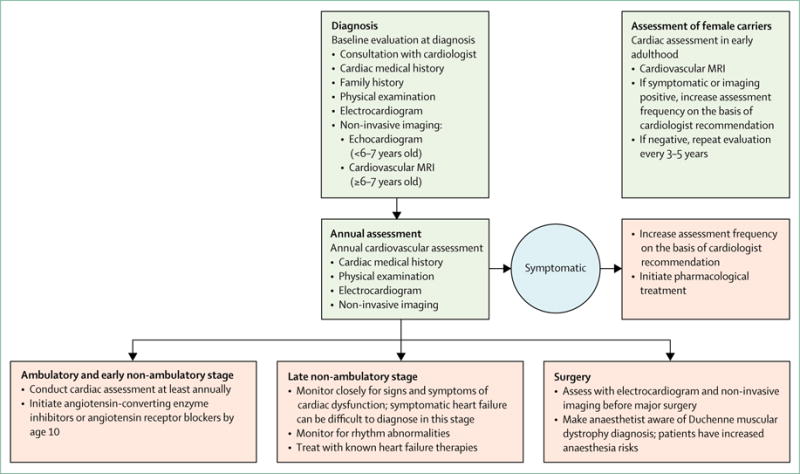
Historically, individuals with DMD have not been referred to a cardiac specialist until late in the disease, contributing to poor clinical outcomes. Furthermore, cardiac management has been challenging because the New York Heart Association classification of heart failure50 relies on reduced exercise tolerance, a feature that in DMD arises from skeletal muscle and cardiac disease combined. The signs and symptoms of heart failure in the non-ambulatory individual are frequently subtle and overlooked. A proactive strategy of early diagnosis and treatment is essential to maximise duration and quality of life. Involvement of a cardiologist who is integrated into a multidisciplinary care team is recommended, given the complex decision making involved in managing DMD cardiomyopathy. Ideally, the cardiologist should have clinical expertise in diagnosing and treating heart failure and the cardiomyopathy associated with neuromuscular disease, and have readily available access to state-of-the-art expertise in noninvasive imaging. A National Heart, Lung, and Blood Institute (NHLBI) expert working group was convened and recently published updated comprehensive DMD cardiac care considerations, including important areas for future research.51 The specific core care considerations are detailed below and summarised in figure 2.
Figure 2.
Cardiac monitoring, diagnosis, and treatment algorithm for patients with Duchenne muscular dystrophy
Ambulatory stage and early non-ambulatory stage
The baseline cardiac assessment includes past and present cardiac medical history, family history, and a physical examination. Electrocardiogram and non-invasive imaging are advised to establish baseline cardiac function and to screen for underlying anatomical abnormalities that could affect long-term cardiovascular health. Cardiovascular MRI (CMR) is the non-invasive imaging modality of choice; however, young individuals might not be able to cooperate for the procedure. Thus, echocardiography is recommended until at least age 6-7 years, when CMR can typically be done without anaesthesia. Until the age of 10 years, individuals should have an annual cardiac assessment, including electrocardiogram and non-invasive imaging. After the age of 10 years, asymptomatic individuals should have a cardiac assessment at least annually because of the increased risk of left ventricular dysfunction. With the onset of heart failure symptoms or when abnormalities are first seen on cardiac imaging—eg, myocardial fibrosis, left ventricular enlargement, or left ventricular dysfunction—the frequency of assessment should increase at the discretion of the cardiologist. An electrocardiogram and non-invasive cardiac imaging should be done before major surgical procedures, such as scoliosis correction. DMD is associated with a particular set of anaesthesia risks, and the anaesthetist should be made aware of the patient’s cardiac history.52
Traditionally, angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) were used as first-line therapy for the treatment of heart disease associated with DMD. Opinion differs on the use of ACE inhibitors in very young (<10 years) asymptomatic individuals without evidence of abnormality on CMR or echocardiogram. After discussing potential benefits and risks with the family, the cardiologist could initiate therapy in this group of individuals. Some evidence suggests that initiation of ACE inhibitors in asymptomatic boys with normal left ventricular systolic function as they approach 10 years of age can improve long-term cardiac outcomes, and the 2014 NHLBI working group recommended use of ACE inhibitors or ARBs by the age of 10 years in boys with DMD.51 Dosing and ACE inhibitor selection are left to the discretion of the cardiologist.53
Irrespective of age, pharmacological therapy should be initiated with the onset of heart failure symptoms or when abnormalities such as depressed left ventricular ejection fraction, abnormal chamber dimensions, or the presence of myocardial fibrosis are noted on imaging studies (CMR or echocardiogram). Given the absence of dystrophin-specific targeted cardiac therapies, traditional treatment strategies for heart failure should be used. β-adrenergic blockade is typically started with evidence of ventricular dysfunction. In a prospective, randomised double-blind placebo-controlled trial in patients aged 7-25 years with DMD, the mineralocorticoid receptor antagonist eplerenone attenuated the decline in cardiac function, as measured by circumferential strain.54 This benefit was supported by findings from a 2 year, open-label extension trial.55 However, although eplerenone might prove to be a useful adjunctive therapy to other heart failure medications, further investigations are needed to establish effectiveness.54,55
Late non-ambulatory stage
Progressive myocardial fibrosis leads to ventricular dysfunction. More frequent cardiac monitoring, as determined by the patient’s cardiologist, is advised in the late, non-ambulatory stage to reduce disease-related morbidity and mortality. The cardiologist should work closely with the multidisciplinary care team to ensure that respiratory care has been optimised, because abnormal pulmonary mechanics affect cardiac function.56,57 Specifically, there is evidence that noninvasive nocturnal ventilation increases long-term survival.8 The NHLBI working group suggested that early initiation of nocturnal ventilation be considered because of potential long-term benefit.51
Symptomatic heart failure can be particularly difficult to diagnose in non-ambulatory patients with DMD. Clinical manifestations of heart failure—fatigue, weight loss, vomiting, abdominal pain, sleep disturbance, and inability to tolerate daily activities—are often unrecognised until late in the disease because of musculoskeletal limitations. The cardiologist should maximise medical therapy for heart failure. Consideration should also be given to thromboembolism prevention in individuals with severe left ventricular dysfunction. Various antithrombotic drugs are available, and should be initiated after discussion with the cardiologist.
People with DMD are at risk of rhythm abnormalities— including atrial fibrillation or flutter, ventricular tachycardia, and ventricular fibrillation—that can be treated with standard antiarrhythmic medications or device management, when indicated. Surveillance should include periodic Holter monitoring. In most circumstances, 24 h Holter monitoring will be sufficient. Event monitors could also be indicated when individuals complain of episodic, non-sustained rhythm disturbances. The optimum frequency of monitoring has not been established and should be directed by the cardiologist, depending on the patient’s clinical course. It is reasonable to initiate annual Holter monitor screening with the onset of abnormal left ventricular function or development of myocardial fibrosis. The benefit of implantable cardioverter defibrillators as primary prevention for ventricular tachycardia or ventricular fibrillation is unknown. These devices can be used for secondary prevention in patients who have had ventricular tachycardia or ventricular fibrillation. At present, placement for primary arrhythmia prevention is on the basis of established adult heart failure guidelines. Among adults with heart failure, placement of implantable cardioverter defibrillators is advised for individuals with an ejection fraction of less than 35%.58 Clearly, patients with DMD have unique issues (eg, chest wall deformities and sedation risk), which might affect this recommendation.
In individuals in whom maximal medical management has failed, the use of mechanical circulatory support is a therapeutic consideration, as illustrated by relevant case reports. A left ventricular assist device could be used as a destination therapy—ie, in individuals for whom a heart transplant is not considered appropriate.59-61 The decision to proceed with a ventricular assist device is complex and involves a deep understanding of all the inherent risks and potential benefits. Risks include, but are not limited to, thromboembolism, bleeding, infection, device malfunction, and right heart failure. In an ideal situation, the device has the potential to improve duration and quality of life. Cardiac transplantation is also a theoretical option, but given the small number of available donors, it needs to be considered on a case-by-case basis.
