

SUMMARY
Highly effective HIV-1-neutralizing antibodies could have utility in the prevention or treatment of HIV-1 infection. To improve the potency of 10E8, an antibody capable of near pan-HIV-1 neutralization, we engineered 10E8-surface mutants and screened for improved neutralization. Variants with the largest functional enhancements involved the addition of hydrophobic or positively charged residues, which were positioned to interact with viral membrane lipids or viral glycan-sialic acids, respectively. In both cases, the site of improvement was spatially separated from the region of antibody mediating molecular contact with the protein component of the antigen, thereby improving peripheral semi-specific interactions while maintaining unmodified dominant contacts responsible for broad recognition. The optimized 10E8 antibody, with mutations to phenylalanine and arginine, retained the extraordinary breadth of 10E8 but with ~10-fold increased potency. We propose surface-matrix screening as a general method to improve antibodies, with improved semi-specific interactions between antibody and antigen enabling increased potency without compromising breadth.
In Brief
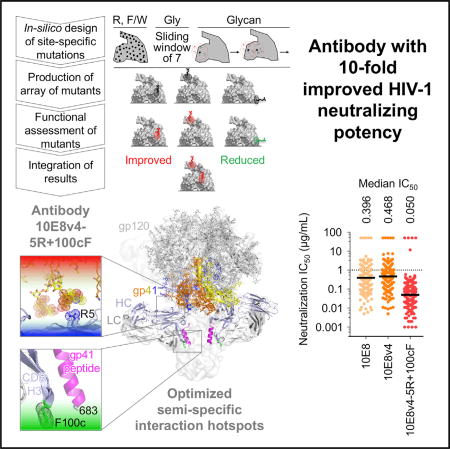
Antibodies could impact the treatment and prevention of HIV-1 if they were sufficiently potent to allow cost-effective delivery. Kwon et al. used a surface-matrix screening approach to improve the potency of antibody 10E8 by ~10-fold. The improved antibody, 10E8v4-5R+100cF, has among the best breadth and potency of current HIV-1-neutralizing antibodies.
INTRODUCTION
With antibody-based therapeutics dominating the biologics pipelines of leading pharmaceutical companies (Ayyar et al., 2016; Chames et al., 2009), methods to improve antibody functionality have been eagerly sought. The payoffs are clear: if antibody efficacy were to increase 10-fold, the amount of delivered antibody required to achieve efficacy might decrease 10-fold, with concomitant decreases in dosing, side effects, and/or costs. Whereas optimization of small-molecule therapeutics is well established, many biologics are utilized as identified by functional screening, without substantial optimization of therapeutic functionality. Indeed, while specific antibody constant-region mutations have been identified for increasing antibody half-life (Roopenian and Akilesh, 2007) or for reducing reactivity (for example, by humanization for non-human antibodies) (Hwang and Foote, 2005), it has been a challenge to improve antibody therapeutic functionality by in vitro optimization. For example, attempts to improve palivizumab (Synagis), the sole monoclonal antibody licensed for an infectious disease (respiratory syncytial virus infection), have met with difficulty, with standard methods for optimizing protein interactions such as phage display showing uncertain translation to in vivo therapeutic efficacy (Ramilo et al., 2014; Wu et al., 2007).
In the case of HIV-1, potent broadly neutralizing antibodies have been sought as therapeutics or for prevention. A cocktail of five antibodies has shown therapeutic potential in animal studies (Klein et al., 2012), and antibodies such as 3BNC117, VRC01, and members of the PGT121 family are showing promise in clinical studies (Bar et al., 2016; Caskey et al., 2015, 2017; Scheid et al., 2016; Schoofs et al., 2016). VRC01, a human antibody identified from an HIV-1-infected donor, is capable of neutralizing over 90% of HIV-1, with a median 50% inhibitory concentration (IC50) of 0.33 µg/mL (Wu et al., 2010), and its preventive efficacy is currently being assessed in a Phase IIb prevention study, with dosing up to ~2 g VRC01 every 2 months for an average adult. At this dosing, long-term use of antibody VRC01 would likely be impractical; however, only ~0.2 g every 2 months might be required for an antibody 10-fold more potent than VRC01, easing issues of practicality.
To improve antibody effectiveness, structure-based design and other hypothesis-based deductive approaches have been undertaken. For example, with antibodies of the VRC01 class (Zhou et al., 2013), antibody NIH45-46 could be significantly improved by a G54W mutation, designed to mimic the recognition of the CD4 receptor (Diskin et al., 2011; Scheid et al., 2011). Unfortunately, the NIH45-46 G54W variant proved to be polyreactive. However, a combination approach succeeded in obtaining VRC07-523LS, an improved VRC01-class antibody containing multiple sequence alterations and displaying increased potency and tolerable polyreactivity (Rudicell et al., 2014). Importantly, the VRC07-523LS antibody required only one-fifth the dose to achieve the same protection as VRC01 in rhesus macaques (Rudicell et al., 2014).
While deductive approaches (Figure 1A, left) can improve antibody functionality, such approaches can only be used with characterized interactions. Here, we sought to test a surface matrix-screening approach (Figure 1A, right) that used virus neutralization as the target readout, incorporated chemical alterations of surface residues, and assessed and integrated the desired properties. We chose antibody 10E8 for improvement, due to its ability to neutralize >97% of HIV strains, with a median IC50 potency of 0.40 µg/mL by recognizing the membrane-proximal external region (MPER) of the HIV-1 envelope (Env) (Huang et al., 2012). We identified single amino-acid mutations that improved neutralization potency and analyzed the chemical preferences of each functional hotspot to reveal mechanistic insights. Identified hotspots were distinct from the region of the antibody mediating molecular contact with the protein component of the antigen and appeared to enhance semi-specific interactions with either the glycan shield or the viral membrane. Notably, the optimized 10E8 antibody, displayed a median IC50 of 0.050 µg/mL, about 2-fold lower than that of antibody N6 (Huang et al., 2016a), a recently described broadly neutralizing antibody targeting the CD4-binding site of HIV-1, which is one of the most potent broadly neutralizing antibodies discovered thus far. Remarkably, the combination of the improved 10E8v4-5R+100cF variant with N6 neutralized all HIV-1 strains in our 208-isolate panel, at less than 1 µg/mL, with a median IC50 of 0.016 µg/mL.
Figure 1. Surface-Matrix Screening to Improve Neutralization Potency of Antibody 10E8.

(A) Hypothesis-driven deductive approach (left) versus hypothesis-independent surface-matrix approach (right). The broad sampling of the matrix approach provides extensive information linking surface chemistry to biological functionalities of interest. Shown is the 10E8 Fab with heavy chain in light blue and light chain in white, and with gp41 peptide in magenta.