Female carriers
In this update, we acknowledge that female carriers of a disease-causing mutation are at risk of not only skeletal muscle disease, but also cardiomyopathy.62 The natural history and incidence of cardiomyopathy in girls and women is not well characterised, but in a 2016 study,63 47% of carriers had at least one positive finding on CMR. We recommend a baseline cardiac assessment in early adulthood that includes an electrocardiogram and non-invasive imaging, preferably CMR, when available. Ongoing surveillance will be required, on the basis of guidance for individuals with cardiomyopathy.64 The optimum frequency has not been established in the DMD carrier population, but our current guidance is for assessment every 3–5 years, on the basis of screening recommendations for other genetic cardiomyopathies.65
Bone health and osteoporosis management
Boys with glucocorticoid-treated DMD frequently develop osteoporosis, which manifests as low-trauma vertebral or long-bone fractures.66 This outcome is not surprising given the potent osteotoxicity of glucocorticoid therapy combined with progressive myopathy, both of which are key risk factors for reduced bone strength. 20–60% of boys with DMD have low-trauma extremity fractures (usually the distal femur, tibia, or fibula), while up to 30% develop symptomatic vertebral fractures.66–68 Vertebral fractures are frequently asymptomatic when identified in children with glucocorticoid-treated illnesses through a monitoring programme that includes a lateral spine radiograph,69-73 so the true prevalence is probably higher than existing reports suggest. Left untreated, vertebral fractures can lead to chronic back pain and spine deformity, while leg fractures can cause premature, permanent loss of ambulation.66 Death due to fat embolism syndrome after long-bone fractures has also been reported in boys with DMD.74,75
The notion that some glucocorticoid agents and dosing regimens are bone-sparing compared with others has arisen from studies of deflazacort versus prednisone or meprednisone (also known as methylprednisone) in children after renal transplant and in those with chronic juvenile arthritis.76-78 The steroid dose equivalences used in these studies were variable, making comparisons difficult; however, disease outcomes were favourable in the deflazacort-treated children, with associated improvements in bone density outcomes, linear growth, weight–height ratios, and lean body mass. By contrast, recent publications cast doubt on the bone-sparing properties of deflazacort, showing that bone fragility (including vertebral fractures) is frequent in deflazacort-treated boys with DMD, probably related partly to the large doses that are used in this condition.73,79 Comparative studies of different steroid regimens in DMD are underway, assessing the effect on final adult height, body composition, and fractures.80
Despite the high prevalence of fractures, no published studies of DMD or any osteoporotic condition of childhood have assessed the safety and efficacy of medical therapy in preventing the first-ever fracture. Therefore, the current standard is to identify and treat early indications of bone fragility (eg, vertebral fractures) in individuals with chronic illnesses who have little possibility of recovery. This secondary prevention approach has the goal of mitigating osteoporosis progression and promoting recovery among patients presenting with early, rather than late, indications of osteoporosis and in those with little potential for medication-unassisted recovery because of persistent risk factors.
We present care considerations for monitoring that will enable timely diagnosis and treatment of osteoporosis in boys and men with DMD (figure 3). We also review specific diagnostic criteria for osteoporosis, along with care considerations for prescription of osteoporosis therapy, including agents, dose, and duration of therapy. Comprehensive reviews of all issues relating to management of osteoporosis therapy (including contraindications and monitoring of safety and efficacy) have been published elsewhere.81,82
Figure 3. Osteoporosis monitoring, diagnosis, and treatment algorithm for patients with Duchenne muscular dystrophy.
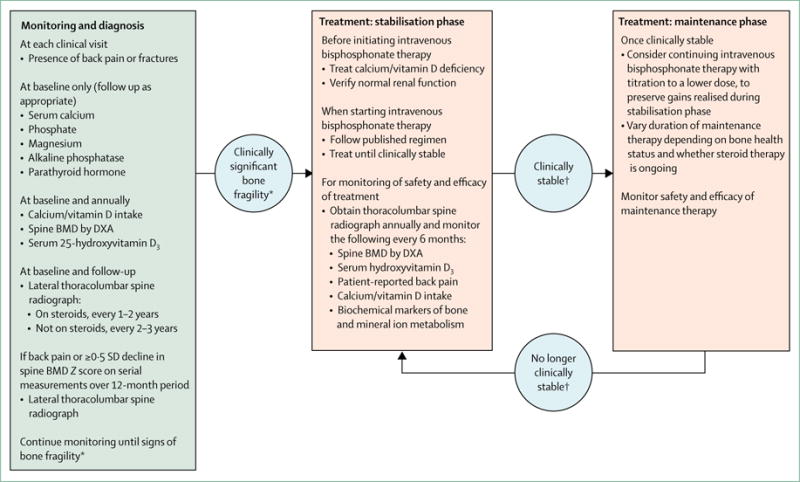
BMD=bone mineral density. DMD=Duchenne muscular dystrophy. DXA=dual-energy x-ray absorptiometry. *Signs of clinically significant bone fragility are low-trauma fractures of long bones or vertebra. †Clinical stability refers to absence of non-vertebral fractures, stable healed vertebral fractures, absence of new vertebral fractures in previously normal vertebral bodies, absence of bone and back pain, and BMD Z score appropriate for height Z score or higher than −2 SD.
Bone health monitoring and diagnosis of osteoporosis
An important development that distinguishes the current guidance from the 2010 care considerations is that bone health monitoring and diagnosis in children no longer focus on bone mineral density (BMD); rather, BMD serves as an adjuvant in an approach that focuses on identification of the earliest signs of bone fragility. Several observations in children with chronic, glucocorticoid-treated illnesses have fuelled this change. First, vertebral fractures—defined according to the Genant method83 as mild (grade 1), moderate (grade 2), or severe (grade 3)—are now understood to be a frequent manifestation of osteoporosis in children with chronic illness, including those with glucocorticoid-treated DMD.73,84 As noted, some individuals with vertebral fractures are relatively asymptomatic, even in more advanced stages of collapse.71 Therefore, to detect vertebral fractures, spine imaging should not be prompted solely by back pain or deformity; rather, people with known risk factors for vertebral fractures, including motor disorders85 or glucocorticoid therapy,72,84 should receive regular spine imaging. The fact that vertebral fractures at any time point in a patient’s clinical course are predictive of future spine fractures even when the initial vertebral fractures are mild or asymptomatic71—a phenomenon known as the vertebral fracture cascade86— underscores the need for early identification.
Vertebral fractures can occur in children who have BMD Z scores higher than −2 SD, an observation that invalidates the use of a BMD Z score threshold to define osteoporosis in children with low-trauma vertebral fractures.87 This observation prompted the International Society for Clinical Densitometry to revise the definition of osteoporosis in a child with a low-trauma vertebral fracture so that cutoff criteria based on BMD Z scores are no longer required to make the diagnosis of osteoporosis.88 Similarly, 15% of children with neuromuscular disorders and extremity fractures will have BMD Z scores for the distal femur higher than −2 SD,89 which again challenges the use of a BMD Z score threshold to define osteoporosis in a child with extremity fractures. Finally, findings from a recent study87 showed that spine BMD Z scores can vary by as much as 2 SD depending on the normative database that is used to generate the Z scores. In view of these findings, the diagnosis of osteoporosis in at-risk children now rests on the presence of evident bone fragility, often manifesting as vertebral fractures, and a BMD Z score above −2 SD does not preclude the diagnosis of osteoporosis.88 Although BMD Z scores are no longer at the forefront of diagnosis, they remain useful to determine the overall trajectory of bone health in an individual child and thereby guide frequency of lateral spine radiographs during the monitoring phase.
The 2010 care considerations recommended a spine radiograph for vertebral fracture detection in patients with a history of back pain or spine deformity on physical examination. In the current care considerations, a baseline spine radiograph for vertebral fracture detection is recommended in all patients, with intermittent follow-up radiographs to assess changes in spine morphology in the face of persistent (ie, glucocorticoid therapy) or permanent (ie, myopathy) risk factors. Given the need for serial spine radiographs, assessment by dual-energy x-ray absorptiometry is an emerging method for at-risk populations, and a validation study in children has shown that this technique compares favourably with detection of Genant-defined vertebral fractures on spine radiographs.90 Overall, spine radiographs should be prioritised over BMD in view of the need to detect the earliest signs of bone fragility.