(B) Schematic flow diagram of surface-matrix approach.
(C) Selection of a 9-virus panel (shown as red in dendrogram) from the 208-isolate panel for neutralization assessment. The clade of each selected strain is displayed in parentheses.
(D) Experimental variation in neutralization. Strains with non-sigmoidal neutralization curves and high assay-replication error are shown in italics.
(E) Minimum observable signal as a function of assay variability and number of variants screened (see Figure S2).
(F) Neutralization IC50 by 10E8 variants, arranged by geometric mean IC50-fold improvement for all viruses shown. “>50” was considered as “50” in geometric mean calculations.
(G) Crystal structure of gp41 peptide (magenta) complexed with improved antibody 10E8v4-5R+100cF (light blue, heavy chain; white, light chain).
See also Figures S1 and S2 and Tables S1C and S2.
RESULTS
Surface-Matrix Screening of Antibody 10E8
For alterations of surface chemistry, we chose types of residue substitutions observed in other cases to improve antibody functionality (Figure 1B). We chose Phe (or Trp, if the wild-type residue was Phe or Tyr), as hydrophobic interactions are common in antibody-antigen recognition (Diskin et al., 2011; Young et al., 1994). We also chose Arg, which can improve solubility, can mediate interactions with phospholipid membranes, or can interfere with antigen interactions through steric clashes (Gorman et al., 2016; McLellan et al., 2011; Robison et al., 2016; Trevino et al., 2007). To assess the effects of amino-acid side-chain removal, we tested substitutions of outwardly facing residues to glycine in windows of 7 residues (2 overlapping). Lastly, we tested the effects of introducing N-linked glycans on the antibody surface, as glycans are hydrophilic, with the potential to increase solubility (Hebert et al., 2014), to introduce steric clashes at sites of interaction and to enhance potency in select cases (Song et al., 2013).
Starting with 10E8, we used the crystal structure (Huang et al., 2012) of the antigen-binding fragment (Fab) to calculate the solvent accessibility of each amino-acid residue. For each residue with greater than 30% solvent accessibility, we produced single point mutants: 105 Phe (or Trp) mutants and 98 Arg mutants. We also produced 48 7-Gly mutants and 47 N-glycan variants (Figure S1; Tables S1A and S1B). In total, we created 298 10E8 variants, each of which we synthesized and expressed.
To assess the impact of each mutation on neutralization, we tested a nine-virus panel, selected to cover a range of potencies and diverse clades (Figure 1C). Assessment of wild-type 10E8 neutralization indicated substantial variation in replicated measurements, ranging from geometric SDs of 1.3 (for strain ZM106.9) to 5.6 (for strain KER2008.12); we proceeded with the five isolates that showed the least replicate variation (Figure 1D). With an error in potency of 1.5-fold, we estimated that assessment of 298 variants should allow for the identification of variants with as little as ~2-fold improvement in potency (Figure 1E; Figure S2).
Two variants showed improvements of over 2.5-fold in potency (Figure 1F; Table S1C). One mutation, S100cFHC (we use Kabat nomenclature for antibody numbering, with residue 100c being the third residue after 100a and subscripts HC and LC to represent heavy and light chains, respectively), increased neutralization potency by almost 4-fold, and a second mutation, V5RHC, increased neutralization potency by just under 3-fold. (Figure 1F). To understand the impact of Arg5HC and Phe100cHC mutations on the structure of 10E8, we added these mutations to antibody 10E8v4 (Kwon et al., 2016), a solubility-improved variant with breadth and neutralization potency similar to those of 10E8, and crystallized the antigen-binding fragment (Fab) of 10E8v4-5R+100cF in complex with a 19-mer peptide encompassing its MPER epitope. Diffraction data extended to 3.1 Å resolution, and structure solution by molecular replacement and refinement yielded an Rwork/Rfree of 0.24/0.28 (Table S2). Overall, the structure revealed both Arg5HC and Phe100cHC mutations to minimally alter the Fab structure and to be distal from one another and peripheral to the main contacts between MPER and 10E8 (Figure 1G).
Delineation of 10E8 Functional Surfaces by Surface-Matrix Screening
Multiple variants showed reductions in neutralization of over 3 SDs (Figure S1, right panels; Table S1C). Many of these deleterious mutations involved the introduction of bulky moieties that could sterically inhibit binding, while others involved substitutions of critical paratope residues. The observed deleterious mutations clustered in coherent patterns to selected surfaces (Figure 2A). To provide biological context, we evaluated these surfaces in the context of the cryoelectron microscopy (cryo-EM) structure of the detergent-solubilized Env trimer bound by antibodies PGT151 and 10E8 (Lee et al., 2016) (Figure 2B). Notably, in addition to epitope-contacting residues, two surfaces—one facing the expected location of the Env trimer and the other facing the expected location of the viral membrane—were identified by clusters of inhibitory mutations.
Figure 2. Interactive Surfaces Identified by Surface-Matrix Screening.

(A) Crystal structure of Fab 10E8 is shown with residues (cyan) that, when altered by surface-matrix screening, decreased neutralization by over 3 SDs (3.4-fold). Residue alteration types are noted above each structure, with all surface-matrix screening results are shown in Figure S1. Notably, two surfaces, one facing the expected position of the Env trimer and the other facing membrane, are identified by clusters of inhibitory N-glycan mutations. These are not seen in Arg mutations, but the putative membrane-interactive surface is identified by both Phe/Trp and 7-glycine mutations. Note that orientations shown in (A) allow better labeling and are slightly different from 10E8 orientation in (B).
(B) Orientation of 10E8 Fab bound to detergent-solubilized Env trimer from a detergent-solubilized complex with PGT151 (PDB: 5FUU and EMDB: 3312).
See also Figures S1 and S2 and Table S1C.
N-linked glycans were the largest steric substitutions introduced, and deleterious N-linked glycan mutations mapped to both of the identified surfaces. We observed strongly deleterious Phe/Trp mutations to cluster to the membrane-facing surface, an unexpected result, as Phe/Trp should enhance membrane interactions. For the 7-glycine mutations, we also observed deleterious mutations to cluster to the membrane-facing surface. The prevalence of the deleterious Phe/Trp and 7-glycine mutations on the expected membrane-facing surface may reflect requirements for specificity in 10E8 interactions with membrane. Arg mutants displayed strong deleterious effects, which mapped mostly to epitope residues. For membrane-facing residues, the lack of deleterious effects (as observed with N-glycan, Phe/Trp, and 7-glycine variants) may relate to the ability of Arg to recognize phospholipid head groups. As for Env-facing residues, a few strong negative mutants were observed, which clustered close to a significantly positive mutation, at residue 5.
Chemical Preferences of 10E8 Hotspots Identified in the Surface-Matrix Screen
To provide insight into the chemistry of the Arg5HC and Phe100cHC mutations that improved neutralization potency, we substituted positions 5HC and 100cHC in the context of 10E8v4 with other amino acids and assessed their neutralization potency. For Val5HC, the positively charged substitutions Lys and Arg were most potent, reducing the median 80% inhibitory concentration (IC80) on a panel of 13 viruses from 4.0 µg/mL to 2.9 and 1.7 µg/mL, respectively (Figure 3A). Modeling this change in the context of the detergent-solubilized Env trimer with antibodies PGT151 and 10E8 (Lee et al., 2016) suggested that positively charged residues at this position interact with negatively charged sialic acid residues in the glycan shield (Figure 3B, left).
Figure 3. Chemical Preferences of Functional Hotspots Identified by Surface-Matrix Screening.

(A) Effects of residue substitutions at heavy chain position 5.
(B) Location of R at position 5 and F at position 100c alteration relative to structure of 10E8 bound to detergent-solubilized Env trimer (insets show residue environments surrounding the two hotspots).
(C) Effects of residue substitutions at heavy chain position 100c.
For S100cHC, the hydrophobic changes Leu, Phe, and Trp were most potent, reducing the median IC80 on a panel of 9 viruses from 0.5 µg/mL to 0.09, 0.02, and 0.01 µg/mL, respectively (Figure 3C). Analysis of 10E8 in the detergent-solubilized Env trimer (Lee et al., 2016) indicated that a Phe at position 100cHC would face the detergent micelle and, in the context of neutralization, may therefore interact with the viral membrane (Figure 3B, right).
Analogous Membrane-Binding Functional Hotspot in MPER-Directed Antibody 4E10
To further define the interaction of Phe100cHC on antibody 10E8 with the viral membrane, we investigated the analogous membrane-binding functional hotspot on antibody 4E10 (Cardoso et al., 2005), which binds to the same region of the MPER as 10E8. Diverse studies ranging from electron paramagnetic resonance (EPR) analysis (Song et al., 2009; Sun et al., 2008) to mutational and partitioning analyses (Ofek et al., 2010; Rujas et al., 2017), to crystallographic analysis of Fab in complex with phospholipid headgroups (Irimia et al., 2016, 2017) indicate antibodies 10E8 and 4E10 to co-recognize membrane and MPER peptide. In particular, the orientation suggested by the structure of 10E8 with 1,2 dihexanoyl-sn-glycerol-3-phospho-(1’-rac-glycerol) and scaffolded MPER (Irimia et al., 2017) would position the indole ring of Trp100cHC to extend into the viral lipid membrane (Figure 4A). Examination of the structure of 4E10 with 1,2 dihexanoyl-sn-glycerol-3-phosphate and MPER peptide (Irimia et al., 2016) suggested that a Trp substitution in 4E10 at position 100aHC would be similarly positioned to interact with the viral lipid membrane (Figure 4B). Indeed, Trp substitutions of different positions at the apex of the third heavy chain complementarity-determining region (CDR H3) of 4E10 showed only the substitution at position 100aHC to increase neutralization potency (Figure 4C). Interestingly, superposition of the MPER peptide to provide a common reference frame indicated the CDR H3s of 10E8v4 and 4E10 to interact with different faces of the MPER helix, with the potency-increasing Trp substitutions in each case placed in an analogous position relative to the plane of the viral membrane but not relative to the face of the MPER helix (Figure 4D). Overall, these results show how hydrophobic substitution can enhance the neutralization potency of both antibodies 10E8v4 and 4E10, with substitutions positioned analogously—not relative to the face of the MPER helix but relative to the viral membrane.
Figure 4. MPER-Directed Antibody 4E10 Shares the Membrane-Binding Functional Hotspot of 10E8.

(A) Structural model of antibody 10E8v4 recognizing lipid headgroups, as defined by Irimia et al. (2017), with hotspot position 100c shown in green.
(B) Structural model of antibody 4E10 recognizing lipid headgroups, as defined by Irimia et al. (2016).
(C) Virus neutralization by 4E10 and variants showing 100aW with improved activity.
(D) Superposition gp41 peptide as recognized by 4E10 and 10E8 identifies 4E10 CDR H3 as a hot-spot of functional enhancement.
To further investigate the Trp-enhanced 10E8v4 and 4E10 interactions with MPER, we used biolayer interferometry (BLI) and surface plasmon resonance (SPR) to measure the interaction between antibody and MPER peptide and between antibody and lipid bilayers containing MPER peptide, respectively. We observed the affinity of 10E8v4 and 10E8v4-100cW to MPER to be virtually identical, as were the affinities of 4E10 and 4E10-100aW (Figure 5A). By contrast, Trp-substituted 10E8v4-100cW and 4E10-100aW appeared to show an increased level of binding to MPER over that of 10E8v4 and 4E10 in the membrane context (Figure 5B). The BLI and SPR measurements thus confirm that the Trp substitutions do not impact MPER affinity but rather impact the co-recognition of MPER and membrane.
Figure 5. Enhanced Binding to gp41 Peptide in a Membrane Context by 10E8v4-100cW and 4E10-100aW IgGs Modeled Membrane-Epitope Co-recognition and Polyreactivity for Several HIV-1-Neutralizing Antibodies.

(A) Binding of 10E8v4, 4E10, 10E8v4-100cW, and 4E10-100aW variants to MPER peptide. IgG, immunoglobulin G. KD, equilibrium dissociation constant.
(B) Binding of 10E8v4, 4E10, 10E8v4-100cW, and 4E10-100aW variants to lipid bilayers containing MPER peptide.
(C) Membrane proximal residues (shown in spheres) for 10E8, 4E10, CAP248-2B, DH511.2, and 2F5. Positions of viral membrane surface approximated by dashed line (Supplemental Experimental Procedures).
(D) Membrane interaction propensity of membrane-proximal residues.
(E) HEp2 cell binding versus lipophilicity. Error bars represent SD.
(F) Cardiolipin binding versus lipophilicity.
See also Figure S3.