Treatment of osteoporosis
Indications for treatment with intravenous bisphosphonate—the presence of low-trauma vertebral fractures or long-bone fractures—generally remain unchanged, but with notable differences in the timing of treatment initiation. Previously, only back pain or spine deformity prompted a radiograph to identify vertebral fractures necessitating bisphosphonate therapy. The current call for routine spine radiographs for all patients with DMD will lead to diagnosis of symptomatic vertebral fractures (mild, moderate, and severe) and asymptomatic moderate and severe vertebral fractures, all of which should prompt referral to an osteoporosis expert for treatment. Because even mild and asymptomatic vertebral fractures are predictive of future fractures in both children71 and adults,91 treatment of asymptomatic moderate (Genant grade 2) and severe (Genant grade 3) vertebral fractures is now recommended. Treatment with intravenous bisphosphonate therapy had a protective effect on spine BMD and vertebral morphology in controlled trials of osteogenesis imperfecta92-94 and in uncontrolled studies of osteogenesis imperfecta95 and DM D.84,96 Additional support for treatment of asymptomatic but nevertheless advanced (ie, moderate and severe) vertebral fractures stems from the fact that no cases of spontaneous (ie, medication unassisted) reshaping of previously fractured vertebral bodies have been reported in boys with DMD;73 however, reshaping has been observed after intravenous bisphosphonate therapy in this population.84 For children with glucocorticoid-treated diseases, such as DMD, including those with minimally symptomatic or asymptomatic mild (grade 1) vertebral fractures, controlled trials are underway to investigate the efficacy of antiresorptive therapy (ClinicalTrials.gov identifiers NCT00799266 and NCT02632916); for now, mild asymptomatic fractures should be closely monitored for symptomatology or progressive height loss that would prompt treatment.
The updated guidance represents a fundamental change in the goals of therapy. The aim is to identify and treat the earliest signs of bone fragility to better preserve the heights of the vertebral bodies.84 We endorse the use of intravenous (and not oral) bisphosphonates as first-line therapy for the treatment of osteoporosis in patients with DMD,81,82 on the basis of an extrapolation from results of controlled trials in osteogenesis imperfecta. Such studies have shown increased vertebral heights in growing patients with osteogenesis imperfecta treated with intravenous bisphosphonate therapy.92-94 By contrast, no controlled studies of oral bisphosphonates in osteogenesis imperfecta have shown an effect on vertebral height.97-99 These data are particularly relevant to patients with glucocorticoid-treated DMD, who have a high frequency of vertebral fractures.73 Recent reviews on the management of children with fractures due to osteoporosis concur with the view that intravenous rather than oral bisphosphonates should be used as first-line therapy.81,82 Because bisphosphonates remain off label for children in most countries and they require judicious prescription, patients with a low-trauma fracture should be referred to an expert in osteoporosis management to ensure proper bisphosphonate dosing, dose titration on longer-term therapy, timing of treatment cessation, and monitoring of treatment safety and efficacy.
Orthopaedic and surgical management
The overall aim of musculoskeletal care is to maintain motor function for as long as possible, minimise joint contractures, maintain a straight spine, and promote bone health. The assessment and treatment of musculoskeletal complications should involve an interdisciplinary team that might include a physical and occupational therapist, rehabilitation physician, neurologist, orthopaedic surgeon, and social worker. When a surgical intervention is recommended, it is crucial to involve the respiratory physician and cardiologist.
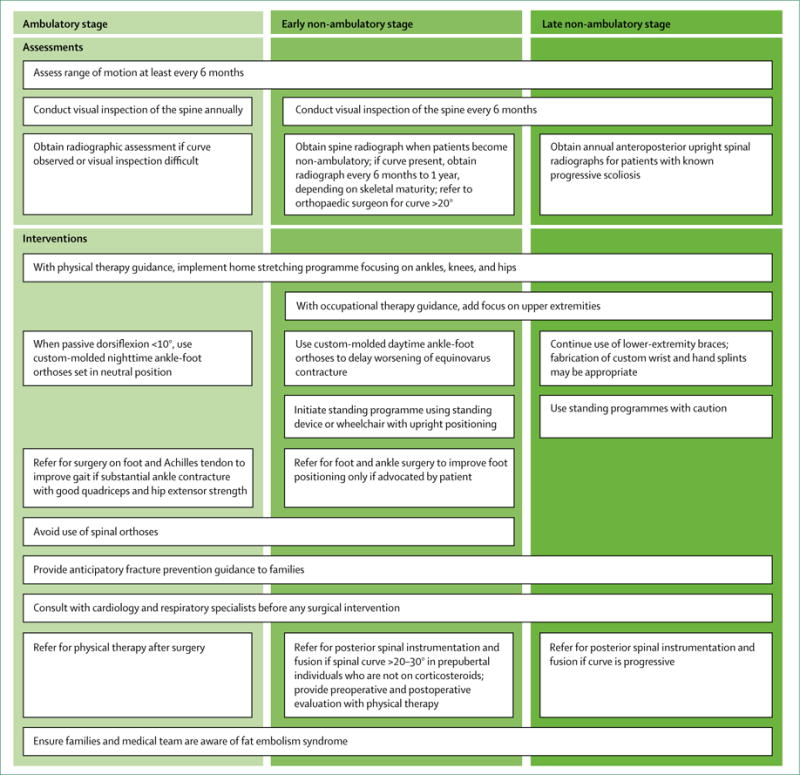
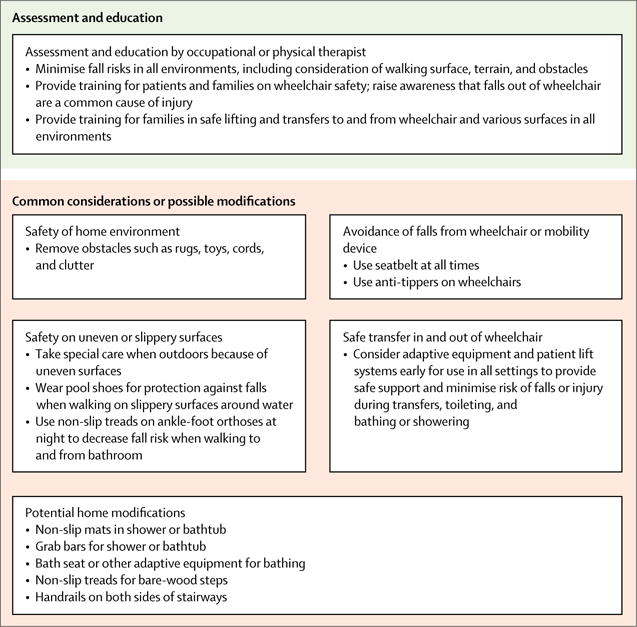
Figure 4 outlines the care considerations for orthopaedic and surgical care related to contracture, spine, and fracture management. Figure 5 provides general guidance for patients and families about fracture prevention. In the absence of RCTs comparing different therapeutic and surgical approaches, this guidance is based on the expert consensus of orthopaedic and rehabilitation specialists, using the methods described in part 1. Care considerations on stretching, orthoses, and adaptive equipment for contracture management are provided in the rehabilitation management section of part 1.
Figure 4.
Considerations for orthopaedic and surgical care of patients with Duchenne muscular dystrophy by stage of disease
Figure 5.
General guidance on fracture prevention for patients with Duchenne muscular dystrophy and their families
Ambulatory stage
Children in the ambulatory stage might benefit most from surgery, but it is recommended less frequently than in the past. Although the 2010 care considerations included recommendations for multilevel surgeries, the consensus of the current panel is that surgery on the foot to improve the varus positioning and on the Achilles tendon to improve dorsiflexion range might be sufficient to improve gait in patients with clinically significant ankle contracture and good quadriceps and hip extensor strength. Interventions related to the hips and knees are not recommended.
Assessment for scoliosis should be done at least annually, although onset is unusual in the ambulatory stage. Visual assessment is appropriate, with radiographic assessment only if a curve is observed on examination or if visual inspection alone is inadequate, such as in children with obesity. The preceding section on bone health and osteoporosis management provides information regarding monitoring and treatment of spinal compression fractures. Use of spinal orthoses is not generally recommended in the setting of a compression fracture.
Anticipatory guidance during routine clinic visits is an important part of a fracture prevention programme throughout all disease stages (figure 5). As noted, corticosteroids have been associated with osteoporosis and subsequent vertebral fractures in DMD.100 In a study of 143 boys with DMD, the long-bone fracture rate in those treated with corticosteroids was 2·6 times greater than in those who had never received steroids.68 A lower-limb fracture during the ambulatory stage might need aggressive management to maintain ambulation. Internal or external fixation allows for early mobilisation compared with casting or splinting.101
There have been case reports of fat embolism syndrome in boys with DMD and acute fracture or trauma to the lower extremities.74,75 Boys with fat embolism syndrome present with altered mental status, respiratory distress, and tachycardia, which should prompt immediate medical attention due to high morbidity and mortality associated with this condition. Current treatment focuses on supportive respiratory care and high-dose corticosteroids.102
Early non-ambulatory stage
Foot and ankle surgery to improve equinovarus foot might help with foot positioning in the wheelchair or for shoe wear, but is typically done only if a patient requests the procedure. After foot and ankle surgery, use of ankle-foot orthoses will be needed during the daytime to prevent a recurrence of the contractures.
Inspection of the spine should be part of every clinical examination. Experienced clinicians should be able to monitor the spine in non-ambulatory boys by visual inspection alone; however, less experienced clinicians should obtain a spine radiograph when a child first becomes non-ambulatory. A spine radiograph is also useful when inspection is unhelpful, such as in children with obesity. Once a curve has been detected with radiography, further surveillance depends on the skeletal maturity of the individual; skeletally immature individuals should undergo radiographs once every 6 months, and skeletally mature individuals should undergo radiographs at least once a year. A curve of 20° or more should warrant involvement of an orthopaedic surgeon. The use of spinal orthoses is not recommended. In contrast to the typical clinical course in untreated boys, patients treated with corticosteroids have milder spinal curvatures and less frequent need for spinal surgeries.68,71,100,103
Despite an absence of RCTs, we advise posterior spinal fusion in young men with DMD, given the positive effect on function, sitting balance and tolerance, pain, and quality of life observed in non-randomised, prospective cohort studies.28,104,105 Posterior spinal instrumentation and fusion are recommended in non-ambulatory individuals who have a spinal curve in the sitting position greater than 20-30°, who have not yet reached puberty, and who have not been treated with corticosteroids because the curve is expected to progress. Although patients treated with corticosteroids can still develop scoliosis, the progression might be less predictable, so observation for clear evidence of progression is a reasonable approach before intervening. An anterior spinal fusion approach is not required as the fusion is generally done in the second decade when little additional longitudinal spine growth is anticipated.
When surgical correction for scoliosis is done, stabilisation into the pelvis and fusion are advised in those with a pelvic obliquity of greater than 15° to assist with seating and positioning. In those without a severe pelvic obliquity, fusion to the lower lumbar vertebra is sufficient. The goal of surgical intervention for the spine is to prevent further progression of scoliosis, improve sitting tolerance, and reduce pain.28
Anticipatory fracture prevention guidance should continue through the non-ambulatory stages (figure 5). A more conservative approach to management of lower-limb fractures is advised in non-ambulatory children because the goal is no longer to bear weight. Internal fixation might be necessary for an unstable fracture, but splinting might be sufficient for bone healing and pain control. Pain management is important for all children, but special monitoring could be necessary in the setting of pulmonary and cardiac compromise. Health-care providers and families should be aware of fat embolism syndrome, as described above.
Late non-ambulatory stage
Surgical interventions to manage contractures involving the upper or lower extremities are not recommended during the late non-ambulatory stage of DMD unless pain, positioning, or skin integrity is the concern.
Clinicians should examine the spine at every clinical visit. Individuals with known scoliosis should have yearly anteroposterior upright spinal radiographs when there is any concern about progression. Posterior spinal fusion is recommended during the late non-ambulatory stage for those with a progressive curve. It is essential to consult with the patient’s respiratory physician and cardiologist to ensure that lung and heart function are sufficient to proceed with this surgical intervention. Some studies indicate that spinal fusion slows the progression of respiratory decline, whereas others show no significant difference in the rate of decline postoperatively.28,106-108
The treatment of an acute fracture during the late non-ambulatory stage is similar to that in the early non-ambulatory stage, with the goals of fracture stabilisation and pain control. Cast or splint management is usually sufficient in the setting of a distal femoral metaphyseal fracture. In the case of a proximal femur fracture, operative stabilisation is necessary. As with any fracture, providers and families should be aware of fat embolism syndrome.
Surgical considerations
Important surgical considerations for individuals with DMD are detailed in figure 6. Young men with DMD are at risk of potentially fatal rhabdomyolysis and hyperkalaemia when exposed to inhalational anaesthetics or when given suxamethonium chloride (succinylcholine).109 A cardiologist and respiratory physician should be consulted before all surgical procedures, and anaesthetists should be aware that individuals with DMD are at risk of cardiac and respiratory decompensation during and after surgery.110 A detailed discussion of surgical considerations is provided in the appendix.
Figure 6. Surgical considerations for patients with Duchenne muscular dystrophy.

DMD=Duchenne muscular dystrophy. FVC=forced vital capacity. *Guidance applies to older teenage and adult patients.
Conclusions and future directions
Improved approaches to respiratory, cardiac, bone health and osteoporosis, and orthopaedic and surgical management can now be offered to children and adults with DMD. However, despite advances in our knowledge and understanding of best approaches to management, progress is needed across these subspecialties to meet the needs of patients.
For respiratory management, diagnostic tools and measures that might have clinical relevance but need further study include assisted cough peak flow, maximum insufflation capacity, the difference between maximum insufflation capacity and FVC, supine FVC, highest flow generated during an inspiratory FVC manoeuvre, the rapid shallow breathing index, and sniff nasal inspiratory pressure. Therapies for which research is needed to establish efficacy and optimum use include high-frequency chest oscillation, intrapulmonary percussive ventilation, and negative-pressure ventilation. Improved understanding of pulmonary phenotypic variability and of the effect of cardiac function and nutritional status on the respiratory system is needed to optimise care and to develop pulmonary outcome measures to assess the efficacy of current and emerging DMD therapies. Prospective studies are needed to assess the criteria recommended in this document for initiation of cough assistance and non-invasive ventilation, with use of clinically relevant outcome measures to develop evidence-based guidelines.
Cardiac outcomes should be included in clinical trials because survival will not improve if emerging therapies do not effectively treat DMD cardiomyopathy. Biomarkers that indicate short-term attenuation of disease-related progression need to be identified. Novel dystrophin-specific cardiac treatments are needed to improve patient outcomes. The natural history of cardiomyopathy in female carriers of a disease-causing mutation needs to be clarified, and studies are needed to identify the best diagnostic and therapeutic strategies for affected girls and women.
Because vertebral fractures are an early manifestation of bone fragility and the adverse effects of glucocorticoids occur rapidly, longitudinal trials addressing osteoporosis prevention should originate with young patients, with vertebral fractures as a key outcome measure. Further studies are also needed to assess the potential of growth-promoting therapies to prevent bone fragility and of anabolic agents, such as parathyroid hormone or antisclerostin antibody, to treat osteoporosis.
Controlled trials of surgical techniques for orthopaedic management, when appropriate, are needed, as is a better understanding of musculoskeletal complications and of the best outcome measures to assess musculoskeletal effects of available and emerging DMD therapies. More studies of patient-reported and family-reported outcomes would help to guide decision making about lower-extremity surgeries and spinal fusion.
Supplementary Material
Acknowledgments
We thank Sharon Barrell and Danielle Hennis (RTI International) for editorial and graphical support, respectively, and Adrienne Herron (US Centers for Disease Control and Prevention [CDC]) for her contributions to the design and conduct of the project and review of the manuscript. We thank Natalie Street (CDC) for contributing to the design and conduct of the project, reviewing the manuscript, and coordinating and editing revisions of the manuscript. This work was supported by CDC contract 200-2007-22644-023. The CDC provided honoraria and travel expenses for committee members to attend an in-person meeting. Funding was provided under the Muscular Dystrophy Care and Treatment Act, legislation that calls for cooperation of the CDC with professional organisations and the patient community in the development, issuance, and periodic review and update of care considerations for Duchenne muscular dystrophy. The findings and conclusions presented in this paper are those of the authors and do not necessarily represent the official position of the CDC.