Polyreactivity for Membrane-Interacting HIV-1 Broadly Neutralizing Antibodies
In addition to 4E10, a number of other HIV-1 broadly neutralizing antibodies have been proposed to interact with viral membrane, including antibodies 2F5 (Ofek et al., 2004), CAP248-2B (Wibmer et al., 2017), and DH511 (Williams et al., 2017) (Figure 5C). As polyreactivity, such as indicated by binding to HEp2 cells or to cardiolipin, can negatively impact the therapeutic efficacy of these antibodies, we assessed the polyreactivity of six membrane-interacting HIV-1 broadly neutralizing antibodies and of Trp-enhanced functional variants for four of the antibodies (Figure 5D; Figure S3). Notably, all of the Trp-enhanced functional variants tested showed strong polyreactivity, except 10E8v4-100cW.
To understand the reduced polyreactivity of 10E8v4-100cW relative to other Trp-enhanced broadly neutralizing antibodies, we examined the lipophilicity of the proposed membrane-interacting residues in each antibody (Figures 5E and 5F). While the introduction of Trp increased hydrophobicity in each case, 10E8v4-100cW showed the lowest propensity to interact with membrane of all of the Trp-enhanced antibodies. Thus, the reduced polyreactivity of 10E8v4-100cW relative to other Trp-enhanced antibodies appears to stem from an overall lower hydrophobicity of the membrane-interacting component of 10E8.
Glycan-Shield-Interacting Variants of 10E8
In addition to an Arg at position 5HC, a number of other Arg substitutions displayed enhanced neutralization potency, including alterations at positions 7HC and 21HC (Table S1C). Modeling these Arg substitutions in the context of the cryo-EM structure of the detergent-solubilized 10E8-bound Env trimer (Lee et al., 2016) indicated that these Arg substitutions cluster on the heavy chain and potentially interact with sialic acids in the HIV-1 glycan shield (Figure 6A).
Figure 6. Semi-specific Interactions with Glycan Shield Observed with Hotspot around Heavy Chain Position 5.

(A) Modeled glycan shield co-recognition of Arg hits identified by surface-matrix screening of 10E8 variants.
(B) Impact of Arg additions on the neutralization (and polyreactivity) of 10E8 variants.
(C) Average improvement in 10E8 variant neutralization upon the addition of 1 or 2 Arg. Error bars represent SD.
We tested these Arg mutations, individually and in combination, for their effects on HIV-1 neutralization as assessed on a panel of nine viruses (Table S1D). Notably, we observed Val5HC Arg to increase neutralization potency in the contexts of 10E8v4, of 10E8v4-100cF, and of 10E8v4-100cW (Figure 6B). Unexpectedly, we also observed several of the Arg substitutions to display substantial polyreactivity (Figures 6B and S4), especially in the context of 100cW, which has heightened polyreactivity of 10E8v4 or 10E8v4-100cF. We note, however, that the mild level of polyreactivity observed for 10E8v4-5R+100cF has been observed with other antibodies, both natural and improved, and that this level of polyreactivity is often tolerated (Rudicell et al., 2014). Interestingly, the largest improvements in neutralization potency came for the introduction of single, not double, Arg residues (Figure 6C). Thus, while Arg substitutions to the Env-facing surface of 10E8 did increase neutralization potency, in some instances, polyreactivity was also increased.
Properties of 10E8v4-5R+100cF
We analyzed, in detail, manufacturing characteristics of three variants, 10E8v4-100cW, 10E8v4-100cF, and 10E8v4-5R+100cF. While 10E8v4-100cW was most potent, it displayed substantial losses during concentration by ultrafiltration, which may be problematic for manufacturing (Figure 7A). While 10E8v4-100cF and 10E8v4-5R+100cF showed acceptable concentration characteristics, 10E8v4-5R+100cF was both more potent and more soluble.
Figure 7. Manufacturing Characteristics, Serum Half-Life, and Neutralization Potency of 10E8 Variants.

(A) Manufacturing properties of optimized 10E8 variants. CD, circular dichroism; DLS, dynamic light scattering; DSC, differential scanning calorimetry; OD350, optical density 350; Tm, melting temperature.
(B) Pharmacokinetics of 10E8 variants in rhesus macaque. Error bars represent SD.
(C) Potency of optimized 10E8v4-5R+100cF on a panel of 208 Env pseudoviruses: antibodies shown are being developed for clinical evaluation. Note that the physical combination of N6 with 10E8v4-5R+100cF neutralizes all strains in the 208-isolate panel at less than 1 µg/mL. The median IC50 based on all 208 viruses (including all resistant strains) for each antibody is displayed.
(D) Completeness of neutralization by single antibodies (10 µg/mL) and antibody combinations with 5 µg/mL of each antibody (predicted using the Bliss Hill model). The numbers on top indicate the percent viruses that were predicted to be neutralized at >95%.
See also Figure S5 and Tables S1E and S1F.
We assessed the half-life in rhesus macaques for each of these three variants, modified by the “LS” half-life-extending mutations (Zalevsky et al., 2010). Notably, all of the variants tracked closely with VRC01-LS through day 14 and appeared to have reduced clearance relative to the parent 10E8 (Figure 7B).
On our 208-isolate panel, 10E8v4-5R+100cF displayed a median IC50 of 0.050 µg/mL (Figure 7C; Table S1E), about 2-fold better than that of the recently described N6 antibody, a broadly neutralizing antibody that targets the CD4-binding site (Huang et al., 2016a). The IC50s of 10E8v4-5R+100cF compared favorably with other HIV-1 broadly neutralizing antibodies being prepared for clinical evaluations, with only PGDM1400 showing a more potent median IC50 (10E8v4-5R+100cF has substantially higher breadth than PGDM1400). We predicted the extent of complete neutralization (defined as >95% neutralization of a pseudovirus) for each antibody (Experimental Procedures; Table S1F) and found that 10E8v4-5R+100cF completely neutralized 96.6% of the viruses at 10 µg/mL, better than all other antibodies (the next best was N6, with 91.8% viruses completely neutralized).
Since combinations of antibodies can increase breadth and potency over individual antibodies (Kong et al., 2015) and might be needed for efficacy in the prevention of diverse HIV-1 infections (Wagh et al., 2016), we explicitly tested the combination of 10E8v4-5R+100cF with N6 (Figure 7C) and observed this Cell combination to neutralize all 208 isolates of our cross-clade panel, with an IC50 < 1 µg/mL of each antibody. We also modeled the performance of the combination of 10E8v4-5R+100cF with other broadly neutralizing antibodies. We used the Bliss-Hill model (Wagh et al., 2016) on individual antibody IC50 and IC80 data for leading antibody candidates under clinical development against the global 208-virus panel to predict neutralization by all two-antibody combinations of antibodies targeting different epitopes (Experimental Procedures). We compared the performance of two-antibody combinations using not only IC80 breadth potency but also completeness of neutralization and simultaneous coverage by both antibodies in the combination (Wagh et al., 2016), and we used target concentrations of 1 µg/mL and 5 µg/mL of each antibody in the combination (Table S1F). We identified the best combination within each epitope class (Table S1F) and compared the best-in-class combinations (Figures 7D and S5). The best overall performance was shown by the combination of 10E8v4-5R+100cF with N6. The main advantages of combining 10E8v4-5R+100cF with N6 were the highest fraction of completely neutralized viruses (96.3%–99.5% viruses, depending on the target concentration as compared to second best of 86.1%–97.6%; p = 0.0002, using Fisher’s exact test for 1 µg/mL of each broadly neutralizing antibody) and substantially improved IC80 coverage with both antibodies to be active (79.3%–92.8% coverage as compared to second best of 60.6%–69.2%, depending on the target concentration; p values = 6.7 × 10−10 to 4.4 × 10−5, using Fisher’s exact test) (Figure S5). These results suggest that the combination of 10E8v4-5R+100cF with N6 has the potential to neutralize completely a majority of global HIV-1 isolates with both antibodies simultaneously active;thus, passive transfer of this combination could have the potential to prevent majority of infections even at low target concentrations.
DISCUSSION
Improvements in functional efficacy may have substantial impact on antibody therapeutic efficacy. However, standard methods of improvement such as phage display have often floundered, because of differences between antibody-mediated neutralization and antibody-probe binding. Having successfully used surface-screening methods such as arginine scanning to identify sites of antibody interaction (Pancera et al., 2013), we decided to alter the antibody surface and to assess neutralization directly. Importantly, the surface-matrix screening approach does not require determination of the antibody-epitope complex structure. Also, the approach allows for the identification of novel sites of interaction.
While identification of sites of substantial improvement in neutralization potency was our original goal (Figure 1), we observed coherent clustering of strong reductions in neutralization to also impart insight into interactive surfaces (Figure 2). Once functional hotspots for improved neutralization were identified, we could further screen their interactive chemistries (Figure 3). Examination of the Phe100cHC change in the 10E8 context suggested that analogous improvements could be made in the context of the related 4E10 antibody. These changes improved the binding of both 10E8 and 4E10 to MPER in the lipid context (Figures 4 and 5), though issues with polyreactivity prevented full optimization of the semi-specific hydrophobic interactions with lipid (Figure 5). Examination of the Arg5HC change, meanwhile, suggested semi-specific electrostatic interactions with sialic acids of the glycan shield; while these could lead to improvements in neutralization potency, considerations of polyreactivity prevented full optimization of these electrostatic interactions (Figure 6). Despite issues of polyreactivity, we nevertheless did achieve significant improvement of 10E8 potency. Overall, optimization of semi-specific interactions provided a means to improve the neutralization potency of antibody 10E8 while maintaining its near pan-neutralization breadth (Figure 7).
We did not observe the neutralization-improving mutations Arg5HC and Phe100cHC in the next-generation sequencing of 10E8 B cell transcripts (Soto et al., 2016), suggesting that the two identified hotspots—one facing Env and the other facing the lipid membrane—may not be fully optimized by in vivo methods of affinity maturation, perhaps because of differences between eliciting antigen and the functional target of antibody neutralization. Moreover, we were unable to corroborate with kifunensine-grown virus, a requirement for sialic acid in mediating Arg enhancements of potency, as the kifunensine impacted neutralization from multiple antibodies (e.g., antibody VRC01) that are not known to have sialic acid interactions. Nevertheless, our surface-matrix data, comprising almost 300 individually assessed antibody variants, did provide a comprehensive chemical/functional mapping of the 10E8 surface and corroborated the biological relevance of the cryo-EM structure of the 10E8-bound-JR-FL trimer complex, indicating similarity between 10E8 binding in the detergent-solubilized context of the cryo-EM structure and the membrane-bound context of the functional spike on infectious virus.
Is there a ceiling to additional improvement? Now that we have identified the mechanistic context of Arg5HC and Phe100cHC improvements, hypothesis-driven means of improvement can be used. Alternatively, it would be fascinating to determine whether a second round of surface-matrix screening could iteratively identify other sites of improvement. It may be helpful in a second round to limit the total number of variants tested, to allow for the identification of changes with only 1.5-fold improvement (Figure S2).
While further improvements are likely possible, the optimized antibody, 10E8v4-5R+100cF, may already have utility in both HIV therapy and prophylaxis, especially in combination with antibody N6. We note that an improved 10E8 antibody has been described that utilized a bi-specific strategy in combination with self-targeting to achieve exquisite potency (Huang et al., 2016b) and that trispecific antibodies engineered with 10E8v4 show substantial protection against simian HIV (SHIV) in rhesus macaques (Huang et al., 2016b; Xu et al., 2017). It will be interesting to see whether the enhancements in neutralization potency—achieved here through optimization of semi-specific interactions—will prove to be a general means to improve antibodies. All broad HIV-1-neutralizing antibodies must accommodate glycan (Stewart-Jones et al., 2016), thereby suggesting that optimization of semi-specific interactions with the glycan shield is likely to allow for improvement of other HIV-1 broadly neutralizing antibodies. Altogether, our results suggest that both surface-matrix screening and optimization of semi-specific interactions provide general strategies for enhancing antibody potency.
EXPERIMENTAL PROCEDURES
In Silico Scanning of Antibody Surface
We performed the following surface matrix screening on 10E8. (1) Phe/Trp scan: for all Fv residues that have a side-chain accessible surface area of greater than 30%, we mutated each Phe/Tyr to Trp and every other non-Cys and non-Trp residue to Phe. (2) Arg scan: for all Fv residues that have a side-chain accessible surface area greater than 30%, we mutated each non-Cys and non-Arg residue to Arg. (3) Glycine scanning: a 7-amino acid window was used to scan through the Fv regions (overlap of 2 amino acids). We mutated each residue in the 7-amino acid window to GLY except for the following: the residue is part of the disulfide bridge, and side-chain accessible surface area is less than 5%. (4) Glycan scanning: for all Fab residues, we applied an in-house computational algorithm, which couples NGlycPred (Chuang et al., 2012) to predict glycan occupancy, to identify suitable positions for introducing N-linked glycosylation sequons. The number of constructs was reduced by clustering based on Cβ distances (r = 10 Å). Accessible surface areas were calculated using NACCESS (Hubbard and Thornton, 1993).