Footnotes
See Online for appendix
Declaration of Interests
DJB was a paid consultant for Hill-Rom Corporation and has US patents (8651107, 8844530, and 9795752) for respiratory devices, as well as related international patents and patent applications. KB was a consultant for Solid Ventures, Catabasis, LGC Ltd, Bristol Myers Squibb, PTC therapeutics, GLC Research, Eli Lilly, and Publicis Life Brands Resolute; she has received grant support from PTC Therapeutics. SDA is a principal investigator for multicentre clinical trials sponsored by PTC Therapeutics and Sarepta Pharmaceuticals. LEC has received personal fees for speaking and participating in research supported by Genzyme Corporation of Sanofi; she has participated in research with CINRG (Cooperative International Neuromuscular Research Group), Enobia Pharma Inc/Alexion, Robertson Foundation, GlaxoSmithKline, Eli Lilly, Valerion, Pfizer, Prosensa, BioMarin, Ionis, Ultragenyx, Roivant Sciences, Therapeutic Research in Neuromuscular Disorders Solutions, NS Pharma, and the Marcus Foundation. DRW is a paid consultant for Health Research Inc and Marathon Pharmaceuticals. LMW has received grant support and honoraria from Novartis and Amgen. All other authors declare no competing interests.
Contributors
DJB, KB, CMB, BAA, SDA, AB, LEC, LC, SH, AKO, DWS, JB, DRW, and LMW provided intellectual expertise in the study design, generation and interpretation of data, review of the literature, writing of the article, and the decision to publish. DJB, aided by CMB, drafted and edited the article and approved the final version.
For more on the Immunization Action Coalition see http://immunize.org/
For more on Parent Project Muscular Dystrophy see http://www.parentprojectmd.org/
For more on the National Heart, Lung, and Blood Institute see https://www.nhlbi.nih.gov/
For more on the International Society for Clinical Densitometry see https://www.iscd.org/
Contributor Information
Prof David J Birnkrant, Department of Pediatrics, MetroHealth Medical Center, Case Western Reserve University, Cleveland, OH, USA.
Prof Katharine Bushby, John Walton Muscular Dystrophy Research Centre, Institute of Genetic Medicine, Newcastle University, Newcastle upon Tyne, UK.
Carla M Bann, RTI International, Research Triangle Park, NC, USA.
Prof Benjamin A Alman, Department of Orthopaedic Surgery, Duke University School of Medicine and Health System, Durham, NC, USA.
Prof Susan D Apkon, Department of Rehabilitation Medicine, Seattle Children’s Hospital, Seattle, WA, USA (S D Apkon MD).
Angela Blackwell, RTI International, Research Triangle Park, NC, USA.
Laura E Case, Doctor of Physical Therapy Division, Department of Orthopaedics, Duke University School of Medicine, Durham, NC, USA.
Prof Linda Cripe, Department of Pediatrics, Nationwide Children’s Hospital, The Ohio State University, Columbus, OH, USA.
Stasia Hadjiyannakis, Division of Endocrinology and Metabolism, Children’s Hospital of Eastern Ontario, and University of Ottawa, Ottawa, ON, Canada.
Aaron K Olson, Department of Pediatrics, Seattle Children’s Hospital, Seattle, WA, USA.
Daniel W Sheehan, John R Oishei Children’s Hospital, University at Buffalo, The State University of New York, Buffalo, NY, USA.
Julie Bolen, Rare Disorders and Health Outcomes Team, National Center on Birth Defects and Developmental Disabilities, Centers for Disease Control and Prevention, Atlanta, GA, USA.
David R Weber, Division of Endocrinology and Diabetes, Golisano Children’s Hospital, University of Rochester Medical Center, Rochester, NY, USA.
Leanne M Ward, Division of Endocrinology and Metabolism, Children’s Hospital of Eastern Ontario, and University of Ottawa, Ottawa, ON, Canada.
References
- 1.Bushby K, Finkel R, Birnkrant DJ, et al. for the DMD Care Considerations Working Group Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 2.Bushby K, Finkel R, Birnkrant DJ, et al. for the DMD Care Considerations Working Group Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 2010;9:177–89. doi: 10.1016/S1474-4422(09)70272-8. [DOI] [PubMed] [Google Scholar]
- 3.Gloss D, Moxley RT, 3rd, Ashwal S, Oskoui M. Practice guideline update summary: Corticosteroid treatment of Duchenne muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2016;86:465–72. doi: 10.1212/WNL.0000000000002337. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Gomez-Merino E, Bach JR. Duchenne muscular dystrophy: prolongation of life by noninvasive ventilation and mechanically assisted coughing. Am J Phys Med Rehabil. 2002;81:411–15. doi: 10.1097/00002060-200206000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Tzeng AC, Bach JR. Prevention of pulmonary morbidity for patients with neuromuscular disease. Chest. 2000;118:1390–96. doi: 10.1378/chest.118.5.1390. [DOI] [PubMed] [Google Scholar]
- 6.Phillips MF, Smith PE, Carroll N, Edwards RH, Calverley PM. Nocturnal oxygenation and prognosis in Duchenne muscular dystrophy. Am J Respir Crit Care Med. 1999;160:198–202. doi: 10.1164/ajrccm.160.1.9805055. [DOI] [PubMed] [Google Scholar]
- 7.Bach JR, Martinez D. Duchenne muscular dystrophy: continuous noninvasive ventilatory support prolongs survival. Respir Care. 2011;56:744–50. doi: 10.4187/respcare.00831. [DOI] [PubMed] [Google Scholar]
- 8.Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord. 2002;12:926–29. doi: 10.1016/s0960-8966(02)00140-2. [DOI] [PubMed] [Google Scholar]
- 9.Ishikawa Y, Miura T, Ishikawa Y, et al. Duchenne muscular dystrophy: survival by cardio-respiratory interventions. Neuromuscul Disord. 2011;21:47–51. doi: 10.1016/j.nmd.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 10.Birnkrant DJ, Ararat E, Mhanna MJ. Cardiac phenotype determines survival in Duchenne muscular dystrophy. Pediatr Pulmonol. 2016;51:70–76. doi: 10.1002/ppul.23215. [DOI] [PubMed] [Google Scholar]
- 11.Birnkrant DJ, Bushby KM, Amin RS, et al. The respiratory management of patients with Duchenne muscular dystrophy: a DMD care considerations working group specialty article. Pediatr Pulmonol. 2010;45:739–48. doi: 10.1002/ppul.21254. [DOI] [PubMed] [Google Scholar]
- 12.Finder JD, Birnkrant D, Carl J, et al. Respiratory care of the patient with Duchenne muscular dystrophy: ATS consensus statement. Am J Respir Crit Care Med. 2004;170:456–65. doi: 10.1164/rccm.200307-885ST. [DOI] [PubMed] [Google Scholar]
- 13.Hull J, Aniapravan R, Chan E, et al. British Thoracic Society guideline for respiratory management of children with neuromuscular weakness. Thorax. 2012;67(suppl 1):i1–40. doi: 10.1136/thoraxjnl-2012-201964. [DOI] [PubMed] [Google Scholar]
- 14.Rahbek J, Steffensen BF, Bushby K, de Groot IJ. 206th ENMC International Workshop: Care for a novel group of patients—adults with Duchenne muscular dystrophy Naarden, The Netherlands, 23-25 May 2014. Neuromuscul Disord. 2015;25:727–38. doi: 10.1016/j.nmd.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 15.LoMauro A, D’Angelo MG, Aliverti A. Assessment and management of respiratory function in patients with Duchenne muscular dystrophy: current and emerging options. Ther Clin Risk Manag. 2015;11:1475–88. doi: 10.2147/TCRM.S55889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katz SL, Barrowman N, Monsour A, Su S, Hoey L, McKim D. Long-term effects of lung volume recruitment on maximal inspiratory capacity and vital capacity in Duchenne muscular dystrophy. Ann Am Thorac Soc. 2016;13:217–22. doi: 10.1513/AnnalsATS.201507-475BC. [DOI] [PubMed] [Google Scholar]
- 17.Miske LJ, Hickey EM, Kolb SM, Weiner DJ, Panitch HB. Use of the mechanical in-exsufflator in pediatric patients with neuromuscular disease and impaired cough. Chest. 2004;125:1406–12. doi: 10.1378/chest.125.4.1406. [DOI] [PubMed] [Google Scholar]
- 18.Melacini P, Vianello A, Villanova C, et al. Cardiac and respiratory involvement in advanced stage Duchenne muscular dystrophy. Neuromuscul Disord. 1996;6:367–76. doi: 10.1016/0960-8966(96)00357-4. [DOI] [PubMed] [Google Scholar]
- 19.Humbertclaude V, Hamroun D, Bezzou K, et al. Motor and respiratory heterogeneity in Duchenne patients: implication for clinical trials. Eur J Paediatr Neurol. 2012;16:149–60. doi: 10.1016/j.ejpn.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 20.Mayer OH, Finkel RS, Rummey C, et al. Characterization of pulmonary function in Duchenne muscular dystrophy. Pediatr Pulmonol. 2015;50:487–94. doi: 10.1002/ppul.23172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rideau Y, Jankowski LW, Grellet J. Respiratory function in the muscular dystrophies. Muscle Nerve. 1981;4:155–64. doi: 10.1002/mus.880040213. [DOI] [PubMed] [Google Scholar]
- 22.Centers for Disease Control and Prevention. Pneumococcal vaccination: information for healthcare professionals. 2016 https://www.cdc.gov/vaccines/vpd/pneumo/hcp/index.html (accessed Aug 3, 2017).