Informatics Considerations of Library Size and Assay Reproducibility
Log-normal distribution was used to estimate the distribution of fold improvement when the neutralization of the wild-type antibody was repeated N times. Different N, assay variance (σg), and different number of variants that can be characterized in detail were investigated to explore the minimal observable signal. All values were calculated using the R statistical package.
Construct Design, Expression and Purification, and Characterization of Antibody Variants
All variant antibodies were constructed, expressed and purified, and characterized as described in the Supplemental Experimental Procedures.
Virus Isolate Panel Selection
The initial 9-virus panel was compiled based on sequence diversity consideration. The panel was further reduced to five viral strains to rank the surface matrix screening variants by moving strains that gave inconsistent results from the wild-type repeat (KER2008.12) and IC50 values that were close to detection limit (6095.V1.C1, TH966.8, and 6101.10).
Neutralization Assays
HIV-1 Env pseudoviruses were prepared by transfecting 293T cells (6 × 106 cells in 50 mL growth medium in a T-175 culture flask) with 10 µg of rev/env expression plasmid and 30 µg of an Env-deficient HIV-1 backbone vector (pSG3ΔEnvelope), using FuGENE 6 Transfection Reagent (Invitrogen). Pseudovirus-containing culture supernatants were harvested 2 days after transfection, filtered (0.45 µm), and stored at −80°C or in the vapor phase of liquid nitrogen. Neutralization was measured using HIV-1 Env pseudoviruses to infect TZM-bl cells, as described previously (Li et al., 2005; Sarzotti-Kelsoe et al., 2014). Briefly, 40 µL of pseudovirus was incubated for 30 min at 37°C with 10 µL of serially diluted test antibody in duplicate wells of a 96-well flat-bottomed culture plate. To keep assay conditions constant, sham medium was used in place of antibody in control wells. The pseudovirus input was set at an MOI of approximately 0.01, which generally results in 100,000 to 400,000 relative light units (RLUs) in a luciferase assay (Bright Glo; Promega, Madison, WI, USA). The same assay was scaled for use in 384-well plates as described previously (Sarzotti-Kelsoe et al., 2014) when testing large numbers of samples; e.g., the panel of 208 viruses. Neutralization curves were fit by nonlinear regression using a 5-parameter hill slope equation, as previously described (Seaman et al., 2010). The IC50s were reported as the antibody concentrations required to inhibit viral entry by 50%.
Prediction of Neutralization by Antibody Combinations
We used previously developed the Bliss-Hill model (Wagh et al., 2016) to predict the IC50 and IC80 titers, completeness of neutralization, and simultaneous neutralization coverage by both antibodies for two-antibody (Ab) combinations using IC50 and IC80 data for individual antibodies, as implemented on the web tool CombiNAber at the Los Alamos HIV Database (http://www.hiv.lanl.gov/content/sequence/COMBINABER/combinaber.html). We used the antibodies 3BNC117 and VRC07-523LS (CD4 binding site); CAP256-VRC26.25 and PGDM1400 (V2 glycan); PGT121 and 10-1074 (V3 glycan); and 10E8 and 10E8v4-5R+100cF (MPER), tested against the 208 virus panel, and we analyzed all two-antibody combinations with different epitope targets. We focused on two target concentrations of 1 µg/mL and 5 µg/mL of each antibody and ranked the combinations using geometric mean IC80, percentage of viruses neutralized at >95%, and percentage of viruses with both antibodies active.
Statistical Analyses
The Mann-Whitney U test and Pearson correlation were applied in Figure 5E and Figure 5F, respectively. All tests were performed using GraphPad Prism 7.
Supplementary Material
Highlights.
Development of a surface-matrix screening approach to improve antibody function
Identified hydrophobic mutations that improved 10E8 interaction with HIV-1 membrane
Identified positively charged mutations that improved interactions with HIV-1 glycan
Optimizing semi-specific interactions can improve potency while maintaining breadth
Acknowledgments
We thank J. Stuckey for assistance with figures and members of the Structural Biology Section and Structural Bioinformatics Core, Vaccine Research Center, for discussions or comments on the manuscript. We thank J. Baalwa, D. Ellenberger, D. Gabuzda, F. Gao, B. Hahn, K. Hong, J. Kim, F. McCutchan, D. Montefiori, L. Morris, J. Overbaugh, E. Sanders-Buell, G. Shaw, R. Swanstrom, M. Thomson, S. Tovanabutra, C. Williamson, and L. Zhang for HIV-1 envelope plasmids used in our neutralization panel. Support for this work was provided by the Intramural Research Programs of the Vaccine Research Center and the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, NIH, and by grants from the International AIDS Vaccine Initiative’s Neutralizing Antibody Consortium and from the Bill and Melinda Gates Foundation. Use of sector 22 (Southeast Region Collaborative Access team) at the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract number W-31-109-Eng-38.
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the atomic coordinate and structure factors for 10E8v4-5R+100cF in complex with HIV-1 gp41 MPER is PDB: 5WDF.
Supplemental Information includes Supplemental Experimental Procedures, five figures, and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.01.023.