- 23.Immunization Action Coalition. Ask the experts: diseases & vaccines: pneumococcal vaccines (PCV13 and PPSV23) 2017 http://www.immunize.org/askexperts/experts_pneumococcal_vaccines.asp (accessed Aug 3, 2017).
- 24.Parent Project Muscular Dystrophy. Vaccination recommendations. 2015 http://www.parentprojectmd.org/site/PageServer?pagename=Care_area_vaccinations (accessed Nov 30, 2017).
- 25.McKim DA, Katz SL, Barrowman N, Ni A, LeBlanc C. Lung volume recruitment slows pulmonary function decline in Duchenne muscular dystrophy. Arch Phys Med Rehabil. 2012;93:1117–22. doi: 10.1016/j.apmr.2012.02.024. [DOI] [PubMed] [Google Scholar]
- 26.Stehling F, Bouikidis A, Schara U, Mellies U. Mechanical insufflation/exsufflation improves vital capacity in neuromuscular disorders. Chron Respir Dis. 2015;12:31–35. doi: 10.1177/1479972314562209. [DOI] [PubMed] [Google Scholar]
- 27.Chiou M, Bach JR, Jethani L, Gallagher MF. Active lung volume recruitment to preserve vital capacity in Duchenne muscular dystrophy. J Rehabil Med. 2017;49:49–53. doi: 10.2340/16501977-2144. [DOI] [PubMed] [Google Scholar]
- 28.Suk KS, Lee BH, Lee HM, et al. Functional outcomes in Duchenne muscular dystrophy scoliosis: comparison of the differences between surgical and nonsurgical treatment. J Bone Joint Surg Am. 2014;96:409–15. doi: 10.2106/JBJS.M.00777. [DOI] [PubMed] [Google Scholar]
- 29.Bianchi C, Baiardi P. Cough peak flows: standard values for children and adolescents. Am J Phys Med Rehabil. 2008;87:461–67. doi: 10.1097/PHM.0b013e318174e4c7. [DOI] [PubMed] [Google Scholar]
- 30.Szeinberg A, Tabachnik E, Rashed N, et al. Cough capacity in patients with muscular dystrophy. Chest. 1988;94:1232–35. doi: 10.1378/chest.94.6.1232. [DOI] [PubMed] [Google Scholar]
- 31.LoMauro A, Romei M, D’Angelo MG, Aliverti A. Determinants of cough efficiency in Duchenne muscular dystrophy. Pediatr Pulmonol. 2014;49:357–65. doi: 10.1002/ppul.22836. [DOI] [PubMed] [Google Scholar]
- 32.Phillips MF, Quinlivan RC, Edwards RH, Calverley PM. Changes in spirometry over time as a prognostic marker in patients with Duchenne muscular dystrophy. Am J Respir Crit Care Med. 2001;164:2191–94. doi: 10.1164/ajrccm.164.12.2103052. [DOI] [PubMed] [Google Scholar]
- 33.Sawnani H, Thampratankul L, Szczesniak RD, Fenchel MC, Simakajornboon N. Sleep disordered breathing in young boys with Duchenne muscular dystrophy. J Pediatr. 2015;166:640–45.e1. doi: 10.1016/j.jpeds.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 34.Bersanini C, Khirani S, Ramirez A, et al. Nocturnal hypoxaemia and hypercapnia in children with neuromuscular disorders. Eur Respir J. 2012;39:1206–12. doi: 10.1183/09031936.00087511. [DOI] [PubMed] [Google Scholar]
- 35.Hamada S, Ishikawa Y, Aoyagi T, Minami R, Bach JR. Indicators for ventilator use in Duchenne muscular dystrophy. Respir Med. 2011;105:625–29. doi: 10.1016/j.rmed.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 36.Hukins CA, Hillman DR. Daytime predictors of sleep hypoventilation in Duchenne muscular dystrophy. Am J Respir Crit Care Med. 2000;161:166–70. doi: 10.1164/ajrccm.161.1.9901057. [DOI] [PubMed] [Google Scholar]
- 37.Clinical indications for noninvasive positive pressure ventilation in chronic respiratory failure due to restrictive lung disease, COPD, and nocturnal hypoventilation—a consensus conference report. Chest. 1999;116:521–34. doi: 10.1378/chest.116.2.521. [DOI] [PubMed] [Google Scholar]
- 38.Mendoza M, Gelinas DF, Moore DH, Miller RG. A comparison of maximal inspiratory pressure and forced vital capacity as potential criteria for initiating non-invasive ventilation in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2007;8:106–11. doi: 10.1080/17482960601030188. [DOI] [PubMed] [Google Scholar]
- 39.Amaddeo A, Moreau J, Frapin A, et al. Long term continuous positive airway pressure (CPAP) and noninvasive ventilation (NIV) in children: initiation criteria in real life. Pediatr Pulmonol. 2016;51:968–74. doi: 10.1002/ppul.23416. [DOI] [PubMed] [Google Scholar]
- 40.Bach JR, Goncalves MR, Hamdani I, Winck JC. Extubation of patients with neuromuscular weakness: a new management paradigm. Chest. 2010;137:1033–39. doi: 10.1378/chest.09-2144. [DOI] [PubMed] [Google Scholar]
- 41.Toussaint M, Steens M, Soudon P. Lung function accurately predicts hypercapnia in patients with Duchenne muscular dystrophy. Chest. 2007;131:368–75. doi: 10.1378/chest.06-1265. [DOI] [PubMed] [Google Scholar]
- 42.McKim DA, Griller N, LeBlanc C, Woolnough A, King J. Twenty-four hour noninvasive ventilation in Duchenne muscular dystrophy: a safe alternative to tracheostomy. Can Respir J. 2013;20:e5–9. doi: 10.1155/2013/406163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katz SL, McKim D, Hoey L, et al. Respiratory management strategies for Duchenne muscular dystrophy: practice variation amongst Canadian sub-specialists. Pediatr Pulmonol. 2013;48:59–66. doi: 10.1002/ppul.22548. [DOI] [PubMed] [Google Scholar]
- 44.Rodger S, Woods KL, Bladen CL, Stringer A, Vry J, Gramsch K, et al. Adult care for Duchenne muscular dystrophy in the UK. J Neurol. 2015;262:629–41. doi: 10.1007/s00415-014-7585-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeppesen J, Green A, Steffensen BF, Rahbek J. The Duchenne muscular dystrophy population in Denmark, 1977–2001: prevalence, incidence and survival in relation to the introduction of ventilator use. Neuromuscul Disord. 2003;13:804–12. doi: 10.1016/s0960-8966(03)00162-7. [DOI] [PubMed] [Google Scholar]
- 46.Bach JR, Goncalves MR, Hon A, et al. Changing trends in the management of end-stage neuromuscular respiratory muscle failure: recommendations of an international consensus. Am J Phys Med Rehabil. 2013;92:267–77. doi: 10.1097/PHM.0b013e31826edcf1. [DOI] [PubMed] [Google Scholar]
- 47.Pathfinders DMD. Ventilation and Duchenne. 2014 http://www.dmdpathfinders.org.uk/wp-content/uploads/2014/11/dmd-ventilation-faq-booklet-small.pdf (accessed June 27, 2017).
- 48.McNally EM. New approaches in the therapy of cardiomyopathy in muscular dystrophy. Annu Rev Med. 2007;58:75–88. doi: 10.1146/annurev.med.58.011706.144703. [DOI] [PubMed] [Google Scholar]
- 49.Chenard AA, Becane HM, Tertrain F, de Kermadec JM, Weiss YA. Ventricular arrhythmia in Duchenne muscular dystrophy: prevalence, significance and prognosis. Neuromuscul Disord. 1993;3:201–06. doi: 10.1016/0960-8966(93)90060-w. [DOI] [PubMed] [Google Scholar]
- 50.New York Heart Association. Classes of heart failure. http://www.heart.org/HEARTORG/Conditions/HeartFailure/AboutHeartFailure/Classes-of-Heart-Failure_UCM_306328_Article.jsp#.WmHCN4CWS9L (accessed Jan 18, 2018).
- 51.McNally EM, Kaltman JR, Benson DW, et al. Contemporary cardiac issues in Duchenne muscular dystrophy. Working Group of the National Heart, Lung, and Blood Institute in collaboration with Parent Project Muscular Dystrophy. Circulation. 2015;131:1590–98. doi: 10.1161/CIRCULATIONAHA.114.015151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cripe LH, Tobias JD. Cardiac considerations in the operative management of the patient with Duchenne or Becker muscular dystrophy. Paediatr Anaesth. 2013;23:777–84. doi: 10.1111/pan.12229. [DOI] [PubMed] [Google Scholar]
- 53.Duboc D, Meune C, Pierre B, et al. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. Am Heart J. 2007;154:596–602. doi: 10.1016/j.ahj.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 54.Raman SV, Hor KN, Mazur W, et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015;14:153–61. doi: 10.1016/S1474-4422(14)70318-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raman SV, Hor KN, Mazur W, et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: results of a two-year open-label extension trial. Orphanet J Rare Dis. 2017;12:39. doi: 10.1186/s13023-017-0590-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Finsterer J, Cripe L. Treatment of dystrophin cardiomyopathies. Nat Rev Cardiol. 2014;11:168–79. doi: 10.1038/nrcardio.2013.213. [DOI] [PubMed] [Google Scholar]
- 57.Wollinsky KH, Kutter B, Geiger PM. Long-term ventilation of patients with Duchenne muscular dystrophy: experiences at the Neuromuscular Centre Ulm. Acta Myol. 2012;31:170–78. [PMC free article] [PubMed] [Google Scholar]
- 58.Epstein AE, DiMarco JP, Ellenbogen KA, et al. ACC/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices). Developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. Circulation. 2008;117:e350–408. doi: 10.1161/CIRCUALTIONAHA.108.189742. [DOI] [PubMed] [Google Scholar]
- 59.Ryan TD, Jefferies JL, Sawnani H, et al. Implantation of the HeartMate II and HeartWare left ventricular assist devices in patients with Duchenne muscular dystrophy: lessons learned from the first applications. ASAIO J. 2014;60:246–8. doi: 10.1097/MAT.0000000000000050. [DOI] [PubMed] [Google Scholar]
- 60.Amodeo A, Adorisio R. Left ventricular assist device in Duchenne cardiomyopathy: can we change the natural history of cardiac disease? Int J Cardiol. 2012;161:e43. doi: 10.1016/j.ijcard.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 61.Iodice F, Testa G, Averardi M, Brancaccio G, Amodeo A, Cogo P. Implantation of a left ventricular assist device as a destination therapy in Duchenne muscular dystrophy patients with end stage cardiac failure: management and lessons learned. Neuromuscul Disord. 2015;25:19–23. doi: 10.1016/j.nmd.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 62.Lang SM, Shugh S, Mazur W, et al. Myocardial fibrosis and left ventricular dysfunction in Duchenne muscular dystrophy carriers using cardiac magnetic resonance imaging. Pediatr Cardiol. 2015;36:1495–501. doi: 10.1007/s00246-015-1192-7. [DOI] [PubMed] [Google Scholar]
- 63.Florian A, Rosch S, Bietenbeck M, et al. Cardiac involvement in female Duchenne and Becker muscular dystrophy carriers in comparison to their first-degree male relatives: a comparative cardiovascular magnetic resonance study. Eur Heart J Cardiovasc Imaging. 2016;17:326–33. doi: 10.1093/ehjci/jev161. [DOI] [PubMed] [Google Scholar]
- 64.Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:2761–96. doi: 10.1161/CIR.0b013e318223e230. [DOI] [PubMed] [Google Scholar]
- 65.Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128:e240–327. doi: 10.1161/CIR.0b013e31829e8776. [DOI] [PubMed] [Google Scholar]
- 66.Larson CM, Henderson RC. Bone mineral density and fractures in boys with Duchenne muscular dystrophy. J Pediatr Orthop. 2000;20:71–74. [PubMed] [Google Scholar]
- 67.McDonald DG, Kinali M, Gallagher AC, et al. Fracture prevalence in Duchenne muscular dystrophy. Dev Med Child Neurol. 2002;44:695–98. doi: 10.1017/s0012162201002778. [DOI] [PubMed] [Google Scholar]
- 68.King WM, Ruttencutter R, Nagaraja HN, et al. Orthopedic outcomes of long-term daily corticosteroid treatment in Duchenne muscular dystrophy. Neurology. 2007;68:1607–13. doi: 10.1212/01.wnl.0000260974.41514.83. [DOI] [PubMed] [Google Scholar]
- 69.Alos N, Grant RM, Ramsay T, et al. High incidence of vertebral fractures in children with acute lymphoblastic leukemia 12 months after the initiation of therapy. J Clin Oncol. 2012;30:2760–67. doi: 10.1200/JCO.2011.40.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rodd C, Lang B, Ramsay T, et al. Incident vertebral fractures among children with rheumatic disorders 12 months after glucocorticoid initiation: a national observational study. Arthritis Care Res. 2012;64:122–31. doi: 10.1002/acr.20589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cummings EA, Ma J, Fernandez CV, et al. Incident vertebral fractures in children with leukemia during the four years following diagnosis. J Clin Endocrinol Metab. 2015;100:3408–17. doi: 10.1210/JC.2015-2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.LeBlanc CM, Ma J, Taljaard M, et al. Incident vertebral fractures and risk factors in the first three years following glucocorticoid initiation among pediatric patients with rheumatic disorders. J Bone Miner Res. 2015;30:1667–75. doi: 10.1002/jbmr.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ma J, McMillan HJ, Karaguzel G, et al. The time to and determinants of first fractures in boys with Duchenne muscular dystrophy. Osteoporos Int. 2017;28:597–608. doi: 10.1007/s00198-016-3774-5. [DOI] [PubMed] [Google Scholar]
- 74.Medeiros MO, Behrend C, King W, Sanders J, Kissel J, Ciafaloni E. Fat embolism syndrome in patients with Duchenne muscular dystrophy. Neurology. 2013;80:1350–52. doi: 10.1212/WNL.0b013e31828ab313. [DOI] [PubMed] [Google Scholar]
- 75.McAdam LC, Rastogi A, Macleod K, Douglas Biggar W. Fat embolism syndrome following minor trauma in Duchenne muscular dystrophy. Neuromuscul Disord. 2012;22:1035–39. doi: 10.1016/j.nmd.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 76.Ferraris JR, Pasqualini T, Alonso G, et al. Effects of deflazacort vs. methylprednisone: a randomized study in kidney transplant patients. Pediatr Nephrol. 2007;22:734–41. doi: 10.1007/s00467-006-0403-0. [DOI] [PubMed] [Google Scholar]
- 77.Ferraris JR, Pasqualini T, Legal S, et al. Effect of deflazacort versus methylprednisone on growth, body composition, lipid profile, and bone mass after renal transplantation. The Deflazacort Study Group. Pediatr Nephrol. 2000;14:682–88. doi: 10.1007/s004670000337. [DOI] [PubMed] [Google Scholar]
- 78.Loftus J, Allen R, Hesp R, et al. Randomized, double-blind trial of deflazacort versus prednisone in juvenile chronic (or rheumatoid) arthritis: a relatively bone-sparing effect of deflazacort. Pediatrics. 1991;88:428–36. [PubMed] [Google Scholar]
- 79.Singh A, Schaeffer EK, Reilly CW. Vertebral fractures in Duchenne muscular dystrophy patients managed with deflazacort. J Pediatr Orthop. 2016 doi: 10.1097/BPO.0000000000000817. published online June 16. [DOI] [PubMed] [Google Scholar]
- 80.Guglieri M, Bushby K, McDermott MP, et al. Developing standardized corticosteroid treatment for Duchenne muscular dystrophy. Contemp Clin Trials. 2017;58:34–39. doi: 10.1016/j.cct.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ward LM, Konji V, Ma J. The management of osteoporsois in children. Osteoporos Int. 2016;27:2147–79. doi: 10.1007/s00198-016-3515-9. [DOI] [PubMed] [Google Scholar]