AUTHOR CONTRIBUTIONS
Conceptualization, Design, & Coordination, Y.D.K., G.-Y.C., B.Z., J.R.M., and P.D.K.; Figure Generation, Manuscript Writing, & Manuscript Revision, Y.D.K., G.-Y.C., B.Z., K. Wagh, B.M.K., L.S., and P.D.K.; Antibody Production, Purification, & Neutralization Assessment, Y.D.K., B.Z., V.J., R.T.B., N.A.D.-R., B.L., M.K.L., K.M., S.O., S.D.S., and J.R.M.; Crystal Structure, Y.D.K.; Antibody Half-Life, A.P., X.C., M.C., K. Wang, J.P.T., and D.G.S.; Polyreactivity Assessment, M.A.; Identification of the S100cW Mutation, T.S.G. and L.S.; Virus Panel Selection, I.S.G.; Arg Screening, M.P.; Glycan Modeling, R.R.; Manufacturing Properties, R.C., L.A.K., and S.D.M.; Antibody-gp41 MPER/ Liposome Interactions, M.K. and E.L.R.; Antibody Combination Neutralization Prediction, K. Wagh and B.M.K.; 10E8 Construct, M.C.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Ayyar BV, Arora S, O’Kennedy R. Coming-of-age of antibodies in cancer therapeutics. Trends Pharmacol. Sci. 2016;37:1009–1028. doi: 10.1016/j.tips.2016.09.005. [DOI] [PubMed] [Google Scholar]
- Bar KJ, Sneller MC, Harrison LJ, Justement JS, Overton ET, Petrone ME, Salantes DB, Seamon CA, Scheinfeld B, Kwan RW, et al. Effect of HIV antibody VRC01 on viral rebound after treatment interruption. N. Engl. J. Med. 2016;375:2037–2050. doi: 10.1056/NEJMoa1608243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso RM, Zwick MB, Stanfield RL, Kunert R, Binley JM, Katinger H, Burton DR, Wilson IA. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity. 2005;22:163–173. doi: 10.1016/j.immuni.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Caskey M, Klein F, Lorenzi JC, Seaman MS, West AP, Jr, Buckley N, Kremer G, Nogueira L, Braunschweig M, Scheid JF, et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature. 2015;522:487–491. doi: 10.1038/nature14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caskey M, Schoofs T, Gruell H, Settler A, Karagounis T, Kreider EF, Murrell B, Pfeifer N, Nogueira L, Oliveira TY, et al. Antibody 10-1074 suppresses viremia in HIV-1-infected individuals. Nat. Med. 2017;23:185–191. doi: 10.1038/nm.4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br. J. Pharmacol. 2009;157:220–233. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang GY, Boyington JC, Joyce MG, Zhu J, Nabel GJ, Kwong PD, Georgiev I. Computational prediction of N-linked glycosylation incorporating structural properties and patterns. Bioinformatics. 2012;28:2249–2255. doi: 10.1093/bioinformatics/bts426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diskin R, Scheid JF, Marcovecchio PM, West AP, Jr, Klein F, Gao H, Gnanapragasam PN, Abadir A, Seaman MS, Nussenzweig MC, Bjorkman PJ. Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science. 2011;334:1289–1293. doi: 10.1126/science.1213782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman J, Soto C, Yang MM, Davenport TM, Guttman M, Bailer RT, Chambers M, Chuang GY, DeKosky BJ, Doria-Rose NA, et al. NISC Comparative Sequencing Program. Structures of HIV-1 Env V1V2 with broadly neutralizing antibodies reveal commonalities that enable vaccine design. Nat. Struct. Mol. Biol. 2016;23:81–90. doi: 10.1038/nsmb.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert DN, Lamriben L, Powers ET, Kelly JW. The intrinsic and extrinsic effects of N-linked glycans on glycoproteostasis. Nat. Chem. Biol. 2014;10:902–910. doi: 10.1038/nchembio.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature. 2012;491:406–412. doi: 10.1038/nature11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Kang BH, Ishida E, Zhou T, Griesman T, Sheng Z, Wu F, Doria-Rose NA, Zhang B, McKee K, et al. Identification of a CD4-binding-site antibody to HIV that evolved near-pan neutralization breadth. Immunity. 2016a;45:1108–1121. doi: 10.1016/j.immuni.2016.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Yu J, Lanzi A, Yao X, Andrews CD, Tsai L, Gajjar MR, Sun M, Seaman MS, Padte NN, Ho DD. Engineered bispecific antibodies with exquisite HIV-1-neutralizing activity. Cell. 2016b;165:1621–1631. doi: 10.1016/j.cell.2016.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard SJ, Thornton JM. NACCESS computer program. Department of Biochemistry and Molecular Biology; University College London: 1993. [Google Scholar]
- Hwang WY, Foote J. Immunogenicity of engineered antibodies. Methods. 2005;36:3–10. doi: 10.1016/j.ymeth.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Irimia A, Sarkar A, Stanfield RL, Wilson IA. Crystallographic identification of lipid as an integral component of the epitope of HIV broadly neutralizing antibody 4E10. Immunity. 2016;44:21–31. doi: 10.1016/j.immuni.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia A, Serra AM, Sarkar A, Jacak R, Kalyuzhniy O, Sok D, Saye-Francisco KL, Schiffner T, Tingle R, Kubitz M, et al. Lipid interactions and angle of approach to the HIV-1 viral membrane of broadly neutralizing antibody 10E8: Insights for vaccine and therapeutic design. PLoS Pathog. 2017;13:e1006212. doi: 10.1371/journal.ppat.1006212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein F, Halper-Stromberg A, Horwitz JA, Gruell H, Scheid JF, Bournazos S, Mouquet H, Spatz LA, Diskin R, Abadir A, et al. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature. 2012;492:118–122. doi: 10.1038/nature11604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong R, Louder MK, Wagh K, Bailer RT, deCamp A, Greene K, Gao H, Taft JD, Gazumyan A, Liu C, et al. Improving neutralization potency and breadth by combining broadly reactive HIV-1 antibodies targeting major neutralization epitopes. J. Virol. 2015;89:2659–2671. doi: 10.1128/JVI.03136-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YD, Georgiev IS, Ofek G, Zhang B, Asokan M, Bailer RT, Bao A, Caruso W, Chen X, Choe M, et al. Optimization of the solubility of HIV-1-neutralizing antibody 10E8 through somatic variation and structure-based design. J. Virol. 2016;90:5899–5914. doi: 10.1128/JVI.03246-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Ozorowski G, Ward AB. Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science. 2016;351:1043–1048. doi: 10.1126/science.aad2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, et al. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 2005;79:10108–10125. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan JS, Pancera M, Carrico C, Gorman J, Julien JP, Khayat R, Louder R, Pejchal R, Sastry M, Dai K, et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature. 2011;480:336–343. doi: 10.1038/nature10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofek G, Tang M, Sambor A, Katinger H, Mascola JR, Wyatt R, Kwong PD. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J. Virol. 2004;78:10724–10737. doi: 10.1128/JVI.78.19.10724-10737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofek G, McKee K, Yang Y, Yang ZY, Skinner J, Guenaga FJ, Wyatt R, Zwick MB, Nabel GJ, Mascola JR, Kwong PD. Relationship between antibody 2F5 neutralization of HIV-1 and hydrophobicity of its heavy chain third complementarity-determining region. J. Virol. 2010;84:2955–2962. doi: 10.1128/JVI.02257-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancera M, Zhu J, O’Dell S, Yang Y, Zhang B, Changela A, McLellan JS, Wu X, Zhou T, Burton DR, et al. Arginine-scanning of PG16 paratope defines quaternary epitope. AIDS Res. Hum. Retroviruses. 2013;29:A59–A60. [Google Scholar]