- 82.Bachrach LK. Diagnosis and treatment of pediatric osteoporosis. Curr Opin Endocrinol Diabetes Obes. 2014;21:454–60. doi: 10.1097/MED.0000000000000106. [DOI] [PubMed] [Google Scholar]
- 83.Genant HK, Wu CY, van Kuijk C, Nevitt MC. Vertebral fracture assessment using a semiquantitative technique. J Bone Miner Res. 1993;8:1137–8. doi: 10.1002/jbmr.5650080915. [DOI] [PubMed] [Google Scholar]
- 84.Sbrocchi AM, Rauch F, Jacob P, et al. The use of intravenous bisphosphonate therapy to treat vertebral fractures due to osteoporosis among boys with Duchenne muscular dystrophy. Osteoporos Int. 2012;23:2703–11. doi: 10.1007/s00198-012-1911-3. [DOI] [PubMed] [Google Scholar]
- 85.Kilpinen-Loisa P, Paasio T, Soiva M, et al. Low bone mass in patients with motor disability: prevalence and risk factors in 59 Finnish children. Dev Med Child Neurol. 2010;52:276–82. doi: 10.1111/j.1469-8749.2009.03464.x. [DOI] [PubMed] [Google Scholar]
- 86.Christiansen BA, Bouxsein ML. Biomechanics of vertebral fractures and the vertebral fracture cascade. Curr Osteoporos Rep. 2010;8:198–204. doi: 10.1007/s11914-010-0031-2. [DOI] [PubMed] [Google Scholar]
- 87.Ma J, Siminoski K, Alos N, et al. The choice of normative pediatric reference database changes spine bone mineral density Z scores but not the relationship between bone mineral density and prevalent vertebral fractures. J Clin Endocrinol Metab. 2015;100:1018–27. doi: 10.1210/jc.2014-3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bishop N, Arundel P, Clark E, et al. Fracture prediction and the definition of osteoporosis in children and adolescents: the ISCD 2013 pediatric official positions. J Clin Densitom. 2014;17:275–80. doi: 10.1016/j.jocd.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 89.Zemel BS, Stallings VA, Leonard MB, et al. Revised pediatric reference data for the lateral distal femur measured by Hologic Discovery/Delphi dual-energy X-ray absorptiometry. J Clin Densitom. 2009;12:207–18. doi: 10.1016/j.jocd.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Crabtree NJ, Chapman S, Högler W, et al. Vertebral fractures assessment in children: evaluation of DXA imaging versus conventional spine radiography. Bone. 2017;97:168–74. doi: 10.1016/j.bone.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 91.Johansson H, Oden A, McCloskey EV, Kanis JA. Mild morphometric vertebral fractures predict vertebral fractures but not non-vertebral fractures. Osteoporos Int. 2014;25:235–41. doi: 10.1007/s00198-013-2460-0. [DOI] [PubMed] [Google Scholar]
- 92.Gatti D, Antoniazzi F, Prizzi R, et al. Intravenous neridronate in children with osteogenesis imperfecta: a randomized controlled study. J Bone Miner Res. 2005;20:758–63. doi: 10.1359/JBMR.041232. [DOI] [PubMed] [Google Scholar]
- 93.Antoniazzi F, Zamboni G, Lauriola S, Donadi L, Adami S, Tato L. Early bisphosphonate treatment in infants with severe osteogenesis imperfecta. J Pediatr. 2006;149:174–79. doi: 10.1016/j.jpeds.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 94.Astrom E, Jorulf H, Soderhall S. Intravenous pamidronate treatment of infants with severe osteogenesis imperfecta. Arch Dis Child. 2007;92:332–38. doi: 10.1136/adc.2006.096552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Palomo T, Fassier F, Ouellet J, et al. Intravenous bisphosphonate therapy of young children with osteogenesis imperfecta: skeletal findings during follow up throughout the growing years. J Bone Miner Res. 2015;30:2150–57. doi: 10.1002/jbmr.2567. [DOI] [PubMed] [Google Scholar]
- 96.Allington N, Vivegnis D, Gerard P. Cyclic administration of pamidronate to treat osteoporosis in children with cerebral palsy or a neuromuscular disorder: a clinical study. Acta Orthop Belg. 2005;71:91–97. [PubMed] [Google Scholar]
- 97.Ward LM, Glorieux FH, Rauch F, Verbruggen N, Heyden N, Lombardi A. A randomized, placebo-controlled trial of oral alendronate in children and adolescents with osteogenesis imperfecta. Bone. 2005;36(S1):0–18. [Google Scholar]
- 98.Rauch F, Munns CF, Land C, Cheung M, Glorieux FH. Risedronate in the treatment of mild pediatric osteogenesis imperfecta: a randomized placebo-controlled study. J Bone Miner Res. 2009;24:1282–89. doi: 10.1359/jbmr.090213. [DOI] [PubMed] [Google Scholar]
- 99.Sakkers R, Kok D, Engelbert R, et al. Skeletal effects and functional outcome with olpadronate in children with osteogenesis imperfecta: a 2-year randomised placebo-controlled study. Lancet. 2004;363:1427–31. doi: 10.1016/S0140-6736(04)16101-1. [DOI] [PubMed] [Google Scholar]
- 100.Houde S, Filiatrault M, Fournier A, et al. Deflazacort use in Duchenne muscular dystrophy: an 8-year follow-up. Pediatr Neurol. 2008;38:200–06. doi: 10.1016/j.pediatrneurol.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 101.Huber H, Andre G, Rumeau F, Journeau P, Haumont T, Lascombes P. Flexible intramedullary nailing for distal femoral fractures in patients with myopathies. J Child Orthop. 2012;6:119–23. doi: 10.1007/s11832-012-0399-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Newbigin K, Souza CA, Torres C, et al. Fat embolism syndrome: state-of-the-art review focused on pulmonary imaging findings. Respir Med. 2016;113:93–100. doi: 10.1016/j.rmed.2016.01.018. [DOI] [PubMed] [Google Scholar]
- 103.Lebel DE, Corston JA, McAdam LC, Biggar WD, Alman BA. Glucocorticoid treatment for the prevention of scoliosis in children with Duchenne muscular dystrophy: long-term follow-up. J Bone Joint Surg Am. 2013;95:1057–61. doi: 10.2106/JBJS.L.01577. [DOI] [PubMed] [Google Scholar]
- 104.Takaso M, Nakazawa T, Imura T, et al. Surgical management of severe scoliosis with high risk pulmonary dysfunction in Duchenne muscular dystrophy: patient function, quality of life and satisfaction. Int Orthop. 2010;34:695–702. doi: 10.1007/s00264-010-0957-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Archer JE, Gardner AC, Roper HP, Chikermane AA, Tatman AJ. Duchenne muscular dystrophy: the management of scoliosis. J Spine Surg. 2016;2:185–94. doi: 10.21037/jss.2016.08.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alexander WM, Smith M, Freeman BJ, Sutherland LM, Kennedy JD, Cundy PJ. The effect of posterior spinal fusion on respiratory function in Duchenne muscular dystrophy. Eur Spine J. 2013;22:411–16. doi: 10.1007/s00586-012-2585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Roberto R, Fritz A, Hagar Y, et al. The natural history of cardiac and pulmonary function decline in patients with Duchenne muscular dystrophy. Spine. 2011;36:E1009–17. doi: 10.1097/BRS.0b013e3181fea1ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chua K, Tan CY, Chen Z, et al. Long-term follow-up of pulmonary function and scoliosis in patients with Duchenne’s muscular dystrophy and spinal muscular atrophy. J Pediatr Orthop. 2016;36:63–69. doi: 10.1097/BPO.0000000000000396. [DOI] [PubMed] [Google Scholar]
- 109.Hayes J, Veyckemans F, Bissonnette B. Duchenne muscular dystrophy: an old anesthesia problem revisited. Paediatr Anaesth. 2008;18:100–06. doi: 10.1111/j.1460-9592.2007.02302.x. [DOI] [PubMed] [Google Scholar]
- 110.Birnkrant DJ, Panitch HB, Benditt JO, et al. American College of Chest Physicians consensus statement on the respiratory and related management of patients with Duchenne muscular dystrophy undergoing anesthesia or sedation. Chest. 2007;132:1977–86. doi: 10.1378/chest.07-0458. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.