- Ramilo O, Lagos R, Sáez-Llorens X, Suzich J, Wang CK, Jensen KM, Harris BS, Losonsky GA, Griffin MP Motavizumab Study Group. Motavizumab treatment of infants hospitalized with respiratory syncytial virus infection does not decrease viral load or severity of illness. Pediatr. Infect. Dis. J. 2014;33:703–709. doi: 10.1097/INF.0000000000000240. [DOI] [PubMed] [Google Scholar]
- Robison AD, Sun S, Poyton MF, Johnson GA, Pellois JP, Jungwirth P, Vazdar M, Cremer PS. Polyarginine interacts more strongly and cooperatively than polylysine with phospholipid bilayers. J. Phys. Chem. B. 2016;120:9287–9296. doi: 10.1021/acs.jpcb.6b05604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- Rudicell RS, Kwon YD, Ko SY, Pegu A, Louder MK, Georgiev IS, Wu X, Zhu J, Boyington JC, Chen X, et al. NISC Comparative Sequencing Program. Enhanced potency of a broadly neutralizing HIV-1 antibody in vitro improves protection against lentiviral infection in vivo. J. Virol. 2014;88:12669–12682. doi: 10.1128/JVI.02213-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rujas E, Caaveiro JM, Insausti S, García-Porras M, Tsumoto K, Nieva JL. Peripheral membrane interactions boost the engagement by an anti-HIV-1 broadly neutralizing antibody. J. Biol. Chem. 2017;292:5571–5583. doi: 10.1074/jbc.M117.775429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarzotti-Kelsoe M, Bailer RT, Turk E, Lin CL, Bilska M, Greene KM, Gao H, Todd CA, Ozaki DA, Seaman MS, et al. Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J. Immunol. Methods. 2014;409:131–146. doi: 10.1016/j.jim.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid JF, Mouquet H, Ueberheide B, Diskin R, Klein F, Oliveira TY, Pietzsch J, Fenyo D, Abadir A, Velinzon K, et al. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science. 2011;333:1633–1637. doi: 10.1126/science.1207227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid JF, Horwitz JA, Bar-On Y, Kreider EF, Lu CL, Lorenzi JC, Feldmann A, Braunschweig M, Nogueira L, Oliveira T, et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature. 2016;535:556–560. doi: 10.1038/nature18929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoofs T, Klein F, Braunschweig M, Kreider EF, Feldmann A, Nogueira L, Oliveira T, Lorenzi JC, Parrish EH, Learn GH, et al. HIV-1 therapy with monoclonal antibody 3BNC117 elicits host immune responses against HIV-1. Science. 2016;352:997–1001. doi: 10.1126/science.aaf0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MS, Janes H, Hawkins N, Grandpre LE, Devoy C, Giri A, Coffey RT, Harris L, Wood B, Daniels MG, et al. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J. Virol. 2010;84:1439–1452. doi: 10.1128/JVI.02108-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Sun ZY, Coleman KE, Zwick MB, Gach JS, Wang JH, Reinherz EL, Wagner G, Kim M. Broadly neutralizing anti- HIV-1 antibodies disrupt a hinge-related function of gp41 at the membrane interface. Proc. Natl. Acad. Sci. USA. 2009;106:9057–9062. doi: 10.1073/pnas.0901474106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song R, Oren DA, Franco D, Seaman MS, Ho DD. Strategic addition of an N-linked glycan to a monoclonal antibody improves its HIV-1-neutralizing activity. Nat. Biotechnol. 2013;31:1047–1052. doi: 10.1038/nbt.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C, Ofek G, Joyce MG, Zhang B, McKee K, Longo NS, Yang Y, Huang J, Parks R, Eudailey J, et al. NISC Comparative Sequencing Program. Developmental pathway of the MPER-directed HIV-1-neutralizing antibody 10E8. PLoS ONE. 2016;11:e0157409. doi: 10.1371/journal.pone.0157409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart-Jones GB, Soto C, Lemmin T, Chuang GY, Druz A, Kong R, Thomas PV, Wagh K, Zhou T, Behrens AJ, et al. Trimeric HIV-1-Env structures define glycan shields from clades A, B, and G. Cell. 2016;165:813–826. doi: 10.1016/j.cell.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZY, Oh KJ, Kim M, Yu J, Brusic V, Song L, Qiao Z, Wang JH, Wagner G, Reinherz EL. HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity. 2008;28:52–63. doi: 10.1016/j.immuni.2007.11.018. [DOI] [PubMed] [Google Scholar]
- Trevino SR, Scholtz JM, Pace CN. Amino acid contribution to protein solubility: Asp, Glu, and Ser contribute more favorably than the other hydrophilic amino acids in RNase Sa. J. Mol. Biol. 2007;366:449–460. doi: 10.1016/j.jmb.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagh K, Bhattacharya T, Williamson C, Robles A, Bayne M, Garrity J, Rist M, Rademeyer C, Yoon H, Lapedes A, et al. Optimal combinations of broadly neutralizing antibodies for prevention and treatment of HIV-1 clade C infection. PLoS Pathog. 2016;12:e1005520. doi: 10.1371/journal.ppat.1005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wibmer CK, Gorman J, Ozorowski G, Bhiman JN, Sheward DJ, Elliott DH, Rouelle J, Smira A, Joyce MG, Ndabambi N, et al. Structure and recognition of a novel HIV-1 gp120-gp41 interface antibody that caused MPER exposure through viral escape. PLoS Pathog. 2017;13:e1006074. doi: 10.1371/journal.ppat.1006074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LTD, Ofek G, Schatzle S, McDaniel JR, Lu X, Nicely NI, Wu L, Lougheed C, Bradley T, Louder M, et al. Potent and broad HIV-neutralizing antibodies in memory B cells and plasma. Sci. Immunol. 2017;2:eaal2200. doi: 10.1126/sciimmunol.aal2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Pfarr DS, Johnson S, Brewah YA, Woods RM, Patel NK, White WI, Young JF, Kiener PA. Development of motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J. Mol. Biol. 2007;368:652–665. doi: 10.1016/j.jmb.2007.02.024. [DOI] [PubMed] [Google Scholar]
- Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010;329:856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Pegu A, Rao E, Doria-Rose N, Beninga J, McKee K, Lord DM, Wei RR, Deng G, Louder M, et al. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science. 2017;358:85–90. doi: 10.1126/science.aan8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young L, Jernigan RL, Covell DG. A role for surface hydrophobicity in protein-protein recognition. Protein Sci. 1994;3:717–729. doi: 10.1002/pro.5560030501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 2010;28:157–159. doi: 10.1038/nbt.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Zhu J, Wu X, Moquin S, Zhang B, Acharya P, Georgiev IS, Altae-Tran HR, Chuang GY, Joyce MG, et al. NISC Comparative Sequencing Program. Multidonor analysis reveals structural elements, genetic determinants, and maturation pathway for HIV-1 neutralization by VRC01-class antibodies. Immunity. 2013;39:245–258. doi: 10.1016/j.immuni.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.